

Abstract
Purpose: Hepatitis B virus (HBV) infection is one main cause of hepatocellular carcinoma (HCC), but the mechanisms of pathogenesis still remain unclear. Methods: We screened the 1351 differentially expressed genes related to HBV-induced HCC by bioinformatics analysis from databases and found that Plasminogen (PLG) may be a key gene in HBV-induced HCC progression. Then, we used a series of experiments in vivo and in vitro to explore the roles of PLG in HBV-HCC progression, such as qRT-PCR, western blot, ELISA, flow cytometry and TUNEL assay, subcutaneous xenografts and histopathological analysis to reveal the underlying mechanisms. Results: PLG was over-expressed in HBV positive hepatocellular carcinoma tissues and cells. PLG silencing promoted HBV-HCC cell apoptosis in vitro and suppressed the growth of HBV-induced HCC xenografts in vivo both through inhibiting HBV replication. Then, GO and KEGG analysis of these differentially expressed genes revealed that the Hippo pathway was the key pathway involved in HBV-induced HCC, and SRC, a downstream target gene of PLG, was highly expressed in HBV-induced HCC and related to the Hippo pathway. Thus, we speculated that PLG promoted HBV-induced HCC progression through up-regulating and activating the expression of SRC and promoting Hippo signaling pathway function on HBV-HCC cell survival. Conclusion: Our study suggests PLG may be an activator of HBV-infected hepatocellular carcinoma development, as a novel prognostic biomarker and therapeutic target for HBV-HCC.
Keywords: PLG, hepatitis B virus, hepatocellular carcinoma, src, hippo signaling pathway, tumor progression
Introduction
Hepatocellular carcinoma (HCC) is the third fatal cancer type in the world [1] and Hepatitis B virus (HBV) is a major and independent risk factor for HCC [2,3]. It has been reported that the incidence of HCC in patients with chronic HBV infection is 5-100 times higher than that in healthy individuals [4]. Despite decades of research have been performing, the underlying mechanism for HBV-induced HCC is still not fully understood [5].
Plasminogen (PLG), also known as plasmatrypsinogen, is an important cell surface-bound zymogen in fibrinolysis, should be activated by fibrinolytic enzymes, such as urokinase plasminogen activator, tissue plasminogen activator and factor VII. Research have reported that plasminogen and fibrinogen were found to be key determinants of tumor growth [6,7]. Soluble urokinase plasminogen is reported as an early screening tool for HCC, especially those with chronic liver disorders [8]. As signal receptors on hepatocellular membrane, urokinase-type plasminogen activator receptor and integrin are identified to exert synergistical effect on malignant transformation of hepatic cells [9]. In subcellular level, hepatocyte endoplasmic reticulum stress induced by plasminogen activator expression is also verified to be related with non-alcoholic steatohepatitis and HCC [10]. However, the role the PLG in HBV-induced HCC still remains unexplored.
SRC proto-oncogene (SRC) family is a non-receptor protein type of intracytoplasmic tyrosine kinases [11]. They are related to cell membranes and could be activated by numerous cell surface receptors, thus involving in cellular signal cascades and regulate cellular biological reactions [12,13]. SRC family kinase proteins are frequently activated in cancer, and are involved in coordinating the growth, survival, invasion and angiogenesis of tumor cells [12,13]. Bouchard et al reported that HBV X protein (HBx) promoted cyclin gene expression and cell proliferation through activating the SRC kinase signaling pathway [14]. Other studies have also shown that HBV-induced HCC cells expressed large numbers of hepatitis B virus Large S protein (LHBs) which could activate the SRC signaling pathways. Besides, the accumulation of LHBs was found to be associated with G1-S cell cycle progression and apoptosis resistance mediated by SRC activation [15]. A few studies have reported the relationship between SRC kinase and HBV-induced HCC, while SRC kinase is previously reported to have certain association with PLG in other disease by interactome analysis [16]. In this study, bioinformatic analysis demonstrated SRC may be a downstream regulatory gene of PLG in HBV-induced HCC.
Hippo signaling pathway is an inhibitory signaling pathway on regulating the homeostasis of various cells and organs, including the intestine [17], cardiovascular system [18], and immune system [19]. Recent studies have shown that Hippo signaling pathway plays an important role in the physiological and pathological processes of mammalian liver cell fate, cell death, liver development, progress of liver cancer, and liver regeneration & remodeling [12,13]. YAP, as one of the transcriptional co-activators in the Hippo signaling pathway, was reported to be linked with cellular proliferation [20]. Deletion of upstream proteins or excessive activation of downstream YAP in Hippo signaling pathway could bring about liver dysplasia, and also lead to the occurrence of liver cancer [21]. Analysis of human hepatocellular carcinoma clinical samples revealed that the disorder of Hippo signaling pathway was closely related to the development of human HCC. YAP overexpression and nuclear localization were observed in about 50% of HCC samples [22]. Moreover, HBx mediated YAP1 transcriptional activation through CREB to promote growth of hepatoma cells [22], indicating that Hippo signaling pathway may also play an important role in HBV-induced HCC development.
In this study, we detected that PLG was obviously up-regulated in HBV-induced HCC, and relevant cell biological behaviors after PLG interference were further confirmed subsequently. Related genes were further identified by gene microarray analysis. The relationship between Hippo signal pathway and PLG was also investigated in vitro and in vivo. In the end, we come to the conclusion that PLG promoted the occurrence and development of liver cancer induced by HBV through activating SRC and inhibiting Hippo signaling pathway.
Materials and methods
Bioinformatics analysis
Through the GEO database, GSE54238 (https://www.ncbi.nlm.nih.gov/geo/) which included data from 10 normal liver samples, 10 chronic inflammatory liver samples, 10 cirrhotic liver samples, 13 early HCC samples, and 13 advanced HCC samples were obtained and standardized by RMA. Protein coding genes were mainly analyzed. All the liver cancers samples in the data were associated with HBV infection. Spearman’s rank correlation was used to analyze liver cancer progression related genes. Gene interactions were performed by https://string-db.org/cgi/input.pl and mapped using Cytoscape software (http://www.cytoscape.org/), GO and KEGG analyses were adopted through online websites (http://geneontology.org/, https://www.kegg.jp/clugg/pathway.html), and TerProfiler package in R language was used for enrichment analysis of GO and pathways.
Tissues and cell lines
40 HBV-positive liver samples and 40 HBV-negative liver samples were collected from HCC patients underwent surgical resection at the Eastern Hepatobiliary Surgery Hospital (Shanghai, China). Written informed consents were acquired from the patients, and all experiments were approved by the Research Ethics Committee of the First Affiliated Hospital of Guangxi Medical University.
Human hepatoma cell lines HepAD38 [23] (HuZhen biology, Shanghai, China), and the HepG2-NTPC cells (GuangZhou Jennio Biotech Co., Ltd, Guangzhou, China) were cultured in high glucose Dulbecco’s Modified Eagle Medium (DMEM) (Biological Industries, Beit Haemek, ISRAEL) with 5% fetal bovine serum (FBS) (Biological Industries, Beit Haemek, ISRAEL) at 37°C incubator with 5% CO2.
Plasmid construction and transfection
PLG-sh1 (target sequence: 5’-GAAGAGGAATGTATGCATTGC-3’), PLG-sh2 (target sequence: 5’-GCCATACAACCAACAGCCAAG-3’), SRC-sh1 (target sequence: 5’-GGCTCCAGATTGTCAACAACA-3’), SRC-sh2 (target sequence: 5’-GCCTCAACGTGAAGCACTACA-3’), and pCDNA3.1-SRC plasmid (SRC-oe) were purchased from AURAgene company (Changsha, China). The control plasmids containing a scramble sequence (shCtrl) or blank gene (Ctrl-oe) were used for negative control. Cells were cultured until the logarithmic growth stage and then seeded into 6-well plates at 1.5×105 cells per well. When cells grew to 80% confluence, the plasmids were transiently transfected into cells as indicated with Lipo2000 and then cells were continue cultured for 48 h. 100 HBV virus genome equivalents (vge) was transfected with HepAD38 for 7 days and HepG2-NTPC for 5 days for preparation of HBV infected HCC cells.
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (MTT) assay
Cells were cultured into 96-well plates (5000 cells/well) for 24 h. 48 and 72 h incubation after starvation, 10 μl MTT solution (Sangon Biotech, China) was added into each well and cultured at 37°C for 4 h, then the medium was removed. 150 μl dimethylsulfoxide (DMSO) solution (MP Biomedicals, USA) was added for 10 min. The absorbance at 490 nm was measured using a microplate reader Multiskan MK (Thermo Scientific, USA).
Flow cytometry and TUNEL staining for cell apoptosis analysis
Cells at the exponential growth phase were treated with trypsinization and resuspended in Annexin V-FITC binding buffer. Annexin V and propidium iodide were added into cell suspension for 15 min. The stained cells were analyzed by flow cytometer (FACScan, BD Biosciences). For TUNEL assay, the cells were fixed by paraformaldehyde for 15 min and treated with immunostaining permeabilization buffer with Triton X-100 (Beyotime, Beijing, China), and then incubated with fluorescent-labeled reagent (45 μl) and terminal deoxynucleotidyl transferase (5 μl) (Beyotime, Beijing, China) at 37°C for 1 h. Nuclear fractions were stained by 4’,6-diamidino-2-phenylindole (DAPI). Cells were observed and photographed using a fluorescence microscope (Leica, Germany).
qRT-PCR
Total RNA was extracted from tissues or cells by the TRIZOL reagent (Invitrogen, Shanghai, China). cDNA was synthesized by the random primers and a Reverse Transcription Kit (Takara, China). qRT-PCR and data collection were performed using an ABI 7300 instrument. Primers (PLG Forward: 5’-CAGGGGGCTTCACTGTTCAG-3’, Reversed: 5’-GCCATTATCACACATTGTTGCTC-3’; HBV Forward: 5’-ACCGACCTTGAGGCATACTT-3’, Reversed: 5’-GCCTACAGCCTCCTAGTACA-3’; HBx Forward, 5’-GGTCGTTGACATTGCAGAGA-3’, Reversed: 5’-GGTCGTTGACA-TTGCAGAGA-3’; β-actin Forward, 5’-AGGGGCCGGACTCGTCATACT-3’, Reversed: 5’-GGCGGCACCACCATGTACCCT-3’) were synthesized by Invitrogen (Shanghai, China) The levels of target genes were normalized with internal reference gene β-actin.
Western blot
Cells were lysed using RIPA lysis buffer (Auragene Bioscience, China). The protein was loaded on a 10-12% SDS-polyacrylamide separating gel and transferred to a PVDF Immobilon-P membrane (Millipore, USA), which was blocked with 3% BSA-TBST for 90 min. The membranes were incubated with primary antibodies at 4°C overnight and incubated with HRP-conjugated secondary antibodies (1:5000, ab62751, Abcam) for another 1 h. β-actin antibody (1:1000, #4967, CST) was used as internal reference. Primary antibodies for PLG (1:1000; PA5-34677, Invitrogen), SRC (1:2000; ab109381, Abcam), p-SRC (1:2000; ab185617, Abcam), YAP (1:1000; #4912, CST), p-YAP (1:1000; #13008, CST), Ki67 (1:1000; ab16667, Abcam), LATS1 (1:1000; #3477, CST), and p-LATS1 (1:1000; #8654, CST) were used. At the end, after washed with TBST for more than three times for 5-10 min, the membranes were then exposed to ECL (Millipore) substrate and visualized by a chemiluminescence detection system (Bio-Rad). Image J software was used to quantify the intensity of bands.
ELISA
The levels of HbsAg and HbeAg in the culture supernatant of the liver cells HepAD38 and HepG2-NTPC after treatment with HBV infection or combined with plasmid transfection as indicated were detected by commercial ELISA kits (HbsAg, HbsAg, Guangrui biology, Shanghai, China; HbeAg, Tong rui biology, Shanghai, China), referring to the standard curve provided by the kits. Procedures of ELISA detection was in strict accordance with protocols provided in the kits. Briefly, the culture supernatant samples with appropriate dilution were added into the wells of the plate, after 30 min incubation at 37°C and then 4 times washing, enzyme-labelled antibodies were added into the wells, and then the reaction plate was incubated for another 30 min at 37°C. Following by 4 times washing, two kind of Color-substrate solutions were added in turn and then incubated in dark for 15 min. At last, after adding with Stop solution, put the plate into a Microplate Reader (SpectraMax Paradigm, Molecular Devices) for OD value analysis at 450 nm.
Xenograft in nude mice
Male BALB/c Mlac-nu mice (aging 4 weeks) from the National Laboratory Animal Center (Mahidol University, Thailand) were acclimatized for 2 weeks under 12 h light-dark cycle in 50% humidity and with free access to food and water. Mice were anesthetized with pentobarbital (100 mg/kg) and received preoperative subcutaneous buprenorphine (0.1 mg/kg). Four mice were used in each group. The animals were positioned in prone to provide better oxygenation. All animal procedures were approved with the instructions of the Institutional Animal Care and Use Committee of the First Affiliated Hospital of Guangxi Medical University. The sh-PLG or/and SRC-oe transfected HepAD38 and HepG2-NTPC cells were trypsinized into cell suspension (1×107 cells/mL). 1 mL cell suspension was centrifuged and resuspended in 100 μl Matrigel (Corning, NY, United States). All procedures with Matrigel were performed on ice. Cell suspension was subcutaneously injected into the back-right flanks of nude mice (n = 4). The tumor volume was measured and calculated for twice every week. The activity and mortality of animals were monitored simultaneously. For weeks later, all the animals were sacrificed after anesthesia, and the tumors were isolated for weighted and subsequent pathologic analysis and molecule detection.
Model establishment
AAV8-HBV1.2 vector was diluted by phosphate-buffered saline (PBS). Mice were anesthetized with pentobarbital (100 mg/kg) and received preoperative subcutaneous buprenorphine (0.1 mg/kg). Four mice were used in each group. The animals were positioned in prone to provide better oxygenation. Next, the mice were injected with 200 μl rAAV8-HBV 1.2 vector [2×1011 vector genome equivalents (vg)] via the tail vein. The controls were injected with PBS. Serum and liver tissues were collected at 1, 3 and 6-months after injection, and were frozen in liquid nitrogen.
Immunohistochemistry
The tissues were fixed in 10% formalin solution, embedded in paraffin, sliced (4 μm), then deparaffinized and rehydrated in xylenes and alcohols followed by retrieval of antigenic epitopes. Antigen retrieval was performed in citrate buffer (pH 6, 100°C for 20 min). The sections were treated with 3% H2O2 for 15 min and blocked with normal serum for 30 min, and then incubated with primary antibody at 4°C overnight. The primary antibodies anti-PLG (1:500; PA5-34677, Invitrogen), anti-SRC (Santa Cruz Biotechnologies; at a dilution of 1:500) and anti-Ki-67 (Ventana Medical Systems, Inc.; AZ, United States; at a dilution of 1:500). ZytoChem Plus (HRP) Polymer anti-Rabbit (Zytomed Systems, Berlin, Germany; at a dilution of 1:500) and rabbit anti-goat immunoglobulin-HRP (Dako; CA, United States; at a dilution of 1:800) were used. The immunoreaction was visualized with ultraView Universal DAB Detection Kit (Ventana Medical Systems, Inc.). The nuclei were counterstained with haematoxylin. Immunoreactions were measured in five microscopic fields (200×) (Nikon Eclipse50i, Japan).
Statistical analysis
Data were demonstrated as the mean ± standard deviation (SD) with repeated independently experiments at least three times. All data were processed using GraphPad Prism 7.0 (GraphPad software, Inc., La Jolla, CA, USA). Student t test with Welch’s correction for comparisons between two groups and One-way analysis of variance (One-way ANOVA) with Tukey test for multiple comparisons more than two groups were used at statistical analysis. P value less than 0.05 was considered as statistically significant.
Results
PLG is overexpressed in HBV positive hepatocellular carcinoma tissues and cells
Significant differences in gene expression between normal tissues and advanced liver cancer tissues were observed (1402 genes FDR < 0.01). There were 3831 genes associated with liver disease progression (rank correlation, FDR < 0.01), among them, 1351 were differentially expressed in advanced HCC. KEGG enrichment analysis of the above genes revealed that Hippo signaling pathway and fatty acid degradation signaling pathway were the two of the most concentrated ones (Figure S1). Through the STRING database, we analyzed the interaction relationship of these 1351 liver cancer related genes in the network (Figure S2). It was found that PLG was highly expressed in HCC induced by HBV, and its high expression was associated with prognosis and disease-free survival (Figure S3).
To verify the results of bioinformatics analysis, PLG expression was detected in some clinical liver samples from 40 HBV-positive HCC (HBV+-HCC) and 40 HBV-negative HCC (HBV--HCC) patients. The results showed that the relative expression levels of PLG in HBV+-HCC liver samples were higher significantly than those in HBV--HCC liver samples (Figure 1A and 1B). In order to investigate the effects of PLG in HBV positive Hepatoma cells in vitro, we used HBV to infect HepG2-NTPC cells for 7 days and HepAD38 cells for 5 days, detecting the secreted form of HBsAg and HBeAg and the detectable levels of HBV RNA (pgRNA), HBx RNA were upregulated as biomarkers of HBV infection of liver cell models (Figure S4), and western blot analysis results also showed PLG was highly expressed in HBV infected HepG2-NTPC and HepAD38 cells, compared to the control groups with no HBV-infection (Figure 1C).
Figure 1.

PLG is overexpressed in HBV positive hepatocellular carcinoma tissues and cells. (A) QPCR analysis for the mRNA level and (B) western blot analysis for protein level of PLG expression in clinical liver samples from 40 HBV-positive HCC (HBV+-HCC) and 40 HBV-negative HCC (HBV--HCC) patients. (C) Western blot analysis for the change of PLG protein expression in two HBV-induced HCC cell models using HBV to infect HepG2-NTPC cells for 7 days and HepAD38 cells for 5 days. *P < 0.05; **P < 0.01; ***P < 0.001.
Silencing of PLG promoted HBV-HCC cell apoptosis through inhibiting HBV replication in vitro
To investigate the roles of PLG in the progression of HBV-HCC, PLG interference plasmids were constructed and confirmed the interference efficiency in HepAD38 and HepG2-NTPC cells using western blot, and results showed that the relative protein level of PLG in the interference groups (shPLG-1 and shPLG-2) was obviously lower than that in the control groups (Con and shCtrl) (Figure 2A), suggesting that both the two interference targets were effective. MTT assay was used to detect the cell proliferation, and flow cytometry and TUNEL assay were used to measure the rate of cell apoptosis after silencing of PLG in HBV-infected HepG2-NTPC and HepAD38 cells. Results showed that the HCC cells proliferation was obviously inhibited after PLG interference in HBV-infected HepAD38 and HepG2-NTPC cells (Figure 2B); the rate of apoptotic cells in HBV-infected HepAD38 and HepG2-NTPC cells were increased markedly after PLG interference (Figure 2C and 2D). All these findings suggested that PLG may be an important factor in HBV-induced HCC progression.
Figure 2.

Silencing of PLG promoted HBV-HCC cell apoptosis through inhibiting HBV replication in vitro. To investigate the roles of PLG in the progression of HBV-HCC, PLG interference plasmids (shPLG-1, shPLG-2) and a negative control plasmids (shCtrl) were respectively transfected into the HBV-infected HepAD38 and HepG2-NTPC cells. (A) Western blot analysis for the changes of PLG protein level to confirmed the interference efficiencies of these interference plasmids. (B) MTT assays for the detection of cell proliferative potential after PLG silencing in HBV-infected HepG2-NTPC and HepAD38 cells. (C) Flow cytometry with Annexin V-FITC/PI staining and (D) TUNEL assays for the cell apoptosis analysis after PLG silencing in HBV-infected HepG2-NTPC and HepAD38 cells. (E) ELISA analysis for the levels of HbsAg and HbeAg in HCC cell supernatants after HBV infection and PLG silencing. (F) QPCR analysis for the mRNA levels of HBV (pgRNA) and HBx in HepG2-NTPC and HepAD38 cells after HBV infection and PLG silencing. */#P < 0.05; **/##P < 0.01; ***/###P < 0.001.
To investigate whether PLG could regulate HBV replication, After silencing treatment by PLG interfering plasmid transfection in HBV-infected HepG2-NTPC and HepAD38 cells, the levels of HbsAg and HbeAg in cell supernatant were detected by ELISA, and the results showed that expression of HbsAg and HbeAg decreased observably in the interferent groups (Figure 2E); moreover, the mRNA levels of HBV (pgRNA) and HBx in the interference groups were reduced markedly (Figure 2F), indicating that silencing of PLG could inhibit the process of HBV replication, and this may be one of the main reasons the silenced PLG promoted the HBV-HCC cell apoptosis.
Silencing of PLG suppressed the growth of HBV-induced HCC xenografts in vivo
To further verify the functions of PLG on HBV-induced HCC progression, experiments of tumorigenesis in nude mice were performed using HBV-induced HCC cells, and the effects of PLG silencing on the growth of xenografts were analyzed (Figure 3A). Results showed that the tumors in the PLG interference group were smaller than those in the control group (Figure 3B and 3C), indicating that PLG interference could suppress the growth of HBV-induced HCC tumorigenesis. Western blot and immunohistochemistry analysis for these tumor tissues showed that the protein expression level of proliferation marker Ki67 in PLG interference group was lower than that in control group (Figure 3D and 3E), suggesting that PLG interference could inhibit HBV-induced HCC progression. To confirm the effects of PLG interference on HBV replication in vivo, ELISA analysis of HbsAg and HbeAg levels in serum samples from mice of the two group were measured, and the mRNA levels of HBV (pgRNA) and HBx in tumor tissues were detected by qPCR. Results showed that not only the levels of serum HbsAg and HbeAg, but also the mRNA levels of tumor HBV (pgRNA) and HBx were downregulated significantly in PLG interference group compared to the control group (Figure 3F and 3G). In vivo experiments have confirmed that silencing of PLG suppressed the growth of HBV-induced HCC xenografts.
Figure 3.
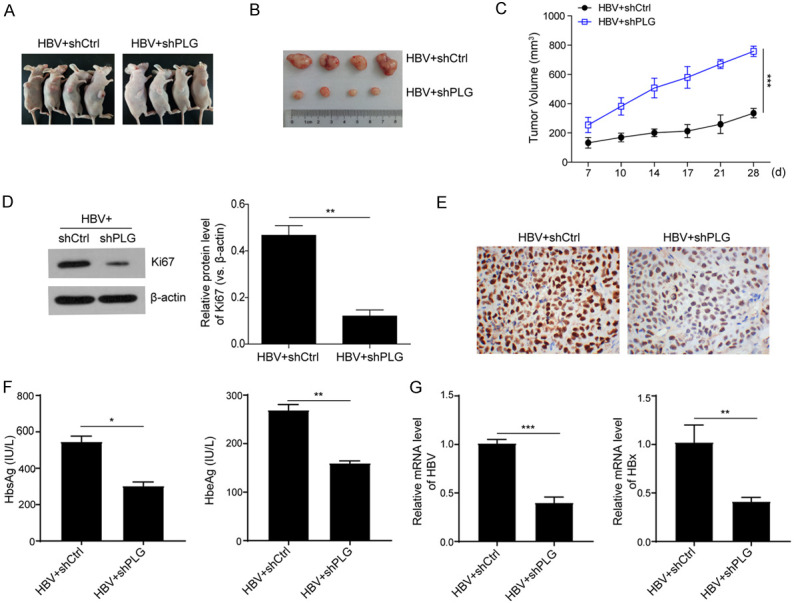
Silencing of PLG suppressed the growth of HBV-induced HCC xenografts in vivo. To further verify the functions of PLG on HBV-induced HCC progression, experiments of tumorigenesis in nude mice were performed using HBV-induced HCC cells. (A) Showcase of subcutaneous xenograft tumors in nude mice and (B) xenograft tumor tissues isolated from the nude mice after about a month growth. (C) The growth curves of xenograft tumor volume in nude mice with PLG silencing or not. (D) Western blot analysis and (E) immunohistochemistry analysis for the protein levels of proliferation marker Ki67 in xenograft tumor tissues with PLG silencing or not. (F) ELISA analysis for the serum levels of HbsAg and HbeAg in xenografts nude mice with PLG silencing or not. (G) QPCR analysis for the mRNA levels of HBV (pgRNA) and HBx in xenograft tumor tissues with PLG silencing or not. *P < 0.05; **P < 0.01; ***P < 0.001.
PLG promoted HBV-induced HCC progression through upregulating and activating the expression of SRC
In order to further explore the mechanisms, bioinformatic analysis for the downstream target genes of PLG using online databases. We screened 1351 HCC differential expression genes and then found there were 84 genes be associated with the progression of HCC and interacted with the Hippo signaling pathway. Some of these 84 genes can interact with multiple genes of the Hippo pathway, including SRC (interacting with 10 Hippo genes), EGFR (7), YWHAH (6), EGR1 (6), ALB (5). These 84 genes are highly expressed in HCC tumor samples compared to liver cirrhosis samples (Table S1), indicating that PLG may play roles in the progression of liver cirrhosis to HCC. Using the online software of String for protein interaction analysis, SRC was found to be identified as one of the downstream-target genes of PLG (Figure 4A; Table S2). To verify this point, confirmatory experiments were performed in vivo and in vitro. Firstly, western blot analysis for SRC protein expression in HepAD38 and HepG2-NTPC cells treated with HBV or not (Figure 4B), and immunohistochemistry staining analysis for PLG and SRC protein levels in different liver samples from normal wild-type mice and HBV-induced mice models of hepatic fibrosis, liver cirrhosis and HCC, respectively (Figure 4C), were demonstrated that the protein level of SRC was upregulated in HBV-induced HCC cells and in the progression from hepatic fibrosis, liver cirrhosis to HCC, as well as PLG. Moreover, silencing of PLG in the tumor tissues of HBV-induced xenografts (Figure 4D) and HBV-induced HepAD38 cells (Figure 4E) also downregulated the levels of phosphorylated SRC protein, resulting in reduced total protein level of SRC, indicating that PLG promoted the SRC protein expression and stabilization by phosphorylation activation. The cell apoptosis analysis by flow cytometry (Figure 4F) and TUNEL staining (Figure 4G) also verified that the PLG silencing-mediated promotion of apoptosis could be reversed by SRC overexpression, further suggesting that PLG regulated HCC cells survival by activating SRC.
Figure 4.

PLG promoted HBV-induced HCC progression through upregulating and activating the expression of SRC. To further explore the mechanisms, bioinformatic analysis for the downstream target genes of PLG were performed with online databases and softwares. (A) The PLG target genes predicted by the String software for protein interaction analysis (https://string db.org/). (B) Western blot analysis for SRC protein expression in HepAD38 and HepG2-NTPC cells treated with HBV or not. (C) Immunohistochemistry staining analysis for PLG and SRC protein levels in different liver samples from normal wild-type mice and HBV-induced different progresses mice models of hepatic fibrosis, liver cirrhosis and HCC. (D) Western blot analysis for the protein levels of PLG, phosphorylated SRC (p-SRC) and total SRC (SRC) in HBV-induced xenografts tumor tissues with PLG silencing or not. (E) Western blot analysis for the protein levels of p-SRC and SRC in HBV-induced HepAD38 cells with PLG silencing or/and SRC overexpression. (F) Flow cytometry analysis and (G) TUNEL staining assays for the cell apoptosis rates in HBV-induced HepAD38 and HepG2-NTPC cells after PLG silencing combined with SRC overexpression or not. *P < 0.05; **P < 0.01; ***P < 0.001.
PLG silencing reversed the effects of SRC overexpression on xenograft growth promotion
For further elucidation for the interaction between PLG and SRC in vivo, experiments of tumorigenesis in nude mice were performed using HBV-induced HCC cells, and the effects of SRC overexpression combined with or without PLG silencing on the growth of xenografts were analyzed (Figure 5A). Results showed that the tumors in the SRC overexpression group were obviously bigger than those in the control group (Figure 5B and 5C), indicating that SRC overexpression could promote the growth of HBV-induced HCC tumorigenesis. However, the promotion effects induced by SRC overexpression were reversed by PLG silencing (Figure 5B and 5C). Furthermore, western blot and immunohistochemistry analysis for the expression changes of proteins Ki67, PLG and both phosphorylated SRC level and total SRC level (Figure 5D and 5E), not only showed that SRC overexpression promoted the HCC development induced by HBV, but also confirmed that the activation of SRC could be regulated by PLG. Expected results of ELISA analysis for HbsAg and HbeAg levels (Figure 5F) and qPCR detection for HBV and HBx mRNA levels (Figure 5G) were also obtained, which were significantly downregulated in combined with PLG silencing group compared to the increase levels by SRC overexpression alone. All these results have verified that SRC was the downstream effector of PLG, and PLG silencing reversed the promotion effects of SRC overexpression on xenograft growth via reducing the phosphorylation level of SRC.
Figure 5.

PLG silencing reversed the effects of SRC overexpression on xenograft growth promotion. To further verify the relationship between PLG and SRC, experiments of tumorigenesis in nude mice were performed using HBV-induced HCC cells. (A) Showcase of subcutaneous xenograft tumors in nude mice and (B) xenograft tumor tissues isolated from the nude mice after about a month growth. (C) The growth curves of xenograft tumor volume in nude mice with SRC overexpresion or not or SRC overexpresion combined with PLG silencing. (D) Western blot analysis and (E) immunohistochemistry analysis for the protein levels of Ki67, PLG and SRC in xenograft tumor tissues with SRC overexpresion or not or SRC overexpresion combined with PLG silencing. (F) ELISA analysis for the serum levels of HbsAg and HbeAg in xenografts nude mice with SRC overexpresion or not or SRC overexpresion combined with PLG silencing. (G) QPCR analysis for the mRNA levels of HBV (pgRNA) and HBx in xenograft tumor tissues with SRC overexpresion or not or SRC overexpresion combined with PLG silencing. *P < 0.05; **P < 0.01; ***P < 0.001.
PLG promoted HBV-induced HCC cell survival via inhibiting the Hippo signaling pathway
As described previously, the 1351 HCC differential expression genes, including PLG and SRC, were found to be mainly concentrated in the Hippo signaling pathway (Figure S1), and closely related to HBV-induced hepatocellular carcinoma. To confirm this point, the expression changes of key proteins of Hippo signaling pathway LATS1, YAP and their phosphorylation levels after PLG silencing or SRC knockdown were analyzed in HBV-induced xenografts (Figure 6A) and HBV-positive HepAD38 cells (Figure 6B and 6C). Results showed that the phosphorylation levels of LATS1 and YAP1 increased obviously after PLG silencing in vivo (Figure 6A) and in vitro (Figure 6B). Similar results were measured after SRC knockdown or overexpression in HBV-positive HepAD38 cells (Figure 6C-E). For further verification, XMU-MP-1, an inhibitor of Hippo signaling pathway, was used to pre-treat the HBV-positive HCC cells before transfected with the PLG interference plasmid. Flow cytometry analysis showed that the elevated cell apoptosis rate induced by HBV and PLG silencing was suppressed dramatically by XMU-MP-1 treatment (Figure 6F), suggesting the inhibition of Hippo signaling pathway promoted HBV-induced HCC cell survival and tumor progression.
Figure 6.

PLG promoted HBV-induced HCC cell survival via inhibiting the Hippo signaling pathway. To verify the relationship of PLG/SRC and Hippo signaling pathway, the expression or activation levels of several key proteins of Hippo signaling pathway were analyzed in vivo and in vitro. (A) Western blot analysis for the total and phosphorylated levels of LATS1 and YAP in HBV-induced xenografts tumor tissues with PLG silencing or not. (B) Western blot analysis for the total and phosphorylated levels of LATS1 and YAP in HBV-positive HepAD38 cells after PLG silencing or (C) SRC silencing. (D) Western blot analysis for the total and phosphorylated levels of LATS1 and YAP in xenograft tumor tissues with SRC overexpresion or not or SRC overexpresion combined with PLG silencing. (E) Western blot analysis for the total and phosphorylated levels of LATS1 and YAP in HBV-positive HepAD38 cells with PLG silencing or/and SRC overexpression. (F) Flow cytometry analysis for changes of cell apoptosis rates in HBV-positive HepAD38 and HepG2-NTPC cells treated with PLG interference plasmids or combined with XMU-MP-1. *P < 0.05; **P < 0.01; ***P < 0.001.
Discussion
Chronic HBV infection is one of the major risk factors for HCC, while the mechanism remains unclear [24]. Our study revealed that PLG and SRC were two of the upregulated note genes and Hippo signaling pathway might be a key signaling pathway in HBV-induced HCC by bioinformatic analysis. Previous studies reported that interactions may be between PLG and SRC [16], as well as SRC and Hippo signaling pathway [12,13]. Therefore, our study designed relevant experiments to prove the role of PLG in HBV-induced HCC and its relationship with SRC and Hippo signaling pathway. The experimental results will help to understand the mechanism of HBV-induced HCC, and provide theoretical basis to reduce the incidence of HCC in patients with HBV.
HBV infection can cause hepatitis, hepatic fibrosis, cirrhosis, and eventually lead to the occurrence of HCC. The persistence of HBV activity also affects the prognosis of patients with HCC even post-hepatectomy. The proteins encoded by HBV genomic DNA may promote the occurrence and development of HCC by promoting cell proliferation and migration, and up-regulating the activities of Ras/Raf/MAPK, PI3K/Akt and other key signaling pathways. In this research, it is found that PLG was highly expressed in HBV-positive HCC tissues and cells, and promoted the progress of HBV-induced HCC. In addition, interfered PLG decreased the expression of SRC and its phosphorylation level, thereby inhibiting the proliferation of HBV-positive HCC cells. Our findings may be contrary to the other studies on the PLG function as a tumor suppressor.
The Hippo signaling pathway regulates cell division, proliferation, differentiation and apoptosis. Upstream of the Hippo signaling pathway includes transmembrane protein FAT and some other membrane-related proteins that sense extracellular signals, and then recruits a core kinase cascade composed of MST1/2 (mammalian Sterile 20-like kinases 1/2) and SAV (protein Salvador homolog) to the cell membrane to activate MOB1 (Mps homolog), further phosphorylating the downstream transcriptional co-activators YAP, TAZ and YKi [12,13,25]. Specific knockout of MST1/2 in the liver can cause severe HCC [26]. Similarly, mixed HCC and cholangiocarcinoma may occur in mice that MOB1A/B was specifically knocked out in the liver [27]. And continuous overexpression of YAP gene in the liver of mice also resulted in lethal HCC [28]. These findings suggested that Hippo signaling pathway was closely related to the occurrence of HCC. In this study, we first found a link between PLG and the Hippo signaling pathway in HCC by bioinformatics analysis. Then we demonstrated that PLG interference can reverse the proliferation of cells induced by Hippo inhibitor, further indicating that PLG interference can activate the Hippo signaling pathway and inhibit the progression of HBV-positive HCC.
The study also demonstrated PLG played a role in HBV-induced HCC by targeting SRC via the Hippo signaling pathway. SRC is a non-receptor protein with tyrosine protein kinase activity. It can be activated by a variety of cell surface receptors and participates in many signaling pathways, including activating MAPK signaling pathway [29], STAT pathway [30], and PI3K/AKT pathway [31]. The activation of SRC family kinase protein is closely related to the occurrence, development and metastasis of multiple malignancies. SRC also plays a role in HBV induced HCC [15]. Butterfield et al [16]. confirmed that PLG interacted with SRC kinase in Alzheimer’s disease. This study demonstrated that SRC overexpression decreased the apoptosis rate, PLG interference can reverse the proliferation of HBV-positive HCC cells induced by SRC, indicating that PLG interference can inhibit the expression of SRC, and then inhibit the proliferation of HBV-positive HCC cells.
Conclusions
This study identified that PLG may be involved in HBV-induced HCC progression through Hippo signaling pathway; however, the specific regulatory mechanism remains to be further explored. Through this study, we discovered a new possible regulatory mechanism in HBV-induced hepatocarcinogenesis and development which might provide theoretical basis for the prevention and treatment.
Acknowledgements
This research was supported in part by the National Natural Science Foundation of China [No. 81771674 and 81660103]; 111 Project [No. D17011]; Guangxi BaGui Scholars.
Disclosure of conflict of interest
None.
Abbreviations
- CHV
Chronic hepatitis virus
- HCC
hepatocellular carcinoma
- HBV
Hepatitis B virus
- SRC
SRC proto-oncogene nonreceptor tyrosine kinase
- PLG
Plasminogen
- FAT
FAT atypical cadherin
- FRMD6
FERM domain-containing protein 6
- NF2
neurofibromin 2
- YAP
Yes-associated protein
- LATS1/2
large tumor suppressor k
- MST1/2
mammalian Sterile 20-like kinases 1/2
- SAV1
protein Salvador homolog 1
- MOB1A/B
Mps one binder kinase activator-like 1A/B
Supporting Information
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 2.Kulik L, El-Serag HB. Epidemiology and management of hepatocellular carcinoma. Gastroenterology. 2019;156:477–491. e471. doi: 10.1053/j.gastro.2018.08.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zoulim F, Levrero M, Testoni B. Hepatitis B. Nat Rev Gastroenterol Hepatol. 2018 doi: 10.1038/s41575-018-0060-3. [DOI] [PubMed] [Google Scholar]
- 4.Farazi PA, DePinho RA. Hepatocellular carcinoma pathogenesis: from genes to environment. Nat Rev Cancer. 2006;6:674–687. doi: 10.1038/nrc1934. [DOI] [PubMed] [Google Scholar]
- 5.Shlomai A, de Jong YP, Rice CM. Virus associated malignancies: the role of viral hepatitis in hepatocellular carcinoma. Semin Cancer Biol. 2014;26:78–88. doi: 10.1016/j.semcancer.2014.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kwaan HC, McMahon B. The role of plasminogen-plasmin system in cancer. Cancer Treat Res. 2009;148:43–66. doi: 10.1007/978-0-387-79962-9_4. [DOI] [PubMed] [Google Scholar]
- 7.Palumbo JS, Talmage KE, Liu H, La Jeunesse CM, Witte DP, Degen JL. Plasminogen supports tumor growth through a fibrinogen-dependent mechanism linked to vascular patency. Blood. 2003;102:2819–2827. doi: 10.1182/blood-2003-03-0881. [DOI] [PubMed] [Google Scholar]
- 8.Chounta A, Ellinas C, Tzanetakou V, Pliarhopoulou F, Mplani V, Oikonomou A, Leventogiannis K, Giamarellos-Bourboulis EJ. Serum soluble urokinase plasminogen activator receptor as a screening test for the early diagnosis of hepatocellular carcinoma. Liver Int. 2015;35:601–607. doi: 10.1111/liv.12705. [DOI] [PubMed] [Google Scholar]
- 9.Zhou YQ, Lv XP, Li S, Bai B, Zhan LL. Synergy of urokinasetype plasminogen activator receptor isomer (D1D2) and integrin alpha5beta1 causes malignant transformation of hepatic cells and the occurrence of liver cancer. Mol Med Rep. 2014;10:2568–2574. doi: 10.3892/mmr.2014.2503. [DOI] [PubMed] [Google Scholar]
- 10.Nakagawa H, Umemura A, Taniguchi K, Font-Burgada J, Dhar D, Ogata H, Zhong Z, Valasek MA, Seki E, Hidalgo J, Koike K, Kaufman RJ, Karin M. ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell. 2014;26:331–343. doi: 10.1016/j.ccr.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Warmuth M, Damoiseaux R, Liu Y, Fabbro D, Gray N. SRC family kinases: potential targets for the treatment of human cancer and leukemia. Curr Pharm Des. 2003;9:2043–2059. doi: 10.2174/1381612033454126. [DOI] [PubMed] [Google Scholar]
- 12.Yu FX, Guan KL. The Hippo pathway: regulators and regulations. Genes Dev. 2013;27:355–371. doi: 10.1101/gad.210773.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang J, Wu S, Barrera J, Matthews K, Pan D. The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell. 2005;122:421–434. doi: 10.1016/j.cell.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 14.Bouchard M, Giannakopoulos S, Wang EH, Tanese N, Schneider RJ. Hepatitis B virus HBx protein activation of cyclin A-cyclin-dependent kinase 2 complexes and G1 transit via a Src kinase pathway. J Virol. 2001;75:4247–4257. doi: 10.1128/JVI.75.9.4247-4257.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu H, Xu J, Zhou L, Yun X, Chen L, Wang S, Sun L, Wen Y, Gu J. Hepatitis B virus large surface antigen promotes liver carcinogenesis by activating the Src/PI3K/Akt pathway. Cancer Res. 2011;71:7547–7557. doi: 10.1158/0008-5472.CAN-11-2260. [DOI] [PubMed] [Google Scholar]
- 16.Butterfield DA, Poon HF, St Clair D, Keller JN, Pierce WM, Klein JB, Markesbery WR. Redox proteomics identification of oxidatively modified hippocampal proteins in mild cognitive impairment: insights into the development of Alzheimer’s disease. Neurobiol Dis. 2006;22:223–232. doi: 10.1016/j.nbd.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 17.Zhou D, Zhang Y, Wu H, Barry E, Yin Y, Lawrence E, Dawson D, Willis JE, Markowitz SD, Camargo FD, Avruch J. Mst1 and Mst2 protein kinases restrain intestinal stem cell proliferation and colonic tumorigenesis by inhibition of Yes-associated protein (Yap) overabundance. Proc Natl Acad Sci U S A. 2011;108:E1312–1320. doi: 10.1073/pnas.1110428108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou Q, Li L, Zhao B, Guan KL. The hippo pathway in heart development, regeneration, and diseases. Circ Res. 2015;116:1431–1447. doi: 10.1161/CIRCRESAHA.116.303311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou D, Medoff BD, Chen L, Li L, Zhang XF, Praskova M, Liu M, Landry A, Blumberg RS, Boussiotis VA, Xavier R, Avruch J. The Nore1B/Mst1 complex restrains antigen receptor-induced proliferation of naive T cells. Proc Natl Acad Sci U S A. 2008;105:20321–20326. doi: 10.1073/pnas.0810773105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, Xie J, Ikenoue T, Yu J, Li L, Zheng P, Ye K, Chinnaiyan A, Halder G, Lai ZC, Guan KL. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007;21:2747–2761. doi: 10.1101/gad.1602907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yi J, Lu L, Yanger K, Wang W, Sohn BH, Stanger BZ, Zhang M, Martin JF, Ajani JA, Chen J, Lee JS, Song S, Johnson RL. Large tumor suppressor homologs 1 and 2 regulate mouse liver progenitor cell proliferation and maturation through antagonism of the coactivators YAP and TAZ. Hepatology. 2016;64:1757–1772. doi: 10.1002/hep.28768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jie L, Fan W, Weiqi D, Yingqun Z, Ling X, Miao S, Ping C, Chuanyong G. The hippo-yes association protein pathway in liver cancer. Gastroenterol Res Pract. 2013;2013:187070. doi: 10.1155/2013/187070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ladner SK, Otto MJ, Barker CS, Zaifert K, Wang GH, Guo JT, Seeger C, King RW. Inducible expression of human hepatitis B virus (HBV) in stably transfected hepatoblastoma cells: a novel system for screening potential inhibitors of HBV replication. Antimicrob Agents Chemother. 1997;41:1715–1720. doi: 10.1128/aac.41.8.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tang LSY, Covert E, Wilson E, Kottilil S. Chronic hepatitis B infection: a review. JAMA. 2018;319:1802–1813. doi: 10.1001/jama.2018.3795. [DOI] [PubMed] [Google Scholar]
- 25.Yin F, Yu J, Zheng Y, Chen Q, Zhang N, Pan D. Spatial organization of Hippo signaling at the plasma membrane mediated by the tumor suppressor Merlin/NF2. Cell. 2013;154:1342–1355. doi: 10.1016/j.cell.2013.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou D, Conrad C, Xia F, Park JS, Payer B, Yin Y, Lauwers GY, Thasler W, Lee JT, Avruch J, Bardeesy N. Mst1 and Mst2 maintain hepatocyte quiescence and suppress hepatocellular carcinoma development through inactivation of the Yap1 oncogene. Cancer Cell. 2009;16:425–438. doi: 10.1016/j.ccr.2009.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nishio M, Sugimachi K, Goto H, Wang J, Morikawa T, Miyachi Y, Takano Y, Hikasa H, Itoh T, Suzuki SO, Kurihara H, Aishima S, Leask A, Sasaki T, Nakano T, Nishina H, Nishikawa Y, Sekido Y, Nakao K, Shin-Ya K, Mimori K, Suzuki A. Dysregulated YAP1/TAZ and TGF-beta signaling mediate hepatocarcinogenesis in Mob1a/1b-deficient mice. Proc Natl Acad Sci U S A. 2016;113:E71–80. doi: 10.1073/pnas.1517188113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford SA, Gayyed MF, Anders RA, Maitra A, Pan D. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell. 2007;130:1120–1133. doi: 10.1016/j.cell.2007.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vindis C, Cerretti DP, Daniel TO, Huynh-Do U. EphB1 recruits c-Src and p52Shc to activate MAPK/ERK and promote chemotaxis. J Cell Biol. 2003;162:661–671. doi: 10.1083/jcb.200302073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tan Q, Wang H, Hu Y, Hu M, Li X, Aodengqimuge , Ma Y, Wei C, Song L. Src/STAT3-dependent heme oxygenase-1 induction mediates chemoresistance of breast cancer cells to doxorubicin by promoting autophagy. Cancer Sci. 2015;106:1023–1032. doi: 10.1111/cas.12712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhu M, Guo J, Li W, Xia H, Lu Y, Dong X, Chen Y, Xie X, Fu S, Li M. HBx induced AFP receptor expressed to activate PI3K/AKT signal to promote expression of Src in liver cells and hepatoma cells. BMC Cancer. 2015;15:362. doi: 10.1186/s12885-015-1384-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.