

Abstract
Chimeric antigen receptor (CAR) T cells have revolutionized blood cancer immunotherapy; however, their efficacy against solid tumors has been limited. A common mechanism of tumor escape from single target therapies is downregulation or mutational loss of the nominal epitope. Targeting multiple antigens may thus improve the effectiveness of CAR immunotherapies. We generated dual CAR-T cells targeting two tumor antigens: TAG-72 (tumor-associated glycoprotein 72) and CD47. TAG-72 is a pan-adenocarcinoma oncofetal antigen, highly expressed in ovarian cancers, with increased expression linked to disease progression. CD47 is ubiquitously overexpressed in multiple tumor types, including ovarian cancer; it is a macrophage “don’t eat me” signal. However, CD47 is also expressed on many normal cells. To avoid this component of the dual CAR-T cells killing healthy tissue, we designed a truncated CD47 CAR devoid of intracellular signaling domains. The CD47 CAR facilitates binding to CD47+ cells, increasing the prospect of TAG-72+ cell elimination via the TAG-72 CAR. Furthermore, we could reduce the damage to normal tissue by monomerizing the CD47 CAR. Our results indicate that the co-expression of the TAG-72 CAR and the CD47-truncated monomer CAR on T cells could be an effective, dual CAR-T cell strategy for ovarian cancer, also applicable to other adenocarcinomas.
Keywords: CAR-T cells, dual specificity, ovarian cancer, TAG-72, CD47
Graphical Abstract
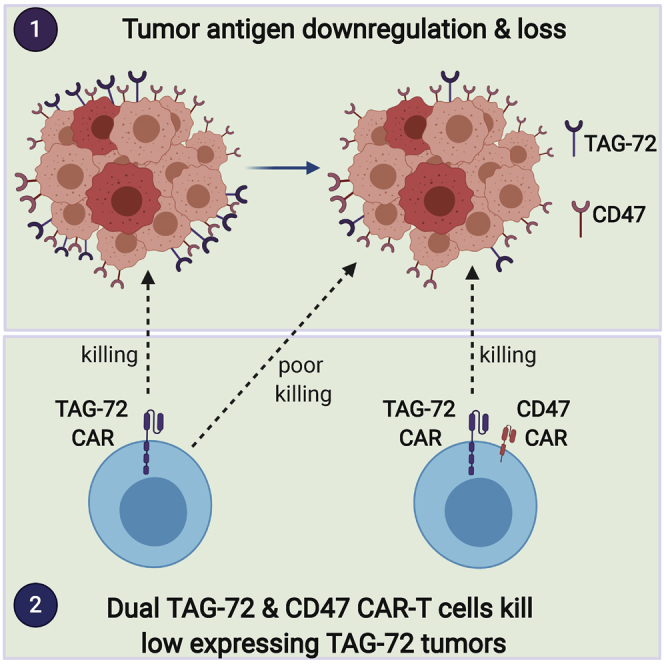
Tumors often escape immunotherapy by mutational downregulation of the nominal target antigen. Use of CAR-T cells with dual antigen specificity can mitigate this problem. Adenocarcinomas expressing low levels of TAG-72 are not killed by single specificity TAG-72 CAR-T cells, but they are killed by dual CAR-T cells targeting TAG-72 and CD47.
Introduction
Patient T cells that are genetically engineered ex vivo to express chimeric antigen receptors (CARs) targeting signature antigens expressed by the patient’s tumor cells eliminate those tumors in a high proportion of patients with acute lymphocytic leukemia or non-Hodgkin’s lymphoma, as evidenced by the 2017 US Food and Drug Administration (FDA) approval of two independent CD19-targeting CAR-T cell products, Yescarta and Kymriah,1 and the recent approval for mantle cell lymphoma.2 In contrast, clinical trials testing CAR-T cell treatment of solid tumors have been disappointing.3,4 The relative lack of efficacy in solid tumors is thought to be due to restricted access to the tumor site, the immunosuppressive tumor microenvironment, and/or modulation of the targeted tumor epitope.3,4
Ovarian cancer is a leading cause of cancer-related death among women, where most (>70%) cases are not diagnosed until the patient presents with advanced disease (stages III and IV) when therapeutic options are limited.5 Of the multiple tumor-associated antigens identified as potential targets for CAR-T cell therapy in ovarian cancer,6, 7, 8, 9 we have selected TAG-72 (tumor-associated glycoprotein 72), an aberrantly glycosylated cell surface glycoprotein overexpressed in adenocarcinomas, particularly of the colon, stomach, breast, prostate, and ovary.10, 11, 12 Numerous considerations render TAG-72 an attractive candidate for CAR-T cell therapy in advanced-stage ovarian cancer.7,13 TAG-72 expression has been documented across all ovarian cancer subtypes, with increased expression being directly correlated with poorer prognosis.14,15 With the exception of limited expression by isolated secretory endometrial tissue and rare duodenal goblet cells, TAG-72 is absent in normal tissues.10,16,17 A recent biodistribution phase I study of TAG-72 in prostate and ovarian cancer metastases using an 124I-labeled diabody showed high levels of TAG-72 specifically in the tumor with no TAG-72-specific uptake in any normal tissue.18 TAG-72 has also been targeted in phase I immunotherapy trials, with one report of a first-generation CAR-T cell, using systemic administration.13,19 While there was some evidence of biological activity, disease relapse ultimately occurred attributable in part to host immune response to immunogenic determinants in the CAR construct. Interestingly, a recent preclinical study using an ovarian cancer xenograft model reported that reduced TAG-72 expression was observed in the recurring ovarian cancer tumors after TAG-72 CAR-T cell treatment.7
Downregulation of tumor antigens is a common immune evasion strategy mounted by many tumors, and one that can sometimes be counteracted by simultaneously targeting multiple tumor antigens expressed by the same tumor.4,20 In this context, we selected CD47, a cell surface protein ubiquitously expressed on ovarian cancer cells,21, 22, 23, 24 as a second target antigen in addition to TAG-72 for the generation of dual antigen-targeting CAR-T cells for ovarian cancer. CD47 is highly expressed on cancer cells and functions as a macrophage “don’t eat me” signal by causing the inhibition of cell phagocytosis via ligation of signal regulatory protein α (SIRPα) on phagocytic cells.25,26 Antibody blockade of CD47 facilitates elimination of cancer cells through restoring the engagement of macrophages.26 While CD47 is expressed at low levels on normal cells,27 this appears inconsequential since clinical trials with B cell lymphoma patients have shown compelling anti-tumor activity of the anti-CD47 monoclonal antibody, Hu5F9, without significant adverse events.28, 29, 30 Additionally, Golubovskaya et al.31 have shown that anti-CD47 CAR-T cells could destroy multiple cancer cell lines in vitro, including ovarian cancer.
In this study, we hypothesize that CAR-T cells with TAG-72/CD47 dual specificity would more effectively target ovarian cancer cells than single-target CAR-T cells, potentially by circumventing escape through downregulation of one of the tumor antigens. Accordingly, we designed a family of TAG-72 and CD47 CAR-T cells including those with bicistronic expression of TAG-72 and CD47 CAR and evaluated their relative expansion potential and cytotoxicity. As CD47 is expressed at low levels on healthy tissue, including on T cells,32 the initial CD47 CAR-T cells endowed with a fully functional intracellular signaling domain resulted in defective T cell expansion in vitro, presumably due to fratricide. We therefore co-expressed a CD47 CAR truncated to lack signal transduction domains, with a second-generation signaling TAG-72 CAR. These dual CAR-T cells compensated the killing of target cells expressing low levels of TAG-72. Furthermore, we found that monomerization of the CD47-truncated CAR, which reduces signaling due to lack of dimer formation, could improve dual CAR-T cell expansion and reduce the cytotoxic impact on normal cells. Finally, we infused the TAG-72.4-1BB and CD47-truncated monomer dual CAR-T cells, which showed the most promising in vitro results, into immune-suppressed mice bearing ovarian cancer xenograft tumors.
Results
Characterization of TAG-72 targeting CAR-T cells
Various anti-TAG-72 monoclonal antibodies, including CC49, have been utilized for radiotherapy and CAR-directed targeting of adenocarcinomas, both in preclinical and clinical studies.7,13,33, 34, 35 In a previous anti-TAG-72 CAR-T cell clinical study, the humanized anti-TAG-72 single-chain variable fragment (scFv), humanized CC49 (huCC49), was utilized to construct first-generation CAR-T cells for solid tumor treatment. However, there was limited tumor response, which may be attributed to the use of huCC49 scFv.13 The huCC49 scFv has been reported to bind with 23- to 30-fold lower affinity compared to that of murine CC49, suggesting this may be the cause of compromised efficacy.36 It also contained foreign epitopes, which the patient’s immune system responded to.13,36 In our study, we implemented a deimmunized version of the murine CC49 scFv for our CAR construct (Figure 1A), which has a high and specific tumor uptake as well as lower immunogenicity.37 We developed second-generation CARs containing this anti-TAG-72-specific scFv coupled to either a CD28 or 4-1BB intracellular co-stimulatory domain linked to a CD3ζ signaling domain and an enhanced green fluorescent protein (EGFP) reporter (Figure 1A). Expansion kinetics, transduction efficiency, prevalent phenotype, and tumor antigen expression were subsequently interrogated. Both TAG-72.CD28 and TAG-72.4-1BB CAR constructs were integrated into T cells isolated from healthy donor peripheral blood mononuclear cells (PBMCs) using lentiviral transduction. Co-expression of CD3 and CAR(GFP) was monitored with comparable transduction efficiencies observed across CAR constructs (Figure 1B). Following isolation, TAG-72.CD28 and TAG-72.4-1BB CAR-T cells displayed similar ex vivo expansion kinetics (Figure 1C). CD4 and CD8 subsets were comparable across groups (Figure 1D). In addition, proportions of T cell subsets remained uniform with effector memory (CCR7−/CD45RO+) cells, the prevalent phenotype.
Figure 1.

Characterization of TAG-72-targeting CAR-T cells containing either a CD28 or 4-1BB intracellular signaling domain
(A) Schematic representation of the lentiviral expression cassette containing a deimmunized anti-TAG-72 scFv, CD8 hinge domain, CD8 or CD28 TM domain, and either a CD28 or 4-1BB intracellular co-stimulatory domain linked to a CD3ζ signaling domain. (B) T cells (no CAR) (left), TAG-72.CD28 CAR (middle), and TAG-72.4-1BB CAR (right) were assessed by flow cytometry to determine transduction efficiency and CD3 expression. (C) Expansion potential of CAR-T cells following transduction from a single representative donor. (D) CD4/CD8 frequency (left) and effector memory, central memory, naive, and effector cell frequency (right). Data are represented as average ± SD; n = 3. (E) Flow cytometric analysis was implemented to determine extracellular expression of TAG-72 on two ovarian cancer cell lines MESOV (left) and OVCAR-3 (right). Gating was based on the fluorescence minus one (FMO) control, where viable, single cells were analyzed; representative pseudo-color contour plots and population frequency (% viable) are presented. (F) Additionally, a panel of ovarian cancer cell lines (Caov-3, Caov-4, OVCAR-429, and OV-90) was assessed in conjunction with Raji cells (negative control) and Jurkat and LS174T cells (positive controls), respectively. Blue, FMO control; red, anti-TAG-72
Previous studies have shown TAG-72 expression in ovarian cancer cells from clinical tissue samples14 and in multiple ovarian cancer cell lines.7,15,38 In particular, the ovarian cancer cell line OVCAR-3 was consistently reported as displaying high expression of TAG-72. Using the anti-TAG-72 diabody and flow cytometry, our data confirmed that the OVCAR-3 cell line has high levels of membrane-associated TAG-72 (Figure 1E). We also showed that the MESOV ovarian cancer cell line has low expression of TAG-72 (Figure 1E). Additionally, we evaluated TAG-72 expression on several other ovarian cancer cell lines in conjunction with Raji (negative control) and Jurkat and LS174T cells (positive controls). Caov-3, Caov-4, and OV-90 cells displayed high expression of TAG-72, whereas OVCAR-429 cells had low expression of TAG-72 (Figure 1F). Thus, from this panel of cell lines with differential TAG-72 expression, OVCAR-3 and MESOV cells were chosen to assess and characterize CAR-T cell activity on tumor cells that display either higher or lower expression of TAG-72, respectively.
TAG-72.CD28 and TAG-72.4-1BB CAR-T cells effectively eliminate ovarian cancer cells in vitro
Both CD28 and 4-1BB costimulatory domains have been used in CD19 CAR-T cell clinical studies and have shown great promise for the treatment of B cell lymphoma.1 However, differences in the kinetics and signaling cascades of the costimulatory domains could affect the clinical efficacy and toxicity of CAR-T cells.39,40 To compare the differences in our TAG-72 CAR-T cells, dose-dependent cytotoxic activity of TAG-72.CD28 and TAG-72.4-1BB CAR-T cells was assessed in vitro. As expected, target cell death was observed within 5 h of CAR-T/OVCAR-3 cell co-culture, with cytotoxicity reaching significance within 10 h (Figures 2A and 2B, effector-to-target [E:T] ratio of 5:1, p < 0.01), and it maintained significance throughout. Whereas cytotoxicity of TAG-72.CD28 CAR-T cells against OVCAR-3 cells was significantly greater (p < 0.05) than for non-CAR-T cells from 5 h of incubation, that of TAG-72.4-1BB CAR-T cells was not above controls until 15 h (Figures 2A and 2B), highlighting the importance of the type of intracellular signaling domain and its impact on the kinetics of CAR-T function. This also indicates that CD28 signaling induces a more rapid CAR-T cell response to cancer antigen exposure compared to CAR-T cells with 4-1BB signaling. Parallel studies were also performed comparing the ability of TAG-72.CD28 and TAG-72.4-1BB CAR-T cells to eliminate low TAG-72-expressing MESOV cells in vitro. The cytotoxic function of CAR-T cells tested did not significantly exceed that of T cell (no CAR) controls (Figures 2A and 2C). These results show that while our TAG-72 CAR-T cells can eliminate TAG-72-positive OVCAR-3 tumor cells, they are less effective against cancer cells with lower TAG-72 expression.
Figure 2.

CD28 and 4-1BB signaling domains endow CAR-T cells with comparable function in vitro
(A) OVCAR-3 (left) and MESOV (right) target cells were co-cultured with T cells (no CAR), TAG-72.CD28 CAR, or TAG-72.4-1BB CAR at an E:T ratio of 5:1, and the response was monitored in real time using xCELLigence where a decrease in the normalized cell index is indicative of target cell death relative to target cells alone (black). (B and C) Additionally, OVCAR-3 (B) or MESOV (C) target cells were co-cultured with effector cells at E:T ratios of 1:1 and 1:5. Cytotoxic functions for all E:T ratios were characterized at 5, 10, 15, and 20 h of co-incubation and are presented here as % cytotoxicity. Biological and intra-assay triplicates were used and are denoted as average ± SD. (D) CD4+ and CD8+ CAR-T cells elicit a cytotoxic response to antigen exposure. CD4+ (left), CD8+ (middle), or a controlled CD4+/CD8+ ratio (right) of TAG-72.4-1BB CAR-T (Δ) cells were co-cultured with OVCAR-3 cells at an E:T ratio of 1:1. CD4+ or CD8+ T cells (no CAR) were cultured in parallel (blue). A reduction in normalized cell index relative to target cells alone (▪) demonstrates effector cell-mediated cytotoxicity. Numerical values embedded within each graph represent average percent cytotoxicity ± SD (n = 5) at 20 h relative to the target cells alone control. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 0.0001.
Both CD4+ and CD8+ CAR-T cells are potent killers in vitro
Given that most current CAR-T cell trials do not distinguish between the relative levels of CD8+ and CD4+ cells in the CAR-T cell population, we compared the functional contribution of these T cell subsets to cytotoxicity in vitro. Purified CD4+ or CD8+ T cells and a defined ratio of CD4+/CD8+ CAR-T cells were co-cultured with OVCAR-3. In parallel, CD4+ and CD8+ T cells (no CAR) were assessed for cytotoxic activity. Our data surprisingly demonstrated that both T cell subsets isolated from the TAG-72.4-1BB CAR have equivalent cytotoxic ability to eliminate TAG-72-expressing target cells in vitro (Figure 2D). There was no significant difference in cytotoxicity observed between CD4+/CAR+ or CD8+/CAR+ or CD4+-CD8+/CAR+ effector cells (Figure 2D).
Pro-inflammatory and regulatory cytokines, as well as growth and chemotactic factors, are considered essential for CAR-T cell longevity and antitumor efficacy.4 Differences in cytokine production between CD28 and 4-1BB CARs were also examined in response to co-culture with OVCAR-3 and MESOV target cells. Both CD28 CAR-T cells and the 4-1BB CAR-T cells cultured with OVCAR-3 cells had increased levels of some pro-inflammatory cytokines, e.g., interferon (IFN)-γ, interleukin (IL)-1β, IL-2, IL-5, IL-9, IL-17A, granulocyte-macrophage colony-stimulating factor (GM-CSF), and tumor necrosis factor (TNF)-α (Figure 3A), relative to the non-CAR-T cells. Regulatory and anti-inflammatory cytokines, e.g., IL-6, IL-1Ra, and IL-4, were also elevated compared to T cell (no CAR) conditions (Figures 3B and 3C). However, this was not universal, as there were no changes in IL-10 and IL-13 (Figure 3C). Interestingly, levels of chemotactic factors macrophage-inflammatory protein (MIP)-1α and MIP-1β were also elevated (Figure 3D). In contrast, CAR-T cells co-cultured with MESOV cells resulted in little change in cytokine production compared to the T cell (no CAR) controls with the exceptions of IFN-γ, IL-5, IL-9, GM-CSF (Figure 3A), IL-1Ra (Figure 3C), and MIP-1α and MIP-1β (Figure 3D), which were significantly elevated following TAG-72.CD28 co-culture (p < 0.05). Overall, these results suggest that while TAG-72.CD28 CAR-T cells may have stronger and faster signaling in response to a cancer antigen, TAG-72.4-1BB CAR-T cells exhibit equivalent levels of beneficial cytokines in response to cancer antigen stimulation.
Figure 3.

The cytokine profile of effector cells was characterized following 20-h exposure to target cells at an E:T ratio of 5:1
(A) Pro-inflammatory, (B) Regulatory, (C) Anti-inflammatory and (D) Chemotactic factors produced by effector cells following co-culture were quantitated. Effector cells are T cells (no CAR) (blue) or TAG-72.CD28 CAR (red) or TAG-72.4-1BB CAR (orange) with the target cell lines OVCAR-3 and MESOV, respectively. Biological and intra-assay triplicates were used and are denoted as average ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
CD47 signaling CAR-T cells have limited expansion capacity in vitro
CD47 is expressed on healthy human cells; however, its upregulation has been documented in multiple cancer indications,27,41 with previous studies highlighting the therapeutic potential of targeting CD47+ tumors. Anti-CD47(B6H12) CAR-T cells have demonstrated the killing of ovarian cancer cell lines,31 while Hu5F9 scFv has progressed to phase I clinical trials with limited adverse events observed.42 We therefore explored the use of CAR-T cells targeting both TAG-72 and CD47.
Second-generation CAR constructs were designed to contain a CD47 signaling (CD47.CD28z) or truncated, non-signaling (ΔCD47) lentiviral CAR cassette, where both constructs contained a CD47(Hu5F9) scFv, FLAG tag, CD28 hinge, and transmembrane (TM) domain. Additionally, the CD47.CD28z CAR contained CD28 and CD3ζ signaling domains (Figure 4A). Levels of plasma membrane expression of CD47 were decreased in expanded CD47.CD28z CAR-T cells compared to ΔCD47 CAR-T cells and unedited T cells (no CAR) (Figure 4B), which were associated with decreased numbers and viability of T cells in some donors (Figures 4C and 4D, respectively), presumably due to fratricide. In order to identify whether the poor expansion was due to the Hu5F9 scFv, we constructed a CD47.CD28z CAR using the B6H12 scFv.31 Comparable results were observed with CD47(B6H12).CD28z CAR-T cells (Figure S1). Taken together, these data suggest that CD47.CD28z CAR alone may not be a viable therapeutic product due to potential CD47-driven fratricide during cell manufacturing.
Figure 4.

CD47 signaling CAR-T cells have reduced proliferative capacity in vitro
(A) Schematic representation of CD47 signaling (CD47.CD28z) or non-signaling (ΔCD47) lentiviral CAR cassette where both constructs contain an anti-CD47 scFv, CD28 hinge, and CD28 TM domain. Additionally, the CD47.CD28z CAR contains CD28 and CD3ζ signaling domains. (B) Retention of endogenous CD47 expression on CD47.CD28z and ΔCD47 CAR-T cells was assessed by flow cytometry 7 days following transduction. (C and D) Expansion (C) and viability (D) of CAR-T cells prepared from three independent healthy donors was tracked for at least 7 days following transduction. T cells (no CAR) and TAG-72.4-1BB CAR-T cells were maintained in parallel. (E) CD47 expression on target cells, including OVCAR-3, MESOV, HLF, and HUF cells, was confirmed by flow cytometry (gray) compared to unstained control (red) where values represent average CD47 expression (percent viable) ± SD (n = 5).
We characterized CD47 expression on OVCAR-3 and MESOV ovarian cancer cell lines, in addition to normal human lung fibroblast (HLF) and normal human uterine fibroblast (HUF) cell lines from healthy adults (Figure 4E). All cells analyzed demonstrated a CD47+ population frequency of >90%, with highest expression observed on OVCAR-3 and MESOV cells (99.9% ± 0.1% and 98.1% ± 2.1%, respectively). These data reinforce a potential safety issue for the implementation of CD47.CD28z CAR-T cells given the ubiquitous expression on both cancerous and non-cancerous cell lines. To overcome this, we generated dual CAR-T cells targeting both TAG-72 and CD47, where the CD47(Hu5F9) scFv was linked to a CD28 hinge and CD28 TM domain but, importantly, was lacking an intracellular signaling domain (Figure 5).
Figure 5.

Construction of dual-specific CARs
(A) Schematic representation of lentiviral CAR construct containing a deimmunized anti-TAG-72 scFv, CD8 hinge domain, CD8 or CD28 TM domain, and either a CD28 or 4-1BB intracellular signaling domain linked to a CD3ζ signaling domain coupled to an anti-CD47 scFv, CD28 hinge domain, CD28 TM domain, and truncated intracellular domain resulting in a non-signaling dimerizing CAR (ΔCD47d). (B) Flow cytometric analysis was performed to ascertain frequency of CAR and CD3 co-expression following transduction (n = 5–8). (C and D) CAR-T cell expansion (C) was tracked for 11 days after transduction, and viability (D) was monitored following 7 days of expansion. (E) Schematic showing constructs similar to those of (A) but with an anti-CD47 scFv and Cys-to-Ser point mutations within the CD28 hinge and TM domain resulting in a non-signaling, monomeric CAR (ΔCD47m). (F) Schematic representation of CAR localization at the cell membrane depicting the difference in TAG-72 CAR + ΔCD47d CAR (left) and TAG-72 CAR + ΔCD47m CAR (right). (G) Western blot analysis of TAG-72 + ΔCD47 dual CAR-T cell lysates in the presence or absence of β-mercaptoethanol (β-met), with reducing (+) and non-reducing (–) conditions. ΔCD47 CAR bands were detected by anti-c-Myc antibody, and TAG-72 CAR bands were detected by anti-CD3ζ antibody. Molecular mass markers (M) are indicated to the right of each panel. (H and I) Transduction efficiency (H) and expansion potential (I) were monitored as previously described. (J) Total viable cells following 11 days of expansion were directly compared for TAG-72.CD28 dual CAR-T cells (left) and TAG-72.4-1BB dual CAR-T cells (right). Data represent average ± SD of biological replicates generated in independent experiments. n.s. not significant, ∗p < 0.05, ∗∗∗p < 0.001.
Design and characterization of TAG-72 + ΔCD47 dual-targeting CAR-T cells
Dual CAR constructs were generated with the aim of increasing CAR-mediated elimination of cells expressing low levels of the target antigen. In the first instance, a TAG-72-specific signaling CAR containing either the CD28 or 4-1BB signaling domain was coupled to a CD47 dimerizing (CD47d) non-signaling (Δ) CAR (Figure 5A). Transduction efficiency of these dual CAR-T cells (Figure 5B) was lower than that observed with the TAG-72 CAR alone (Figure 1B). However, it was comparable between the two different ΔCD47 dual CAR constructs (TAG-72.CD28 and TAG-72.4-1BB). In vitro expansion was tracked for 11 days following transduction (Figure 5C). TAG-72.CD28 CAR + ΔCD47d CAR and TAG-72.4-1BB CAR + ΔCD47d CAR had reduced proliferation compared to TAG-72 single CAR-T cells; this may, in part, be due to reduced viability at least for TAG-72.CD28 CAR + ΔCD47d CAR (Figure 5D). We postulated that CD47d may contribute to restricted growth and reduced dual CAR-T cell viability. Disulfide bridges mediated by cysteine (Cys) residues are essential for the dimerization and signaling of cell surface proteins including CD28.43 This is also true for CARs that dimerize through the hinge and TM region to enhance function.44 Accordingly, we introduced point mutations in the CD28 hinge and TM region of ΔCD47 CAR replacing the Cys with serine (Ser) residues, resulting in monomerization of CD47 CAR (ΔCD47m) (Figure 5E). This prevents dimerization of ΔCD47 CAR co-expressed with TAG-72.CD28 or TAG-72.4-1BB CAR (Figure 5F). Western blot results confirmed that Cys-to-Ser mutations in the ΔCD47 CAR hinge and TM region showed a single band under both reducing and non-reducing conditions equivalent to the monomeric form, as opposed to the wild-type, which showed dimers under non-reducing conditions (Figure 5G). To clarify whether the TAG-72 CAR could dimerize with the ΔCD47d CAR, TAG-72 CAR dimeric bands were blotted as well. We did not detect extra bands that indicated the presence of any dimerization between the TAG-72 CAR and ΔCD47d CAR. (Figure 5G). The TAG-72 + ΔCD47m dual CARs could be transduced and expressed on T cells with comparable efficiency to the TAG-72 + ΔCD47d dual CARs (Figure 5H). Unlike ΔCD47d constructs, the expansion kinetics of ΔCD47m CARs closely mimicked T cell (no CAR) controls (Figures 5I and 5J). As the differing dimer/monomer status of the CD47 CAR between the two constructs may result in differences in CD47 binding affinity and ultimately impact their function, we quantitatively monitored the ability of dual specific CARs to eliminate cancerous and non-cancerous target cells in vitro.
In vitro elimination of TAG-72low target cells is enhanced by TAG-72 CAR + ΔCD47 CAR-T cells
To evaluate whether TAG-72 + ΔCD47 dual CAR-T cells were able to eliminate low TAG-72-expressing cells over time, ΔCD47d CAR and ΔCD47m single CAR-T cells were tested in parallel. When co-cultured with target cells, ΔCD47d CAR-T cells and ΔCD47m CAR-T cells were unable to elicit a cytotoxic effect greater than T cell (no CAR) controls, demonstrating that binding of the ΔCD47 scFv alone presented by a T cell is insufficient to result in target cell death. As expected, signaling TAG-72 CAR-T cells in all their iterations were potent killers of TAG-72high/CD47+ OVCAR-3 cells with near complete target cell elimination achieved within 10 h of co-culture (Figure 6A). Importantly, whereas TAG-72 CAR alone T cells (either TAG-72.CD28 CAR or TAG-72.4-1BB CAR) were unable to kill TAG-72low/CD47+ MESOV cancer cells, the dual CAR-T cells of both CD28 and 4-1BB were now able to eliminate these cells efficiently at 20 h (Figure 6B). Moreover, TAG-72.CD28 CAR + ΔCD47d CAR-T cells induced significant killing of MESOV cells at 10 h (p < 0.05), indicating they had a faster killing response compared to the 4-1BB variant, which was consistent with our observations using TAG-72 CD28 and 4-1BB single CAR-T cells (Figure 2). The dual CAR technology therefore enhanced the ability of CAR-T cells to effectively eliminate cancer cells that express low levels of the target antigen, even though the CD47 CAR does not contain intracellular signaling domains. We presume the increased killing is due to higher binding avidity of the dual CAR to the target cell.
Figure 6.

In vitro, dual-specific CARs demonstrate enhanced ability to eliminate cancer cells expressing low levels of the target antigen
(A–D) Mono-specific TAG-72 CAR-T cells and dual-specific TAG-72 CAR + ΔCD47 CAR-T cells were co-cultured with either OVCAR-3 (A) or MESOV (B) cancer cell lines or HLF (C) or HUF (D) non-cancerous fibroblast cell lines at an E:T ratio of 5:1. Target cell response kinetics were measured in real time using xCELLigence (left panels) where data represent the average normalized cell index of 4–11 biological replicates ± SEM. Efficiency of target cell killing was quantitated at 10 and 20 h of co-culture and is presented as average percent cytotoxicity ± SD (right panels). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 0.0001.
To ascertain whether this increased potency of the dual CAR-T cells would lead to killing of CD47-expressing cells from non-cancerous tissues, two healthy fibroblast cell lines (TAG-72−/CD47+), HLF and HUF, were used as targets in vitro. Single antigen-targeting TAG-72 CAR-T cells, co-cultured with HLF (Figure 6C) and HUF (Figure 6D) cell lines, showed no significant killing relative to T cell (no CAR) controls. However, TAG-72.CD28 CAR + ΔCD47d or ΔCD47m CAR-T cells did show cytotoxicity within 10 h of co-culture with HLF cells and within 20 h of co-culture with HUF cells. TAG-72.4-1BB CAR + ΔCD47d only showed cytotoxicity of HLF cells within 20 h of co-culture (Figure 6C). Importantly, TAG-72.4-1BB CAR + ΔCD47m CAR-T cells, which were able to kill both TAG-72high (OVCAR-3) and TAG-72low (MESOV) cancer targets within 20 h, had no effect on both HLF and HUF cell lines at 10 and 20 h (Figures 6A–6D). Given these data, we explored the in vivo efficacy of TAG-72.4-1BB dual CAR-T cells.
In vivo elimination of TAG-72low target cells is enhanced by TAG-72.4-1BB CAR + ΔCD47 CAR-T cells
To evaluate the potential therapeutic value of TAG-72.4-1BB CAR + ΔCD47d or + ΔCD47m CAR-T cells in vivo, we established TAG-72high OVCAR-3 (Figures 7A and 7B) and TAG-72low MESOV (Figures 7C and 7D) subcutaneous (s.c.) models in non-obese diabetic (NOD)/severe combined immunodeficiency (SCID) gamma (NSG) mice. We observed a sustained response in OVCAR-3- bearing animals (Figure 7B) when treated with the single and dual TAG-72 CAR-T cells compared to T cell (no CAR) controls, with the latter approaching the experimental endpoint (tumor volume of 1,000 mm3) within 28 days. As expected, single antigen-targeting TAG-72.4-1BB CAR-T cells were unable to delay tumor growth in the MESOV model (Figure 7D), which occurred in all recipients within 15 days of treatment commencement. However, tumor growth was delayed with TAG-72.4-1BB CAR + ΔCD47d CAR and TAG-72.4-1BB CAR + ΔCD47m CAR treatment groups.
Figure 7.

Dual specific CARs demonstrate ability to eliminate cancer cells in vivo
(A) NSG mice bearing OVCAR-3 derived tumors were treated at 5-day intervals with either 2 × 106 T cells (no CAR), TAG-72.4-1BB CAR-T cells, TAG-72.4-1BB CAR + ΔCD47d CAR, or TAG-72.4-1BB CAR + ΔCD47m CAR by intravenous (i.v.) injection as soon as tumor volume (TV) was approximately 100mm3. (B) Tumor volume was monitored during 28 days. Data were generated from three independent experiments (and therefore three independent T cell donors) and are presented as raw tumor volume. (C) The ability for the aforementioned CAR-T cells to eliminate MESOV cells in vivo was also monitored. NSG mice bearing MESOV-derived tumors were treated as soon as palpable tumors were detected. (D) Tumor volume was monitored regularly during 28 days.
Discussion
The landmark approvals of three CD19 CAR-T cell products have sparked a new era of adoptive T cell immunotherapy for cancer. It is possible that the success of these CAR-T cell products is due in part to the choice of target antigen, CD19, a B cell marker expressed highly and uniformly on all B cells, including leukemic B cells. Ideally, however, from a safety perspective the molecule for CAR targeting should have no or low expression on normal cells to prevent toxicity.45, 46, 47 Accordingly, we selected TAG-72, an oncofetal antigen, which has a high degree of pan-specificity for adenocarcinomas but is negative on normal healthy cells. TAG-72 has previously been safely targeted by first-generation CAR-T cells in a phase I clinical trial.13 However, as for most solid tumors, ovarian cancer tends to have a heterogeneous expression of nominal tumor antigens such as TAG-72. Therefore, targeting multiple antigens may be required to overcome antigen-low escape.14
To overcome these limitations, multiple approaches have been explored to develop CAR-T cells that recognize two antigens. Simultaneous co-administration or co-transfection of different CAR constructs is a simple approach to give multiple specificities to CAR-T cells, but these have inherent disadvantages. For example, mixed cell products, complicated pre-clinical and clinical analysis, as well as higher cost for multiple vector manufacturing make them infeasible for further development.20 An alternative approach utilizes tandem CARs, which link two antigen-binding moieties in one bicistronic CAR cassette.48,49 This approach, however, doubles the potential for off-tumor activity raising safety concerns. A third strategy is splitting the CD3ζ signaling domain and the CD28 or 4-1BB co-stimulatory domains into two separate CARs with different antigen specificities. Thus, these dual CAR-T cells only eliminate tumor cells that express both antigens.50,51 As a result, this approach avoids the side effects associated with off-target reactivity by limiting the CAR-T function to tumor cells co-expressing both target antigens.
The dual targeting approach in the present study differs significantly from those previously described.20,48, 49, 50, 51 Fully functional second-generation CARs with both the CD28 or 4-1BB and the CD3ζ signaling domains were utilized to target the main tumor antigen of interest, TAG-72. An additional, truncated CAR, devoid of the signaling domain and targeting CD47, the secondary tumor antigen, was bicistronically expressed to enhance the binding affinity of CAR-T cells for TAG-72low cancer cells in tumor lesions. As CD47 is also normally expressed on T cells, we found that the full signaling CD47 CAR using the 5F9 scFv could interact with CD47 molecules on the T cell surface, leading to poor T cell expansion and subsequent potential fratricide. We noticed a discrepancy between the previous CD47 CAR-T cell study and ours. In the previous study, the CD47 CAR-T cells utilizing the B6H12 scFv could be expanded, but the CD47 expression on CAR-T cells and its potential related fratricide was not addressed.31 Our results showed that a signaling CD47 CAR could lead to poor expansion not only with a HuF9 scFv but also with a B6H12 scFv. The degree of this inhibition may be influenced by the differing endogenous CD47 expression levels between individuals and the anti-CD47 CAR on T cells. These results suggest that greater caution is required in the clinical application of signaling CD47 CAR-T cells, even when a carefully selected CD47 antibody can be well tolerated clinically.28,52
Hinge and TM regions in CAR design are implemented to extend the scFv various lengths beyond the cell membrane, providing flexibility and ultimately ensuring ligand-receptor interaction and facilitating signal transduction. Additionally, hinge and TM regions are crucial for CAR dimerization.44 In our study, the ΔCD47 CAR and TAG-72.CD28 CAR have the same CD28 TM region, allowing for potential self-association; however, our western blot analysis confirmed that they did not form heterodimers. We detected two ΔCD47 CAR dimeric bands in both iterations of TAG-72 + ΔCD47d dual CAR-T cell lysates, but not in iterations of TAG-72 + ΔCD47m dual CAR-T cell lysates under non-reducing conditions, indicating that the ΔCD47m CAR was monomeric. Importantly, no additional TAG-72 CAR dimeric bands were detected in both iterations of TAG-72 + ΔCD47d dual CAR-T cell lysates compared to TAG-72 + ΔCD47m dual CAR-T cell lysates under non-reducing conditions, respectively (Figure 5G). Early studies utilizing a CD3ζ TM domain showed that a Cys residue mutation within this region could disrupt dimerization and reduce CAR activity.53 The CD28 TM region contains conserved dimerization motifs,54 and CD28 oligomerization reduces the binding affinity of CD28 with its ligand.43 In our study, the two Cys-to-Ser point mutations in the hinge and TM region of CD47-truncated CAR could improve the CAR-T cell in vitro expansion and reduce the potential off-tumor targeting mediated by CD47. Other targets with expression patterns comparable to CD47, such as the epidermal growth factor receptor,55 CD166,56 and CD71,57 have a similar problem of normal tissue toxicity. Our dual CAR strategy with attenuated signaling, but which facilitates enhanced binding to the target cell, has the potential to expand the tumor antigen spectrum.
Recent clinical studies of the combined CD47(5F9) antibody with rituximab or azacitidine treatment in patients with non-Hodgkin’s lymphoma or acute myeloid leukemia, respectively, showed beneficial activity against tumors.28,29 Additional clinical studies investigating CD47(5F9) in colorectal (ClinicalTrials.gov: NCT02953782) and ovarian cancer (ClinicalTrials.gov: NCT03558139) are ongoing. Overall, combining CD47 blockade with other anticancer therapies is logical.52 However, the broad distribution of CD47 in normal tissues, especially on cells of the hematopoietic lineage, creates a large antigen sink that could constrain CD47 targeting agents from reaching tumor cells.30 We observed a delay in tumor growth in our low TAG-72-expressing MESOV model, suggesting that these dual CAR-T cells accumulate in tumor lesions; however, further studies are warranted to confirm their presence. Furthermore, the CAR-T cell anti-tumor activity could be boosted through the anti-CD47 scFv blocking SIRPα, thereby negating don’t eat me signals and facilitating macrophage attack. Unlike the scFv secreting strategy,58 our approach further retains the 5F9 CD47 interaction within the tumor tissue. Whereas the CD47 interaction with SIRPα on macrophages modulates phagocytosis, ligation of thrombospondin-1 or anti-CD47 antibodies with CD47 on T cells triggers CD47 intracellular signaling and anti-inflammatory effects, and it results in reduced proliferation or even T cell death.59 Our results also indicated that the tumor suppression by TAG-72.4-1BB + ΔCD47 dual CAR-T cells relapsed faster than did the single antigen targeting TAG-72.4-1BB CAR-T cells in the TAG-72high (OVCAR-3 xenograft), even though the CD47 CAR is truncated with no signaling domains. Since the dual CAR-T cells are more effective on the lower antigen-expressing cancer cell line, the combined binding of the two CARs may result in hyperactivation-induced senescence when killing the TAG-72high cells. This requires further investigation.
In conclusion, this proof-of-principle study provides support for the implementation of bicistronic expression of a monomerized, truncated CAR targeting an antigen that has elevated expression on tumors but is broadly expressed on normal tissues, coupled to a functional CAR targeting a tumor-specific antigen such as TAG-72 to create more effective dual specific CAR-T cells. This approach could potentially overcome tumor resistance elicited by tumor antigen downregulation or varying levels of antigen expression to improve the clinical outcome of CAR-T cell therapy for solid tumors, while restricting destruction of normal cells and tissues.
Materials and methods
Cell lines
All cell lines were acquired from the American Type Culture Collection (ATCC, Manassas, VA, USA) and maintained using recommended culture conditions. Ovarian cancer cell lines were routinely maintained where OVCAR-3 (HTB-161) was maintained in RPMI 1640 (Sigma-Aldrich, St. Louis, MO, USA) supplemented with 20% (v/v) fetal bovine serum (FBS, Bovogen, Keilor East, VIC, Australia), 0.01 mg/mL bovine insulin (Sigma-Aldrich, St. Louis, MO, USA), and 1× penicillin-streptomycin (1× Pen/Strep; Gibco, Waltham, MA, USA). The ovarian cancer cell line derived from ascites MESOV (CRL-3272, herein referred to as MESOV) was cultured in McCoy’s 5A medium (modified) (Gibco, Waltham, MA, USA) containing 10% (v/v) FBS and 1× Pen/Strep. The ovarian cancer cell line Caov-3 (HTB-75) was maintained in Dulbecco’s modified Eagle’s medium (Gibco, Waltham, MA, USA) supplemented with 10% (v/v) FBS and 1× Pen/Strep. Caov-4 (HTB-76) was maintained in Leibovitz’s L-15 medium (ATCC, Manassas, VA, USA) supplemented with 20% (v/v) FBS. The OV-90 (CRL-11732) cell line was maintained in a 1:1 mixture of MCDB-105 medium (Sigma-Aldrich, St. Louis, MO, USA) and medium 199 (Life Technologies, Carlsbad, CA, USA) supplemented with 15% (v/v) FBS and 1× Pen/Strep. LS174T (Dukes’ type B, colorectal adenocarcinoma, CL-188) cells were maintained in Eagle’s minimum essential medium (Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% (v/v) FBS and 1× Pen/Strep. Finally, the ovarian cancer cell line OVCAR-429, the Burkitt’s lymphoma cell line Raji (CCL-86), and the T cell leukemia line Jurkat (TIB-152) were maintained in RPMI 1640 (Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% (v/v) FBS and 1× Pen/Strep. With the exception of Caov-4, which was maintained in 100% air at 37°C, all cell lines were maintained in 5% CO2 at 37°C.
The human primary uterine (HUF, PCS-460-010) and lung (HLF, PCS-201-013) fibroblast cell lines were cultured in fibroblast basal medium supplemented with fibroblast growth kit components (ATCC, Manassas, VA, USA) comprised of 5 ng/mL recombinant human basic fibroblast growth factor β (rhFGFβ), 7.5 mM l-glutamine, 50 μg/mL ascorbic acid, 1 μg/mL hydrocortisone hemisuccinate, 5 μg/mL recombinant human-insulin, and 2% (v/v) FBS.
DNA constructs and lentivirus production
CAR constructs have been developed with an scFv for either TAG-7237 or CD4737,42 as described in Boyd et al.37 Following a conventional human secretion signal leader, both scFvs were constructed in a VH-linker-VL orientation with a 15-residue (Gly4Ser ×3) linker. The CAR constructs used either human CD8 or CD28 as hinge or TM regions, and used CD28, 4-1BB, and CD3ζ cytoplasmic signaling domains. The P2A sequence, a signal sequence directing proteolytic cleavage, was used to direct bicistronic expression of EGFP or the CD47 CAR. The second-generation lentiviral packaging system was used to produce the lentiviral vectors for CAR transduction. Briefly, 293T cells were plated onto poly-l-lysine-coated tissue culture plates (Sigma-Aldrich, St. Louis, MO, USA). The lentiviral transfer vector DNA, together with packaging and envelope plasmid DNA, was transfected with Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). Viral supernatant was collected after 48 h and cleared by centrifugation followed by 0.45-μm filtration (Millipore, Burlington, MA, USA). Concentration of lentivirus using ultracentrifugation was performed with a SORVALL Discovery 100 SE centrifuge (90 min at 20,000 × g). Virus pellets were resuspended in PBS and stored at −80°C until use.
CAR-T cell production
Primary human T cells were isolated from healthy human donors either from fresh whole blood or from buffy coats obtained from the Australian Red Cross Lifeblood. All healthy donors provided informed consent. PBMCs were isolated by Ficoll-Paque (GE Healthcare, IL, USA)-mediated centrifugation using Leucosep tubes (Greiner Bio-One, Kremsmünster, Austria) as per the manufacturer’s instructions. PBMCs were cryopreserved prior to use. For use in transductions, PBMCs were thawed and isolated using Dynabeads human T-expander CD3/CD28 beads (Thermo Fisher Scientific, Waltham, MA, USA) and subsequently activated for 72 h at a bead-to-cell ratio of 3:1 in the presence of 200 U/mL IL-2 (Miltenyi Biotec, Bergisch Gladbach, Germany). Following activation, bead-free T cells were incubated with lentiviral particles in RetroNectin (Takara Bio, Kusatsu, Japan)-coated plates for 48 h at a multiplicity of infection (MOI) of 50. T cell cultures were transferred to complete T cell expansion media comprising of IL-2, IL-7, IL-15, IL-21 (Miltenyi Biotec, Bergisch Gladbach, Germany), human AB (hAb) serum (Sigma-Aldrich, St. Louis, MO, USA) and Stemulate (Cook Regentec, Indianapolis, IN, USA) in TexMACS (Miltenyi Biotec, Bergisch Gladbach, Germany) for continued expansion. Activated but non-transduced T cells (T cells [no CAR]) were maintained in parallel for all donors analyzed.
Flow cytometry
Transduction efficiency was evaluated by flow cytometry using either GFP or goat anti-mouse F(ab′)2 allophycocyanin (APC, Abacus dx, Meadowbrook, QLD, Australia) or anti-FLAG M2-fluorescein isothiocyanate (FITC, Sigma-Aldrich, St. Louis, MO, USA). Frequency of T cell subsets was determined by staining for CD3 phycoerythrin (PE, clone REA613), CD4 VioBlue (clone VIT4), CD8 VioGreen (clone BW135-80), CCR7 PE-Vio770 (clone REA675), and CD45RO APC (clone REA611). T cell activation was characterized by staining for histocompatibility leukocyte antigen (HLA)-DR PE-Vio615 (clone REA805) and CD137 APC (clone REA765). Antigen expression on target cell lines OVCAR-3, MESOV, HUF, and HLF was characterized using the Alexa Fluor 488-conjugated anti-TAG-72 diabody (Avipep, Parkville, VIC, Australia) or CD47 antibody (clone REA220). All antibodies were acquired from Miltenyi Biotec (Bergisch Gladbach, Germany) unless otherwise stated. Staining was performed at 4°C for 15–30 min. Cells were then washed with 3% (v/v) FBS in PBS. Either Viobility 405/520 dye (Miltenyi Biotec, Bergisch Gladbach, Germany) or propidium iodide (PI, Sigma-Aldrich, St. Louis, MO, USA) was used to select for viable cells. Data were acquired using the MACSQuant analyzer 10 (Miltenyi Biotec, Bergisch Gladbach, Germany), and analysis was subsequently performed using FlowLogic (Inivai Technologies, Mentone, VIC, Australia).
Fluorescent-activated cell sorting
CAR-positive cells were isolated following 10–13 days in culture using either GFP or F(ab′)2-APC or FLAG-FITC as a reporter for CAR expression. Viable cells were selected for using Viobility 405/452 dye (Miltenyi Biotec, Bergisch Gladbach, Germany). Staining was performed at 4°C for 15 min in 3% (v/v) FBS in PBS. Samples were washed once before transferring to Tyto buffer (Miltenyi Biotec, Bergisch Gladbach, Germany). Cell sorting was performed using the MACSQuant Tyto (Miltenyi Biotec, Bergisch Gladbach, Germany).
CD4+ and CD8+ CAR-T cells were isolated following at least 13 days in culture. T cell subsets from matched T cells (no CAR) were isolated in parallel. Briefly, cells were resuspended in 3% (v/v) FBS in PBS and incubated with primary antibodies as previously described. Sorts were performed using the FACSAria Fusion droplet sorter (BD Biosciences, San Jose, CA, USA). Resultant cells were maintained in complete T cell expansion media until required.
In vitro T cell cytotoxicity assay
The real-time cell analysis instrument xCELLigence (ACEA Biosciences, San Diego, CA, USA) was utilized for the assessment of T cell function in vitro. Before use, effector cells were transferred to fresh T cell expansion media or activation media comprising 200 U/mL IL-2 or recovery media comprising 5 ng/mL IL-7 in basal media for 12–24 h before use. In this instance, basal media refer to TexMACS supplemented with 5% (v/v) hAb serum. T cells were co-cultured with cancer cell lines or healthy primary fibroblast lines at an E:T ratio of either 5:1, 1:1, or 1:5 unless otherwise stated. Target cells were plated for 6–24 h in 96-well electronic microtiter plates (ACEA Biosciences, San Diego, CA, USA) before addition of sorted effector cells. Cell impedance was monitored at 15-min intervals during 20 h from the addition of effector cells. All data were normalized to the time of addition of effector cells unless otherwise stated and are presented herein as the arbitrary unit normalized cell index (CI). CAR-T cell function was calculated as % cytotoxicity = [(normalized CItarget cells alone – normalized CItest) ÷ normalized CItarget cells alone] × 100.
Cytokine array
Supernatant samples from T cell functional assays were retained following 20-h co-culture and frozen at −80°C until required for further use. Supernatants were subsequently analyzed for secreted human IL-1β, IL-1Ra, IL-2, IL-4, IL-5, IL-6, IL-9, IL-10, IL-13, IL-17A, GM-CSF, IFN-γ, MIP-1α, MIP-1β, and TNF-α according to the Bio-Plex Pro assay manufacturer’s instructions (Bio-Rad, Hercules, CA, USA). Plates were read using the Bio-Plex MAGPIX system (Bio-Rad, Hercules, CA, USA). Data were acquired using Bio-Plex Manager MP software and processed using Bio-Plex Data Pro Plus (both Bio-Rad, Hercules, CA, USA).
In vivo tumor studies
All animal experiments were pre-approved by the Monash Medical Centre Animal Ethics Committee (MMCA/2016/61, MMCA/2018/04), and all procedures followed the National Health and Medical Research Council of Australia guidelines for the use and care of experimental animals. In vivo models were established using female 6- to 12-week-old NSG mice either purchased from Australian BioResources or bred in in-house colonies (Monash Animal Research Platform). OVCAR-3 (1 × 107) and MESOV (5 × 104) cells were prepared in 100 μL of PBS combined with an equal volume of Matrigel (Corning Life Sciences, NY, USA) and injected s.c. into the back flank. OVCAR-3 tumors were allowed to reach at least 100 mm3 in size before treatment was commenced. MESOV tumors are rapidly growing, and therefore treatment commenced once a tumor mass was palpable. Mice were randomized into experimental groups (four mice/group). Three injections of 2 × 106 CAR-positive T cells/injection were administered intravenously (i.v.) at 5-day intervals. Control mice received T cells (no CAR) at comparable dosages. Tumor volume and clinical parameters of animal health were monitored regularly until the experimental endpoint (day 28 following initial CAR treatment). Tumors were measured using digital calipers, and volumes were calculated using (width2 × length)/2 (mm3). At the end of experiments, mice were euthanized by CO2 inhalation.
Western blot
Transduced T cells were lysed in radioimmunoprecipitation assay (RIPA) buffer (Sigma-Aldrich, St. Louis, MO, USA) with protease inhibitor cocktail (Roche, Basel, Switzerland) on ice for 10 min. After centrifuging at 10,000 × g for 10 min at 4°C, supernatants were incubated at 70°C in 4× loading buffer under non-reducing or reducing (in the presence of β-mercaptoethanol) conditions. Samples were separated by Mini-PROTEAN TGX precast gels (Bio-Rad, Hercules, CA, USA) and transferred to a polyvinylidene fluoride (PVDF) membrane (Millipore, Burlington, MA, USA) using the Mini-PROTEAN Tetra cell system (Bio-Rad, Hercules, CA, USA) according to the manufacturer’s instructions. Antibodies targeting c-Myc (Sigma-Aldrich, St. Louis, MO, USA) and CD3ζ (BD Biosciences, San Jose, CA, USA) were used as probes to detect ΔCD47 and TAG-72 CAR proteins respectively. Immune complexes were detected using IRDye secondary antibodies and the Odyssey imaging system (LI-COR Biosciences).
Statistical analysis
Data represent mean ± SD from at least three biological replicates unless otherwise stated. GraphPad Prism 8.0 was used to perform statistical analysis throughout. Results were analyzed using an unpaired Welch’s t test (two-tailed) or Mann-Whitney t test for two group comparisons. A multiple t test with the Holm-Sidak method for determining statistical significance was implemented for multi-group comparisons. One-way ANOVA analysis was implemented for multi-group analysis with the Brown-Forsythe test applied to datasets with unequal variance. Statistical significance was defined as p ≤ 0.05 (∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001, ∗∗∗∗p ≤ 0.0001).
Acknowledgments
This work was supported by Cartherics Pty, Ltd, Australia. The authors acknowledge the facilities and scientific and technical assistance of the Monash Histology Platform, Department of Anatomy and Developmental Biology, Monash University and the MHTP Cell Therapy and Regenerative Medicine Platform. The authors acknowledge FlowCore for sorting of cells using FACS. We thank staff members of the Monash Medical Centre Animal Facility. We also thank Drs. Michael Kershaw and Irvine Weissman for scientific feedback. Additionally, the authors thank Dr. Thao Nguyen and Callum Docherty for technical support. The graphical abstract was created with BioRender.com.
Author contributions
Conceptualization and design, V.J.E., R.S., P.J.H., M.C.H., A.O.T., and R.L.B.; development of methodology, V.J.E., R.S., and M.V.H.; acquisition of data, V.J.E., R.S., M.V.H., N.N.N., and J.Z.; analysis and interpretation of data, V.J.E., R.S., and R.L.B.; writing, review, and/or revision of the manuscript, V.J.E., R.S., N.N.N., M.C.H., A.P., A.O.T., and R.L.B.; study supervision, A.O.T. and R.L.B.
Declaration of interests
The research described in this paper was funded by Cartherics Pty Ltd. All authors are paid employees or advisors of Cartherics, and A.O.T., R.L.B., and P.J.H. hold equity in the company. Additionally, P.J.H. is a consultant to Avipep Pty, Ltd. and a Director of IsoClide Medical Pty, Ltd., whose products are entirely unrelated to Cartherics.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omto.2021.01.002.
Contributor Information
Runzhe Shu, Email: runzhe.shu@hudson.org.au.
Vera J. Evtimov, Email: vera.evtimov@hudson.org.au.
Supplemental information
References
- 1.Sadelain M., Rivière I., Riddell S. Therapeutic T cell engineering. Nature. 2017;545:423–431. doi: 10.1038/nature22395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang M., Munoz J., Goy A., Locke F.L., Jacobson C.A., Hill B.T., Timmerman J.M., Holmes H., Jaglowski S., Flinn I.W. KTE-X19 CAR T-cell therapy in relapsed or refractory mantle-cell lymphoma. n. engl. j. med. 2020;382:1331–1342. doi: 10.1056/NEJMoa1914347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.newick k., o’brien s., moon e., Albelda S.M. CAR T cell therapy for solid tumors. Annu. Rev. Med. 2017;68:139–152. doi: 10.1146/annurev-med-062315-120245. [DOI] [PubMed] [Google Scholar]
- 4.Martinez M., Moon E.K. CAR T cells for solid tumors: new strategies for finding, infiltrating, and surviving in the tumor microenvironment. Front. Immunol. 2019;10:128. doi: 10.3389/fimmu.2019.00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Torre L.A., Trabert B., DeSantis C.E., Miller K.D., Samimi G., Runowicz C.D., Gaudet M.M., Jemal A., Siegel R.L. Ovarian cancer statistics, 2018. CA Cancer J. Clin. 2018;68:284–296. doi: 10.3322/caac.21456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haas A.R., Tanyi J.L., O’Hara M.H., Gladney W.L., Lacey S.F., Torigian D.A., Soulen M.C., Tian L., McGarvey M., Nelson A.M. Phase I study of lentiviral-transduced chimeric antigen receptor-modified T cells recognizing mesothelin in advanced solid cancers. Mol. Ther. 2019;27:1919–1929. doi: 10.1016/j.ymthe.2019.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murad J.P., Kozlowska A.K., Lee H.J., Ramamurthy M., Chang W.C., Yazaki P., Colcher D., Shively J., Cristea M., Forman S.J., Priceman S.J. Effective targeting of TAG72+ peritoneal ovarian tumors via regional delivery of CAR-engineered T cells. Front. Immunol. 2018;9:2268. doi: 10.3389/fimmu.2018.02268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhu X., Cai H., Zhao L., Ning L., Lang J. CAR-T cell therapy in ovarian cancer: from the bench to the bedside. Oncotarget. 2017;8:64607–64621. doi: 10.18632/oncotarget.19929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ritchie D.S., Neeson P.J., Khot A., Peinert S., Tai T., Tainton K., Chen K., Shin M., Wall D.M., Hönemann D. Persistence and efficacy of second generation CAR T cell against the LeY antigen in acute myeloid leukemia. Mol. Ther. 2013;21:2122–2129. doi: 10.1038/mt.2013.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thor A., Viglione M.J., Muraro R., Ohuchi N., Schlom J., Gorstein F. Monoclonal antibody B72.3 reactivity with human endometrium: a study of normal and malignant tissues. Int. J. Gynecol. Pathol. 1987;6:235–247. doi: 10.1097/00004347-198709000-00005. [DOI] [PubMed] [Google Scholar]
- 11.Johnson V.G., Schlom J., Paterson A.J., Bennett J., Magnani J.L., Colcher D. Analysis of a human tumor-associated glycoprotein (TAG-72) identified by monoclonal antibody B72.3. Cancer Res. 1986;46:850–857. [PubMed] [Google Scholar]
- 12.Genega E.M., Hutchinson B., Reuter V.E., Gaudin P.B. Immunophenotype of high-grade prostatic adenocarcinoma and urothelial carcinoma. Mod. Pathol. 2000;13:1186–1191. doi: 10.1038/modpathol.3880220. [DOI] [PubMed] [Google Scholar]
- 13.Hege K.M., Bergsland E.K., Fisher G.A., Nemunaitis J.J., Warren R.S., McArthur J.G., Lin A.A., Schlom J., June C.H., Sherwin S.A. Safety, tumor trafficking and immunogenicity of chimeric antigen receptor (CAR)-T cells specific for TAG-72 in colorectal cancer. J. Immunother. Cancer. 2017;5:22. doi: 10.1186/s40425-017-0222-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chauhan S.C., Vinayek N., Maher D.M., Bell M.C., Dunham K.A., Koch M.D., Lio Y., Jaggi M. Combined staining of TAG-72, MUC1, and CA125 improves labeling sensitivity in ovarian cancer: antigens for multi-targeted antibody-guided therapy. J. Histochem. Cytochem. 2007;55:867–875. doi: 10.1369/jhc.7A7213.2007. [DOI] [PubMed] [Google Scholar]
- 15.Ponnusamy M.P., Venkatraman G., Singh A.P., Chauhan S.C., Johansson S.L., Jain M., Smith L., Davis J.S., Remmenga S.W., Batra S.K. Expression of TAG-72 in ovarian cancer and its correlation with tumor stage and patient prognosis. Cancer Lett. 2007;251:247–257. doi: 10.1016/j.canlet.2006.11.025. [DOI] [PubMed] [Google Scholar]
- 16.Nagle J.A., Wilbur D.C., Pitman M.B. Cytomorphology of gastric and duodenal epithelium and reactivity to B72.3: a baseline for comparison to pancreatic lesions aspirated by EUS-FNAB. Diagn. Cytopathol. 2005;33:381–386. doi: 10.1002/dc.20343. [DOI] [PubMed] [Google Scholar]
- 17.Xu M., Real F.X., Welt S., Schüssler M.H., Oettgen H.F., Old L.J. Expression of TAG-72 in normal colon, transitional mucosa, and colon cancer. Int. J. Cancer. 1989;44:985–989. doi: 10.1002/ijc.2910440607. [DOI] [PubMed] [Google Scholar]
- 18.Scott A.M., Akhurst T., Lee F.-T., Ciprotti M., Davis I.D., Weickhardt A.J., Gan H.K., Hicks R.J., Lee S.T., Kocovski P. First clinical study of a pegylated diabody 124I-labeled PEG-AVP0458 in patients with tumor-associated glycoprotein 72 positive cancers. Theranostics. 2020;10:11404–11415. doi: 10.7150/thno.49422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rosenblum M.G., Verschraegen C.F., Murray J.L., Kudelka A.P., Gano J., Cheung L., Kavanagh J.J. Phase I study of 90Y-labeled B72.3 intraperitoneal administration in patients with ovarian cancer: effect of dose and EDTA coadministration on pharmacokinetics and toxicity. Clin. Cancer Res. 1999;5:953–961. [PubMed] [Google Scholar]
- 20.Majzner R.G., Mackall C.L. Tumor antigen escape from CAR T-cell therapy. Cancer Discov. 2018;8:1219–1226. doi: 10.1158/2159-8290.CD-18-0442. [DOI] [PubMed] [Google Scholar]
- 21.Li Y., Lu S., Xu Y., Qiu C., Jin C., Wang Y., Liu Z., Kong B. Overexpression of CD47 predicts poor prognosis and promotes cancer cell invasion in high-grade serous ovarian carcinoma. Am. J. Transl. Res. 2017;9:2901–2910. [PMC free article] [PubMed] [Google Scholar]
- 22.Liu R., Wei H., Gao P., Yu H., Wang K., Fu Z., Ju B., Zhao M., Dong S., Li Z. CD47 promotes ovarian cancer progression by inhibiting macrophage phagocytosis. Oncotarget. 2017;8:39021–39032. doi: 10.18632/oncotarget.16547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang C.L., Lin M.J., Hsu C.Y., Lin H.Y., Tsai H.P., Long C.Y., Tsai E.M., Hsieh T.H., Wu C.H. CD47 promotes cell growth and motility in epithelial ovarian cancer. Biomed. Pharmacother. 2019;119:109105. doi: 10.1016/j.biopha.2019.109105. [DOI] [PubMed] [Google Scholar]
- 24.Brightwell R.M., Grzankowski K.S., Lele S., Eng K., Arshad M., Chen H., Odunsi K. The CD47 “don’t eat me signal” is highly expressed in human ovarian cancer. Gynecol. Oncol. 2016;143:393–397. doi: 10.1016/j.ygyno.2016.08.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jaiswal S., Jamieson C.H., Pang W.W., Park C.Y., Chao M.P., Majeti R., Traver D., van Rooijen N., Weissman I.L. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell. 2009;138:271–285. doi: 10.1016/j.cell.2009.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Majeti R., Chao M.P., Alizadeh A.A., Pang W.W., Jaiswal S., Gibbs K.D., Jr., van Rooijen N., Weissman I.L. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell. 2009;138:286–299. doi: 10.1016/j.cell.2009.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Willingham S.B., Volkmer J.P., Gentles A.J., Sahoo D., Dalerba P., Mitra S.S., Wang J., Contreras-Trujillo H., Martin R., Cohen J.D. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc. Natl. Acad. Sci. USA. 2012;109:6662–6667. doi: 10.1073/pnas.1121623109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Advani R., Flinn I., Popplewell L., Forero A., Bartlett N.L., Ghosh N., Kline J., Roschewski M., LaCasce A., Collins G.P. CD47 blockade by Hu5F9-G4 and rituximab in non-Hodgkin’s lymphoma. N. Engl. J. Med. 2018;379:1711–1721. doi: 10.1056/NEJMoa1807315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sallman D.A., Donnellan W.B., Asch A.S., Lee D.J., Malki M.A., Marcucci G., Pollyea D.A., Kambhampati S., Komrokji R.S., Elk J.V. The first-in-class anti-CD47 antibody Hu5F9-G4 is active and well tolerated alone or with azacitidine in AML and MDS patients: initial phase 1b results. J. Clin. Oncol. 2019;37(15, Suppl):7009. [Google Scholar]
- 30.Ingram J.R., Blomberg O.S., Sockolosky J.T., Ali L., Schmidt F.I., Pishesha N., Espinosa C., Dougan S.K., Garcia K.C., Ploegh H.L., Dougan M. Localized CD47 blockade enhances immunotherapy for murine melanoma. Proc. Natl. Acad. Sci. USA. 2017;114:10184–10189. doi: 10.1073/pnas.1710776114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Golubovskaya V., Berahovich R., Zhou H., Xu S., Harto H., Li L., Chao C.C., Mao M.M., Wu L. CD47-CAR-T cells effectively kill target cancer cells and block pancreatic tumor growth. Cancers (Basel) 2017;9:139. doi: 10.3390/cancers9100139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oldenborg P.A. CD47: a cell surface glycoprotein which regulates multiple functions of hematopoietic cells in health and disease. ISRN Hematol. 2013;2013:614619. doi: 10.1155/2013/614619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shen S., Forero A., Meredith R.F., LoBuglio A.F. Biodistribution and dosimetry of In-111/Y-90-HuCC49ΔCh2 (IDEC-159) in patients with metastatic colorectal adenocarcinoma. Cancer Biother. Radiopharm. 2011;26:127–133. doi: 10.1089/cbr.2010.0864. [DOI] [PubMed] [Google Scholar]
- 34.Muraro R., Kuroki M., Wunderlich D., Poole D.J., Colcher D., Thor A., Greiner J.W., Simpson J.F., Molinolo A., Noguchi P. Generation and characterization of B72.3 second generation monoclonal antibodies reactive with the tumor-associated glycoprotein 72 antigen. Cancer Res. 1988;48:4588–4596. [PubMed] [Google Scholar]
- 35.Pavlinkova G., Booth B.J.M., Batra S.K., Colcher D. Radioimmunotherapy of human colon cancer xenografts using a dimeric single-chain Fv antibody construct. Clin. Cancer Res. 1999;5:2613–2619. [PubMed] [Google Scholar]
- 36.Kashmiri S.V., Shu L., Padlan E.A., Milenic D.E., Schlom J., Hand P.H. Generation, characterization, and in vivo studies of humanized anticarcinoma antibody CC49. Hybridoma. 1995;14:461–473. doi: 10.1089/hyb.1995.14.461. [DOI] [PubMed] [Google Scholar]
- 37.Boyd R., Trounson A., Kawamoto H. 2017. Genetically modified cells and uses thereof. Australian patent WO2017/088012. First filings/priority dates: 27 November 2015, 11 April 2016 International filing date: 23 November 2016 Publication of the international patent application: 1 June 2017. [Google Scholar]
- 38.Kelly F.J., Miller C.R., Buchsbaum D.J., Gomez-Navarro J., Barnes M.N., Alvarez R.D., Curiel D.T. Selectivity of TAG-72-targeted adenovirus gene transfer to primary ovarian carcinoma cells versus autologous mesothelial cells in vitro. Clin. Cancer Res. 2000;6:4323–4333. [PubMed] [Google Scholar]
- 39.Salter A.I., Ivey R.G., Kennedy J.J., Voillet V., Rajan A., Alderman E.J., Voytovich U.J., Lin C., Sommermeyer D., Liu L. Phosphoproteomic analysis of chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function. Sci. Signal. 2018;11:eaat6753. doi: 10.1126/scisignal.aat6753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ying Z., He T., Wang X., Zheng W., Lin N., Tu M., Xie Y., Ping L., Zhang C., Liu W. Parallel comparison of 4-1BB or CD28 co-stimulated CD19-targeted CAR-T cells for B cell non-Hodgkin’s lymphoma. Mol. Ther. Oncolytics. 2019;15:60–68. doi: 10.1016/j.omto.2019.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oldenborg P.A., Zheleznyak A., Fang Y.F., Lagenaur C.F., Gresham H.D., Lindberg F.P. Role of CD47 as a marker of self on red blood cells. Science. 2000;288:2051–2054. doi: 10.1126/science.288.5473.2051. [DOI] [PubMed] [Google Scholar]
- 42.Liu J., Wang L., Zhao F., Tseng S., Narayanan C., Shura L., Willingham S., Howard M., Prohaska S., Volkmer J. Pre-clinical development of a humanized anti-CD47 antibody with anti-cancer therapeutic potential. PLoS ONE. 2015;10:e0137345. doi: 10.1371/journal.pone.0137345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lazar-Molnar E., Almo S.C., Nathenson S.G. The interchain disulfide linkage is not a prerequisite but enhances CD28 costimulatory function. Cell. Immunol. 2006;244:125–129. doi: 10.1016/j.cellimm.2007.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gacerez A.T., Arellano B., Sentman C.L. How chimeric antigen receptor design affects adoptive T cell therapy. J. Cell. Physiol. 2016;231:2590–2598. doi: 10.1002/jcp.25419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu B., Yan L., Zhou M. Target selection of CAR T cell therapy in accordance with the TME for solid tumors. Am. J. Cancer Res. 2019;9:228–241. [PMC free article] [PubMed] [Google Scholar]
- 46.Junghans R.P. Is it safer CARs that we need, or safer rules of the road? Mol. Ther. 2010;18:1742–1743. doi: 10.1038/mt.2010.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brentjens R., Yeh R., Bernal Y., Riviere I., Sadelain M. Treatment of chronic lymphocytic leukemia with genetically targeted autologous T cells: case report of an unforeseen adverse event in a phase I clinical trial. Mol. Ther. 2010;18:666–668. doi: 10.1038/mt.2010.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ruella M., Barrett D.M., Kenderian S.S., Shestova O., Hofmann T.J., Perazzelli J., Klichinsky M., Aikawa V., Nazimuddin F., Kozlowski M. Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. J. Clin. Invest. 2016;126:3814–3826. doi: 10.1172/JCI87366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hegde M., Mukherjee M., Grada Z., Pignata A., Landi D., Navai S.A., Wakefield A., Fousek K., Bielamowicz K., Chow K.K. Tandem CAR T cells targeting HER2 and IL13Rα2 mitigate tumor antigen escape. J. Clin. Invest. 2016;126:3036–3052. doi: 10.1172/JCI83416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kloss C.C., Condomines M., Cartellieri M., Bachmann M., Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat. Biotechnol. 2013;31:71–75. doi: 10.1038/nbt.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wilkie S., van Schalkwyk M.C., Hobbs S., Davies D.M., van der Stegen S.J., Pereira A.C., Burbridge S.E., Box C., Eccles S.A., Maher J. Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J. Clin. Immunol. 2012;32:1059–1070. doi: 10.1007/s10875-012-9689-9. [DOI] [PubMed] [Google Scholar]
- 52.Sikic B.I., Lakhani N., Patnaik A., Shah S.A., Chandana S.R., Rasco D., Colevas A.D., O’Rourke T., Narayanan S., Papadopoulos K. First-in-human, first-in-class phase I trial of the anti-CD47 antibody Hu5F9-G4 in patients with advanced cancers. J. Clin. Oncol. 2019;37:946–953. doi: 10.1200/JCO.18.02018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Romeo C., Amiot M., Seed B. Sequence requirements for induction of cytolysis by the T cell antigen/Fc receptor ζ chain. Cell. 1992;68:889–897. doi: 10.1016/0092-8674(92)90032-8. [DOI] [PubMed] [Google Scholar]
- 54.Leddon S.A., Fettis M.M., Abramo K., Kelly R., Oleksyn D., Miller J. The CD28 transmembrane domain contains an essential dimerization motif. Front. Immunol. 2020;11:1519. doi: 10.3389/fimmu.2020.01519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nicholson R.I., Gee J.M., Harper M.E. EGFR and cancer prognosis. Eur. J. Cancer. 2001;37(Suppl 4):S9–S15. doi: 10.1016/s0959-8049(01)00231-3. [DOI] [PubMed] [Google Scholar]
- 56.Weichert W., Knösel T., Bellach J., Dietel M., Kristiansen G. ALCAM/CD166 is overexpressed in colorectal carcinoma and correlates with shortened patient survival. J. Clin. Pathol. 2004;57:1160–1164. doi: 10.1136/jcp.2004.016238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shen Y., Li X., Dong D., Zhang B., Xue Y., Shang P. Transferrin receptor 1 in cancer: a new sight for cancer therapy. Am. J. Cancer Res. 2018;8:916–931. [PMC free article] [PubMed] [Google Scholar]
- 58.Rafiq S., Yeku O.O., Jackson H.J., Purdon T.J., van Leeuwen D.G., Drakes D.J., Song M., Miele M.M., Li Z., Wang P. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat. Biotechnol. 2018;36:847–856. doi: 10.1038/nbt.4195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wiersma V.R., van Bommel P.E., de Bruyn M., Helfrich W., Bremer E. CD47, a multi-facetted target for cancer immunotherapy. Atlas Genet. Cytogenet. Oncol. Haematol. 2015;19:417–431. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.