

Summary
Celastrol, a compound extracted from traditional Chinese medicine, has been reported as a potent anti-obesity agent with controversial mechanisms. Here both C57BL/6J and leptin-deficient (ob/ob) mice fed a high-fat diet (HFD) displayed body weight loss after celastrol therapy, opposing the previous viewpoint that celastrol improves obesity by sensitizing leptin signaling. More importantly, celastrol downregulated lipid transporters in the intestine, increased lipid excretion in feces, and reduced body weight gain in HFD mice. Meanwhile, analysis of gut microbiota revealed that celastrol altered the gut microbiota composition in HFD-fed mice, and modulating gut microbiota by antibiotics or fecal microbiota transplantation blocked the celastrol effect on intestinal lipid transport and body weight gain, suggesting a critical role of the gut microbiota composition in mediating the anti-obesity role of celastrol under HFD. Together, the findings revealed that celastrol reduces intestinal lipid absorption to antagonize obesity by resetting the gut microbiota profile under HFD feeding.
Subject areas: Biological Sciences, Endocrinology, Microbiology, Microbiome Physiology
Graphical abstract
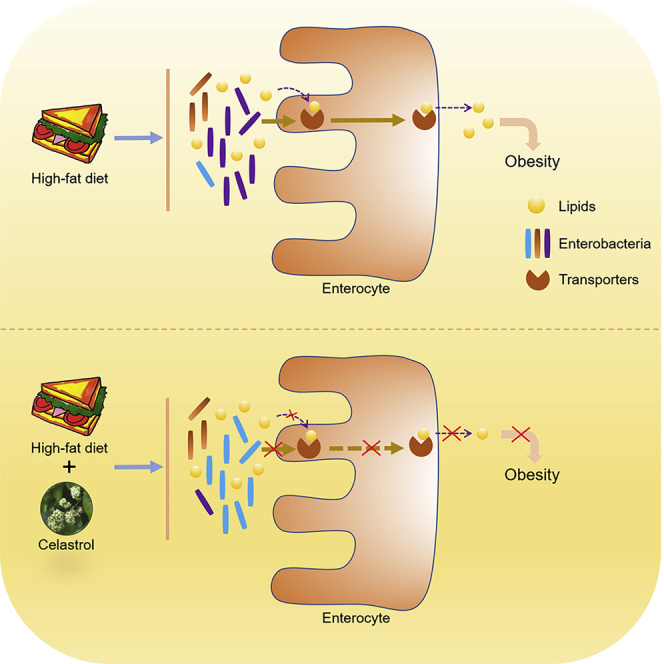
Highlights
-
•
Celastrol reduced intestinal lipid transporters and lipids absorption
-
•
Celastrol reset gut microbiota profile to modulate intestinal lipid transport
-
•
Celastrol attenuated obesity in leptin-deficient (ob/ob) mice fed high fat diet
Biological Sciences; Endocrinology; Microbiology; Microbiome Physiology
Introduction
In recent decades, changes in the amount and composition of dietary fat have led to the current prevalence of obesity worldwide (Collaborators et al., 2017; Cordain et al., 2005). Obesity is a major risk factor for type 2 diabetes, cardiovascular disease, high blood pressure, nonalcoholic steatohepatitis, and even cancers (Haslam and James, 2005; Olshansky et al., 2005). Although some weight-loss drugs have been developed, they often do not work well or have serious side effects (Padwal and Majumdar, 2007). Therefore, it is urgent to develop new drugs for the treatment of obesity and its complications.
Celastrol (also known as tripterine), a compound isolated from Tripterygium wilfordii or other members of the Celastraceae family, has been reported to have antiinflammatory and anticancer effects (Allison et al., 2001; Kashyap et al., 2018; Lee et al., 2006; Pang et al., 2010). In recent years, celastrol has been proven to have a strong anti-obesity effect and has the potential to be an anti-obesity drug (Chellappa et al., 2019; Feng et al., 2019; Kyriakou et al., 2018; Liu et al., 2015; Ma et al., 2015; Pfuhlmann et al., 2018; Saito et al., 2019; Wang et al., 2014). The possible mechanisms of its weight-loss effect have also been reported. Liu and colleagues reported that celastrol can act as a leptin sensitizer to reduce body weight (BW) in diet-induced obese (DIO) mice by reducing hypothalamic ER stress and thereby inhibiting food intake, as evidenced by studies from wild-type and leptin-deficient (ob/ob)/leptin-receptor-deficient (db/db) mouse models (Liu et al., 2015). Later, several possible targets of celastrol, including interleukin-1 receptor 1 (IL1R1), tyrosine phosphatase (PTP) 1B (PTP1B), and T cell PTP (TCPTP), in the hypothalamus were reported in subsequent studies (Feng et al., 2019; Kyriakou et al., 2018). In addition, altered adipocyte differentiation and increased lipolysis (Choi et al., 2016; Hong et al., 2018), increased expression of liver Sirt1 (Zhang et al., 2017), induced interactions of Nur77 and TRAF2 to alleviate inflammation (Hu et al., 2017), and increased energy expenditure by increasing muscle and brown adipose tissue (BAT) thermogenesis and inguinal fat depot (iWAT) browning (Ma et al., 2015) were also reported as potential mechanisms of the anti-obesity effect of celastrol. However, the data from Katrin Pfuhlmann et al. showed that global PTP1B KO and wild-type (WT) mice and uncoupling protein 1 (UCP1) WT and KO mice also have comparable weight loss after high-fat diet challenge upon celastrol treatment. In addition, they reported that celastrol had little impact on hypothalamic ER stress and hypothalamic inflammation (Pfuhlmann et al., 2018). Moreover, the data from Kenji Saito and colleagues showed that the anti-obesity effect of celastrol is independent of melanocortin 4 receptor (MC4R), a G-protein-coupled receptor involved in inhibiting food intake and lowering BW (Saito et al., 2019). These results are discordant with previously proposed underlying mechanisms. Thus, more investigations on the mechanism of celastrol in antagonizing obesity are required.
It is well known that there are many factors affecting diet-induced obesity, including food intake (Liu et al., 2015), nutrient absorption through the gastrointestinal tract (Li et al., 2016), the composition of intestinal microorganisms (Martinez-Guryn et al., 2018), lipid deposition and lipolysis (Wei et al., 2019), and energy expenditure (Sidsosis et al., 2015). In our present study, both in vivo and in vitro data did not support that celastrol exerts its weight loss function by regulating energy expenditure. However, we found a striking phenotype that the intestinal lipid excretion of high-fat diet (HFD)-fed mice was increased by celastrol, accompanied by changes in intestinal microbial composition and decreased expression of transporters related to intestinal lipid absorption, suggesting that celastrol may achieve its weight loss effect through regulation of the intestinal microbial profile and intestinal lipid absorption, which reveals a mechanism of celastrol in the treatment of obesity.
Results
Celastrol protects C57BL/6 mice from HFD-induced obesity
We first confirmed the effect of celastrol as a weight loss agent. In our study, HFD-fed C57BL/6 mice with an average BW of 46 g were orally administered 3 mg/kg/day celastrol by mixing the drug into high-fat chow for 21 days. Consistent with the reported phenotype, the mice in the experimental group lost approximately 22% of BW compared with the control group (Figures 1A and 1B). The size and weight of the epididymal fat depots (eWAT), inguinal fat depots (iWAT), perirenal fat depots (pWAT) and BAT, as well as the size of adipocytes of eWAT and BAT in the celastrol intervention group were less than those in the control group (Figures 1C–1H). We also observed that celastrol intervention reduced liver volume in DIO mice, significantly improved liver steatosis evidenced by H&E and Oil red O (ORO) staining (Figure S3A), and reduced liver weight and triglycerides (TG) levels (Figures S3B and S3C). The GTT results showed that the glucose metabolism of the mice treated with celastrol was significantly improved (Figures 1I and 1J). In this model, we also observed that the food intake of the mice treated with celastrol was reduced, but the level of leptin in blood was not changed significantly (Figures 1K and 1L), which was consistent with the report of Liu and colleagues (Liu et al., 2015). To our surprise, we did not detect a significant upregulation of mitochondrial and energy-expenditure-related genes in brown fat, which have been reported to be upregulated by celastrol (Ma et al., 2015) (Figure 1M). All these data confirmed a potent anti-obesity effect of celastrol.
Figure 1.

Celastrol protects C57BL/6 mice from HFD-induced obesity
(A–M) HFD-fed obese C57BL/6 mice were subjected to oral administration of celastrol (3 mg/kg/day) for 3 weeks. (A) Representative pictures of male DIO C57BL/6 mice after 3 weeks of celastrol treatment. (B) BW of DIO mice during the treatment (n = 8 for each group). (C) Macroscopic view of representative sections of epididymal fat depots (eWAT), inguinal fat depots (iWAT), perirenal fat depots (pWAT), and brown adipose tissue (BAT) of mice treated with celastrol or not. (D) Representative H&E-stained images of eWAT (200×) (Scale bar, 50 μm) and BAT (400×) (Scale bar, 20 μm). (E–H) Weights of (E) eWAT, (F) iWAT, (G) pWAT, and (H) BAT of mice treated with celastrol or not (n = 8 for each group). (I–J) Results for (I) intraperitoneal glucose tolerance tests (GTT) and (J) the area under the curve (AUC) for GTTs (n = 8 for each group). (K) Three-day average food intake of DIO mice during celastrol treatment (n = 8 for each group). (L) Serum leptin levels of mice treated with celastrol or not (n = 8 for each group). (M) Gene expression analysis of mitochondrial and brown fat gene programs in BAT of mice treated with celastrol or not (n = 8 for each group). Error bars represent the mean ± SEM. p values were determined by two-way ANOVA (B) or Student's t test. ∗p < 0.05, ∗∗∗p < 0.001; n.s., not significant (p > 0.05).
Celastrol-induced weight loss is independent of the upregulation of UCP1 and energy expenditure
Celastrol protects against obesity and metabolic dysfunction by increasing energy expenditure (Ma et al., 2015). The results from our experiments showed that celastrol-induced weight loss was not accompanied by significant upregulation of the mitochondrial and brown fat genes, prompting us to further verify the impact of celastrol on energy expenditure using a combined indirect calorimetry system. Two groups of male mice aged 8 weeks were fed a HFD or HFD supplemented with celastrol (3 mg/kg/day) for two weeks, and then the energy expenditure and home-cage activity of these mice were examined using the TSE system. The results showed that administration of celastrol had little effect on O2 consumption, CO2 production, the respiratory exchange rate, and heat production (Figures S1A–S1G), as well as the cumulative moving distance (Figure S1H). Next, we evaluated the effect of celastrol on brown fat at the cellular level. Primary brown fat precursor cells were isolated, and the safe doses of celastrol ranging from 0 to 600 nM were determined based on the cell viability results (Figure S2A). First, we studied the effects of celastrol on differentiated mature brown adipocytes induced in vitro. The qRT-PCR results showed that 50 nM or 100 nM celastrol treatment for 48 h or 96 h did not further upregulate the expression of the mitochondrial and brown fat genes but downregulated the expression of the most important thermogenic gene UCP1 (Figures S2B and S2C). The protein levels of UCP1 were confirmed by WB (Figure S2D). Given that obesity is often accompanied by whitening of brown fat, leading to a decline of brown fat function (Shimizu et al., 2014), we then mimicked the process of whitening of brown fat in vitro by stimulating differentiated mature brown fat cells with free fatty acids (FFAs). The WB results showed that FFA (200 μM) significantly downregulated the protein expression level of UCP1 in mature brown adipocytes, but treatment with 50 nM and 100 nM celastrol for 24, 48, 72, or 96 h aggravated the FFA-induced downregulation of UCP1 at the protein level (Figure S2E). Overall, these results suggest that the weight loss effect of celastrol was possibly not due to the increment of UCP1 and energy expenditure.
Celastrol reduces intestinal lipid absorption in DIO C57BL/6 mice
Although we have not confirmed that celastrol works by enhancing energy expenditure, we noticed that the intestines of the DIO mice treated with celastrol orally were filled with a large amount of green content similar to the color of the high-fat food we used, but this phenomenon was not observed in mice from the control group (Figure 2A). From the isolated intestines of mice, we observed more clearly that there was more food residue in the intestine of the celastrol-treated DIO mice. These food residues did not form granular feces but instead appeared mushy (Figure 2B). This prompted us to analyze the fecal components, especially the TG and FFA levels, and we found that the TG and FFA levels in the feces of mice from the celastrol-treated group were significantly higher than those in the control group (Figures 2D and 2F), which is consistent with our observations. Similarly, based on the 24-h defecation volume, celastrol-treated mice also showed higher TG and FFA excretion (Figures 2E and 2G). The pathological observation of intestinal tissues revealed that the intestines of the mice in the celastrol-treated group were denser and contained fewer vesicular structures than the intestines of the control group (Figure 2C). These phenomena suggested that celastrol may have an effect on mouse intestinal absorption of lipids from high-fat food. Next, we evaluated the effect of celastrol on intestinal lipid absorption through an acute experiment that examined the changes in plasma TG levels in mice administered oral olive oil (Li et al., 2016). After pretreatment of the DIO mice with celastrol for 6 days, the BW of the mice was significantly decreased compared with that of the control group (Figure 2H). In these mice, 2 h after olive oil gavage, the plasma of mice in the drug-administration group looked more transparent, and the plasma TG levels of the DIO mice treated with celastrol were significantly lower than those in the control group (Figures 2I and 2J). However, in ND-fed mice, 6 days of administration of celastrol did not alter the BW (Figure 2K), and the plasma TG levels of the two groups were not significantly different 2 h after olive oil gavage (Figure 2L).
Figure 2.

Celastrol reduces intestinal lipid absorption in DIO C57BL/6 mice
(A–G) HFD-fed obese C57BL/6 mice were subjected to oral administration of celastrol (3 mg/kg/day) for 3 weeks. (A and B) Representative pictures for (A) the peritoneal cavity and (B) isolated intestines of mice treated with celastrol or not. (C) Representative H&E-stain images of the intestine (400×). Scale bar, 20 μm. (D and E) Relative fecal TG and 24 h TG excretion levels of mice treated with celastrol or not (n = 8 for each group). (F and G) Relative fecal FFA and 24 h FFA excretion levels of mice treated with celastrol or not (n = 8 for each group).
(H) BW of mice on HFD, either without (HFD, n = 7) or treated with celastrol at doses of 3 mg/kg/day (HFD + CEL, n = 7).
(I) Plasma TG levels of mice on HFD, either without (HFD, n = 7) or treated with celastrol at doses of 3 mg/kg/day (HFD + CEL, n = 7) before and 2 h after administration of olive oil by gavage.
(J) Macroscopic view of representative plasma of mice on HFD, either without (HFD) or treated with celastrol at doses of 3 mg/kg/day (HFD + CEL) 2 h after administration of olive oil by gavage.
(K) BW of mice on ND, either without (ND, n = 6) or treated with celastrol at doses of 3 mg/kg/day (ND + CEL, n = 6).
(L) Plasma TG levels of mice on ND, either without (ND, n = 6) or treated with celastrol at doses of 3 mg/kg/day (ND + CEL, n = 6) before and 2 h after administration of olive oil by gavage.
Error bars represent the mean ± SEM. p values were determined by two-way ANOVA (H, I, K, and L) or Student's t test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001; n.s., not significant (p > 0.05).
In a separate experiment, we treated the HFD-fed mice with celastrol by intraperitoneal injection instead of oral administration, which ensures the absorption of the drug into the circulation and avoids the direct contact between celastrol and lipids in food. The results showed that intraperitoneal administration of celastrol also decreased body weight of HFD-fed mice without significant reduction in food intake (Figures S4A and S4B), accompanied by a significant reduction of the weight of eWAT and liver in HFD + CEL group compared with HFD group (Figures S4C and S4D). Meanwhile, the mice with i.p. injection of celastrol exhibited the same intestinal phenotype as the mice treated with celastrol orally (Figure S4E). The levels of TG and FFA in feces of mice injected with celastrol were significantly increased (Figures S4F and S4G), which was accompanied by the significantly downregulated expression of intestinal lipid transporter genes of CD36, FATP4, and FATP2 (Figures S4H–S4J). These results demonstrated that celastrol does affect the absorption of lipids in the intestine under high-fat food challenge.
Celastrol inhibits the expression of intestinal lipid transporters in DIO C57BL/6 mice
The intestinal absorption of lipids requires the participation of a series of proteins, such as lipid transporters (Abumrad and Davidson, 2012). Because celastrol inhibits the intestinal absorption of lipids, it is reasonable to perform a comprehensive study to explore whether celastrol has an effect on lipid-absorption-related proteins of the intestine by TMT-based LC-MS/MS analysis. The results showed that a total of 4,248 proteins were identified by one or more unique peptides. The protein ratio was obtained by comparing the control (HFD) group with the celastrol-treated (CEL) group. Among the differentially expressed proteins, 115 proteins were upregulated and 297 proteins were downregulated in the CEL group compared with the HFD group (Data S1 and Figure 3A). We found that a large number of proteins related to lipid absorption, including apolipoprotein B-100 (ApoB), very-long-chain acyl-CoA synthetase (FATP2), long-chain fatty acid transport protein 4 (FATP4), and fatty acid-binding protein (Fabp2), were downregulated by administration of celastrol. Some of the differential concentrations of proteins between the two groups were visualized using a global heatmap analysis (Figure 3B), and the protein ID, protein name, gene name, and relative abundance ratios of these differentially expressed proteins are listed in Table 1. We next performed KEGG analysis to investigate the enriched pathways in which the upregulated and downregulated proteins participated in response to celastrol treatment. Among the enriched pathways evaluated by p value, the fat digestion and absorption pathways were significantly affected (Figure 3C). Next, we verified the nutrient-absorption-related genes in the small intestine by qRT-PCR and/or WB as follows: ApoB, FATP2 (encoded by the Slc27a2 gene), and FATP4 (encoded by the Slc27a4 gene) for lipid absorption (Losacco et al., 2018) and SGLT1, GLUT2, and GLUT5 for carbohydrate absorption. The results showed that celastrol treatment significantly downregulated ApoB expression at both the mRNA and protein levels (Figures 3F, 3L, and 3M). The expression of FATP2 and FATP4 was downregulated at the protein level rather than at the mRNA level (Figures 3E, 3G, 3L, and 3M). This is consistent with the proteomics analysis. In addition, we examined CD36, an important lipid transporter, which was also significantly downregulated at both the mRNA and protein levels (Figures 3D, 3L, and 3M). However, the transporters for carbohydrates, namely, SGLT1, GLUT2, and GLUT5, were not significantly altered by celastrol treatment at either the mRNA or the protein level (Figures 3I–3K, 3L, and 3M). The transcription factor peroxisome proliferator-activated receptor (PPAR) can regulate the expression of some nutrient transporters (Lynes and Widmaier, 2011). PPARα, the major isoform expressed in the intestine (de Vogel-van den Bosch et al., 2008), was decreased significantly in the celastrol-treated group (Figures 3H, 3L, and 3M). In order to explore whether celastrol affected digestive functions as well as absorptive functions, we detected enteroendocrine-related genes of cholecystokinin (Cck), secretin (Sct), and gastric inhibitory polypeptide (Gip) by qRT-PCR, and the results showed that celastrol significantly downregulated Sct and Gip mRNA levels without affecting Cck (Figures S5A–S5C). We next checked the data of proteomics of intestine from HFD and HFD + CEL mice and found that the level of GIP protein was significantly downregulated with a trend reduction of SCT protein in the HFD + CEL group (Figures S5D and S5E). However, CCK protein was not detected in this proteomics assay. These results suggest that the inhibition of intestinal lipid absorption by celastrol is related to the downregulation of lipid digestion- and transport-related genes.
Figure 3.

Celastrol inhibits the expression of intestinal lipid transport-related genes in DIO C57BL/6 mice
(A–C) The intestinal protein of mice was identified and analyzed by proteomics. (A) The volcano plot is used to show significant differences between the two sets of sample data (n = 3 for each group). Red dots represent significantly differentially expressed proteins (fold changes greater than 1.2-fold and p value <0.05), with black dots representing proteins with no differential changes. (B) Heatmaps representing the selected 21 significantly differentially expressed proteins. Red represents significantly upregulated proteins, and blue represents significantly downregulated proteins. (C) KEGG enrichment was used to analyze the effect of celastrol on protein expression in the intestine of DIO C57BL/6 mice treated with celastrol or not (n = 3 for each group). The number on the right of the column on the histogram represents the enrichment factor. The enriched KEGG pathways with p < 0.05 are shown.
(D–K) mRNA levels of intestinal lipid and glucose-transport-related genes of mice treated with celastrol or not (n = 8 for each group). The genes included CD36 (D), FATP4 (E), ApoB (F), FATP2 (G), PPARα (H), GLUT2 (I), GLUT5 (J), and SGLT1 (K).
(L and M) (L) Lipid- and glucose-transport-related protein levels and (M) quantified signal intensity.
Error bars represent the mean ± SEM. p values were determined by Student's t test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001; n.s., not significant (p > 0.05).
Table 1.
Key differentially expressed proteins identified by tandem mass tag-based proteomics analysis between DIO mice fed with or without celastrol
| Protein ID | Protein name (Gene name) | FC valuea | p-valueb |
|---|---|---|---|
| Q99JY8 | Phospholipid phosphatase 3 (Plpp3) | 0.81294 | 0.0069 |
| E9Q414 | Apolipoprotein B-100 (Apob) | 0.802762 | 0.0344 |
| Q61469 | Phospholipid phosphatase 1 (Plpp1) | 0.776152 | 0.0087 |
| Q91VE0 | Long-chain fatty acid transport protein 4 (Slc27a4) | 0.663703 | 0.0116 |
| Q80W94 | 2-Acylglycerol O-acyltransferase 2 (Mogat2) | 0.602726 | 0.0417 |
| P06728 | Apolipoprotein A-IV (Apoa4) | 0.544414 | 0.0208 |
| O08601 | Microsomal triglyceride transfer protein large subunit (Mttp) | 0.513655 | 0.0155 |
| P55050 | Fatty acid-binding protein, intestinal (Fabp2) | 0.400546 | 0.0031 |
| P12710 | Fatty acid-binding protein, liver (Fabp1) | 0.334806 | 0.0063 |
| P41216 | Long-chain fatty acid CoA ligase 1 (Acsl1) | 0.82888 | 0.0351 |
| P45952 | Medium-chain specific acyl-CoA dehydrogenase, mitochondrial (Acadm) | 0.814053 | 0.0038 |
| P52825 | Carnitine O-palmitoyltransferase 2, mitochondrial (Cpt2) | 0.788776 | 0.0138 |
| O35488 | Very-long-chain acyl-CoA synthetase (Slc27a2) | 0.715861 | 0.0489 |
| Q921H8 | 3-Ketoacyl-CoA thiolase A, peroxisomal (Acaa1a) | 0.702893 | 0.0254 |
| P06801 | NADP-dependent malic enzyme (Me1) | 0.698114 | 0.0125 |
| Q9DBM2 | Peroxisomal bifunctional enzyme (Ehhadh) | 0.675939 | 0.0135 |
| Q9R0H0 | Peroxisomal acyl-coenzyme A oxidase 1 (Acox1) | 0.652622 | 0.0139 |
| Q64516 | Glycerol kinase (Gk) | 0.649668 | 0.0087 |
| P32020 | Nonspecific lipid-transfer protein (Scp2) | 0.544846 | 0.0245 |
| Q9Z2V4 | Phosphoenolpyruvate carboxykinase, cytosolic [GTP] (Pck1) | 0.483453 | 0.0010 |
| Q8JZR0 | Long-chain fatty acid CoA ligase 5 (Acsl5) | 0.42567 | 0.0156 |
FC, fold change; HFD, high-fat diet; CEL, celastrol. Related to Figure 3.
The FC value was calculated as the ratio of the average mass response (area) between the two groups (FC value = CEL/HFD). Thus, FC values > 1 indicate significantly higher levels in the CEL group than in the HFD group, whereas FC values < 1 indicate significantly lower levels in the CEL group.
Only proteins with FC values greater than ±1.2 and p values less than 0.05 were deemed statistically significant.
Celastrol reduces the BW of ob/ob mice fed an HFD
Previous reports have shown that celastrol is a sensitizer for leptin and has no weight loss effect on ob/ob or db/db mice (Feng et al., 2019; Liu et al., 2015). In general, the obesity of ob/ob or db/db mice was caused by an increase in food intake of ND but not HFD chow due to the lack of leptin or leptin receptor. If celastrol reduces BW by inhibiting intestinal lipid absorption, as suggested by our data above, it should also reduce the BW of ob/ob mice when they are fed HFD chow. To explore this issue, we fed 4-week-old ob/ob mice with an average BW of 19.95 ± 0.2846 g with HFD chow, and 3 weeks later, the average BW of the mice reached 46.06 ± 0.4831 g. These mice were randomly divided into two groups: one group of mice was fed HFD chow containing the same concentration of celastrol (55.2 ppm) as used in DIO C57BL/6 mice, and another group of mice continuing on the HFD were used as the controls. At the end of the sixth week, the body size of the mice in the administration group appeared to be smaller than that of the control group (Figure 4A), and the BW of mice in the control group reached 61.05 ± 1.489 g, but the mice in the drug-administered group weighed only 53.47 ± 1.081 g, which was approximately 12.4% less than that in the control group (Figure 4B). Figure 4D shows the gross appearance of eWAT, iWAT, pWAT, and BAT. Both iWAT and pWAT were significantly reduced in size, which is consistent with the weight of these tissues (Figures 4E–4H). The effect of celastrol on these mice was not accompanied by significant changes in food intake (Figure 4C). In addition, the gross appearance of the liver showed that HFD exposure caused an increase in liver volume accompanied by a lightening of the color in ob/ob mice. Histologically, the H&E staining and ORO staining results showed ballooning degeneration of liver cells, and massive accumulation of large lipid droplets was found in the livers of these mice. All these phenotypes were clearly reversed by celastrol treatment (Figure S3D). The significant decrease in liver weight and TG level of celastrol-treated ob/ob mice matched the change in liver gross appearance and pathological examination (Figure S3E and S3F). The GTT results showed that celastrol-treated ob/ob mice demonstrated faster glucose clearance than the control mice (Figure 4I), and the total systemic glucose (represented by the area under the curve (AUC)) significantly decreased by 25.5% (Figure 4J). To assess whether the effects on mice were from drug toxicity under this condition, we tested the main biochemical indicators in the plasma of ob/ob mice. The results showed that celastrol did not significantly change the levels of BUN, SCr, and TG. At the same time, the levels of ALT, AST, LDH, and TC in plasma were significantly decreased (Figures S6A–S6G), which suggested that the BW-lowering effect of celastrol on ob/ob mice was not from drug toxicity, and the liver function of the mice was even improved based on reduced ALT and AST levels. All these phenotypes were consistent with the effects of celastrol on DIO C57BL/6 mice, further verifying that the weight loss effect of celastrol is food-type dependent but not leptin signaling dependent.
Figure 4.

Celastrol reduces the BW of ob/ob mice fed an HFD
(A–J) HFD-fed obese ob/ob mice were subjected to oral administration of celastrol (55.2 ppm) for 6 weeks. (A) Representative pictures of male obese ob/ob mice after 6 weeks of celastrol treatment. (B) BW of ob/ob mice during the treatment (n = 6 for each group). (C) The average 3-day food intake of ob/ob mice during celastrol treatment (n = 6 for each group). (D) Macroscopic view of representative sections of eWAT, iWAT, pWAT, and BAT of ob/ob mice treated with celastrol or not. (E–H) Weights of (E) eWAT, (F) iWAT, (G) pWAT, and (H) BAT of ob/ob mice treated with celastrol or not (n = 6 for each group). (I and J) Results for (I) GTT and (J) the AUC for GTTs (n = 6 for each group). Error bars represent the mean ± SEM. p values were determined by two-way ANOVA (B) or Student's t test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001; n.s., not significant (p > 0.05).
Celastrol downregulates the expression of lipid-absorption-related genes in the intestine of ob/ob mice fed an HFD
Consistent with the DIO C57BL/6 mice treated with celastrol, we also observed that there was more food residue in the intestine of the HFD-fed ob/ob mice administered celastrol, and these mice could not form granular feces in the colon (Figure 5A). The pathological morphology of the jejunum also became denser compared with the control mice (Figure 5B). Meanwhile, the fecal TG levels of ob/ob mice were significantly increased by celastrol administration (Figure 5C). Next, we tested the transcriptional levels of lipid transporters, including CD36, FATP2, and FATP4, in the small intestine of ob/ob mice and found that all these genes were also significantly downregulated (Figures 5D–5F), and the protein levels of CD36, FATP2, and FATP4 were confirmed by WB (Figures 5G and 5H). Combined with the reported fact that celastrol had no effect on the BW of ob/ob mice on an ND, our data further demonstrate that celastrol inhibits lipid absorption of the intestinal tract by inhibiting the expression of lipid-transport-related genes.
Figure 5.

Effect of celastrol on the intestinal tract and its lipid transporters in HFD-fed ob/ob mice
(A–H) HFD-fed obese ob/ob mice were subjected to oral administration of celastrol (55.2 ppm) for 6 weeks. (A) Representative pictures of isolated intestines of mice treated with celastrol or not. (B) Representative H&E-stained images of the intestine (400×). Scale bar, 20 μm. (C) Relative fecal TG levels of ob/ob mice treated with celastrol or not (n = 6 for each group). (D–F) mRNA levels of intestinal lipid-transport-related genes of mice treated with celastrol or not (n = 6 for each group). (G) Lipid-transport-related protein levels and (H) quantified signal intensity. Error bars represent the mean ± SEM. p values were determined by Student's t test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Early intervention with celastrol prevents HFD-induced weight gain in C57BL/6 mice
Studies have shown that changes in many indicators, including glucose tolerance and the leptin pathway, occur secondarily to weight loss (Adams et al., 2017; Guven et al., 1999). Therefore, we performed a short-term celastrol intervention on lean C57BL/6 mice in the early period to observe whether the early celastrol intervention had the same effect on reducing diet-induced obesity in lean mice on an ND and mildly obese mice on an HFD. We treated 10-week-old lean mice on an ND or HFD with celastrol. Within 24 days of dosing, we observed that celastrol significantly inhibited HFD-induced increases in BW but had little effect on the BW of mice fed an ND (Figure 6A). Consistent with the BW phenotype, drug intervention did not affect the weight of the eWAT, iWAT, pWAT, and BAT in ND-fed mice but significantly inhibited the increase in the weight of these tissues induced by HFD feeding (Figures 6B–6F). No significant differences in the food intake of mice fed an ND or HFD were detected in the presence and absence of celastrol (Figure 6G). Meanwhile, this early celastrol intervention did not upregulate the expression of most of the mitochondrial and brown fat genes of BAT in either ND-fed or HFD-fed mice (Figure S7), which provided more evidence that celastrol may not work by enhancing the function of brown fat.
Figure 6.

Early intervention with celastrol inhibits intestinal lipid absorption and prevents HFD-induced weight gain in C57BL/6 mice
(A–K) ND- or HFD-fed C57BL/6 mice were subjected to oral administration of celastrol (3 mg/kg/day) for 24 days. (A) The BW of each group of mice during the treatment (n = 10 for each group). (B) Macroscopic view of representative sections of eWAT, iWAT, pWAT, and BAT of each group of mice. (C–F) Weights of (C) eWAT, (D) iWAT, (E) pWAT, and (F) BAT of each group of mice (n = 10 for each group). (G) The average 3-day food intake of each group of mice during celastrol treatment (n = 10 for each group). (H) Relative fecal TG levels of each group of mice during celastrol treatment (n = 10 for each group). (I–J) Representative pictures of isolated intestines of celastrol-treated mice (I) on an ND or (J) HFD. (K) Representative H&E-stained images of the intestine (200×). Scale bar, 50 μm. Error bars represented the mean ± SEM. p values were determined by two-way ANOVA. ∗∗∗p < 0.001, ∗∗p < 0.01, ∗p < 0.05; n.s., not significant (p > 0.05).
Early intervention with celastrol inhibits intestinal lipid absorption in C57BL/6 mice fed an HFD
Consistent with the effect of celastrol on the intestines of DIO mice, we still observed that early celastrol intervention significantly increased the fecal TG levels of mice on an HFD (Figure 6H), with no significant changes in food intake (Figure 6G). The intestinal appearance and pathological changes in celastrol-treated HFD rather than ND-fed mice were consistent with the DIO mice treated with celastrol (Figures 6I–6K). In view of the abnormalities in the mouse intestines, we further investigated whether the metabolism of the intestinal tissue was changed by LC-MS-based metabolomics analysis. The orthogonal partial least squares-discriminant analysis (OPLS-DA) result showed a clear separation between the HFD and HFDC groups (R2X = 0.366, R2Y = 0.993, and Q2 = 0.9) (Figure 7A). Moreover, a permutation test with 200 iterations confirmed that the constructed OPLS-DA model was valid and not overfitted, as the original right R2 and Q2 values were significantly higher than the corresponding permutated left values [R2 = (0.0, 0.9421), Q2 = (0.0, −0.167)] (Figure 7B). As shown in Figure 7C, the volcanic maps obtained from the univariate analyses of fold change analysis and t test intuitively showed the significance of the changes in metabolites between the two groups of samples (the red dots in the figure are the metabolites with FC > 1.5 and p value <0.05, that is, the differential metabolites screened by univariate statistical analysis). Hierarchical clustering analysis showed that the significantly different metabolites shown in Figure 7D can represent the differences between different samples. The results of KEGG pathway enrichment analysis of differentially expressed metabolites in the HFD and HFDC groups showed that ABC transporters, arginine biosynthesis, central carbon metabolism in cancer, and other important pathways underwent significant changes (Figure 7E). These results suggest that celastrol may have value in altering intestinal metabolism and provide additional evidence that the drug has an impact on the intestines. The results of these early celastrol interventions indicated that the effect of celastrol on the intestines was not secondary to the enhanced leptin sensitivity and weight loss of mice.
Figure 7.

Metabolomics analysis of the intestines of HFD-fed mice treated with or without celastrol
(A–E) HFD-fed C57BL/6 mice were subjected to oral administration of celastrol (3 mg/kg/day) for 24 days. (A) OPLS-DA score plot derived from metabolomics analysis of mice in the HFD and HFDC groups. (B) Statistical validation of the OPLS-DA model by permutation testing. (C) Volcano plot. The FC value was calculated as the ratio of the average mass response (area) between the two groups (FC value = HFDC/HFD). The p value was calculated using the Mann-Whitney U-test. (D) Hierarchical clustering of groups of samples using qualitatively significant metabolite expression levels. Red means up, blue means down. (E) KEGG pathway enrichment analysis of differentially expressed metabolites in the HFD and HFDC groups by Fisher's exact test. HFD, high-fat diet; HFDC, high-fat diet supplemented with celastrol; FC, fold change. n = 6 for each group.
Dietary celastrol beneficially alters the gut microbiota
Studies have shown that the intestinal flora itself not only affects the digestion of lipids but also regulates transporters related to intestinal lipid absorption (Chang and Martinez-Guryn, 2019; Kuang et al., 2019; Martinez-Guryn et al., 2018; Semova et al., 2012). To define the effects of dietary celastrol on the gut microbiome, we analyzed the composition, abundance, and function of the gut microbiota. The rarefaction curve analysis results showed that the OTU rarefaction curves reached a steady level, indicating that the libraries were large enough to obtain the major information of the bacterial diversity of all samples (Figures S8A and S8B). In the feces of DIO mice treated with celastrol for 5 weeks, principal coordinate analyses (PCoA) revealed a distinct clustering of microbiota composition between the ND, HFD, and celastrol-supplemented HFD (HFDC) groups along the first principal component (PC1) (Figure 8A). To determine whether these microbial changes are secondary to improvements in obesity or direct effects of the drug, we treated HFD-fed lean mice with celastrol at an early stage for 24 days and still observed a distinct clustering of microbiota composition between the ND, HFD, and celastrol-supplemented HFD (HFDC) groups along PC1 (Figure 8G). Histograms of the community structure at the genus level illustrate the significant changes in the relative composition of the fecal microbiota, whether in DIO mice treated with celastrol (Figure 8B and Data S2) or HFD-fed lean mice treated early with celastrol (Figure 8H and Data S3). At the genus level, a direct comparison of relative abundance showed that among the top 20 changed genera, celastrol intervention restored the changes in 13 genera caused by HFD in DIO mice (Figure 8C) and 13 genera caused by HFD in early celastrol-treated HFD-fed lean mice (Figure 8I). Notably, the abundance of the Akkermansia genus, which was reported to be negatively correlated with waist circumference, body weight, body mass index, overall fat content, ovarian fat, and leptin concentrations in the circulation (Axling et al., 2012; Kim et al., 2014), was significantly upregulated by celastrol to more than eight times compared with the ND group in DIO mice (Figure 8C). Fourteen identical genera were altered by celastrol intervention in both DIO mice (Figure 8C) and early celastrol-treated HFD-fed lean mice (Figure 8I). At the species level, of the top 20 species with significant changes in DIO mice (Figures 8D–8F) and early celastrol-treated HFD-fed lean mice (Figures 8J–8L), nine species such as Lachnospiraceae_bacterium_609, Parabacteroides_ goldsteinii, and others were changed simultaneously in both early and late intervention groups. (Figures 8D–8F and 8J–8L). In addition to HFD-fed mice, celastrol intervention also changed the composition of the gut microbiome of ND-fed mice at the genus (Figure S9A) and species levels (Figure S9B).
Figure 8.

Dietary celastrol beneficially alters the gut microbiota
(A–I) DIO mice (n = 7 for each group) or HFD-fed lean C57BL/6 mice (n = 10 for each group) were subjected to oral administration of celastrol (3 mg/kg/day). Mouse fecal samples were taken and subjected to 16S rRNA gene sequencing. (A and G) PCoA of Bray-Curtis dissimilarity for all samples from (A) DIO mice treated with celastrol or (G) early celastrol-treated HFD-fed lean mice. (B and H) Stacked bar graph showing the relative abundance of bacterial taxa (genus level) of samples from (B) DIO mice treated with celastrol or (H) early celastrol-treated HFD-fed lean mice. (C and I) Significantly different intestinal microbiota at the genus level (C) among lean, DIO, and celastrol-treated DIO mice (n = 7 for each group) or (I) among lean, HFD and early celastrol-treated mice on an HFD (treated at the beginning of HFD) (n = 10 for each group). (D–F and J–L) The top 20 microbial species with significant changes in (D–F) DIO mice treated with celastrol or (J–L) early celastrol-treated HFD-fed lean mice. (M–N) Principal coordinate analysis (PCoA) of all KOs that were significantly altered by celastrol treatment in (M) DIO mice or (N) early celastrol-treated HFD-fed lean mice. (O–P) Pathway enrichment analysis of all significantly altered KOs in (O) DIO mice treated with celastrol or (P) early celastrol-treated HFD-fed lean mice. Error bars represent the mean ± SEM. p values were determined by ANOVA. 0.01<∗p < 0.05, 0.001<∗∗p < 0.01, 0.0001< ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. ND, normal diet; HFD, high-fat diet; HFDC, high-fat diet supplemented with celastrol.
To further investigate the functional changes in the gut microbiome after celastrol treatment, we annotated genes to the Kyoto Encyclopedia of Genes and Genomes (KEGG) Orthology (KO) database (Kanehisa and Goto, 2000). Principal coordinate analysis (PCoA) of the relative abundance of all of the significantly altered KOs revealed significant shifts after celastrol treatment in DIO mice (Figure 8M) or early celastrol-treated HFD-fed lean mice (Figure 8N). The relative abundance of KEGG pathways predicted by phylogenetic reconstruction of unobserved states (PICRUSt) revealed that in both celastrol-treated DIO mice (Figures S10A and 8O) and HFD-fed lean mice treated early with celastrol (Figures S10B and 8P), changes in the gut microbiota were linked mainly to the enrichment of genes related to transporters, ATP-binding cassette (ABC) transporters, and transcription factors. These results indicated that the decrease of intestinal lipid transporters caused by celastrol might come from changes of the gut microbiota.
Gut microbiota plays an important role in mediating the regulation of intestinal lipid transport by celastrol
To determine whether the gut microbiota is necessary for the decrease in intestinal lipid transporters caused by celastrol, we treated HFD-induced obesity mice with antibiotics and celastrol. The data showed that mice in AH group had a rapid body weight gain (about 3 g) during first 3 days, then all the mice from the antibiotics-treated groups (AN, AH, and AHC groups) gained similar amount of body weight from day 3 to day 24 (Figure 9A). Compared with HFD mice with 34% body weight gain, antibiotics strikingly blocked body weight gain by around 50%, suggesting an important role of gut microbiota in high-fat diet-induced obesity (Figure 9A). During this period, we did not detect a significant change in the food intake of HFD-fed mice by celastrol, regardless of antibiotics intervention (Figure 9B). Consistent with the body weight change, the antibiotics enhanced the excretion of TG from the feces of HFD-fed mice (Figure 9C), which was not further affected by celastrol, indicating that celastrol-caused lipid excretion in feces could be depended on the modulation on gut microbiota. At the same time, the pathological differences in the intestine observed between celastrol-treated and untreated mice with normal intestinal commensal bacteria almost disappeared after antibiotics treatment (Figure 9D). Finally, we observed a similar effect of antibiotics as celastrol on suppressing intestinal lipid transport-associated transporters and proteins such as CD36, ApoB, FATP4, and FABP2, which were not further affected by celastrol (Figures 9E and 9F).
Figure 9.

Modification of the gut microbiota by antibiotics blocks the celastrol-induced decrease in intestinal lipid absorption
(A–F) Eight-week-old C57BL/6J mice fed an ND or HFD supplemented with celastrol and/or antibiotics. (A) Body weight changes. (B) Food intake. (C) Fecal TG levels of mice during the treatments. (D) Representative H&E-stained images of the jejunum (200×). Scale bar, 50 μm. (E) Intestinal lipid-transport-related protein levels and (F) quantified signal intensity (n = 4–6 for each group). The dose of celastrol is 3 mg/kg/day. n = 10 in each group of ND, NC, HFD, and HC, and n = 6–7 in each group of AN, AH, and AHC. Error bars represent the mean ± SEM. p values were determined by two-way ANOVA (A) or one-way ANOVA. ∗∗∗p < 0.001, ∗∗p < 0.01, ∗p < 0.05; n.s., not significant (p > 0.05). ND, normal diet; NC, normal diet supplemented with celastrol; HFD, high-fat diet; HC, high-fat diet plus celastrol; AN, antibiotics treatment plus normal diet; AH, antibiotics treatment plus high-fat diet; AHC, antibiotics treatment plus high-fat diet plus celastrol.
Furthermore, we performed fecal microbiota transplantation (FMT) experiment to better clarify the role of gut microbiota in mediating the celastrol effect on lowering the body weight gain in HFD-induced obesity. The mice transplanted with fecal bacteria derived from HFD + CEL mice gained less body weight than the mice transplanted with fecal bacteria derived from HFD mice along with the reduced weight of fat tissues (Figures S11A and S11C–S11E), and there was no significant difference in the food intake between these two groups of mice with FMT (Figure S11B). Importantly, the mice transplanted with celastrol-conditioned intestinal microbes showed higher levels of fecal TG and FFA compared with the mice transplanted with fecal bacteria derived from HFD mice (Figures S11F and S11G). These data indicated that the intestinal flora contributed to the weight loss effect of celastrol on HFD-fed mice.
Discussion
Growing evidence indicates that celastrol could improve obesity and related comorbidities, suggesting that celastrol could be a new promising therapeutic strategy. As a natural pharmaceutical compound most likely to be developed into modern medicines, the mechanism of action of celastrol must be studied before it is finally deployed to the clinic (Corson and Crews, 2007). Several studies have been conducted on the mechanism of the weight loss effect of celastrol. Celastrol may exert its weight-loss effect by increasing the sensitivity of leptin signaling in the hypothalamus to reduce food intake (Feng et al., 2019; Kyriakou et al., 2018; Liu et al., 2015) or increasing energy expenditure by enhancing the function of brown fat (Ma et al., 2015). In addition, some studies have suggested that celastrol can reduce weight by mitigating oxidative stress (Wang et al., 2014) or inhibiting the differentiation of adipocytes (Hong et al., 2018). However, the subsequent research results did not fully support the previous conclusions (Pfuhlmann et al., 2018; Saito et al., 2019). Thus, the mechanism of the weight loss effect of celastrol is still controversial. In our study, we revealed that celastrol regulates intestinal flora to inhibit intestinal lipid absorption.
White adipose tissue (WAT) and brown adipose tissue (BAT) are the two principal types of adipose tissues in mammals. WAT stores energy in the form of TG (Gesta et al., 2007), whereas classical BAT is specialized at dissipating energy directly as heat through the high contents of mitochondrial UCP1 (Rosen and Spiegelman, 2014) and has attracted considerable research interest because of its therapeutic potential to treat obesity and obesity-associated metabolic diseases (Liu et al., 2013; Stanford et al., 2013). All our data from both cells and animals suggest that celastrol does not improve the function of brown fat directly and does not increase the energy expenditure of DIO mice by indirect calorimetry system. Recent reports suggested that the indirect calorimetry was not the gold standard to evaluate energy expenditure (Burnett and Grobe, 2014; Speakman, 2014). Especially, no difference in respirometry of our study might be affected by the altered gut microbiota, which was reported to result in decreased energy expenditure (Bahr et al., 2015). Thus, the effect of celastrol on energy consumption needs further in-depth study.
It is well known that in the process of evolution, the gut gained highly effective mechanisms for fat absorption. Intestinal lipid transport plays a central role in fat homeostasis. The fat content of the Western diet averages ∼35% of energy intake and is mostly in the form of triglycerides (TGs). Dietary TG is hydrolyzed in the intestinal lumen by pancreatic lipases and generates two fatty acids (FAs) and one sn-2-monoacylglycerol (2-MG). These products are absorbed through the apical membrane of intestinal cells (Goldberg et al., 2009). In our study, we were surprised to observe that there was more food residue in the intestine of HFD-fed mice administered celastrol, and these mice could not form granular feces in the colon. In addition, the TG and FFA levels in the feces of mice treated with celastrol were higher. Celastrol has been reported to cause injuries to the anus, colon, and intestine (Zhao et al., 2019), but we did not observe this phenomenon in our experiment (data not shown). All these phenomena prompted us to put forth the idea that celastrol may inhibit lipid absorption in the intestinal tract of mice and thus protect against obesity caused by an HFD. The acute lipid absorption experiment confirmed our speculation based on the phenotype we observed.
Proteomic analysis of the intestine of DIO mice treated with celastrol revealed that celastrol significantly changed the abundance of a large number of proteins in the intestinal tissues. The KEGG pathway analysis of the proteome showed that the fat digestion and absorption pathways had changed significantly, with ApoB, FABP2, FATP2, and FATP4 being identified as important signaling molecules of that pathway. Small intestinal cells uptake long-chain FAs (LCFAs) via both passive diffusion and protein-facilitated FA transfer. ApoB is the requisite surface structural protein that facilitates the formation of lipoproteins within the ER (Cartwright and Higgins, 2001; Fisher and Ginsberg, 2002; Pariyarath et al., 2001). The current concept is that ApoB is an obligate requirement for intestinal lipoprotein biogenesis (Abumrad and Davidson, 2012). FABP2 is reported to contribute to the uptake of dietary fatty acids in intestine (Besnard et al., 2002; Levy et al., 2001). Fatty acid transport proteins (FATPs) are classified as members of the Solute Carrier 27 (Slc27) family of proteins based on their ability to function in the transport of exogenous fatty acids. Results from both knockdown and overexpression studies of several members of the FATP family are consistent with the conclusion that they function in the process governing the transport of exogenous fatty acids into the cell in a highly regulated manner (Black et al., 2016). It has been confirmed that FATP2 expression in the small intestinal mucosa is significantly increased in mice fed an HFD and plays an important role in the transport of exogenous fatty acids (van den Bosch et al., 2007; Wisniewski et al., 2015). One member of this family, FATP4, located at the ER and subapical membranes, also mediates the apical uptake of long-chain fatty acids (Milger et al., 2006; Stahl et al., 1999). In addition to the proteins identified by the proteome, we also detected a significant decrease in the expression of CD36 in the intestinal tract of mice in the celastrol intervention group via WB. A large body of in vivo evidence from rodents and humans indicates that CD36 plays an important role in facilitating tissue FA uptake (Coburn et al., 2000; Hajri et al., 2002; Hirano et al., 2003; Watanabe et al., 1998). PPARα is a transcription factor and has been shown to be expressed at a high level in the small intestine (Bunger et al., 2007). In the small intestine, FATP2 expression levels are increased 4- to 5-fold in response to PPARα (Hirai et al., 2007). In our study, the decreased expression of PPARα by celastrol treatment may be an important cause of the downregulation of FATP2 expression. Overall, the significantly decreased expression of the proteins associated with intestinal lipid absorption in the intestine of celastrol-treated mice further demonstrated that intestinal lipid absorption was inhibited by celastrol.
Ob/ob mice have a large increase in food intake due to the lack of leptin, which in turn causes obesity even when fed an ND. It has been reported that the weight gain of ob/ob mice is not affected by celastrol (Liu et al., 2015). However, when we fed ob/ob mice an HFD supplemented with celastrol, the increase in BW was significantly inhibited with little change in food intake and a significant increase in fecal TG content. This phenomenon was consistent with the results of DIO C57BL/6 mice treated with celastrol, indicating that the effect of celastrol on BW is food-type dependent and that celastrol is more suitable for protecting against obesity caused by HFD. Previous findings from ob/ob mice showing the resistance to celastrol in treating obesity could be due to the diet difference.
Consistent with the reported phenomenon (Ma et al., 2015; Zhang et al., 2017), celastrol significantly improved the degree of fatty liver in DIO C57BL/6 mice in our study. It cannot be ruled out that the improvement in fatty liver was due to a decrease in food intake caused by administration of celastrol. However, what is amazing is that the same phenomenon occurred in ob/ob mice fed an HFD, yet the food intake of these mice was not reduced by celastrol treatment. This indicates that the decrease in lipid absorption in mice may be the main reason for the improvement in fatty liver in these celastrol-treated obese mice.
The intestinal flora promotes the absorption of fat from the diet and therefore plays an important role in the development of obesity (Gentile and Weir, 2018), and antibiotics can suppress HFD-induced body weight gain (Liu et al., 2019). Studies have shown that the intestinal flora can regulate the expression of intestinal lipid transporters (Kuang et al., 2019). In our research, we found that celastrol significantly changed the composition of intestinal microbes in HFD-fed mice and reversed some of the changes caused by HFD. The functional prediction of the changed flora showed that the change in the flora mainly affected the lipid transporters. Moreover, continuous antibiotic treatment depleting these changed gut bacteria abrogated the beneficial effects of celastrol supplementation on HFD-fed mice. Further analysis found that the intervention of antibiotics abolished the regulation of celastrol on intestinal transporters in HFD-fed mice. We noticed that the use of antibiotics weakened the capability of celastrol in inhibiting HFD-induced obesity and intestinal lipid transport, suggesting that the regulation on intestinal flora could be a critical mechanism of celastrol in attenuating HFD-induced obesity.
Our data were not consistent with previous findings in leptin signaling modulation and energy expenditure regulation (Fang et al., 2019; Kyriakou et al., 2018; Liu et al., 2015; Ma et al., 2015). Actually, recent publications also opposed each other on above mechanisms (Pfuhlmann et al., 2018; Saito et al., 2019). Our explanations could be as follows: (1) the secondary effect of body weight loss could affect leptin signaling and mitochondrial function in metabolic tissues. It is known that lean mice could have improved leptin signaling sensitivity and mitochondrial function in metabolic tissues. The long-term celastrol treatment led to a better metabolic condition due to the loss of fat mass, which may subsequently affect the leptin signaling sensitivity and mitochondrial metabolism. (2) The use of different animal models: as we know, the body weight gain of db/db or ob/ob mice is caused by the enhanced food intake but not high-fat diet. Our data strongly suggested that celastrol inhibited intestinal lipid absorption to improve obesity. Thus, under the obese conditions not related to high-fat diet, an anti-obesity effect of celastrol should be disappeared. (3) Other unexpected factors such as animal genetic background and the length and time points of celastrol treatment during the setup of experiments might also affect the data and conclusion.
In conclusion, we demonstrated that celastrol attenuates HFD-induced obesity by inhibiting intestinal lipid absorption and intestinal flora resetting rather than by increasing leptin signaling sensitivity and brown fat function. Our studies reveal a mechanism contributing to the celastrol effect on treating obesity.
Limitations of the study
In this study, we clarified that celastrol inhibited the HFD-induced obesity by lowering the intestinal lipid digestion and absorption. Further evidence showed that the changes in the composition of intestinal microbes could mediate celastrol effect on the intestinal lipid transport. However, this study had a couple of limitations: (1) we did not define the specific bacteria mediating the celastrol effect on lowering the intestinal lipid transport and digestion. (2) We did not define the specific metabolites released from the bacteria and more detailed mechanisms that contributed to the regulatory effect of celastrol on intestinal lipid transport and digestion in obesity induced by high-fat diet.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Zhanjun Jia (jiazj72@hotmail.com).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Data supporting the findings of this study are available within the paper and its Supplemental information and also from the authors upon reasonable request.
Methods
All methods can be found in the accompanying Transparent methods supplemental file.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (81873599 to Z.J., 82000642 to H.H., 81570616 to Y.Z., and 81830020to A.Z.).
Author contributions
H. H., Y. Z., F. Z., K. C., T. W., and Q. L. conducted the experiments. Z. J., H. H., Y. Z., A. Z., and S. H. designed the experiments and wrote the paper. Data were analyzed by H. H., Y. Z., and Z. J., A. Z., and S. H. supervised whole research work. All authors approved the final version of the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: February 19, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.102077.
Contributor Information
Songming Huang, Email: smhuang@njmu.edu.cn.
Aihua Zhang, Email: zhaihua@njmu.edu.cn.
Zhanjun Jia, Email: jiazj72@hotmail.com.
Supplemental information
References
- Abumrad N.A., Davidson N.O. Role of the gut in lipid homeostasis. Physiol. Rev. 2012;92:1061–1085. doi: 10.1152/physrev.00019.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams T.D., Davidson L.E., Litwin S.E., Kim J., Kolotkin R.L., Nanjee M.N., Gutierrez J.M., Frogley S.J., Ibele A.R., Brinton E.A. Weight and metabolic outcomes 12 Years after gastric Bypass. N. Engl. J. Med. 2017;377:1143–1155. doi: 10.1056/NEJMoa1700459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allison A.C., Cacabelos R., Lombardi V.R., Alvarez X.A., Vigo C. Celastrol, a potent antioxidant and anti-inflammatory drug, as a possible treatment for Alzheimer's disease. Prog. Neuropsychopharmacol. Biol. Psychiatry. 2001;25:1341–1357. doi: 10.1016/s0278-5846(01)00192-0. [DOI] [PubMed] [Google Scholar]
- Axling U., Olsson C., Xu J., Fernandez C., Larsson S., Strom K., Ahrne S., Holm C., Molin G., Berger K. Green tea powder and Lactobacillus plantarum affect gut microbiota, lipid metabolism and inflammation in high-fat fed C57BL/6J mice. Nutr. Metab. (Lond) 2012;9:105. doi: 10.1186/1743-7075-9-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahr S.M., Weidemann B.J., Castro A.N., Walsh J.W., deLeon O., Burnett C.M., Pearson N.A., Murry D.J., Grobe J.L., Kirby J.R. Risperidone-induced weight gain is mediated through shifts in the gut microbiome and suppression of energy expenditure. EBioMedicine. 2015;2:1725–1734. doi: 10.1016/j.ebiom.2015.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besnard P., Niot I., Poirier H., Clement L., Bernard A. New insights into the fatty acid-binding protein (FABP) family in the small intestine. Mol. Cell Biochem. 2002;239:139–147. [PubMed] [Google Scholar]
- Black P.N., Ahowesso C., Montefusco D., Saini N., DiRusso C.C. Fatty acid transport proteins: targeting FATP2 as a gatekeeper involved in the transport of exogenous fatty acids. Medchemcomm. 2016;7:612–622. doi: 10.1039/C6MD00043F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Bosch H.M., Bunger M., de Groot P.J., van der Meijde J., Hooiveld G.J., Muller M. Gene expression of transporters and phase I/II metabolic enzymes in murine small intestine during fasting. BMC Genomics. 2007;8:267. doi: 10.1186/1471-2164-8-267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunger M., van den Bosch H.M., van der Meijde J., Kersten S., Hooiveld G.J., Muller M. Genome-wide analysis of PPARalpha activation in murine small intestine. Physiol. Genomics. 2007;30:192–204. doi: 10.1152/physiolgenomics.00198.2006. [DOI] [PubMed] [Google Scholar]
- Burnett C.M., Grobe J.L. Dietary effects on resting metabolic rate in C57BL/6 mice are differentially detected by indirect (O2/CO2 respirometry) and direct calorimetry. Mol. Metab. 2014;3:460–464. doi: 10.1016/j.molmet.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartwright I.J., Higgins J.A. Direct evidence for a two-step assembly of ApoB48-containing lipoproteins in the lumen of the smooth endoplasmic reticulum of rabbit enterocytes. J. Biol. Chem. 2001;276:48048–48057. doi: 10.1074/jbc.M104229200. [DOI] [PubMed] [Google Scholar]
- Chang E.B., Martinez-Guryn K. Small intestinal microbiota: the neglected stepchild needed for fat digestion and absorption. Gut Microbes. 2019;10:235–240. doi: 10.1080/19490976.2018.1502539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chellappa K., Perron I.J., Naidoo N., Baur J.A. The leptin sensitizer celastrol reduces age-associated obesity and modulates behavioral rhythms. Aging Cell. 2019;18:e12874. doi: 10.1111/acel.12874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S.K., Park S., Jang S., Cho H.H., Lee S., You S., Kim S.H., Moon H.S. Cascade regulation of PPARgamma(2) and C/EBPalpha signaling pathways by celastrol impairs adipocyte differentiation and stimulates lipolysis in 3T3-L1 adipocytes. Metabolism. 2016;65:646–654. doi: 10.1016/j.metabol.2016.01.009. [DOI] [PubMed] [Google Scholar]
- Coburn C.T., Knapp F.F., Jr., Febbraio M., Beets A.L., Silverstein R.L., Abumrad N.A. Defective uptake and utilization of long chain fatty acids in muscle and adipose tissues of CD36 knockout mice. J. Biol. Chem. 2000;275:32523–32529. doi: 10.1074/jbc.M003826200. [DOI] [PubMed] [Google Scholar]
- Collaborators G.B.D.O., Afshin A., Forouzanfar M.H., Reitsma M.B., Sur P., Estep K., Lee A., Marczak L., Mokdad A.H., Moradi-Lakeh M. Health effects of overweight and obesity in 195 countries over 25 years. N. Engl. J. Med. 2017;377:13–27. doi: 10.1056/NEJMoa1614362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordain L., Eaton S.B., Sebastian A., Mann N., Lindeberg S., Watkins B.A., O'Keefe J.H., Brand-Miller J. Origins and evolution of the Western diet: health implications for the 21st century. Am. J. Clin. Nutr. 2005;81:341–354. doi: 10.1093/ajcn.81.2.341. [DOI] [PubMed] [Google Scholar]
- Corson T.W., Crews C.M. Molecular understanding and modern application of traditional medicines: triumphs and trials. Cell. 2007;130:769–774. doi: 10.1016/j.cell.2007.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang P., He B., Yu M., Shi M., Zhu Y., Zhang Z., Bo P. Treatment with celastrol protects against obesity through suppression of galanin-induced fat intake and activation of PGC-1alpha/GLUT4 axis-mediated glucose consumption. Biochim. Biophys. Acta Mol. Basis Dis. 2019;1865:1341–1350. doi: 10.1016/j.bbadis.2019.02.002. [DOI] [PubMed] [Google Scholar]
- Feng X., Guan D., Auen T., Choi J.W., Salazar Hernandez M.A., Lee J., Chun H., Faruk F., Kaplun E., Herbert Z. IL1R1 is required for celastrol's leptin-sensitization and antiobesity effects. Nat. Med. 2019;25:575–582. doi: 10.1038/s41591-019-0358-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher E.A., Ginsberg H.N. Complexity in the secretory pathway: the assembly and secretion of apolipoprotein B-containing lipoproteins. J. Biol. Chem. 2002;277:17377–17380. doi: 10.1074/jbc.R100068200. [DOI] [PubMed] [Google Scholar]
- Gentile C.L., Weir T.L. The gut microbiota at the intersection of diet and human health. Science. 2018;362:776–780. doi: 10.1126/science.aau5812. [DOI] [PubMed] [Google Scholar]
- Gesta S., Tseng Y.H., Kahn C.R. Developmental origin of fat: tracking obesity to its source. Cell. 2007;131:242–256. doi: 10.1016/j.cell.2007.10.004. [DOI] [PubMed] [Google Scholar]
- Goldberg I.J., Eckel R.H., Abumrad N.A. Regulation of fatty acid uptake into tissues: lipoprotein lipase- and CD36-mediated pathways. J. Lipid Res. 2009;50(Suppl):S86–S90. doi: 10.1194/jlr.R800085-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guven S., El-Bershawi A., Sonnenberg G.E., Wilson C.R., Hoffmann R.G., Krakower G.R., Kissebah A.H. Plasma leptin and insulin levels in weight-reduced obese women with normal body mass index: relationships with body composition and insulin. Diabetes. 1999;48:347–352. doi: 10.2337/diabetes.48.2.347. [DOI] [PubMed] [Google Scholar]
- Hajri T., Han X.X., Bonen A., Abumrad N.A. Defective fatty acid uptake modulates insulin responsiveness and metabolic responses to diet in CD36-null mice. J. Clin. Invest. 2002;109:1381–1389. doi: 10.1172/JCI14596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haslam D.W., James W.P. Obes. Lancet. 2005;366:1197–1209. doi: 10.1016/S0140-6736(05)67483-1. [DOI] [PubMed] [Google Scholar]
- Hirai T., Fukui Y., Motojima K. PPARalpha agonists positively and negatively regulate the expression of several nutrient/drug transporters in mouse small intestine. Biol. Pharm. Bull. 2007;30:2185–2190. doi: 10.1248/bpb.30.2185. [DOI] [PubMed] [Google Scholar]
- Hirano K., Kuwasako T., Nakagawa-Toyama Y., Janabi M., Yamashita S., Matsuzawa Y. Pathophysiology of human genetic CD36 deficiency. Trends Cardiovasc. Med. 2003;13:136–141. doi: 10.1016/s1050-1738(03)00026-4. [DOI] [PubMed] [Google Scholar]
- Hong W., Park J., Yun W., Kang P.J., Son D., Jang J., Kim I.Y., You S. Inhibitory effect of celastrol on adipogenic differentiation of human adipose-derived stem cells. Biochem. Biophys. Res. Commun. 2018;507:236–241. doi: 10.1016/j.bbrc.2018.11.014. [DOI] [PubMed] [Google Scholar]
- Hu M., Luo Q., Alitongbieke G., Chong S., Xu C., Xie L., Chen X., Zhang D., Zhou Y., Wang Z. Celastrol-induced Nur77 interaction with TRAF2 alleviates inflammation by promoting mitochondrial ubiquitination and autophagy. Mol. Cell. 2017;66:141–153.e46. doi: 10.1016/j.molcel.2017.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M., Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashyap D., Sharma A., Tuli H.S., Sak K., Mukherjee T., Bishayee A. Molecular targets of celastrol in cancer: recent trends and advancements. Crit. Rev. Oncol. Hematol. 2018;128:70–81. doi: 10.1016/j.critrevonc.2018.05.019. [DOI] [PubMed] [Google Scholar]
- Kim B.S., Song M.Y., Kim H. The anti-obesity effect of Ephedra sinica through modulation of gut microbiota in obese Korean women. J. Ethnopharmacol. 2014;152:532–539. doi: 10.1016/j.jep.2014.01.038. [DOI] [PubMed] [Google Scholar]
- Kuang Z., Wang Y., Li Y., Ye C., Ruhn K.A., Behrendt C.L., Olson E.N., Hooper L.V. The intestinal microbiota programs diurnal rhythms in host metabolism through histone deacetylase 3. Science. 2019;365:1428–1434. doi: 10.1126/science.aaw3134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyriakou E., Schmidt S., Dodd G.T., Pfuhlmann K., Simonds S.E., Lenhart D., Geerlof A., Schriever S.C., De Angelis M., Schramm K.W. Celastrol promotes weight loss in diet-induced obesity by inhibiting the protein tyrosine phosphatases PTP1B and TCPTP in the hypothalamus. J. Med. Chem. 2018;61:11144–11157. doi: 10.1021/acs.jmedchem.8b01224. [DOI] [PubMed] [Google Scholar]
- Lee J.H., Koo T.H., Yoon H., Jung H.S., Jin H.Z., Lee K., Hong Y.S., Lee J.J. Inhibition of NF-kappa B activation through targeting I kappa B kinase by celastrol, a quinone methide triterpenoid. Biochem. Pharmacol. 2006;72:1311–1321. doi: 10.1016/j.bcp.2006.08.014. [DOI] [PubMed] [Google Scholar]
- Levy E., Menard D., Delvin E., Stan S., Mitchell G., Lambert M., Ziv E., Feoli-Fonseca J.C., Seidman E. The polymorphism at codon 54 of the FABP2 gene increases fat absorption in human intestinal explants. J. Biol. Chem. 2001;276:39679–39684. doi: 10.1074/jbc.M105713200. [DOI] [PubMed] [Google Scholar]
- Li J., Song J., Zaytseva Y.Y., Liu Y., Rychahou P., Jiang K., Starr M.E., Kim J.T., Harris J.W., Yiannikouris F.B. An obligatory role for neurotensin in high-fat-diet-induced obesity. Nature. 2016;533:411–415. doi: 10.1038/nature17662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Zheng Z., Zhu X., Meng M., Li L., Shen Y., Chi Q., Wang D., Zhang Z., Li C. Brown adipose tissue transplantation improves whole-body energy metabolism. Cell Res. 2013;23:851–854. doi: 10.1038/cr.2013.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Lee J., Salazar Hernandez M.A., Mazitschek R., Ozcan U. Treatment of obesity with celastrol. Cell. 2015;161:999–1011. doi: 10.1016/j.cell.2015.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D., Wen B., Zhu K., Luo Y., Li J., Li Y., Lin H., Huang J., Liu Z. Antibiotics-induced perturbations in gut microbial diversity influence metabolic phenotypes in a murine model of high-fat diet-induced obesity. Appl. Microbiol. Biotechnol. 2019;103:5269–5283. doi: 10.1007/s00253-019-09764-5. [DOI] [PubMed] [Google Scholar]
- Losacco M.C., de Almeida C.F.T., Hijo A.H.T., Bargi-Souza P., Gama P., Nunes M.T., Goulart-Silva F. High-fat diet affects gut nutrients transporters in hypo and hyperthyroid mice by PPAR-a independent mechanism. Life Sci. 2018;202:35–43. doi: 10.1016/j.lfs.2018.03.053. [DOI] [PubMed] [Google Scholar]
- Sidsosis L.S., Porter C., Saraf M.K., Børsheim E., Radhakrishnan R.S., Chao T., Ali A., Chondronikola M., Micak R., Finnerty C.C. Browning of subcutaneous white adipose tissue in humans after severe adrenergic stress. Cell Metab. 2015;22:219–227. doi: 10.1016/j.cmet.2015.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynes M.D., Widmaier E.P. Involvement of CD36 and intestinal alkaline phosphatases in fatty acid transport in enterocytes, and the response to a high-fat diet. Life Sci. 2011;88:384–391. doi: 10.1016/j.lfs.2010.12.015. [DOI] [PubMed] [Google Scholar]
- Ma X., Xu L., Alberobello A.T., Gavrilova O., Bagattin A., Skarulis M., Liu J., Finkel T., Mueller E. Celastrol protects against obesity and metabolic dysfunction through activation of a HSF1-PGC1alpha transcriptional Axis. Cell Metab. 2015;22:695–708. doi: 10.1016/j.cmet.2015.08.005. [DOI] [PubMed] [Google Scholar]
- Martinez-Guryn K., Hubert N., Frazier K., Urlass S., Musch M.W., Ojeda P., Pierre J.F., Miyoshi J., Sontag T.J., Cham C.M. Small intestine microbiota regulate host digestive and absorptive adaptive responses to dietary lipids. Cell Host Microbe. 2018;23:458–469.e55. doi: 10.1016/j.chom.2018.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milger K., Herrmann T., Becker C., Gotthardt D., Zickwolf J., Ehehalt R., Watkins P.A., Stremmel W., Fullekrug J. Cellular uptake of fatty acids driven by the ER-localized acyl-CoA synthetase FATP4. J. Cell Sci. 2006;119:4678–4688. doi: 10.1242/jcs.03280. [DOI] [PubMed] [Google Scholar]
- Olshansky S.J., Passaro D.J., Hershow R.C., Layden J., Carnes B.A., Brody J., Hayflick L., Butler R.N., Allison D.B., Ludwig D.S. A potential decline in life expectancy in the United States in the 21st century. N. Engl. J. Med. 2005;352:1138–1145. doi: 10.1056/NEJMsr043743. [DOI] [PubMed] [Google Scholar]
- Padwal R.S., Majumdar S.R. Drug treatments for obesity: orlistat, sibutramine, and rimonabant. Lancet. 2007;369:71–77. doi: 10.1016/S0140-6736(07)60033-6. [DOI] [PubMed] [Google Scholar]
- Pang X., Yi Z., Zhang J., Lu B., Sung B., Qu W., Aggarwal B.B., Liu M. Celastrol suppresses angiogenesis-mediated tumor growth through inhibition of AKT/mammalian target of rapamycin pathway. Cancer Res. 2010;70:1951–1959. doi: 10.1158/0008-5472.CAN-09-3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pariyarath R., Wang H., Aitchison J.D., Ginsberg H.N., Welch W.J., Johnson A.E., Fisher E.A. Co-translational interactions of apoprotein B with the ribosome and translocon during lipoprotein assembly or targeting to the proteasome. J. Biol. Chem. 2001;276:541–550. doi: 10.1074/jbc.M007944200. [DOI] [PubMed] [Google Scholar]
- Pfuhlmann K., Schriever S.C., Baumann P., Kabra D.G., Harrison L., Mazibuko-Mbeje S.E., Contreras R.E., Kyriakou E., Simonds S.E., Tiganis T. Celastrol-induced weight loss is driven by hypophagia and independent from UCP1. Diabetes. 2018;67:2456–2465. doi: 10.2337/db18-0146. [DOI] [PubMed] [Google Scholar]
- Rosen E.D., Spiegelman B.M. What we talk about when we talk about fat. Cell. 2014;156:20–44. doi: 10.1016/j.cell.2013.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito K., Davis K.C., Morgan D.A., Toth B.A., Jiang J., Singh U., Berglund E.D., Grobe J.L., Rahmouni K., Cui H. Celastrol reduces obesity in MC4R deficiency and stimulates sympathetic nerve activity affecting metabolic and cardiovascular functions. Diabetes. 2019;68:1210–1220. doi: 10.2337/db18-1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semova I., Carten J.D., Stombaugh J., Mackey L.C., Knight R., Farber S.A., Rawls J.F. Microbiota regulate intestinal absorption and metabolism of fatty acids in the zebrafish. Cell Host Microbe. 2012;12:277–288. doi: 10.1016/j.chom.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu I., Aprahamian T., Kikuchi R., Shimizu A., Papanicolaou K.N., MacLauchlan S., Maruyama S., Walsh K. Vascular rarefaction mediates whitening of brown fat in obesity. J. Clin. Invest. 2014;124:2099–2112. doi: 10.1172/JCI71643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speakman J.R. Should we abandon indirect calorimetry as a tool to diagnose energy expenditure? Not yet. Perhaps not ever. Commentary on Burnett and Grobe (2014) Mol. Metab. 2014;3:342–344. doi: 10.1016/j.molmet.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl A., Hirsch D.J., Gimeno R.E., Punreddy S., Ge P., Watson N., Patel S., Kotler M., Raimondi A., Tartaglia L.A. Identification of the major intestinal fatty acid transport protein. Mol. Cell. 1999;4:299–308. doi: 10.1016/s1097-2765(00)80332-9. [DOI] [PubMed] [Google Scholar]
- Stanford K.I., Middelbeek R.J., Townsend K.L., An D., Nygaard E.B., Hitchcox K.M., Markan K.R., Nakano K., Hirshman M.F., Tseng Y.H. Brown adipose tissue regulates glucose homeostasis and insulin sensitivity. J. Clin. Invest. 2013;123:215–223. doi: 10.1172/JCI62308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vogel-van den Bosch H.M., Bunger M., de Groot P.J., Bosch-Vermeulen H., Hooiveld G.J., Muller M. PPARalpha-mediated effects of dietary lipids on intestinal barrier gene expression. BMC Genomics. 2008;9:231. doi: 10.1186/1471-2164-9-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C., Shi C., Yang X., Yang M., Sun H., Wang C. Celastrol suppresses obesity process via increasing antioxidant capacity and improving lipid metabolism. Eur. J. Pharmacol. 2014;744:52–58. doi: 10.1016/j.ejphar.2014.09.043. [DOI] [PubMed] [Google Scholar]
- Watanabe K., Ohta Y., Toba K., Ogawa Y., Hanawa H., Hirokawa Y., Kodama M., Tanabe N., Hirono S., Ohkura Y. Myocardial CD36 expression and fatty acid accumulation in patients with type I and II CD36 deficiency. Ann. Nucl. Med. 1998;12:261–266. doi: 10.1007/BF03164911. [DOI] [PubMed] [Google Scholar]
- Wei C., Wang X., Yao X., Xi F., He Y., Xu Y., Ma L., Chen X., Zhao C., Du R. Bifenthrin induces fat deposition by improving fatty acid uptake and inhibiting lipolysis in mice. J. Agric. Food Chem. 2019;67:14048–14055. doi: 10.1021/acs.jafc.9b06727. [DOI] [PubMed] [Google Scholar]
- Wisniewski J.R., Friedrich A., Keller T., Mann M., Koepsell H. The impact of high-fat diet on metabolism and immune defense in small intestine mucosa. J. Proteome Res. 2015;14:353–365. doi: 10.1021/pr500833v. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Geng C., Liu X., Li M., Gao M., Liu X., Fang F., Chang Y. Celastrol ameliorates liver metabolic damage caused by a high-fat diet through Sirt1. Mol. Metab. 2017;6:138–147. doi: 10.1016/j.molmet.2016.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J., Luo D., Zhang Z., Fan N., Wang Y., Nie H., Rong J. Celastrol-loaded PEG-PCL nanomicelles ameliorate inflammation, lipid accumulation, insulin resistance and gastrointestinal injury in diet-induced obese mice. J. Control Release. 2019;310:188–197. doi: 10.1016/j.jconrel.2019.08.026. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data supporting the findings of this study are available within the paper and its Supplemental information and also from the authors upon reasonable request.