

Summary
Tumors comprise cancer cells and the associated stromal and immune/inflammatory cells, i.e., tumor microenvironment (TME). Here, we identify a metabolic signature of human and mouse model of gastric cancer and show that vagotomy in the mouse model reverses the metabolic reprogramming, reflected by metabolic switch from glutaminolysis to OXPHOS/glycolysis and normalization of the energy metabolism in cancer cells and TME. We next identify and validate SNAP25, mTOR, PDP1/α-KGDH, and glutaminolysis as drug targets and accordingly propose a therapeutic strategy to target the nerve-cancer metabolism. We demonstrate the efficacy of nerve-cancer metabolism therapy by intratumoral injection of BoNT-A (SNAP25 inhibitor) with systemic administration of RAD001 and CPI-613 but not cytotoxic drugs on overall survival in mice and show the feasibility in patients. These findings point to the importance of neural signaling in modulating the tumor metabolism and provide a rational basis for clinical translation of the potential strategy for gastric cancer.
Subject Areas: Cancer, Cancer Systems Biology
Graphical Abstract
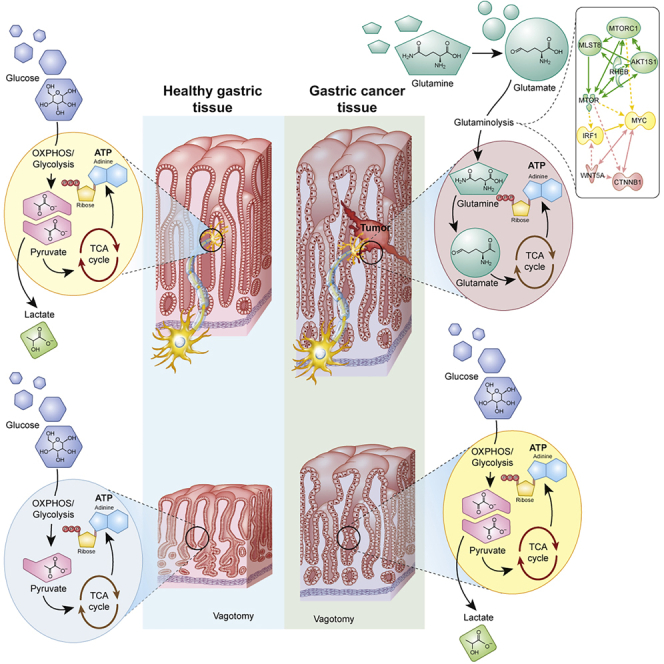
Highlights
-
•
Metabolic reprogramming in gastric cancer cells and tumor microenvironment
-
•
SNAP25, mTOR, PDP1/α-KGDH, and glutaminolysis as potential drug targets
-
•
Combination of botulinum toxin type A, RAD001, and CPI-613 as a potential treatment
Cancer; Cancer Systems Biology
Introduction
Cancer is considered a genetic disease with tumor characteristics and metabolic reprogramming (Hanahan and Weinberg, 2011; Wishart, 2015; Whiteside, 2008). The tumor mass consists of primary tumor (i.e. cancer cells) and the associated stromal cells and immune/inflammatory cells, i.e. tumor microenvironment (TME) that usually is different from normal stroma. As cancer cells continue proliferation, the tumor increases in size with an associated remodeling of the TME that determines whether the primary tumor is eradicated, metastasizes, or establishes dormant micrometastases (Loponte et al., 2019; Yoshida, 2015; Vander Heiden and Deberardinis, 2017). The cancer metabolic reprogramming is reflected by alterations in the metabolic profiles of both cancer cells as well as TME. Cancer cells can activate glycolysis in the presence of adequate oxygen levels (aerobic glycolysis or the so-called Warburg effect) within TME, whereas cells of normally differentiated tissues obtain energy through the oxygen-dependent pathway of oxidative phosphorylation (OXPHOS) as well as through the oxygen-independent pathway of glycolysis (Liberti and Locasale, 2016). Emerging evidence suggests that the cancer metabolic reprogramming exhibits the following hallmarks: (1) deregulated uptake of glucose and amino acids, (2) use of opportunistic modes of nutrient acquisition, (3) use of glycolysis/tricarboxylic acid (TCA) cycle intermediates for biosynthesis and nicotinamide adenine dinucleotide phosphate (NADPH) production, (4) increased demand for nitrogen, (5) alterations in metabolite-driven gene regulation, and (6) metabolic interactions between cancer cells and the TME (Pavlova and Thompson, 2016). However, development of treatment targeting the cancer metabolic reprogramming has not yet been successful due to the large differences between tumor types and TME, thus this area is ripe for the strategic development of future targeted treatments for individual cancer types (Schulze and Harris, 2012; Coller, 2014; Liberti and Locasale, 2016; Wishart, 2015; Seyfried et al., 2014).
Gastric cancer (GC) is the fifth most common malignant disease worldwide with the third highest incidence and mortality rate among all cancers (Rawla and Barsouk, 2019). The 5-year overall survival rate for gastric cancer is 10%–30% except for Japan (50%–70%) (Parkin et al., 2005; Matsuda and Saika, 2013). Previously, we and others have demonstrated that vagotomy suppressed gastric tumorigenesis, suggesting a “nerve-cancer cell crosstalk” (Zhao et al., 2014; Hayakawa et al., 2017; Jobling et al., 2015; Wang et al., 2018). In the present study, we used the approaches of in vitro, in vivo, in silico, clinical evaluation, and pilot clinical trial, and employed the omics technology including comparative transcriptomics (human versus mouse), t-SNE, multi-omics (transcriptomics versus metabolomics), drug-target interaction prediction, and computational drug repositioning (Figure S1). We showed that the animal model we used mimicked GC patients in terms of tumor characteristics and metabolic reprogramming. We found that GC exhibited a metabolic reprogramming, i.e., the use of glutaminolysis for biosynthesis, that differed from other cancer types (Hanahan and Weinberg, 2011; Schulze and Harris, 2012) and that vagotomy reversed the metabolic reprogramming from glutaminolysis to OXPHOS/glycolysis in cancer cells as well as TME of the GC mice. We also identified the metabolic signature and validated the drug-target signaling interactions targeting nerve-cancer metabolism in human cancer cell lines and GC mice and developed a therapeutic strategy using a combination of denervation and cytotoxic free chemotherapy. This treatment appeared effective, particularly with regard to overall survival rate in aged GC mice, and showed potential in a pilot clinical trial in aged GC patients, suggesting a possible “metabolism-based” approach for GC (Figure S1. Study design, Related to Figure 1).
Figure 1.

Signaling pathways in gastric cancer (GC)
Waterfall diagrams showing signaling pathways that were differentially activated (marked in red) and inhibited (blue) in human gastric cancer (GC) versus normal adjacent tissue (A) and mouse GC versus WT (B) or after vagotomy (C). Note: names of signaling pathways are listed in the same order in A–C. For detailed information, see Data S1.
Correlation between human and mouse GC (D) and mouse GC after vagotomy (E) in terms of Z-score. Z-scores were generated in IPA using datasets with differently expressed genes (p < 0.05). Pearson's test was used for correlation, and a linear regression line was drawn using GraphPad Prism v6. UVT in E: unilateral vagotomy (which results in innervated and denervated sides within the same stomach).
For study groups, see Table S12 (Study groups, Related to Figures 7K and 8G–8L).
Results
Human and mouse GC display similar metabolic reprogramming profile
Many studies on cancer metabolic reprogramming have been performed primarily in cancer cell lines to link “oncometabolites” to specific mutations of oncogenes. However, there has been a paucity of mechanistic studies in animal models of cancer investigating metabolic reprogramming, particularly any studies that have been closely linked with human studies or clinical trials (Deberardinis and Chandel, 2016). In the present study, we performed comparative transcriptomics using surgical biopsies of patients diagnosed with gastric adenocarcinoma and stomach samples from GC mice, i.e., the transgenic INS-GAS mice, which is a well-known model of spontaneous GC (Wang et al., 1996; Zhao et al., 2014; Fox and Wang, 2007). Human GC samples comprised of intestinal, diffuse, and mixed type adenocarcinoma, whereas mouse GC were predominantly of intestinal type. We compared transcriptomics profiles of the human GC tumor versus benign tissue in the same stomach and the mouse GC tumor versus normal tissue of wild-type (WT) mice. We found that the expression profile of signaling pathways was similar between human and mouse GC (Figures 1A, 1B, and 1D; Data S1. Canonical pathways in gastric cancer, Related to Figure 1). It should be noticed that the upregulated signaling pathways in both human and mouse GC included WNT/β-catenin; mTOR, PI3K/Akt, neuroinflammation, ERK/MAPK, HIPPO, and the CCK/gastrin-mediated pathway (which is specific for the stomach), and the downregulated signaling pathways included AMPK signaling, which is associated with OXPHOS, glycolysis, and fatty acid β-oxidation (Data S1). The expression profile of signaling pathways was confirmed by real-time PCR in which 89 genes related to WNT signaling pathway were measured (Table S1. Gene detected by real-time PCR and RNAseq, Related to Figure 1).
We then constructed a “GC metabolic gene expression profile” consisting of 140 genes that are involved in OXPHOS (37 genes), fatty acid β-oxidation (8 genes), carbohydrate metabolism (62 genes), and energy metabolism including the TCA cycle and glutaminolysis (34 genes). The GC metabolic gene expression profile was characterized by dysregulations of glutaminolysis and associated transporters of amino acids, the TCA cycle, carbohydrate metabolism, and fatty acid β-oxidation and displayed a positive correlation between human and mouse GC (Figures 2A, 2B, and 2D; Data S2. Metabolic genes in gastric cancer, Related to Figures 2 and 3). These results suggested that the mouse model of GC used in the present study would be useful for studying the molecular mechanisms of human GC metabolic reprogramming. Upstream regulator analysis of mouse RNA sequencing data from neoplastic lesions versus normal/healthy tissue revealed 144 regulators with increased activation whereof 8 regulators within the WNT/β-catenin signaling pathway—Tgf beta, WNT1, CD44, JUN, TGFB1, TGFBR1, TGFB2, and CTNNB1 (z-scores>2, p < 0.05)—were activated upstream of the mTOR pathway intermediates EIF3C, MRAS, PDPK1, RHOB, PPP2CA, PRKCG, and RHOA (Figure 2F; Table S2. Upstream regulators in mouse GC, Related to Figure 2F).
Figure 2.

Metabolic gene expression profiles in gastric cancer (GC)
Waterfall diagrams showing metabolic gene expression profiles of human gastric cancer (GC)(A), mouse GC (B), and mouse GC (6 months after vagotomy (VT)(C). Note: names of genes are listed in the same order in A–C and differentially expressed genes in upregulation (marked in red) and downregulation (blue). For detailed information, see Data S2. Pearson's test was used for correlation, and a linear regression line was drawn using GraphPad Prism v6. UVT in E: unilateral vagotomy (which results in innervated and denervated sides within the same stomach). Upstream regulator analysis of mouse GC transcriptomics performed in IPA revealed regulators of the WNT/β-catenin signaling pathway being upstream of mTOR signaling (F).
Orange: predicted activated; Blue: predicted inhibition; Green: downregulated; Red: upregulated; Gray: did not pass p value cut-off. Annotated with log2 fold change, p value, and z-scores, see Table S2.
Figure 3.

Glutamine-dependent gene expression profile of gastric cancer (GC) according to the single-cell atlas
tSNE plot of single-cell data released by the study of premalignant lesions and gastric cancer (Zhang et al., 2019) (A). The expression patterns of 34 TCA/glutaminolysis/gln uptake genes according to Figure 2 (B). Single-cell data were processed using Seurat v3 (https://doi.org/10.1016/j.cell.2019.05.031). Data was normalized for each of the 13 samples independently, followed by the functions FindIntegrationAnchors, IntegrateData, ScaleData, and RunPCA with default parameters. As in the original study, cells with number of expressed genes lower than 400 or larger than 7,000 were removed, and 20% or more of UMIs were mapped to mitochondrial or ribosomal genes. 50 PCs were utilized to visualize the single-cell atlas with a tSNE plot.
A single-cell atlas of gastric antral premalignant and early malignant mucosa has been recently constructed using single-cell mRNA sequencing data and visualized with t-distributed stochastic neighbor embedding (t-SNE) algorithm (Zhang et al., 2019). We performed t-SNE utilizing the gene expression data that are deposited at GEO (accession number GSE134520) (Zhang et al., 2019) and found that TCA cycle and glutaminolysis-dependent gene expression profile, particularly MDH1, MDH2, GLUL, IDH3B, DLD, SDHB, SLC17A5, SLC12A8, and OGDH, overlaid on the single-cell atlas of both cancer cells and TME (such as other proliferative cells, neck-like cells, pit mucous cells, enteroendocrine cells, T cells, and fibroblasts) (Figures 3A and 3B).
Vagotomy reverses the metabolic reprogramming of GC
Vagal innervation is known to regulate epithelial cell proliferation in the stomach and has recently been implicated in GC development and progression (Hayakawa et al., 2017; Wang et al., 2018; Zhao et al., 2014; Zahalka and Frenette, 2020). Vagal denervation can be achieved surgically, pharmacologically, or genetically. The surgery includes bilateral truncal vagotomy or unilateral truncal vagotomy (UVT). UVT takes advantage of the fact that each (anterior or posterior) vagal trunk innervates only one-half of the stomach, and consequently, UVT does not impair the overall function of the stomach. In a previous study, we showed that vagotomy during the pre-neoplastic stage of tumorigenesis diminished tumor incidence and size, and attenuated tumor cell proliferation specifically in the denervated portion of the stomach, suggesting that the vagus nerve promotes gastric cancer growth. Consistent with this idea, pharmacologic denervation via local injection of botulinum toxin A (BoNT-A) into the gastric wall similarly impaired pre-neoplastic growth. Furthermore, vagotomy or BoNT-A treatment at later stages of tumorigenesis suppressed GC progression and augmented the antitumor effect of cytotoxic chemotherapy in tumor-bearing mice, resulting in prolonged survival (Zhao et al., 2014). In the present study, we further examined the effect of vagotomy on the metabolic reprogramming of GC. In a comparison between the innervated and denervated GC mouse stomachs after UVT, the expression profiles of signaling pathways as well as the metabolic genes were reversed after vagotomy activities and displayed negative correlations between the two sides of stomach after UVT (Figure 1B versus 1C; 2B versus 2C; 1E, 2E; Data S1 and S2).
We then performed metabolomics analysis of gastric tissues in GC and WT mice that underwent UVT. By comparison of GC between innervated tumors and denervated tumors, we identified 48 dysregulated metabolites representing a “metabolic signature” of GC, and furthermore we found that the levels of metabolites, regardless of the direction of change in individual metabolites, were reversed after vagotomy to the normal levels of WT mice (Figure 4A; Table S3. Metabolite signature, Related to Figure 4; Table S13. Metabolite involved with DNA/protein syntheses, Related to Figure 4). We suggest that the metabolic signature of GC reflects the changes in both cancer cells and TME rather than specific mutations of oncogenes (i.e. “oncometabolites”). Among the metabolites in the metabolic signature of GC, some metabolites such as prostaglandin E2, methionine, and glycine are known to be abundant in GC (Uefuji et al., 2000; Wang and Dubois, 2018; Sanderson et al., 2019; Hirayama et al., 2009). Of note, the effects of vagotomy on the metabolites were different between WT and GC mice, suggesting a different response of denervation on normal tissue compared with tumor tissue (Figure 4A; Table S3; Data S3. Metabolites measured by LC/MS and GC/MS by Metabolon, Related to Figures 4 and S2A–S2D). These results corresponded well to changes in the metabolic gene expression profile, suggesting that vagotomy reversed the metabolic reprogramming of GC at both transcript and metabolite levels.
Figure 4.

Effects of vagotomy on metabolite levels in wild-type (WT) and gastric cancer (GC) mice
Heatmap showing metabolite fold changes of mouse gastric cancer (GC) versus wild-type (WT), mouse GC after unilateral vagotomy (VT), i.e. innervated versus denervated sides within the same stomach, mouse GC after VT versus WT mice, and mouse WT after VT, i.e. innervated versus denervated sides within the same stomach (A). Color key shows fold change in red (increase) or blue (decrease), generated using differently expressed metabolites (p < 0.05) in mouse GC versus WT (1) and mouse GC after VT (2) with the heatmap.2 function in RStudio version 3.5.2. For detailed information, see Table S3 and Data S3. Energy metabolism after VT (B–N): levels of metabolites in energy metabolism encompassing OXPHOS/glycolysis/Warburg effect, glutaminolysis, and TCA (B-N). Metabolites in mouse gastric cancer (GC) (marked in black), wild-type WT (blue), GC after VT (red), and WT after VT (purple). Glu: L-glutamate; Gln: L-glutamine; GSH: reduced glutathione; GSSG: oxidized glutathione; Gly: glycine; Thr: threonine; Oxo: 5-oxoproline; C-at: cis-aconitate; Glc: glucose; G6P: glucose-6-phosphate; F6P: fructose-6-phosphate.
Bars represent means of n = 6 (GC) or n = 10 (WT) relative scaled intensities with SEM and one-way ANOVA p values. For detailed information, see Table S4 and Figures S2A–S2D.
We next focused on the effects of vagotomy on tumor energy metabolism encompassing the OXPHOS/glycolysis (including the Warburg effect), glutaminolysis, and the TCA cycle (Figures 4B–4N; Table S4. Energy metabolites, Related to Figures 4B–4N). Metabolic flexibility of the tumor involves anaplerotic steps in energy metabolism (Smith et al., 2018). In comparison with WT mice, GC mice displayed an increased glutaminolytic flux through the TCA cycle, which was reflected by higher levels of glycine, oxidized glutathione (GSSG), citrate, 5-oxoproline, cis-aconitate, L-glutamate, L-glutamine, and threonine (Figures 4B, 4C, 4E, 4F, 4I, 4J, 4K, and 4N) but not the glycolysis (represented by glucose, lactate, and fructose-6-phosphate) (Figures 4G, 4L, and 4M). After vagotomy, the glutaminolysis, but not glycolysis, intermediates were reduced in GC mice (Figure 4). Comparison of GC after vagotomy versus WT without vagotomy revealed no difference in the energy metabolism (Figures 4B–4N and S2A–S2D), suggesting that vagotomy in GC mice led to a normalization of the energy metabolism. However, WT mice responded to vagotomy differently compared with GC mice, namely having reduced glutaminolysis as well as glycolysis (Figures 4B–4N and S2A–S2D. Gastric cancer is glutamine-dependent, Related to Figure 4; Table S4).
To confirm that GC was dependent on glutamine and/or pyruvate, we measured endogenous L-glutamine and L-glutamate levels in human gastric adenocarcinoma cell line (AGS cells). We found stable levels of L-glutamine and L-glutamate during a culture period of 1–24 h (Figure S2E). We further performed in vitro experiments in AGS and MKN45 cells (both human gastric adenocarcinoma cell lines). The proliferation rates of both cells were time and concentration dependent on glutamine (Figures S2, S2F, and S2G). Moreover, the cell proliferation was reduced and eventually stopped 24–72 h after depletion of glutamine but not pyruvate (Figures S2, S2H, and S2I). These results confirmed that GC cells were glutamine dependent.
Vagotomy alters neuronal, metabolic, and WNT-mTOR signaling pathways in GC
To explore the signaling pathways by which vagotomy reverses metabolic reprogramming in GC mice, we performed integrative omics (multi-omics) of transcriptomics versus metabolomics and found the signaling pathways associated with metabolism, such as synaptogenesis signaling pathway, endocannabinoid neuronal synapse pathway, role of MAPK signaling, neuroinflammation signaling pathway, glutamate receptor signaling, glutathione biosynthesis, glutamate degradation II, and UDP-N-acetyl-D-glucosamine biosynthesis II (Figure 5A; Table S5. Signaling pathways involved in mouse gastric cancer, Related to Figure 5A). We also found that metabolite-related signaling pathways, such as synaptic long-term depression, triacylglycerol biosynthesis, and CDP-diacylglycerol biosynthesis I were attenuated after vagotomy, whereas antioxidant action of vitamin C and purine nucleotides de novo biosynthesis II were activated, suggesting compensatory responses after vagotomy (Figure 5B; Table S6. Signaling pathways involved in mouse gastric cancer after vagotomy, Related to Figure 5B). Furthermore, vagotomy in GC mice reversed or restored the signaling pathways of “WT” phenotype, including TCA cycle II (eukaryotic), protein kinase A signaling, calcium signaling, gap junction signaling, and phospholipases (Figure 5C; Table S7. Signaling pathways involved in mouse gastric cancer with and without vagotomy, Related to Figure 5C). The results were in line with the signaling pathways revealed by transcriptomics showing that WNT/β-catenin signaling and mTOR signaling were inhibited after vagotomy (Data S1) and in agreement with our previous study showing that vagotomy reduced the expression of WNT-regulated stem cell markers and decreased the expansion of leucine-rich repeat containing G-protein-coupled receptor 5-positive (LGR5+) stem cells in the gastric mucosa (Zhao et al., 2014). Other reports showed that mTORC1/2 activity was associated with glutamine-dependent anaplerosis (Liao et al., 2019; Duran et al., 2012). Taken together, the results suggested that glutaminolysis, neuronal signaling, WNT/β-catenin signaling, and mTOR signaling of GC were altered by vagotomy, and thus might represent potential therapeutic targets.
Figure 5.

Multi-omics in gastric cancer (GC)
“Butterfly” diagrams showing signaling pathways (in center) that overlap with a significant Fisher's test (p < 0.05) between transcriptomics (left panel) and metabolomics datasets (right panel) in comparison between mouse gastric cancer (GC) versus wild-type (A), GC with versus without innervation (after VT) (B), and both GC versus WT and GC after VT (C).
Diagram plots were created with JavaScript library D3.js v4. For detailed information, see Tables S5–S7.
Drug-target interaction prediction shows SNAP25, mTOR, PDP1/α-KGDH, and glutaminolysis as drug targets
We next performed drug-target interaction prediction and computational drug repositioning of approved and investigational drugs/compounds (e.g. existing at the website of ClinicalTrials.gov) in GC mice and patients. We identified the network nodes (i.e. drug targets) at the levels of proteins, mRNAs, microRNA, and metabolites with special focus on the following four targets with potential drugs: SNAP25 with BoNT-A, mTOR with RAD001 (also known as Everolimus), PDP1/α-KGDH with CPI-613, and GLS with DON, 968, CB839, or BPTES in both GC mice and patients (Figures 6 and S3. Drug target prediction, Related to Figure 6). In our previous study, we have demonstrated that either local vagotomy or local injection of BoNT-A suppresses GC (Zhao et al., 2014). This was most likely because BoNT-A binds selectively to synaptosomal nerve-associated protein 25 (SNAP25), which is an integral protein required for docking and release of acetylcholine from vesicles situated in the vagal nerve endings (Naumann and Jankovic, 2004; Dressler et al., 2005). RAD001 is a rapamycin analog that specifically inhibits the mTORC1 complex by binding to FKBP12 (Faivre et al., 2006). The enzymes pyruvate dehydrogenase (PDH/PDP1) and α-ketoglutarate dehydrogenase (α-KGDH) control acetyl-CoA/pyruvate and glutamine/glutamate anaplerotic steps to the TCA cycle, respectively. The lipoate analog CPI-613 (6,8-Bis[(phenylmethyl)thio-octanoic acid) inhibits both enzymes (Dorsam and Fahrer, 2016; Lee et al., 2014; Pardee et al., 2014; Stuart et al., 2014; Zachar et al., 2011). Glutaminase inhibitors, such as CB-839, BPTES, DON, and 968, have been tested in a variety of cancers (Fung and Chan, 2017) but have limited efficacy and considerable adverse effects. It should also be noticed that the WNT signaling pathway did not appear as drug-target per se, as there are no drugs yet developed (Kahn, 2014). Thus, we tested neither the glutaminase inhibitors nor any inhibitors of the WNT signaling pathway in the present study. Next, we performed in vitro experiments to validate the efficacies of these potential metabolic-targeted therapies. Treatment of human GC cells with either RAD001 or CPI-613 reduced cell proliferation in dose-dependent manners (Figures 7A–7D). Combination of both inhibitors at IC50 doses for either 24 or 48 h resulted in synergistic inhibition (Figures 7E–7G). We found that BoNT-A alone was without any significant concentration-dependent inhibition on cell proliferation and did not enhance the inhibitory effects of neither 5-FU and/or oxaliplatin nor RAD001 and/or CPI-613 in any range of concentration responses (Figure S4. In vitro drug screening, Related to Figure 7), suggesting that the cytotoxic effect of BoNT-A on the cells does not take place in vitro. We also found that combination of RAD001 and CPI-613 had similar inhibitory effect with or without adding 5-FU and/or oxaliplatin (Figure S4).
Figure 6.

Drug target prediction and repurposing
“Waterdrop” diagrams showing drug-target. Interaction prediction and computational drug repositioning from transcriptomics data in mouse GC created in Ingenuity Pathway Analysis (IPA). Note: nodes of RAD001-targeted mTOR (marked in red), CPI-613-targeted PDP1 and/or α-KGDH (purple), BoNT-A-targeted SNAP25 (yellow), and DON/968/CB-839/BPTES-targeted GLS (light blue). Lines represent biological relationships between molecules that include proteins, genes, mRNAs, microRNAs, and metabolites, generated from differentially expressed drug target genes (only drug targets differentially expressed between mouse GC versus WT at p < 0.05, q < 0.05 are shown, light gray). Molecule nodes are fetched from RNA sequencing data in mouse GC versus WT, edges are generated based on causal information in the Ingenuity Knowledge Base. The overlay-tool in IPA is used to predict drugs for indicated nodes.
See also Figure S3.
Figure 7.

Validation of BRC treatment in vitro and in vivo
Dose- and time-dependent inhibition of proliferation in response to RAD001 and CPI-613 in MKN74 (A, C) and KATO-III cells (B, D).
Proliferation inhibition of MKN74 cells in response to either RAD001 (25 μM), CPI-613 (200 μM or 250 μM), or combinations at 24–48 h (E-F) with BLISS synergy score for each combination (G).
BLISS score >10 indicates synergistic effect. Mean of n = 3–12 replicates/treatment with SD. Two-way ANOVA between treatments (time x dose). Proliferation was measured using CCK-8 kit at 450 nm. See also Figures S2 and S4. Timeline for in vivo treatment of BoNT-A, RAD001, CPI-613 with and without FUOX over a period of two months (H).
Kaplan-Meier survival curves (I) and body weight change (J) in mouse GC (age 9–15 months) that received more than 1 cycle of treatments of BoNT-A (0.1 U/month) + RAD001 (1.5 mg/kg/day) + CPI-613 (20 mg/kg/week) (BRC), 5-fluorouracil (5 mg/kg/week) + Oxaliplatin (25 mg/kg/week) (FUOX), or BRC + FUOX or no treatment (age-matched control, AMC). Log rank (Mantel-Cox) post hoc test between groups (two-tailed). GraphPad Prism v6.
Tumor size (expressed as volume density in % of glandular area of stomach occupied by tumor) of mouse GC after 2 months treatment with indicated drugs (K). Mean ± SEM with paired t test (AMC: two-tailed, treatments: one-tailed) between anterior (denervated) and posterior (innervated) side of the stomach or non-parametric test as appropriate. Sham: Laparotomy procedure without denervation surgery.
Preclinical trial shows therapeutic effects of nerve-cancer metabolism therapy for GC
Previously, we demonstrated that gastric denervation by either vagotomy or local BoNT-A injection had similar anti-tumor effects (Zhao et al., 2014). Vagotomy can be performed at open surgery (laparotomy) or using minimally invasive surgery (laparoscopy), whereas pharmacological denervation by BoNT-A injection into the gastric wall can be achieved through gastroscopy, which is much less invasive in comparison with laparoscopic vagotomy.
Many GC patients are elderly who have poor tolerance to the current therapeutic options including subtotal or total gastrectomy with radical lymph node dissection, adjuvant chemoradiotherapy, or perioperative chemotherapy. Systemical use of cytotoxic drug treatment in elderly patients is usually associated with concerns regarding quality of life and overall survival (OS). Thus, the therapeutic strategy should be focused on the clinical endpoints, including OS and quality of life and, to lesser extent, tumor size. Thus, we performed different treatments of GC mice at 9–15 months of age and followed-up as long as the mice lived (maximal 14 months after starting treatment). Treatments included 5-FU plus oxaliplatin (named FUOX) and combinations of gastric injection of BoNT-A, RAD001, and CPI-613 (named BRC) with or without FUOX for 2 months (Figure 7H), and mice were followed-up by measuring OS, median survival (MS) time, body weight changes, and tumor size. We found that OS and MS were 33% and 148 days, respectively, in GC mice without any treatment (age-matched controls, AMC), 40% and 40 days in GC mice that received either FUOX or BRC + FUOX, but 90% and 249 days in GC mice that received BRC in comparisons with AMC (Figure 7I). Of note, the survival rates in mice with cytotoxic drugs per se (i.e. FUOX) or in combination with BRC were worse than mice without any treatment. Quality of life in mice can be measured by body weight change. FUOX induced body weight loss to the human endpoint (i.e. 25% of initial weight or less than 25% but with poor physical appearance) during the treatment period. BRC induced about 10% weight loss during the treatment and attenuated the weight loss by FUOX (Figure 7J). Within the treatment period of 2 months, 5-FU and oxaliplatin given either alone or as FUOX did not reduce the tumor size, RAD001 and CPI-613 given either alone or in combination also did not reduce the tumor size, whereas BoNT-A alone reduced the tumor size and had synergic effects when given together with FUOX or as BRC + FUOX (Figure 7K). There was no difference in tumor size between FUOX and BRC + FUOX (Figure 7K). Thus, these results suggested that BoNT-A per se had no cytotoxic effect in vivo and that BRC (BoNT-A + RAD001+CPI-613 without 5-FU and/or oxaliplatin) increased OS and MS, suggesting a potential cytotoxic chemo-free therapy for GC.
In order to verify the mechanism of action, we performed transcriptomic profiling with focus on the gene expression profile of glutaminolysis-WNT-mTOR-c-MYC signaling pathways and found that vagotomy and treatment with metabolic inhibitors with or without pharmacological denervation, i.e., RC or BRC, in GC mice for 2 months reversed the gene expression profile of glutaminolysis-WNT-mTOR-c-MYC signaling pathway, suggesting a possible mechanism of “nerve-cancer metabolism therapy” (Figures 8 and S5A–S5D. Nerve-cancer metabolism in gastric cancer, Related to Figure 8A), supporting that BRC is a nerve-cancer metabolism therapy for GC. We further analyzed the gene expression pattern of glutaminolysis-WNT-mTOR-c-MYC signaling pathway in GC mice based on the single-cell transcriptome atlas of (human) stomach (Zhang et al., 2019) and found that the upregulated gene expression was reversed after vagotomy, RC or BRC in both cancer cells and TME (e.g. T cells, B cells, macrophages, fibroblast, mast cells, and endothelial cells) (Figure 8B), suggesting that the nerve-cancer metabolism therapy acts on both cancer cells and TME in GC. Furthermore, computational network modeling revealed intensive connections across the genes within the cell type and with the genes involved in glutaminolysis (Figures 8C, 8D, and S6A–S6E. Single-cell atlas and glutamine pathways, Related to Figures 8C and 8D).
Figure 8.

Transcriptome profiling of nerve-cancer metabolism pathways and cellular compartments, computational network modeling, in silico testing, clinical analysis, and trial in GC
Heatmap of the gene expression profile of pathways including WNT/β-catenin signaling, mTOR signaling, synaptogenesis pathway, and TCA cycle in mouse gastric cancer (GC), GC after VT, RC, or BRC (A). See also Figure S5.
Heatmap of the single-cell transcriptome atlas in mouse GC, GC after VT, RC, or BRC (B). The expression levels of marker genes were analyzed in mouse GC versus WT for each representative cell type according to the single-cell atlas (Zhang et al., 2019, PMID: 31067475, GSE134520). EC, endothelial cell; GMC, antral basal gland mucous cell; EEC, enteroendocrine cell; MSC, metaplastic stem-like cell; PC, proliferative cell; PMC, pit mucous cell; NLC, neck-like cell. Percentages of total number of genes in each cell type are displayed under each cell name (smooth muscle cell not included). Gene expression on log2 fold scale; blue: downregulated; red: upregulated.
Heatmaps in A–B created in RStudio version 3.5.2 using heatmap.2-function. Computational network modeling (IPA) showing interactions within cancer cell gene cluster (C) and T cell gene cluster (D) and connections between the cell type-specific genes and genes involved in WNT/mTOR/glutaminolysis (C,D).
See also Figure S6. In silico modeling showing WNT/β-catenin signaling (left) and mTOR signaling (right) clusters of mouse GC utilizing the MAP function in IPA (E).
In silico data inhibition to predict effects of inhibition of WNT/β-catenin signaling pathway intermediates on mTOR network cluster (F, black bars) and vice versa (F, white bars). Percentages (in F) are calculated based on semi-quantitative reference values derived from predicted downstream effects generated in IPA. Means of n = 7–14 in silico tests/group.
See also Figure S7. Kaplan-Meier survival curves of 17 gastric cancer (GC) patients with low GHAI score (0–4, MS: 2420 days) and high GHAI score (5–11, MS: 949.5 days) (G).
Correlation of number of upregulated metabolic genes in patients diagnosed with gastric adenocarcinoma versus GHAI score (H), and correlation of number of upregulated metabolic genes (corresponding to Figure 2) in patients diagnosed with gastric adenocarcinoma versus survival days (I).
Note: metabolic genes are the same as in Figures 2A–2C. Volcano plots of global gene expression profiles in metaplasia (J) and neoplasia (K) in GC patients. Metabolic genes and interactions are highlighted according to regulation status (blue: downregulated; red: upregulated). Note: metabolic genes are the same as presented in Figures 2A–2C and Data S2. Tumor growth was stabilized after 20 weeks post-BoNT-A endoscopic injection in one patient as shown by CT scan (L).
WNT-signaling induces activation of mTORC1 signaling through the inhibition of GSK3β (Shimobayashi and Hall, 2014) or through induction of MYC in a CTNNB1-dependent manner (Zhang et al., 2012; Grigoryan et al., 2013). We next performed an in silico experiment to predict the effects of inhibition of nodes/genes in WNT/β-catenin network on mTOR network and vice versa, which are based on both experimental data, the Ingenuity Knowledge Base, and peer-reviewed literature. We first constructed two functional cluster networks of WNT/β-catenin and mTOR signaling based on the gene expression profile of GC mice (Figure 8E), and the in silico testing by inhibition of β-catenin (CTNNB1), c-MYC (MYC), or WNT7B either alone or in combination with other genes/nodes within the cluster showed that the inhibition of WNT/β-catenin network led to inhibition of the mTOR cluster, whereas inhibition of the mTOR kinase or mTORC1 complex either alone or in combination with other genes/nodes within the cluster was without inhibition on the WNT/β-catenin cluster (Figures 8F and S7A. In silico modeling, Related to Figure 8), probably suggesting a downstream signal flow in WNT-mTOR signaling pathways. Of note, inhibition of either Frizzled, GSK3β, or DVL nodes alone was without effect on mTOR cluster. As expected, functional cluster networks of both WNT/β-catenin and mTOR as predicted through in silico testing were inhibited 2 months after RC in GC mice (Figure S7B. In silico modeling, Related to Figure 8).
Metabolic gene expression profile in neoplasia is a target site for BRC in clinical trial
Pathogenesis of GC is believed to involve the following cascade: gastritis, atrophy, intestinal metaplasia, dysplasia, and ultimately malignant neoplasms, known as the Correa pathway (Correa, 1992). However, it remains unclear whether metaplasia is a direct precursor of GC, and if so, it should be taken together with neoplasia as targets for treatment of GC (Kinoshita et al., 2017). We have followed-up 17 patients who underwent subtotal or total gastrectomy with radical lymph node dissection, adjuvant chemoradiotherapy, or perioperative chemotherapy for 5 years. We found that patients with high scores of gastric histology activation index (GHAI) had shorter MS than those with low scores, and there was positive correlation between upregulated gene expression and GHAI score and negative correlation between upregulated gene expression profile and OS (Figures 8G–8I). Furthermore, we found distinct expression profiles in signaling pathways in general and the metabolic gene expression profiles in particular between metaplasia and neoplasia (Figures 8J and 8K), suggesting that the two pathological phenotypes harbored distinct metabolic profiles and that the network of the metabolic genes within the neoplasia should be the potential target. These results supported the rationale of BRC clinical trial.
We next carried out a pilot phase II clinical trial (https://clinicaltrials.gov/ct2/show/NCT01822210?term=BoNT-A+and+gastric+cancer&draw=2&rank=1) in which BoNT-A injection was performed through gastroscopy (without systemic administration of RAD001 plus CPI-613). The purpose of this initial clinical trial was to obtain data needed to calculate sample size in a larger controlled trial. Six enrolled patients were diagnosed as gastric adenocarcinomas with locally non-resectable and/or with distant metastasis and lack of response or non-tolerance to second-line chemotherapy (Table S8. Baseline patient data, Related to Figure 8L). We found that the procedure with BoNT-A injections was well tolerated, without any immediate surgical complications or adverse effects. Injections directly into the tumor were associated with a small amount of bleeding from the injection sites, but the bleeding was self-limited and none of the patients required surgical or endoscopic intervention or blood transfusions. We found that the tumor size was reduced during the first 8 weeks and the tumor growth was stabilized afterward in one of three patients (Figure 8L; Table S9. Primary outcome measures, Related to Figure 8L). All patients were without adverse effects or complications and discharged from hospital the first day after the procedure (Tables S10. Secondary outcome measures (short term), Related to Figure 8L; Table S11. Secondary outcome measures (long-term), Related to Figure 8L). Due to aggressive progression at advanced late-stage disease, four out of six patients did not survive until eight weeks after the BoNT-A injection. Two out of six patients were followed for eight weeks and one patient was followed for 20 weeks after receiving BoNT-A treatment. These results suggested that endoscopic injection of BoNT-A could be safe and BRC can be further tested in GC patients that failed second-line chemotherapy.
Discussion
Vagotomy was used extensively in 70s–80s as a surgical treatment for peptic ulcer, due to its inhibitory effects on gastric acid secretion (Rabben et al., 2016). Inhibition of cholinergic signaling has proved to be a possible therapeutic modality (Magnon et al., 2013) and epidemiological, animal, and clinical studies have shown that vagotomy reduces the risk of GC and suppresses gastric tumorigenesis, most likely through muscarinic cholinergic/acetylcholine receptor 3 (M3R)-mediated WNT signaling, proposed as a nerve-cancer crosstalk (Zhao et al., 2014; Hayakawa et al., 2017; Wang et al., 2018).
Cancer cells exhibit a high rate of glycolysis even in the presence of oxygen, the so-called Warburg effect, which has been well recognized as a form of metabolic reprogramming (Hanahan and Weinberg, 2011; Liberti and Locasale, 2016; Schulze and Harris, 2012; Lunt and Vander Heiden, 2011). In line with our recent understanding of tumor heterogeneity, it seems unlikely that there exists a common “metabolic reprogramming” that describes all cancer cell types and/or tumor types (including both cancer cells and TME) (Cluntun et al., 2017). It has been a dogma that GC is associated with the Warburg effect (Vander Heiden et al., 2009). In the present study, we found that the mouse GC model was well representative of human GC, particularly regarding the metabolic reprogramming, which was not associated with the Warburg effect. By comparisons between WT versus GC mice, which also included a comparison of the innervated side versus denervated sides of the same stomach of WT or GC mice, we found that inhibition of glutaminolysis and restoration of OXPHOS/glycolysis after vagotomy were the likely mechanisms underlying vagotomy-induced suppression of GC tumorigenesis. Thus, the lack of glycolytic metabolite elevations, along with a notable increase of glutaminolytic metabolites and inactivated AMPK signaling in the mouse GC model, led us to suggest that GC is glutamine dependent rather than glucose dependent, given the fact that AMPK signaling is considered to be a demand-driven regulator of glucose uptake and glycolysis (Ye and Medzhitov, 2019). This was in line with previous reports in several other cancer types including triple negative breast cancer (Sherwood et al., 2014; Sethi and Vidal-Puig, 2010; Li and Zhang, 2016; Minkler et al., 2005; Berg et al., 2002). This was also in line with reports of fluorine 18-fluorodeoxyglucose positron tomography (FDG-PET) for GC patients showing a limited value in diagnosis and evaluation, as it is designed based on increased glucose metabolism in tumor (Morgagni et al., 2020; Matzinger et al., 2009; Sprinz et al., 2018).
Through a multi-omics approach, we identified common signaling pathways that were shared between transcriptomics and metabolomics analyses. We found that among nerve-related signaling pathways, synaptogenesis signaling pathway was activated in GC and inhibited after vagotomy. This signaling pathway consists of several components involved with the nerve-cancer axis, including WNT signaling-related molecules, neurexins, neuroligins, EphB, and Trk receptors (Biederer and Stagi, 2008; Rosso et al., 2013). By transcriptomics analysis, we found that vagotomy inhibited WNT/β-catenin signaling and mTOR signaling. The WNT/β-catenin signaling pathway has been demonstrated to play prominent roles during embryonic development and adult tissue homeostasis by maintaining somatic stem cell functions (Fu et al., 2013). The mTORC1 signaling pathway has also been implicated in regulating stem cell functions in multiple tissue types (Zoncu et al., 2011). In homeostatic conditions, these pathways show a fine regulation through feedback mechanisms and are connected at multiple levels involving both upstream and downstream common effectors. For instance, activation of mTORC1 signaling could lead to suppression of WNT/β-catenin signaling through downregulating the FZD level in normal mouse intestines (Zeng et al., 2018). However, the interconnection (or feedback loop) between these two signaling pathways could be dysregulated in the case of cancer. The results of the present study suggested that the WNT/β-catenin signaling regulated the mTOR pathway in GC and might be an upstream driver of the mTOR pathway in GC. Both pathways were suppressed by vagotomy in GC and even more so by RAD001 (inhibition of mTOR) and CPI-613 (inhibition of PDH and α-KGDH). This is in line with the hypothesis that cancer is driven by dysregulated WNT/β-catenin signaling, and the relationship between WNT/β-catenin and mTORC1 pathways is so close that they should be considered as a unique therapeutic target (Prossomariti et al., 2020).
The phenotype of gastric tissue after vagotomy in GC mice appeared to be “normal” in terms of histology (Zhao et al., 2014) and metabolic profile (this study), which was associated with the signaling pathways such as the TCA cycle, protein kinase A signaling, calcium signaling, gap junction signaling, and phospholipases. Thus, these signaling pathways presented in “normalized” tissues would likely not be considered as potential therapeutic targets. Using a network integration approach for drug-target interaction prediction and computational drug repositioning, we predicted that targeting mTOR with RAD001 (everolimus), PDP1/α-KGDH with CPI-613, and SNAP25 with BoNT-A as BRC therapy would inhibit the downstream factors of signaling pathways including proteins, microRNA, and metabolites and lead to therapeutic outcomes. Indeed, the results of the present study showed that the therapeutic effects of either RC or BRC were associated with downregulation of glutaminolysis-WNT-mTOR-c-MYC signaling pathway in the cancer cells as well as the TME. It should be noticed that the metabolic reprogramming took place in the neoplasia but not in the metaplasia in patients, supporting that (1) the metabolic reprogramming is reflected by cancer cells as well as TME; (2) that the metabolic properties evolve during tumor progression, and (3) that the site of neoplasia is ideal location for injection of BoNT-A.
Recent findings in immunometabolism have shown that the effects of cancer cell metabolism on the TME may involve direct modulation of essential T cell metabolic pathways and activities and suggested a “metabolic checkpoint” for tumor immunotherapy, in which effector T cells responded to glutamine antagonism by markedly upregulating oxidative metabolism and adopting a long-lived, highly activated phenotype (Leone et al., 2019). It was also reported that inhibiting glutamine metabolism of myeloid-derived suppressor cells (MDSCs) led to activation-induced cell death and conversion of MDSCs to inflammatory macrophages and suggested that myeloid cells comprised a major component of TME, promoting tumor growth and immune evasion (Oh et al., 2020). The success of immunotherapy in GC has to date been limited in part by the lack of knowledge on gastric-specific TME (Subhash et al., 2015). A recent study showed that a gastric-specific TME atlas consisted of the gene expression pattern in connection with a variety of resident and infiltrating host cells (such as endothelial cells, enterocytes, chief cells, antral basal gland cells, metaplastic stem-like cells, pit mucous cells, enteroendocrine cells, fibroblasts, T cells, B cells, mast cells, and microphages) (Zhang et al., 2019). According to the atlas, the results of the present study further showed that the gene expression of immune/inflammatory cells, such as T cells, B cells, macrophages, and mast cells in TME of GC was reversed together with the metabolic reprogramming after vagotomy or RC or BRC. Presumably, the therapeutic strategy should be to enhance the robustness of GC immunotherapy by the “nerve-cancer metabolism therapy” that was presented in the present study.
GC accounts for the third highest cancer-related disability-adjusted life-years (DALYs) after lung and liver cancers (Collaborators, 2020), in addition to its high incidence and mortality. Although H. pylori infection is declining, the trends toward increased obesity and aging of the population will likely result in a continued high incidence of GC. Thus, less invasive and better tolerated therapies need to be developed for the treatment of elderly patients with GC. Based on the successful progress in the treatment of gastric cancer in Japan over the last 50 years, it was suggested that endoscopic submucosal dissection (ESD) combined with “gentler” chemotherapy or immunotherapy could be applied to more than half of the GC patients (Sasako, 2020). The results of the present study indicated that endoscopic submucosal/intratumoral injection of BoNT-A combined with non-cytotoxic chemotherapy could be an ideal therapy for the elderly patients. In the present study, we choose the non-cytotoxic drugs, RAD001 (also known as everolimus) and CPI-613 (also known as devimistat), as they have been well tested in clinical trials for other types of cancer (Kim et al., 2017, 2018; Chung et al., 2016; Pardee et al., 2018; Alistar et al., 2017).
In addition to GC, nerve-cancer crosstalk takes place in other types of cancer, e.g. prostate cancer, colorectal cancer, pancreatic cancer, and breast cancer (Chen and Ayala, 2018; Zahalka et al., 2017; Renz et al., 2018; Dubeykovskaya et al., 2016; Kamiya et al., 2019; Mauffrey et al., 2019). More studies are needed to investigate the underlying mechanisms, along with the metabolic reprogramming and immunometabolism, and to develop the nerve-cancer metabolism therapy.
In conclusion, the nature of the present study was translational in order to develop new treatment that is closely linked with clinical trials. Based on the results of the present study, we suggested that GC (including both the cancer cells and TME) was glutamine dependent with altered neuronal and metabolic signaling pathways; vagotomy and metabolic inhibitors reversed the metabolic reprogramming in GC; WNT-mTOR signaling pathway played an important role in the metabolic switch between oxidative phosphorylation/glycolysis and glutaminolysis in GC; SNAP25, mTOR, PDP1/α-KGDH, and glutaminolysis were potential drug-targets for treatment of GC; and intratumoral injection of BoNT-A with systemic administration of RAD001(everolimus) and CPI-613 (devimistat) can be a potential therapy for GC. The potential therapy was particular for elderly patients with clinical endpoints of increased OS and QoF, which have been established according to the regulation of European Commission. The treatment methods used in the present study were commonly considered having no/little stress and abdominal pain, i.e. injections of BoNT-A (through gastroscopy in patients and laparotomy in mice) and RAD001 and CPI-613 (i.v. in patients and i.p. in mice). In the present study, the long-term follow-up (14 months after starting treatment in mice) showed that OS was 33% without any treatment, 40% with chemotherapy (cytotoxic drugs), but 90% with the new treatment (without cytotoxic drugs).
Limitations of the study
The so-called “metabolic escape” has been suggested as a mechanism by cancer cells to avoid cell death in response to inhibited glutaminolysis. Thus, tumors that suffer from glucose/glutamine starvation frequently activate fatty acid catabolism for survival (Halama et al., 2018; Wise et al., 2008; Li and Zhang, 2016). The results of the present study might suggest that the metabolic escape takes place after vagotomy, leading to an activation of Acetyl-CoA with increased levels of lysolipids and polyunsaturated fatty acids in GC but not in WT mice. Furthermore, acyl carnitine oleoylcarnitine, a long-chain acyl carnitine that accumulates during certain metabolic conditions, such as fasting and nutrient deficiency (Minkler et al., 2005), was increased after vagotomy along with its transporter SLC25A20, probably supporting the notion that acyl carnitines serves to deliver fatty acids to the mitochondria for β-oxidation to produce acetyl-CoA (Berg et al., 2002). Monoacylglycerol 1-stearoylglycerol (1-monostearin) was increased after vagotomy in GC but not in WT mice, probably further suggesting that vagotomy-induced suppression of tumorigenesis was mediated in part through accelerated degradation of diacyl- or triacylglyserols, as well as deoxycarnitine, succinylcarnitine, and 3-dehydrocarnitine. These assumptions need to be further investigated.
Resource availability
Lead contact
Information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Chun-Mei Zhao (chun-mei.zhao@ntnu.no).
Materials availability
This study did not generate new unique reagents.
Data and code availability
All relevant data are available from the Lead Contact upon request. The mouse RNA seq/microarray data (related to Data S1–S3) have been deposited in the NCBI Bioproject database under the accession number PRJNA690520, which can be accessed using the following link: http://www.ncbi.nlm.nih.gov/bioproject/690520, and in the GEO under accession number GSE30295, respectively. The human microarray data (related to Data S1 and S2) are available online via Mendeley Data repository with DOI link at https://doi.org/10.17632/hzmfshy7hp.1.
Methods
All methods can be found in the accompanying Transparent methods supplemental file.
Acknowledgments
The authors thank the grant supports by the Liaison Committee between the Central Norway Regional Health Authority (Helse-Midt Norge RHF) and Norwegian University of Science and Technology (NTNU), Norway (grant numbers 46056636/46056928/90061700/90061701), Joint Program of the Medical Faculty of NTNU and St. Olavs University Hospital, the Cancer Foundation of St. Olavs Hospital (Kreftfondet ved St. Olavs hospital), and the technical support by Genomics Core Facility (GCF), which is funded by the Faculty of Medicine and Health Sciences at NTNU and RHF.
Authors contribution
H-L.R. and G.T.A.: in vitro, in vivo, and in silico experiments, sample/data collection and preparation, data analysis and interpretation, writing manuscript. M.K.O.: in vivo, experiments, writing manuscript. A.I. and D.K.: data visualization and drug synergy prediction, writing manuscript. A.Ø.: in vitro experiments, writing manuscript. T.C.W.: data acquisitioning, writing manuscript. S.L.: clinical interpretation, writing manuscript. J.E.G.: clinical samples and data collection, writing manuscript. D.C.: project concept, study idea and design, data analysis and interpretation, writing manuscript. C-M.Z.: project concept, study idea and design, lab and clinical experiments, data interpretation, writing manuscript.
Declaration of interests
The authors declare no competing interests.
Published: February 19, 2021
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2021.102091.
Supplemental information
References
- Alistar A., Morris B.B., Desnoyer R., Klepin H.D., Hosseinzadeh K., Clark C., Cameron A., Leyendecker J., D'agostino R., Jr., Topaloglu U. Safety and tolerability of the first-in-class agent CPI-613 in combination with modified FOLFIRINOX in patients with metastatic pancreatic cancer: a single-centre, open-label, dose-escalation, phase 1 trial. Lancet Oncol. 2017;18:770–778. doi: 10.1016/S1470-2045(17)30314-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg J.M., Tymoczko J.L., Stryer L. W.H.Freeman & Co Ltd; 2002. Biochemistry: The Utilization of Fatty Acids as Fuel Requires Three Stages of Processing. [Google Scholar]
- Biederer T., Stagi M. Signaling by synaptogenic molecules. Curr. Opin. Neurobiol. 2008;18:261–269. doi: 10.1016/j.conb.2008.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D., Ayala G.E. Innervating prostate cancer. N. Engl. J. Med. 2018;378:675–677. doi: 10.1056/NEJMcibr1714003. [DOI] [PubMed] [Google Scholar]
- Chung V., Frankel P., Lim D., Yeon C., Leong L., Chao J., Ruel N., Luevanos E., Koehler S., Chung S. Phase Ib trial of mFOLFOX6 and everolimus (NSC-733504) in patients with metastatic gastroesophageal adenocarcinoma. Oncology. 2016;90:307–312. doi: 10.1159/000445297. [DOI] [PubMed] [Google Scholar]
- Cluntun A.A., Lukey M.J., Cerione R.A., Locasale J.W. Glutamine metabolism in cancer: understanding the heterogeneity. Trends Cancer. 2017;3:169–180. doi: 10.1016/j.trecan.2017.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaborators G.B.D.S.C. The global, regional, and national burden of stomach cancer in 195 countries, 1990-2017: a systematic analysis for the Global Burden of Disease study 2017. Lancet Gastroenterol. Hepatol. 2020;5:42–54. doi: 10.1016/S2468-1253(19)30328-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coller H.A. Is cancer a metabolic disease? Am. J. Pathol. 2014;184:4–17. doi: 10.1016/j.ajpath.2013.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correa P. Human gastric carcinogenesis: a multistep and multifactorial process--first American cancer society award lecture on cancer epidemiology and prevention. Cancer Res. 1992;52:6735–6740. [PubMed] [Google Scholar]
- Deberardinis R.J., Chandel N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016;2:e1600200. doi: 10.1126/sciadv.1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsam B., Fahrer J. The disulfide compound alpha-lipoic acid and its derivatives: a novel class of anticancer agents targeting mitochondria. Cancer Lett. 2016;371:12–19. doi: 10.1016/j.canlet.2015.11.019. [DOI] [PubMed] [Google Scholar]
- Dressler D., Saberi F.A., Barbosa E.R. Botulinum toxin: mechanisms of action. Arq Neuropsiquiatr. 2005;63:180–185. doi: 10.1590/s0004-282x2005000100035. [DOI] [PubMed] [Google Scholar]
- Dubeykovskaya Z., Si Y., Chen X., Worthley D.L., Renz B.W., Urbanska A.M., Hayakawa Y., Xu T., Westphalen C.B., Dubeykovskiy A. Neural innervation stimulates splenic TFF2 to arrest myeloid cell expansion and cancer. Nat. Commun. 2016;7:10517. doi: 10.1038/ncomms10517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran R.V., Oppliger W., Robitaille A.M., Heiserich L., Skendaj R., Gottlieb E., Hall M.N. Glutaminolysis activates Rag-mTORC1 signaling. Mol. Cell. 2012;47:349–358. doi: 10.1016/j.molcel.2012.05.043. [DOI] [PubMed] [Google Scholar]
- Faivre S., Kroemer G., Raymond E. Current development of mTOR inhibitors as anticancer agents. Nat. Rev. Drug Discov. 2006;5:671–688. doi: 10.1038/nrd2062. [DOI] [PubMed] [Google Scholar]
- Fox J.G., Wang T.C. Inflammation, atrophy, and gastric cancer. J. Clin. Invest. 2007;117:60–69. doi: 10.1172/JCI30111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y., Huang B., Shi Z., Han J., Wang Y., Huangfu J., Wu W. SRSF1 and SRSF9 RNA binding proteins promote Wnt signalling-mediated tumorigenesis by enhancing beta-catenin biosynthesis. EMBO Mol. Med. 2013;5:737–750. doi: 10.1002/emmm.201202218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung M.K.L., Chan G.C.-F. Drug-induced amino acid deprivation as strategy for cancer therapy. J. Hematol. Oncol. 2017;10:144. doi: 10.1186/s13045-017-0509-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoryan T., Stein S., Qi J., Wende H., Garratt A.N., Nave K.A., Birchmeier C., Birchmeier W. Wnt/Rspondin/beta-catenin signals control axonal sorting and lineage progression in Schwann cell development. Proc. Natl. Acad. Sci. U S A. 2013;110:18174–18179. doi: 10.1073/pnas.1310490110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halama A., Kulinski M., Dib S.S., Zaghlool S.B., Siveen K.S., Iskandarani A., Zierer J., Prabhu K.S., Satheesh N.J., Bhagwat A.M. Accelerated lipid catabolism and autophagy are cancer survival mechanisms under inhibited glutaminolysis. Cancer Lett. 2018;430:133–147. doi: 10.1016/j.canlet.2018.05.017. [DOI] [PubMed] [Google Scholar]
- Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hayakawa Y., Sakitani K., Konishi M., Asfaha S., Niikura R., Tomita H., Renz B.W., Tailor Y., Macchini M., Middelhoff M. Nerve growth factor promotes gastric tumorigenesis through aberrant cholinergic signaling. Cancer Cell. 2017;31:21–34. doi: 10.1016/j.ccell.2016.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirayama A., Kami K., Sugimoto M., Sugawara M., Toki N., Onozuka H., Kinoshita T., Saito N., Ochiai A., Tomita M. Quantitative metabolome profiling of colon and stomach cancer microenvironment by capillary electrophoresis time-of-flight mass spectrometry. Cancer Res. 2009;69:4918–4925. doi: 10.1158/0008-5472.CAN-08-4806. [DOI] [PubMed] [Google Scholar]
- Jobling P., Pundavela J., Oliveira S.M., Roselli S., Walker M.M., Hondermarck H. Nerve-cancer cell cross-talk: a novel promoter of tumor progression. Cancer Res. 2015;75:1777–1781. doi: 10.1158/0008-5472.CAN-14-3180. [DOI] [PubMed] [Google Scholar]
- Kahn M. Can we safely target the WNT pathway? Nat. Rev. Drug Discov. 2014;13:513–532. doi: 10.1038/nrd4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiya A., Hayama Y., Kato S., Shimomura A., Shimomura T., Irie K., Kaneko R., Yanagawa Y., Kobayashi K., Ochiya T. Genetic manipulation of autonomic nerve fiber innervation and activity and its effect on breast cancer progression. Nat. Neurosci. 2019;22:1289–1305. doi: 10.1038/s41593-019-0430-3. [DOI] [PubMed] [Google Scholar]
- Kim H.S., Shaib W.L., Zhang C., Nagaraju G.P., Wu C., Alese O.B., Chen Z., Brutcher E., Renfroe M., El-Rayes B.F. Phase 1b study of pasireotide, everolimus, and selective internal radioembolization therapy for unresectable neuroendocrine tumors with hepatic metastases. Cancer. 2018;124:1992–2000. doi: 10.1002/cncr.31192. [DOI] [PubMed] [Google Scholar]
- Kim S.T., Lee J., Park S.H., Park J.O., Park Y.S., Kang W.K., Lim H.Y. Prospective phase II trial of everolimus in PIK3CA amplification/mutation and/or PTEN loss patients with advanced solid tumors refractory to standard therapy. BMC Cancer. 2017;17:211. doi: 10.1186/s12885-017-3196-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita H., Hayakawa Y., Koike K. Metaplasia in the stomach-precursor of gastric cancer? Int. J. Mol. Sci. 2017;18:2063. doi: 10.3390/ijms18102063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K.C., Maturo C., Perera C.N., Luddy J., Rodriguez R., Shorr R. Translational assessment of mitochondrial dysfunction of pancreatic cancer from in vitro gene microarray and animal efficacy studies, to early clinical studies, via the novel tumor-specific anti-mitochondrial agent, CPI-613. Ann. Transl Med. 2014;2:91. doi: 10.3978/j.issn.2305-5839.2014.05.08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leone R.D., Zhao L., Englert J.M., Sun I.M., Oh M.H., Sun I.H., Arwood M.L., Bettencourt I.A., Patel C.H., Wen J. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science. 2019;366:1013–1021. doi: 10.1126/science.aav2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z., Zhang H. Reprogramming of glucose, fatty acid and amino acid metabolism for cancer progression. Cell Mol. Life Sci. 2016;73:377–392. doi: 10.1007/s00018-015-2070-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao G.Y., Lee M.T., Fan J.J., Hsiao P.W., Lee C.S., Su S.Y., Hwang J.J., Ke F.C. Blockage of glutamine-dependent anaplerosis affects mTORC1/2 activity and ultimately leads to cellular senescence-like response. Biol. Open. 2019;8:bio038257. doi: 10.1242/bio.038257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberti M.V., Locasale J.W. The Warburg effect: how does it benefit cancer cells? Trends Biochem. Sci. 2016;41:211–218. doi: 10.1016/j.tibs.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loponte S., Lovisa S., Deem A.K., Carugo A., Viale A. The many facets of tumor heterogeneity: is metabolism lagging behind? Cancers (Basel) 2019;11:1574. doi: 10.3390/cancers11101574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunt S.Y., Vander Heiden M.G. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011;27:441–464. doi: 10.1146/annurev-cellbio-092910-154237. [DOI] [PubMed] [Google Scholar]
- Magnon C., Hall S.J., Lin J., Xue X., Gerber L., Freedland S.J., Frenette P.S. Autonomic nerve development contributes to prostate cancer progression. Science. 2013;341:1236361. doi: 10.1126/science.1236361. [DOI] [PubMed] [Google Scholar]
- Matsuda T., Saika K. The 5-year relative survival rate of stomach cancer in the USA, Europe and Japan. Jpn. J. Clin. Oncol. 2013;43:1157–1158. doi: 10.1093/jjco/hyt166. [DOI] [PubMed] [Google Scholar]
- Matzinger O., Gerber E., Bernstein Z., Maingon P., Haustermans K., Bosset J.F., Gulyban A., Poortmans P., Collette L., Kuten A. EORTC-ROG expert opinion: radiotherapy volume and treatment guidelines for neoadjuvant radiation of adenocarcinomas of the gastroesophageal junction and the stomach. Radiother. Oncol. 2009;92:164–175. doi: 10.1016/j.radonc.2009.03.018. [DOI] [PubMed] [Google Scholar]
- Mauffrey P., Tchitchek N., Barroca V., Bemelmans A., Firlej V., Allory Y., Romeo P.H., Magnon C. Progenitors from the central nervous system drive neurogenesis in cancer. Nature. 2019;569:672–678. doi: 10.1038/s41586-019-1219-y. [DOI] [PubMed] [Google Scholar]
- Minkler P.E., Kerner J., North K.N., Hoppel C.L. Quantitation of long-chain acylcarnitines by HPLC/fluorescence detection: application to plasma and tissue specimens from patients with carnitine palmitoyltransferase-II deficiency. Clin. Chim. Acta. 2005;352:81–92. doi: 10.1016/j.cccn.2004.02.004. [DOI] [PubMed] [Google Scholar]
- Morgagni P., Bencivenga M., Colciago E., Tringali D., Giacopuzzi S., Framarini M., Saragoni L., Mura G., Graziosi L., Marino E. Limited usefulness of 18F-FDG PET/CT in predicting tumor regression after preoperative chemotherapy for noncardia gastric cancer: the Italian research group for gastric cancer (GIRCG) experience. Clin. Nucl. Med. 2020;45:177–181. doi: 10.1097/RLU.0000000000002911. [DOI] [PubMed] [Google Scholar]
- Naumann M., Jankovic J. Safety of botulinum toxin type A: a systematic review and meta-analysis. Curr. Med. Res. Opin. 2004;20:981–990. doi: 10.1185/030079904125003962. [DOI] [PubMed] [Google Scholar]
- Oh M.H., Sun I.H., Zhao L., Leone R.D., Sun I.M., Xu W., Collins S.L., Tam A.J., Blosser R.L., Patel C.H. Targeting glutamine metabolism enhances tumor specific immunity by modulating suppressive myeloid cells. J. Clin. Invest. 2020;130:3865–3884. doi: 10.1172/JCI131859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardee T.S., Anderson R.G., Pladna K.M., Isom S., Ghiraldeli L.P., Miller L.D., Chou J.W., Jin G., Zhang W., Ellis L.R. A phase I study of CPI-613 in combination with high-dose cytarabine and mitoxantrone for relapsed or refractory acute myeloid leukemia. Clin. Cancer Res. 2018;24:2060–2073. doi: 10.1158/1078-0432.CCR-17-2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardee T.S., Lee K., Luddy J., Maturo C., Rodriguez R., Isom S., Miller L.D., Stadelman K.M., Levitan D., Hurd D. A phase I study of the first-in-class antimitochondrial metabolism agent, CPI-613, in patients with advanced hematologic malignancies. Clin. Cancer Res. 2014;20:5255–5264. doi: 10.1158/1078-0432.CCR-14-1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkin D.M., Bray F., Ferlay J., Pisani P. Global cancer statistics, 2002. CA Cancer J. Clin. 2005;55:74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- Pavlova N.N., Thompson C.B. The emerging hallmarks of cancer metabolism. Cell Metab. 2016;23:27–47. doi: 10.1016/j.cmet.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prossomariti A., Piazzi G., Alquati C., Ricciardiello L. Are Wnt/beta-catenin and PI3K/AKT/mTORC1 distinct pathways in colorectal cancer? Cell Mol. Gastroenterol. Hepatol. 2020;10:491–506. doi: 10.1016/j.jcmgh.2020.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabben H.L., Zhao C.M., Hayakawa Y., Wang T.C., Chen D. Vagotomy and gastric tumorigenesis. Curr. Neuropharmacol. 2016;14:967–972. doi: 10.2174/1570159X14666160121114854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawla P., Barsouk A. Epidemiology of gastric cancer: global trends, risk factors and prevention. Prz Gastroenterol. 2019;14:26–38. doi: 10.5114/pg.2018.80001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renz B.W., Takahashi R., Tanaka T., Macchini M., Hayakawa Y., Dantes Z., Maurer H.C., Chen X., Jiang Z., Westphalen C.B. beta2 adrenergic-neurotrophin feedforward loop promotes pancreatic cancer. Cancer Cell. 2018;33:75–90.e7. doi: 10.1016/j.ccell.2017.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosso S., Inestrosa N., Rosso S. WNT signaling in neuronal maturation and synaptogenesis. Front. Cell Neurosci. 2013;7:103. doi: 10.3389/fncel.2013.00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson S.M., Gao X., Dai Z., Locasale J.W. Methionine metabolism in health and cancer: a nexus of diet and precision medicine. Nat. Rev. Cancer. 2019;19:625–637. doi: 10.1038/s41568-019-0187-8. [DOI] [PubMed] [Google Scholar]
- Sasako M. Progress in the treatment of gastric cancer in Japan over the last 50 years. Ann. Gastroenterol. Surg. 2020;4:21–29. doi: 10.1002/ags3.12306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze A., Harris A.L. How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature. 2012;491:364–373. doi: 10.1038/nature11706. [DOI] [PubMed] [Google Scholar]
- Sethi J.K., Vidal-Puig A. Wnt signalling and the control of cellular metabolism. Biochem. J. 2010;427:1–17. doi: 10.1042/BJ20091866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seyfried T.N., Flores R.E., Poff A.M., D'agostino D.P. Cancer as a metabolic disease: implications for novel therapeutics. Carcinogenesis. 2014;35:515–527. doi: 10.1093/carcin/bgt480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherwood V., Chaurasiya S.K., Ekstrom E.J., Guilmain W., Liu Q., Koeck T., Brown K., Hansson K., Agnarsdottir M., Bergqvist M. WNT5A-mediated beta-catenin-independent signalling is a novel regulator of cancer cell metabolism. Carcinogenesis. 2014;35:784–794. doi: 10.1093/carcin/bgt390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimobayashi M., Hall M.N. Making new contacts: the mTOR network in metabolism and signalling crosstalk. Nat. Rev. Mol. Cell Biol. 2014;15:155–162. doi: 10.1038/nrm3757. [DOI] [PubMed] [Google Scholar]
- Smith R.L., Soeters M.R., Wüst R.C.I., Houtkooper R.H. Metabolic flexibility as an adaptation to energy resources and requirements in health and disease. Endocr. Rev. 2018;39:489–517. doi: 10.1210/er.2017-00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprinz C., Altmayer S., Zanon M., Watte G., Irion K., Marchiori E., Hochhegger B. Effects of blood glucose level on 18F-FDG uptake for PET/CT in normal organs: a systematic review. PLoS One. 2018;13:e0193140. doi: 10.1371/journal.pone.0193140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart S.D., Schauble A., Gupta S., Kennedy A.D., Keppler B.R., Bingham P.M., Zachar Z. A strategically designed small molecule attacks alpha-ketoglutarate dehydrogenase in tumor cells through a redox process. Cancer Metab. 2014;2:4. doi: 10.1186/2049-3002-2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subhash V.V., Yeo M.S., Tan W.L., Yong W.P. Strategies and advancements in harnessing the immune system for gastric cancer immunotherapy. J. Immunol. Res. 2015;2015:308574. doi: 10.1155/2015/308574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uefuji K., Ichikura T., Mochizuki H. Cyclooxygenase-2 expression is related to prostaglandin biosynthesis and angiogenesis in human gastric cancer. Clin. Cancer Res. 2000;6:135–138. [PubMed] [Google Scholar]
- Vander Heiden M.G., Cantley L.C., Thompson C.B. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden M.G., Deberardinis R.J. Understanding the intersections between metabolism and cancer biology. Cell. 2017;168:657–669. doi: 10.1016/j.cell.2016.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D., Dubois R.N. Role of prostanoids in gastrointestinal cancer. J. Clin. Invest. 2018;128:2732–2742. doi: 10.1172/JCI97953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Xu J., Xia Y., Yin K., Li Z., Li B., Wang W., Xu H., Yang L., Xu Z. Muscarinic acetylcholine receptor 3 mediates vagus nerve-induced gastric cancer. Oncogenesis. 2018;7:88. doi: 10.1038/s41389-018-0099-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T.C., Koh T.J., Varro A., Cahill R.J., Dangler C.A., Fox J.G., Dockray G.J. Processing and proliferative effects of human progastrin in transgenic mice. J. Clin. Invest. 1996;98:1918–1929. doi: 10.1172/JCI118993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteside T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene. 2008;27:5904–5912. doi: 10.1038/onc.2008.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise D.R., Deberardinis R.J., Mancuso A., Sayed N., Zhang X.Y., Pfeiffer H.K., Nissim I., Daikhin E., Yudkoff M., Mcmahon S.B. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. U S A. 2008;105:18782–18787. doi: 10.1073/pnas.0810199105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart D.S. Is cancer a genetic disease or a metabolic disease? EBioMedicine. 2015;2:478–479. doi: 10.1016/j.ebiom.2015.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J., Medzhitov R. Control strategies in systemic metabolism. Nat. Metab. 2019;1:947–957. doi: 10.1038/s42255-019-0118-8. [DOI] [PubMed] [Google Scholar]
- Yoshida G.J. Metabolic reprogramming: the emerging concept and associated therapeutic strategies. J. Exp. Clin. Cancer Res. 2015;34:111. doi: 10.1186/s13046-015-0221-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachar Z., Marecek J., Maturo C., Gupta S., Stuart S.D., Howell K., Schauble A., Lem J., Piramzadian A., Karnik S. Non-redox-active lipoate derivates disrupt cancer cell mitochondrial metabolism and are potent anticancer agents in vivo. J. Mol. Med. (Berl) 2011;89:1137–1148. doi: 10.1007/s00109-011-0785-8. [DOI] [PubMed] [Google Scholar]
- Zahalka A.H., Arnal-Estape A., Maryanovich M., Nakahara F., Cruz C.D., Finley L.W.S., Frenette P.S. Adrenergic nerves activate an angio-metabolic switch in prostate cancer. Science. 2017;358:321–326. doi: 10.1126/science.aah5072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahalka A.H., Frenette P.S. Nerves in cancer. Nat. Rev. Cancer. 2020;20:143–157. doi: 10.1038/s41568-019-0237-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng H., Lu B., Zamponi R., Yang Z., Wetzel K., Loureiro J., Mohammadi S., Beibel M., Bergling S., Reece-Hoyes J. mTORC1 signaling suppresses Wnt/beta-catenin signaling through DVL-dependent regulation of Wnt receptor FZD level. Proc. Natl. Acad. Sci. U S A. 2018;115:E10362–E10369. doi: 10.1073/pnas.1808575115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P., Yang M., Zhang Y., Xiao S., Lai X., Tan A., Du S., Li S. Dissecting the single-cell transcriptome network underlying gastric premalignant lesions and early gastric cancer. Cell Rep. 2019;27:1934–1947 e5. doi: 10.1016/j.celrep.2019.04.052. [DOI] [PubMed] [Google Scholar]
- Zhang S., Li Y., Wu Y., Shi K., Bing L., Hao J. Wnt/beta-catenin signaling pathway upregulates c-Myc expression to promote cell proliferation of P19 teratocarcinoma cells. Anat. Rec. (Hoboken) 2012;295:2104–2113. doi: 10.1002/ar.22592. [DOI] [PubMed] [Google Scholar]
- Zhao C.M., Hayakawa Y., Kodama Y., Muthupalani S., Westphalen C.B., Andersen G.T., Flatberg A., Johannessen H., Friedman R.A., Renz B.W. Denervation suppresses gastric tumorigenesis. Sci. Transl. Med. 2014;6:250ra115. doi: 10.1126/scitranslmed.3009569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoncu R., Efeyan A., Sabatini D.M. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All relevant data are available from the Lead Contact upon request. The mouse RNA seq/microarray data (related to Data S1–S3) have been deposited in the NCBI Bioproject database under the accession number PRJNA690520, which can be accessed using the following link: http://www.ncbi.nlm.nih.gov/bioproject/690520, and in the GEO under accession number GSE30295, respectively. The human microarray data (related to Data S1 and S2) are available online via Mendeley Data repository with DOI link at https://doi.org/10.17632/hzmfshy7hp.1.