

Abstract
The focus of this review is the human de novo purine biosynthetic pathway. The pathway enzymes are enumerated, as well as the reactions they catalyze and their physical properties. Early literature evidence suggested that they might assemble into a multi-enzyme complex called a metabolon. The finding that fluorescently-tagged chimeras of the pathway enzymes form discrete puncta, now called purinosomes, is further elaborated in this review to include: a discussion of their assembly; the role of ancillary proteins; their locus at the microtubule/mitochondria interface; the elucidation that at endogenous levels, purinosomes function to channel intermediates from phosphoribosyl pyrophosphate to AMP and GMP; and the evidence for the purinosomes to exist as a protein condensate. The review concludes with a consideration of probable signaling pathways that might promote the assembly and disassembly of the purinosome, in particular the identification of candidate kinases given the extensive phosphorylation of the enzymes. These collective findings substantiate our current view of the de novo purine biosynthetic metabolon whose properties will be representative of how other metabolic pathways might be organized for their function.
Keywords: metabolism, de novo purine biosynthesis, purinosome, metabolon, substrate channeling, condensate, signaling
Purines are ubiquitous biomolecules that sustain life. Purines are incorporated into DNA and RNA, found as the energy currency of cells (ATP and GTP), used as signaling molecules (ATP, cAMP, and cGMP), and integrated into coenzymes (FAD, NAD+, NADP+, and coenzyme A). These purines are generated by either or both of the two pathways: de novo purine biosynthesis (DNPB) or purine salvage (Lane and Fan, 2015; Pedley and Benkovic, 2017) (Fig 1A). The demand for purines oftentimes exceeds the capacity of the complementary salvage pathway and might result in activating DNPB. Dysregulation of purine metabolism has been associated with many cancers, gout, neuropathologies, and immunological defects (Garcia-Gil et al., 2018; Jinnah, 2009; Jurecka et al., 2015; Marie et al., 2004; Vander Heiden and DeBerardinis, 2017; Villa et al., 2019); thus, propelling the development of effective anti-cancer (Christopherson, Lyons, and Wilson, 2002; Parker, 2009; Yin et al., 2018), anti-infective (Hanrahan and Hutchinson, 1992; Thomson and Lamont, 2019), and anti-inflammatory therapeutics (Hartsel et al., 1997). Rather than a broad overview of the many aspects of purine biochemistry, we will narrow the focus of this review to the human de novo biosynthesis of purines and eschew discussion of the complementary salvage pathway that draws on the recycling of degraded DNA/RNA to regenerate purines.
Figure 1. Purine salvage and de novo purine biosynthesis (DNPB).
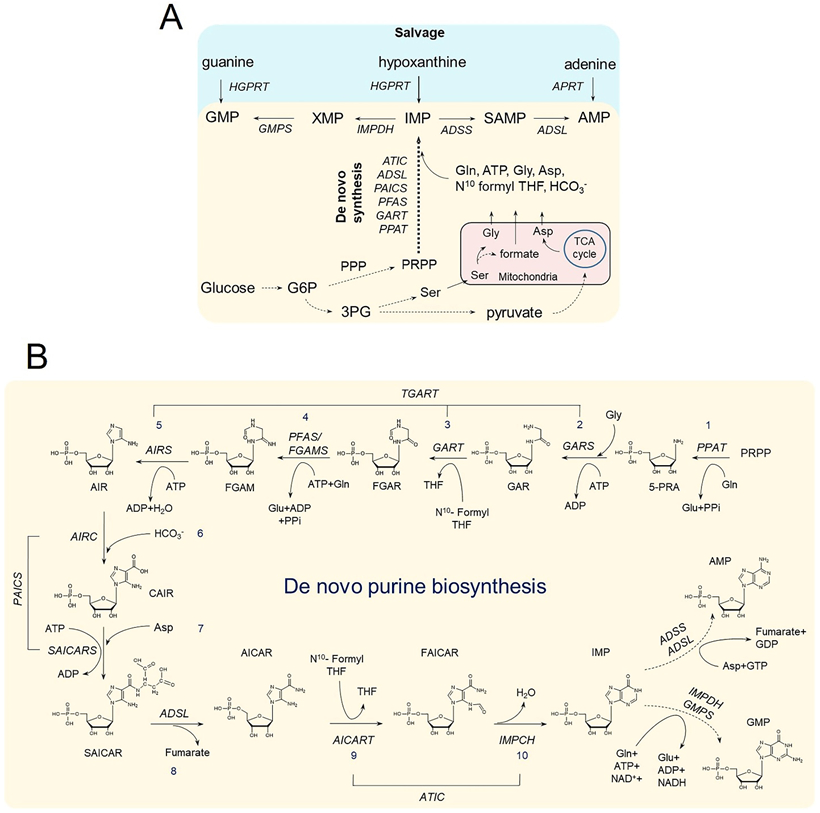
(A) Purines are regenerated either by salvage from the bases hypoxanthine, guanine, and adenine; or synthesized de novo from PRPP, utilizing building block substrates generated by other metabolic processes. Glucose metabolism generates- 1) Glucose-6-phosphate (G6P) that can generate PRPP via the pentose phosphate pathway (PPP); 2) 3-phosphoglycerate (3PG) that can generate Ser via the serine biosynthesis pathway; and 3) pyruvate that fuels the TCA cycle inside mitochondria, which in turn maintains a stable supply of aspartic acid. Conversion of Ser to Gly and formate is carried out inside mitochondria and requires the involvement of one carbon metabolism enzymes. Enzyme names are represented in italicized letters. (B) Schematic shows the step by step conversion of PRPP to AMP and GMP constituting the DNPB pathway. A cascade of 10 reactions catalyzed by the six DNPB enzymes produces IMP, which is either converted to AMP or GMP in two additional each way. It is an energy intensive pathway that requires three amino acids- Gly, Asp, Gln- and the cofactor N10-formyl tetrahydrofolate (N10- formyl THF). All the purine intermediate and enzyme acronym expansions are defined in the text.
Human de novo Purine Biosynthesis
In humans, the de novo synthesis of inosine 5′-monophosphate (IMP) is catalyzed by six enzymes that sequentially assemble a purine base onto phosphoribosyl pyrophosphate (PRPP) in ten steps (Fig 1B). This process is referred to as DNPB. IMP is a branch point purine intermediate that then is readily converted to either adenosine 5′-monophosphate (AMP) or guanosine 5′-monophosphate (GMP) by four enzymes. These enzymes are not exclusive to the DNPB pathway as the salvage synthesis of purine monophosphates also relies on their activities. DNPB is an energy intensive process and requires many of its substrates and cofactors to be generated from other metabolic pathways such as the glycolytic and pentose phosphate pathways (PRPP), serine biosynthesis (glycine), one carbon metabolism (formate), the tricarboxylic acid cycle coupled with amino acid metabolism (aspartate, glutamine). For glycine (Gly), aspartate (Asp), and formate, mitochondria are the primary production site, and their utilization in the DNPB pathway requires their transport to the cytoplasm where DNPB is performed (Palmieri, 2013; Vettore, Westbrook, and Tennant, 2020; Tibbetts and Appling, 2010; Ducker and Rabinowitz, 2017; Kory et al., 2018). Shuttling of these critical biomolecules out of the mitochondria is aided by several mitochondrial transporters. Transporters for Gly and Asp are known (Amoedo et al., 2016; Lunetti et al., 2016); however the presence of mitochondrial formate transporters have not yet been confirmed. One recent study suggests that the mitochondrial inner membrane anion channels have some formate transport functionality (Misak et al., 2013). Once in the cytoplasm, formate is converted into N10-formyltetrahydrofolate by methylenetetrahydrofolate dehydrogenase 1 (MTHFD1) and used as a cofactor in DNPB.
In the first committed step of DNPB, 5-phosphoribosylamine (5-PRA) is produced by the addition of a glutamine (Gln)-derived amine group onto PRPP (Zalkin and Dixon, 1992; Zhang, Morar, and Ealick, 2008; Hove-Jensen et al., 2017). This reaction is catalyzed by glutamine phosphoribosyl amidotransferase (PPAT/GPAT/PRAT). This enzyme has a glutaminase domain, where Gln is hydrolyzed to produce ammonia, and a phosphoribosyl transferase domain, where ammonia is added to PRPP. As inferred from the bacterial enzymes, substrate binding to the first domain leads to conformational changes priming the second domain for the binding of PRPP and subsequent 5-PRA production (Kim et al., 1996; Bera et al., 1999; Bera, Smith, and Zalkin, 2000; Krahn et al., 1997). Given the sequence and structural similarities between the human and the Bacillus subtilis enzyme, a similar behavior may be expected (Zhang, Morar, and Ealick, 2008; Batool, Nawaz, and Kamal, 2013). PPAT exists in a dynamic equilibrium between dimeric and tetrameric states and subject to feedback inhibition by AMP when in its proposed active tetrameric state (Smith et al., 1994; Wong et al., 1981). For mammalian PPAT to attain and stabilize its active structure, cleavage of an N-terminal propeptide and formation of an iron sulfur cluster [4Fe-4S] are required (Smith et al., 1994; Zhou et al., 1992). The effect of prolonged oxidative stress on the stability of PPAT’s iron-sulfur cluster was revealed in the human neuroblastoma cell line SH-SY5Y, where mitochondrial complex I inhibition by rotenone significantly reduced the fraction of mature enzyme (Mena et al., 2011).
The second enzyme in the DNPB pathway is trifunctional GART (TGART), whose domains and activities include: glycinamide ribonucleotide synthase (GARS) that catalyzes the ATP-dependent process that uses 5-PRA and Gly to make glycinamide ribonucleotide (GAR); glycinamide ribonucleotide transformylase (GART) that transfers the formyl group of N10-formyltetrahydrofolate to GAR, generating formylglycinamide ribonucleotide (FGAR); and aminoimidazole ribonucleotide synthase (AIRS) that converts formylglycinamidine ribonucleotide (FGAM) to aminoimidazole ribonucleotide (AIR) in an ATP-dependent manner. The first two reactions catalyzed by TGART are sequential and produce FGAR, which is then acted upon by the third enzyme in the pathway, formylglycinamidine synthase (PFAS/FGAMS). Similar to PPAT (Batool, Nawaz, and Kamal, 2013), FGAMS also has a glutaminase domain and based on structure and sequence similarity to bacterial enzymes, is inferred to form a channel that directly transfers ammonia produced in its active site to the amidotransferase domain (Sharma et al., 2020). Due to this, the ammonia produced by the glutaminase domain does not equilibrate with the bulk aqueous cytosolic volume and remains available for the next reaction in its neutral form. The transferred ammonia is then used to convert FGAR to FGAM. The FGAMS protein exhibits interesting biophysical properties and will be covered later in this review. The FGAM produced by FGAMS is then converted into AIR by the AIRS domain of TGART, resulting in a five membered ring closure.
The next two reactions (steps 6 and 7) involve carboxylation of AIR to 4-carboxy-5-aminoimidazole ribonucleotide (CAIR) and ligation of the carboxy group of CAIR with an amide group derived from Asp in an ATP-dependent reaction forming 4-(N-succinylcarboxamide)-5-aminoimidazole ribonucleotide (SAICAR). These reactions are catalyzed by the bifunctional enzyme phosphoribosylaminoimidazole carboxylase/phosphoribosylaminoimidazole succinocarboxamide synthetase (PAICS). PAICS has an N-terminal 4-(N-succinylcarboxamide)-5-aminoimidazole ribonucleotide synthetase (SAICARS) domain and a C-terminal 5-aminoimidazole ribonucleotide carboxylase (AIRC) domain. This enzyme has been shown to form homo-octamers by gel filtration and X-ray crystallography (Li et al., 2007; Skerlova et al., 2020). Crystal structure analysis also revealed a tunnel system in the octamer, leading to the hypothesis that this might act to channel CAIR from the AIRC domain to the SAICARS domain and restrict the diffusion of CAIR into the bulk cytosol (Li et al., 2007). These structural details have recently been confirmed in a pair of substrate bound octameric structures for the human enzyme (Skerlova et al., 2020).
The eighth step in the DNPB pathway is catalyzed by adenylosuccinate lyase (ADSL) and involves a β-elimination reaction on SAICAR that releases fumarate and aminoimidazole-4-carboxamide ribonucleotide (AICAR). ADSL can also catalyze the reverse reaction. The active site of this homotetrameric enzyme is constituted by residues from the three other subunits (Ray et al., 2012). The last two steps in the pathway are catalyzed by the bifunctional AICAR transformylase/IMP cyclohydrolase (ATIC). The transformylase domain of the enzyme first catalyzes the conversion of AICAR to formylaminoimidazole-4-carboxamide ribonucleotide (FAICAR) using the N10-formyltetrahydrofolate. Then, the cyclohydrolase domain closes the purine ring to form IMP. The first reaction is reversible; however, action of the cyclohydrolase domain promotes an overall forward reaction (Wall, Shim, and Benkovic, 2000). ATIC has been crystallized as a dimer, and a dimer:monomer equilibrium was observed in solution (Greasley et al., 2001; Vergis et al., 2001). While the presence of a molecular tunnel for direct transfer of the intermediate between the two domains was anticipated, kinetic and structural analysis of the enzyme did not reveal channeling in the isolated enzyme in vitro (Greasley et al., 2001; Bulock, Beardsley, and Anderson, 2002).
In summary, human DNPB requires the coordinated actions of six enzymes to catalyze ten sequential reactions converting PRPP into IMP. Many of these enzymes have multiple activities: TGART is trifunctional and PAICS and ATIC are bifunctional, while PPAT, FGAMS and ADSL each catalyze a single DNPB step. These reactions require the use of numerous substrates and cofactors that are generated by other metabolic processes, many of which are synthesized in and transported from mitochondria. More detailed discussion on the structural information, allosteric regulation, and changes in expression of these enzymes has previously been reviewed by Zhang, Morar, and Ealick (Zhang, Morar, and Ealick, 2008), Hartman and Buchanan (Hartman and Buchanan, 1959), and Lane and Fan (Lane and Fan, 2015; Villa et al., 2019), respectively.
Metabolons: The Supramolecular Metabolic Enzyme Assemblies
In the simplest terms, enzymes are viewed as chemical catalysts that bring down activation barriers to speed up a biochemical reaction, sometimes by a factor of 1012-1023 compared to the uncatalyzed reaction (Miller and Wolfenden, 2002). Application of sophisticated in vitro kinetic analyses, high resolution structure determination, and high resolution imaging has led to the discovery of intra- and inter-enzyme substrate channeling (Schmitt and An, 2017; Sweetlove and Fernie, 2018; Kastritis and Gavin, 2018; Lynch, Kollman, and Webb, 2020; Raushel, Thoden, and Holden, 2003). Based on the evidence gathered on the organization of Krebs cycle enzymes in the mitochondrial matrix, Paul Srere first coined the term “metabolon” and defined it as a “supramolecular complex of sequential metabolic enzymes and cellular structural elements” (Srere, 1985). He envisaged metabolons to arise as a result of interactions between sequential enzymes of a metabolic pathway and between different cellular structural elements that guide their subcellular localization. Therefore, metabolons can conceivably provide conditions for substrate channeling and pathway flux enhancement and regulation by their assembly/disassembly (Ovadi and Saks, 2004; Srere, 1987).
On the Trail of a DNPB Metabolon: Searching for Clues
Extensive work on the DNPB pathway enzymes over the last 50 years has not only revealed regulation of the pathway at the level of transcription, translation, and allostery (Lane and Fan, 2015; Villa et al., 2019; Zalkin and Dixon, 1992; Gassmann, Stanzel, and Werner, 1999), but also has hinted at the possibility of a purine biosynthetic metabolon. The economy of product formation as the advantage of physical proximity of sequential reactions of a pathway can be inferred from the evolution of large multi-domain DNPB enzymes in several organisms. For example, TGART harbors three catalytic domains in a single polypeptide chain; however, in bacteria, each of those domains are separate gene products. The same holds true for the bifunctional enzymes PAICS and ATIC. In addition, TGART catalyzes two sequential steps and a third non-sequential step, which requires the action of another enzyme, FGAMS. This argument raises the possibility for the coevolution of an interaction between the two enzymes for efficient metabolite processing.
A metabolon also would mitigate the loss of unstable substrates. The product of the first committed step for the DNPB pathway, 5-PRA, under physiological conditions has a half-life of 5 seconds (Mueller et al., 1994). Kinetic analyses of the first two steps in the DNPB pathway, using E. coli PPAT and GART, found a direct uptake of 5-PRA by GART, limiting its complete equilibration with the bulk solvent (Rudolph and Stubbe, 1995). Although a physical interaction between the two enzymes was not established. The instability of 5-PRA was also evident from a recent metabolic profiling of a HeLa mutant cell line lacking the expression of GART (Mazzarino et al., 2020). Unlike the substrate accumulation observed when ADSL or ATIC were knocked out in HeLa cells (Mazzarino et al., 2019; Mazzarino et al., 2020), the GART knockout HeLa cell line showed no accumulation of the 5-PRA substrate.
In addition to their roles in DNPB, two key regulatory molecules, SAICAR and AICAR, signal nutrient and energy imbalances in cancer cells by allosterically modulating other metabolic enzymes. SAICAR has been identified as an allosteric activator of the inactive cancer-specific pyruvate kinase isoform M2 (PKM2) dimer under glucose starvation conditions (Keller, Tan, and Lee, 2012; Keller et al., 2014; Yan et al., 2016). The SAICAR-induced activity of PKM2 has been linked to the promotion of the pentose phosphate pathway needed for sustained tumor growth (Keller et al., 2014; Zahra et al., 2020). Additionally, elevated AICAR levels lead to allosteric activation of AMP-activated protein kinase (AMPK) to regulate the intracellular AMP:ATP ratio (Asby et al., 2015). AICAR levels can also inhibit mTOR activity and block cell cycle progression via degradation of the G2/M phosphatase, cdc25 (Liu et al., 2014; Racanelli et al., 2009). Through sequestration of pathway intermediates, a metabolon might adjust the levels of these molecules commensurate with cellular demands.
The Case for a DNPB Metabolon
The lack of experimental evidence for complexation by enzyme copurification or in vitro reconstitution was not overcome until the spatial arrangement of the enzymes inside of human cancer cells was explored by confocal microscopy. Plasmids encoding the DNPB enzymes as fluorescent protein chimeras were transfected and expressed in HeLa cells, and their intracellular localizations determined (An et al., 2008). Under pathway activating conditions assisted by purine-depletion, FGAMS showed non-homogenous cytosolic distribution, marked by discrete punctate structures. FGAMS also showed colocalization with other DNPB enzymes upon cotransfection with the respective fluorescently tagged chimeras (Fig 2A). This coclustering of pathway enzymes was designated as purinosome and interpreted to represent a metabolon (An et al., 2008). Purinosome formation has been further confirmed on the endogenous protein level by visualizing DNPB enzymes in fixed cells by immunofluorescence (Baresova et al., 2016; Baresova et al., 2012; Baresova et al., 2018; Chan et al., 2015; Zhao et al., 2015) and more recently, by proximity ligation assays (Doigneaux et al., 2020) (Fig 2B). Since its discovery, assemblies of FGAMS, ATIC, and PAICS have also been observed in neuronal cell body and axons and implicated in neuronal differentiation and signaling (Williamson et al., 2017; Mangold et al., 2018; Yamada, Sato, and Sakakibara, 2020). Additionally, PPAT, GART, FGAMS, ADSL and ATIC puncta and/or their pairwise colocalizations have been observed in breast cancer cells (An et al., 2008; Doigneaux et al., 2020; Schmitt et al., 2018), hepatocarcinoma liver cancer cells (Baresova et al., 2012; French et al., 2013), and primary dermal fibroblasts from patients diagnosed with Lesch-Nyhan Syndrome (Chan et al., 2015; Chan et al., 2018; Fu et al., 2015).
Figure 2. Detection of Purinosomes on the Overexpressed and Endogenous Protein Levels.
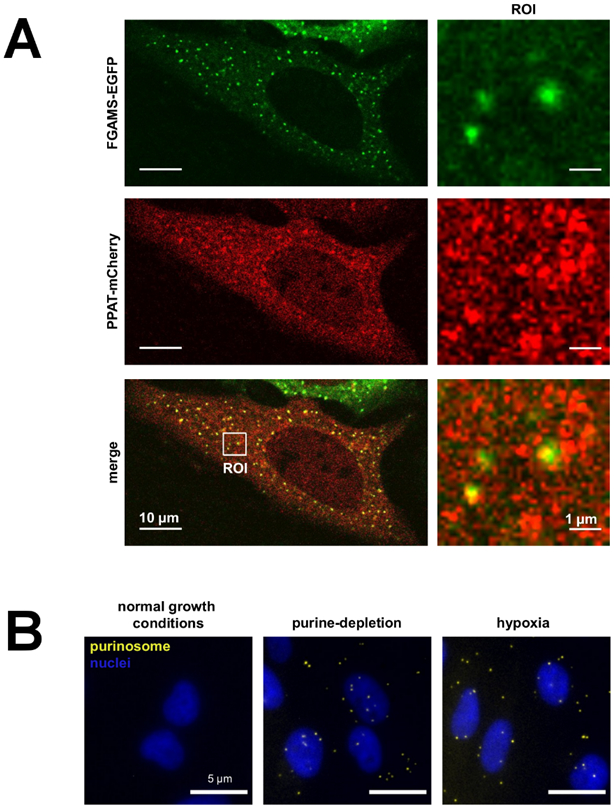
(A) A purine-depleted HeLa cell showing the colocalization of FGAMS-EGFP (green) and PPAT-mCherry (red) identifying purinosomes (yellow puncta) in a transient, overexpressed system. The image was modified from Pedley and Benkovic (Pedley and Benkovic, 2018). (B) The colocalization of endogenous FGAMS and ADSL by proximity ligation assays to identify purinosomes (yellow puncta) under purine-depleted and hypoxic growth conditions. The image was modified from the research originally published in the Journal of Biological Chemistry (Doigneaux et al., 2020) and used with permission from C. Doigneaux and A. Tavassoli.
The Properties of Purinosomes
The formation of purinosomes is a dynamic and reversible process driven by an imbalance in purine supply and demand triggered by the unavailability of extracellular purines or by a defective purine salvage (An et al., 2008; Chan et al., 2015; Fu et al., 2015). Purinosomes, as denoted by cytosolic FGAMS clusters that quickly dissolve upon supplementation of purines to the culturing medium and re-emerge within a few hours after purine depletion (An et al., 2008). Early studies challenged the nature of the purinosome by prescribing it as a non-functional stress body (Zhao et al., 2014; Zhao et al., 2013). Since then, its functional organization has been inferred from a combination of subcellular proteomic fractionations, protein proximity assays, diffusion coefficients of the individual enzymes, and activity assays (Deng et al., 2012; Kyoung et al., 2015; Wan et al., 2015; Pareek et al., 2020). The latter were based on fluorescence recovery after photobleaching of fluorescently-tagged enzymes in human breast carcinoma Hs578T cells (Kyoung et al., 2015). The diffusion coefficients for each of the six pathway enzymes analyzed were derived from their respective fluorescent recovery curves. The first three enzymes of the pathway (PPAT, GART, and FGAMS) showed similar, slow diffusion rates, whereas PAICS and ADSL were found to diffuse twice as fast, and ATIC faster yet. Overall, a significant reduction in the diffusion of the DNPB enzymes under purine-depleted conditions was observed when compared to non-purinosome forming (purine supplemented) growth conditions. Combined with protein proximity assays, a model was generated suggesting a stepwise assembly mechanism where PPAT, GART, and FGAMS establish the core scaffold and PAICS, ADSL and ATIC act as more peripheral partners (Pedley and Benkovic, 2017; Deng et al., 2012; Kyoung et al., 2015).
Although the free exchange of pathway enzymes between the purinosome and the bulk solvent is greatest for the peripheral enzymes, impairment in their expression, activity, and/or oligomerization of those enzymes greatly hinders purinosome formation. HeLa cells deficient in a single pathway enzyme greatly reduced or completely eliminated purinosome formation relative to normal HeLa cells (Baresova et al., 2016). Similarly, primary dermal fibroblasts derived from patients with rare metabolic disorders associated with mutations in PAICS, ADSL, or ATIC showed reduced purinosome formation consistent with a decrease or loss in enzymatic activity (Baresova et al., 2012; Pelet et al., 2019). The oligomerization and activity of ATIC can also be disrupted by the use of Cpd14, a cyclic peptide-derived inhibitor, concomitantly affecting purinosome formation (Spurr et al., 2012; Tavassoli and Benkovic, 2005).
The Identification and Roles of Ancillary Interactions
Attempts at identifying the ancillary proteins that regulate purinosome formation have remained a challenge. Immunoprecipitation of FGAMS under purine-depleted growth conditions, in the presence or absence of chemical crosslinkers, confirmed interactions with pathway enzymes, ADSL and PAICS, and revealed potential interactions with glycolytic enzymes, pyruvate kinase isozymes M1/M2 and transketolase, and components of molecular chaperone complexes (French et al., 2013). Molecular chaperones HSP90 and HSP70 were shown to colocalize with purinosomes in purine-depleted HeLa cells (French et al., 2013), and their direct interactions were validated biochemically in an HSP90-client interaction assay (Pedley et al., 2018). Two of the pathway enzymes, PPAT and FGAMS, were identified as clients of HSP90. Inhibition of HSP90 activity by STA9090 decreased its interactions with the client proteins and resulted in reduced levels of soluble protein to sustain purinosome formation (Pedley et al., 2018). More recently, Nwd1, a member of the signal transduction ATPases with numerous domains (STAND) protein superfamily, was shown to interact with FGAMS, PAICS, and HSP90 in neurons, and these interactions have been proposed to be involved in purinosome assembly (Yamada, Sato, and Sakakibara, 2020).
By super-resolution fluorescence microscopy, purinosomes were shown to colocalize with both microtubules and mitochondria in purine-depleted and HPRT-deficient dermal fibroblasts (Chan et al., 2018; French et al., 2016). In HPRT-deficient fibroblasts, 91% of purinosomes colocalized with microtubules (Chan et al., 2018). Disruption of microtubule polymerization with nocodazole in purine-depleted HeLa cells reduced the number of purinosome-positive cells and decreased purine production by 36%, suggesting that microtubules likely have a role in the stability of the purinosome (An et al., 2010). Two-color imaging of purinosomes (FGAMS as the marker) and mitochondria also showed that 65% and 81% of purinosomes colocalized with mitochondria in purine-depleted HeLa cells and HPRT-deficient fibroblasts, respectively (Chan et al., 2018; French et al., 2016). Recently, three-color spatiotemporal analyses were performed to investigate purinosome (FGAMS) dynamics with respect to microtubules and mitochondria (Fig 3) (Chan et al., 2018). These studies concluded that purinosomes traffic along microtubules in a directed manner, suggestive of active transport towards mitochondria. Nocodazole treatment reduced the number of purinosomes displaying directed motion by about 65% with a 30% reduction in the number of purinosomes colocalized with mitochondria (Chan et al., 2018). Taken together, these studies show the importance of an intact microtubular network and the interplay between microtubules and mitochondria for purinosome dynamics.
Figure 3. Interplay of Purinosomes with Mitochondria and Microtubules.
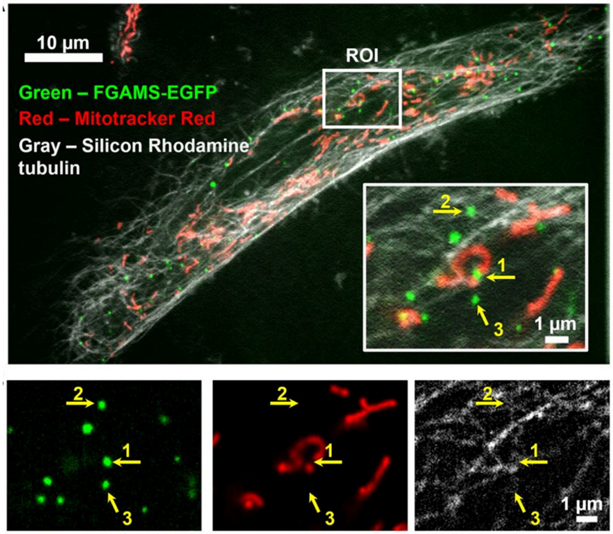
Colocalization of purinosomes (FGAMS-EGFP, green), mitochondria (Mitotracker Red, red), and microtubules (silicon rhodamine tubulin, gray) in an HPRT-deficient fibroblast by high-resolution confocal microscopy (VT-iSIM). Inset shows a magnified region of interest (ROI) with individual purinosome colocalizations annotated with respect to the mitochondrion (1), microtubule (2), or neither subcellular structure-of-interest (3). Individual channels for purinosomes, mitochondria, and microtubules of the ROI are shown underneath. Image was modified from Chan et al., 2018 (Chan et al., 2018).
Many studies have used cytosolic FGAMS puncta as the marker for identifying purinosomes. Characterization of these puncta with fluorescence microscopy revealed broad distributions in size (sub-micron diameters), heterogeneity in subcellular location, and varying degrees of colocalization with other DNPB enzymes (An et al., 2008; Chan et al., 2015; Chan et al., 2018; French et al., 2016; Schmitt et al., 2016). An accurate identification and characterization of purinosomes is also complicated by the observation that FGAMS can self-assemble upon AMPK activation and over-expression (Schmitt et al., 2018; Schmitt et al., 2016). Such complexities are not unique to the study of purinosomes but are a recurring feature in studies involving ectopic overexpression of proteins. Thus, formation of punctate by enzymes, or multi-enzyme colocalization does not provide unequivocal evidence of functionality. Mathews has warned against these misinterpretations clouding the research in this field and argues for the merits of application of multiple experimental approaches to overcome the limitations (Mathews, 1993).
The Purinosome as a Biomolecular Condensate
Recent research suggests a liquid-liquid phase separation ensues upon the condensation of proteins involved in metabolon formation and represents a means of cytosolic protein organization (Kastritis and Gavin, 2018; Prouteau and Loewith, 2018; Strom and Brangwynne, 2019; Banani et al., 2017). Due to technical limitations, simultaneous biophysical and biochemical characterization of metabolites and enzymes in their endogenous intracellular state has not been achieved. However, the observation of distinct purinosome puncta under purine-depleted conditions fits the literature criteria of a liquid condensate (Prouteau and Loewith, 2018; Bracha, Walls, and Brangwynne, 2019). The purinosome has repeatedly been shown to be distinct from other well characterized cytoplasm-localized RNA-containing condensates, namely stress granules and processing bodies (Chan et al., 2015; French et al., 2013). Owing to the fact that most of the DNPB enzymes exist as oligomers in their catalytically competent state, we hypothesize that oligomerization might play a role in DNPB enzyme condensation into the purinosomes. Disruption of the purinosome assembly by an ATIC oligomerization inhibitor, Cmpd14, supports this view (Doigneaux et al., 2020).
Biomolecular condensates exist in a continuum from transient, reversible interactions to stronger, irreversible interactions. Oftentimes, protein nucleation, leading to condensate formation, has been observed upon upregulation of the target protein expression and/or the participation of various protein modifying enzymes such as kinases or heat shock proteins resulting in altered surface properties of the modified protein(s) (Soding et al., 2020; Mateju et al., 2017; Rai et al., 2018; Wippich et al., 2013). The collection of weak and transient interactions between the DNPB pathway enzymes and requisite ancillary proteins likely play a role in condensate formation (Banani et al., 2017; Bracha, Walls, and Brangwynne, 2019; Alberti, Gladfelter, and Mittag, 2019) and could justify the difficulties in isolating intact purinosomes from cells. Recently, purinosomes, as marked by FGAMS puncta, are shown to be spherical and tend to coalesce to form larger liquid droplet-like structures (Pedley et al., to be submitted) (Movie 1). To maintain these liquid-like properties, the heat shock proteins, HSP90 and HSP70, have been assigned a role in the process through the refolding of client proteins, PPAT and FGAMS (Pedley et al., 2018). Thus, disruption of HSP90 activity induced intracellular FGAMS aggregates, distinct from purinosomes, suggesting that the purinosomes might have transitioned from a liquid-like state to a more solid, rigid intracellular body (Pedley et al., to be submitted). Other alterations to DNPB enzymes such as post-translational modifications might also contribute to condensate formation and active regulation of its liquid-like properties.
Catching a Metabolon in Action
For the functional characterization of purinosomes and to test the hypothesis that the DNPB pathway is carried out by the mitochondria-associated purinosomes, metabolomics and mass spectrometry imaging were performed (Pareek et al., 2020). Taking advantage of the mitochondria-compartmentalized serine (Ser) to Gly conversion and formate production by one-carbon metabolism (Ducker and Rabinowitz, 2017; Labuschagne et al., 2014), an isotope incorporation study was designed to trace the flow of labeled Gly and formate into DNPB intermediates and product nucleotides after [13C3,15N]Ser supplementation in the media (Pareek et al., 2020) (Fig 4A). Cells were grown under a limited abundance of Ser to promote preferential access to the labeled substrates by the enzyme pool proximal to mitochondria. Evidence for metabolic channeling in the pathway, as would be expected to arise from a metabolon, came from the comparison of the observed isotope incorporation pattern to a model that assumes a completely diffusive mode of PRPP to AMP and GMP conversion (i.e., all fourteen steps are mutually independent events). Our results disproved the assumptions built into the operation of a diffusive model that postulates complete equilibration of all the substrates, cofactors, and pathway intermediates and a homogeneous distribution of the pathway enzymes in the bulk cytosolic pool. As per the diffusive model a homogenous isotopic incorporation in all the DNPB intermediates and end products is to be expected. Conversely, the results showed that the synthesis of AMP and GMP is accomplished in a highly channeled manner, preventing pathway intermediates from equilibrating with their bulk cytosolic pools (Fig 4B). The higher isotope enrichment in AMP and GMP compared to IMP required the physical proximity or plausible direct association of the DNPB metabolon with the mitochondrial metabolite transporters, to facilitate the capture of mitochondria generated substrates (Gly, and formate) by the purinosomes. This provides a functional basis for the active migration of purinosomes along the microtubule towards mitochondria. The total abundance of newly synthesized AMP and GMP is approximately sevenfold higher than that of the total IMP synthesized by a minor diffusive pathway constituted by enzymes acting outside the purinosome (Fig 6). Contrary to the expected partitioning based on the activities of pathway enzymes for IMP to synthesize more GMP than AMP (Zhao et al., 2015), the channeled pathway favors the synthesis of AMP over GMP (Pareek et al., 2020).
Figure 4. HeLa cells show channeled DNPB.
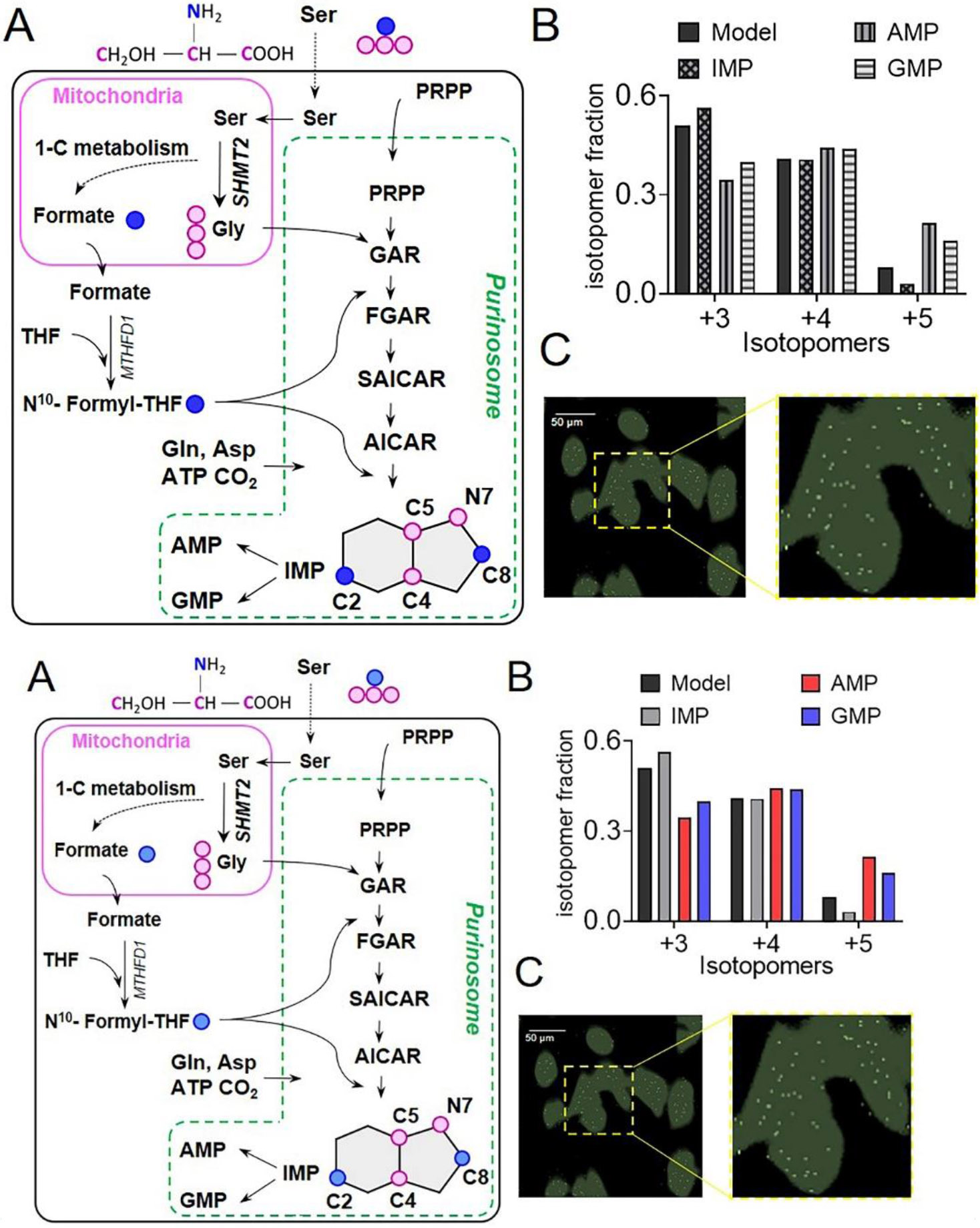
(A) In three separate steps upstream of IMP, 13C3, 15N Ser derived labeled 13C2, 15N Gly and 13C formate get incorporated into the intermediates and the end-product purines synthesized by DNPB, allowing monitoring of cellular DNPB flux. The unique isotopomer species in IMP, AMP, and GMP- +3, +4, and +5- all have one 13C2, 15N Gly; and 0, 1 or 2 13C formate incorporations, respectively. For clarity, only the relevant steps and intermediates are shown. Blue circles: 13C labeled β-carbon of Ser; pink circles: 13C, 15N labeled Ser backbone atoms. (B) The fractional abundance of the de novo synthesized purine isotopomers, +3, +4, and +5 can be predicted assuming uniform cytosolic abundance of the labeled substrates; a similar distribution of the three isotopomers is to be expected for IMP and all the downstream purines, including AMP and GMP. While the isotopomeric distribution of IMP closely resembles the model, AMP and GMP show a different isotopomeric distribution. The underlying reason being a higher labeled Gly and formate enrichment in AMP and GMP compared to that predicted by the model. Data from one representative experiment are shown. (C) A representative GCIB-SIMS image of purine depleted HeLa cells grown in 15N Ser supplemented medium to allow 15N label incorporation in purines. Overlay of the pixels with high 15N AICAR abundance (white) and cell image generated using total ion current (green) is shown. A zoomed-in view shows the spatial distribution of AICAR ‘metabolic hotspots’ in a region of interest highlighted with yellow box in (C). Figures are prepared from the data originally reported and presented in (Pareek et al., 2020).
Figure 6. A model of purinosomes, the DNPB metabolon.

Results from high resolution fluorescence imaging, biochemical, proteomic, metabolomic, and chemical imaging studies taken together suggest that purinosomes are constituted by at least nine enzyme and plausibly more ancillary proteins that assist in stabilization and dynamics of the complex. The HSP90/70 machinery and signaling pathways involving kinases and other post translationally modifying enzymes regulate the process of assembly of an active purinosome. A majority of purinosomes reside proximal to mitochondria-microtubule junction and act as cellular metabolic hot-spots that synthesize AMP and GMP in a highly channeled manner. Purinosome assisted AMP+GMP synthesis has ~7 times higher flux than the diffusive synthesis of IMP.
To validate the presence of the purinosomes at the single cell level, in situ, sub-micron chemical imaging by gas cluster ion beam-time-of-flight-secondary ion mass spectrometry (GCIB-SIMS) was leveraged (Pareek et al., 2020). A multi-layer analysis of frozen hydrated HeLa cells revealed regions of high abundance of the DNPB intermediate AICAR, which are congruent with sites of higher ATP synthesis. These “metabolic hotspots” identified the sites of active purinosomes situated proximal to mitochondria (Fig 4C). Our analysis captured 10–30 active purinosomes per cell; although due to the technical limitations, it is likely to be an underestimate of the number of purinosomes per cell. Together with the isotope labeling studies, we propose that the definition of the DNPB metabolon must be expanded to represent an assembly of nine enzymes localized proximal to mitochondria, capable of catalyzing the conversion of PRPP to AMP and GMP in a sequence of fourteen highly channeled steps to achieve greater pathway flux (Fig 6).
Signaling Pathways and Their Plausible Role in Purinosome Regulation
The regulation of DNPB can be driven by an increase in protein expression, substrate availability, and/or enzymatic activity. Additional level of regulation could arise from the DNPB enzyme modifications underlying the formation and dissolution of purinosomes. Deciphering these regulatory mechanisms can expose potential metabolic vulnerabilities that can be exploited for therapeutic applications.
Early studies had largely attributed the upregulation of purine biosynthesis to upregulation of DNPB pathway enzymes’ gene expression by the mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinase (PI3K)/AKT signaling pathways. Stimulation of quiescent human HaCaT keratinocytes by keratinocyte or epidermal growth factors, which activates PI3K and AKT, increased the mRNA expression of ADSL 4-fold (Gassmann, Stanzel, and Werner, 1999). Further, epidermal growth factor (EGF) stimulated DNPB (Ali et al., 2020) (Fig 5A). In C2C12 mouse mesenchymal cells, inhibition of PI3K with LY294002, decreased the formate incorporation into purines by over 75% resulting from a 31% decrease in PRPP availability and a 20% reduction in the enzymatic activity of ATIC (Wang et al., 2009). PI3K inhibition also abolished the downstream substrate AKT activation, as noted by a lack of Thr308 and Ser473 phosphorylation. Consistent with PI3K inhibition, AKT inhibition by MK2296 resulted in a 73% reduction in purine production in HeLa cells (Saha et al., 2014). More recently, a putative AKT phosphorylation site on PPAT (Thr397) was shown to be present only in normal growth conditions and absent upon pathway activation (purine-depletion) which suggests that the phosphorylation event might impact the activity of the enzyme and influence the pathway (Liu et al., 2019). AKT phosphorylated a peptide-based substrate mimic of PPAT as well as recombinant protein expressed and isolated from 293T cells. Further, computational predictions of substrate motifs for canonical kinases suggest that AKT might also phosphorylate PFAS on Ser215 (Ali et al., 2020; Liu et al., 2019) (Fig 5B). All these data suggest that PI3K and AKT are key regulators of purine biosynthesis.
Figure 5. The Regulation of de novo Purine Biosynthesis and Purinosome Formation.

(A) Pathway activation and/or purinosome formation has been associated with signaling through the GPCR, MAPK, and PI3K/AKT signaling pathways. Downstream of AKT is mTOR, whose activity assists in the colocalization of purinosomes with mitochondria. Dashed lines suggest an association or correlation, whereas solid lines represent a direct interaction or consequence. (B) The phosphorylation sites identified on PFAS/FGAMS as determined our recent post-translational modification study (Liu et al., 2019) and other global phosphoproteomic studies recorded on PhosphoSite. Solid red circles and white circles represent those phosphorylation events identified under purine supplemented and purine-depleted growth conditions, respectively. Red stripped circles were either present in both conditions or the site preference is unknown. Kinase predictions are a combination from ScanSite and/or Liu et al., 2019.
Other proteomics studies implicated casein kinase 2 (CK2) in the post-translational modifications of TGART and FGAMS, hinting at their involvement in the signaling pathways. Live cell imaging showed that CK2 inhibitors (DMAT and TBB) could modulate purinosome formation (An et al., 2010). Treatment of HeLa cells with DMAT induced purinosome formation, whereas TBB at high concentrations dissociated DMAT or purine-depletion promoted purinosomes. In addition, signaling through G-protein coupled receptors have also been implied in the regulation of DNPB (Fig 5A). To deconvolute this association, a label-free dynamic mass redistribution (DMR) assay was developed (Verrier et al., 2011). This screening platform was calibrated for the de novo purine biosynthetic pathway by using inhibitors of CK2. A screen of 113 GPCR agonists identified adrenergic receptor (AR) agonists such as epinephrine, clonidine, and dopamine as perturbing the mass redistribution that occurs upon purinosome formation (Verrier et al., 2011). Further investigations of these agonists unveiled that the α2A-AR is responsible for purinosome formation and provided a mechanism by which the Gαi signaling pathway is important for purinosome formation.
The framework for the DMR assay also served to identify the kinases that might be involved in the regulation of the purinosome. A short hairpin RNA (shRNA) loss-of-function kinome screen was performed to identify those kinases that change the biomass redistribution, a label-free measure of purinosome assembly or disassembly, in purine-depleted HeLa cells (French et al., 2016). Among the kinases identified, mammalian target of rapamycin (mTOR) showed the ability to maintain purinosome localization near mitochondria (Fig 5A). An inhibitor of mTOR, rapamycin, reduced activating transcription factor (ATF) 4-mediated transcription of MTHFD2 and purine production through the de novo pathway (Ben-Sahra et al., 2016). MTHFD2 is important for the generation of the one-carbon units that are used to synthesize the N10-formyltetrahydrofolate cofactor needed for GART and ATIC transformylase activities. Taken together, these results suggest that deficiencies in one-carbon metabolism might impact DNPB by reducing the availability of formate needed for cofactor generation and the functionally important colocalization of purinosomes with mitochondria (French et al., 2016).
A post-translational modification (PTM) study was performed to investigate the extent of DNPB enzyme modification upon pathway activation. 293T cells transiently expressing each of the pathway enzymes were grown in the presence and absence of purines, and seven PTMs present on each of the enzymes were catalogued (Liu et al., 2019). One of the most striking observations was the heavy phosphorylation on FGAMS with many of the sites only present under purine-depleted, or purinosome forming, growth conditions (Fig 5B). One of the sites, Thr619 was recently shown to be phosphorylated by EGF-stimulated or oncogenic RAS or RAF activation of ERK2 (Ali et al., 2020). This phosphorylation event enhances cell and tumor growth, whereby expression of a nonphosphorylatable FGAMS (T619A) in RAS-dependent cancer cell lines resulted in a 30% - 40% decrease in colony formation and reduced tumor growth in athymic nude mice compared to cells expressing wild type FGAMS enzyme. Other phosphorylation sites were queried against the substrate motifs for the kinome (Ali et al., 2020; Liu et al., 2019). In addition to the likely AKT phosphorylation of FGAMS, aurora kinases and other MAPKs have also been predicted to be its modifiers (Fig 5B). Much remains unknown about the importance of the many phosphorylation sites and the identity of their respective kinases on the regulation of purinosome activity
A recent study of purinosome formation under hypoxic conditions revealed further complexities in purinosome assembly and function. Although the purinosomes form and locate to the mitochondria, they are devoid of activity (Doigneaux et al., 2020) (Fig 2B). Metabolite analysis under hypoxia found a down-regulation in the mitochondrial one-carbon metabolism needed for purinosome function in HeLa cells. Purinosome formation was abolished in hypoxic HeLa cells upon treatment with Cpd14 or by a genetic knockdown of glucose-6-phosphate dehydrogenase suggesting that ATIC oligomerization and PRPP availability via the pentose phosphate pathway are necessary for purinosome formation. These findings underscore the many unknown intricacies at play connected to the formation and properties of this metabolon.
Concluding Remarks
The pathway for human DNPB converts phosphoribosyl pyrophosphate first to IMP followed by its partitioning to form AMP and GMP. The nine enzymes responsible for the transformation typically assemble into a metabolon, termed a purinosome. Srere presciently described the advantages of a supramolecular complex such as increased pathway flux, sequestration of unstable metabolic intermediates, and increased opportunities for pathway regulation (Srere, 1985; Srere, 1987). Although early literature suggested that a cellular metabolon might be operative in purine biosynthesis, the proof required the development and application of high-resolution intracellular fluorescence imaging, metabolomics, and biomolecular imaging by GCIB-SIMS techniques that are capable of revealing the subcellular distribution and function of enzymes at endogenous cellular levels (Fig 6). The purinosome was found to channel all the pathway intermediates in this multistep pathway, providing a sevenfold increase in product formation relative to the enzymes functioning independently in a diffusive pathway. Moreover, it actively migrates on microtubules to dock at a microtubule/mitochondria interface where it is positioned to capture the metabolites derived from mitochondrial one carbon metabolism for its substrates (e.g., Gly and Asp) and cofactors (e.g., N10-formyl THF). When cells are transiently transfected with chimeric pathway enzymes fused with fluorescent probes, the purinosomes appear as cytosolic punta and exhibit the properties of a liquid condensate. This observation implies that metabolons in general might prove to be examples of protein condensates. Assembly of a purinosome is initiated by an α2A-AR and linked to the P13K/AKT signaling pathways. Proteomic studies have found extensive phosphorylation of the individual enzymes with the identification of the relevant kinases currently in progress. One can imagine that their identification and potential role in condensate formation will present opportunities for novel therapeutic modalities.
Supplementary Material
Movie 1. Intracellular Transport of Purinosomes along Microtubules. Two purinosomes (FGAMS-EGFP, green) are shown to move along microtubules (silicon rhodamine tubulin, gray) and merge into a larger purinosome at 260 seconds. Scale bar = 1 μm. Movie was generated from time-lapse images reported in Chan et al., 2018 (Chan et al., 2018).
ACKNOWLEDGEMENTS
We wish to thank Cyrielle Doigneaux and Ali Tavassoli for use of the proximity ligation assay images (Fig 2B) and Chung Yu Chan for collecting the time-lapse images for the associated movie (Movie 1). VP acknowledges financial support from the Huck Institutes of Life Sciences, PSU.
FUNDING
This work was supported by the National Institutes of Health (grant number 5R01GM024129 to S.J.B.).
Footnotes
DISCLOSURE STATEMENT
No potential conflict of interest was reported by the authors.
REFERENCES
- 1.Lane AN, Fan TW. (2015).Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res, 43, 2466–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pedley AM, Benkovic SJ. (2017).A New View into the Regulation of Purine Metabolism: The Purinosome. Trends Biochem Sci, 42, 141–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Garcia-Gil M, Camici M, Allegrini S, Pesi R, Petrotto E, Tozzi MG. (2018).Emerging Role of Purine Metabolizing Enzymes in Brain Function and Tumors. Int J Mol Sci, 19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jinnah HA. (2009).Lesch-Nyhan disease: from mechanism to model and back again. Dis Model Mech, 2, 116–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jurecka A, Zikanova M, Kmoch S, Tylki-Szymanska A. (2015).Adenylosuccinate lyase deficiency. J Inherit Metab Dis, 38, 231–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marie S, Heron B, Bitoun P, Timmerman T, Van Den Berghe G, Vincent MF. (2004).AICA-ribosiduria: a novel, neurologically devastating inborn error of purine biosynthesis caused by mutation of ATIC. Am J Hum Genet, 74, 1276–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vander Heiden MG, DeBerardinis RJ. (2017).Understanding the Intersections between Metabolism and Cancer Biology. Cell, 168, 657–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Villa E, Ali ES, Sahu U, Ben-Sahra I. (2019).Cancer Cells Tune the Signaling Pathways to Empower de Novo Synthesis of Nucleotides. Cancers (Basel), 11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Christopherson RI, Lyons SD, Wilson PK. (2002).Inhibitors of de novo nucleotide biosynthesis as drugs. Acc Chem Res, 35, 961–71 [DOI] [PubMed] [Google Scholar]
- 10.Parker WB. (2009).Enzymology of purine and pyrimidine antimetabolites used in the treatment of cancer. Chem Rev, 109, 2880–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yin J, Ren W, Huang X, Deng J, Li T, Yin Y. (2018).Potential Mechanisms Connecting Purine Metabolism and Cancer Therapy. Front Immunol, 9, 1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hanrahan JR, Hutchinson DW. (1992).The enzymatic synthesis of antiviral agents. J Biotechnol, 23, 193–210 [DOI] [PubMed] [Google Scholar]
- 13.Thomson JM, Lamont IL. (2019).Nucleoside Analogues as Antibacterial Agents. Front Microbiol, 10, 952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hartsel SA, Chen C, Kennedy J, Bendele A, Wright C, Marshall WS. (1997).Anti-Inflammatory Activity of Purine Nucleoside Analogs. Nucleosides and Nucleotides, 16, 1291–94 [Google Scholar]
- 15.Palmieri F (2013).The mitochondrial transporter family SLC25: identification, properties and physiopathology. Mol Aspects Med, 34, 465–84 [DOI] [PubMed] [Google Scholar]
- 16.Vettore L, Westbrook RL, Tennant DA. (2020).New aspects of amino acid metabolism in cancer. Br J Cancer, 122, 150–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tibbetts AS, Appling DR. (2010).Compartmentalization of Mammalian folate-mediated one-carbon metabolism. Annu Rev Nutr, 30, 57–81 [DOI] [PubMed] [Google Scholar]
- 18.Ducker GS, Rabinowitz JD. (2017).One-Carbon Metabolism in Health and Disease. Cell Metab, 25, 27–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kory N, Wyant GA, Prakash G, Uit de Bos J, Bottanelli F, Pacold ME, Chan SH, Lewis CA, Wang T, Keys HR, Guo YE, Sabatini DM. (2018).SFXN1 is a mitochondrial serine transporter required for one-carbon metabolism. Science, 362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Amoedo ND, Punzi G, Obre E, Lacombe D, De Grassi A, Pierri CL, Rossignol R. (2016).AGC1/2, the mitochondrial aspartate-glutamate carriers. Biochim Biophys Acta, 1863, 2394–412 [DOI] [PubMed] [Google Scholar]
- 21.Lunetti P, Damiano F, De Benedetto G, Siculella L, Pennetta A, Muto L, Paradies E, Marobbio CM, Dolce V, Capobianco L. (2016).Characterization of Human and Yeast Mitochondrial Glycine Carriers with Implications for Heme Biosynthesis and Anemia. J Biol Chem, 291, 19746–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Misak A, Grman M, Malekova L, Novotova M, Markova J, Krizanova O, Ondrias K, Tomaskova Z. (2013).Mitochondrial chloride channels: electrophysiological characterization and pH induction of channel pore dilation. Eur Biophys J, 42, 709–20 [DOI] [PubMed] [Google Scholar]
- 23.Zalkin H, Dixon JE. (1992).De novo purine nucleotide biosynthesis. Prog Nucleic Acid Res Mol Biol, 42, 259–87 [DOI] [PubMed] [Google Scholar]
- 24.Zhang Y, Morar M, Ealick SE. (2008).Structural biology of the purine biosynthetic pathway. Cell Mol Life Sci, 65, 3699–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hove-Jensen B, Andersen KR, Kilstrup M, Martinussen J, Switzer RL, Willemoes M. (2017).Phosphoribosyl Diphosphate (PRPP): Biosynthesis, Enzymology, Utilization, and Metabolic Significance. Microbiol Mol Biol Rev, 81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim JH, Krahn JM, Tomchick DR, Smith JL, Zalkin H. (1996).Structure and function of the glutamine phosphoribosylpyrophosphate amidotransferase glutamine site and communication with the phosphoribosylpyrophosphate site. J Biol Chem, 271, 15549–57 [DOI] [PubMed] [Google Scholar]
- 27.Bera AK, Chen S, Smith JL, Zalkin H. (1999).Interdomain signaling in glutamine phosphoribosylpyrophosphate amidotransferase. J Biol Chem, 274, 36498–504 [DOI] [PubMed] [Google Scholar]
- 28.Bera AK, Smith JL, Zalkin H. (2000).Dual role for the glutamine phosphoribosylpyrophosphate amidotransferase ammonia channel. Interdomain signaling and intermediate channeling. J Biol Chem, 275, 7975–9 [DOI] [PubMed] [Google Scholar]
- 29.Krahn JM, Kim JH, Burns MR, Parry RJ, Zalkin H, Smith JL. (1997).Coupled formation of an amidotransferase interdomain ammonia channel and a phosphoribosyltransferase active site. Biochemistry, 36, 11061–8 [DOI] [PubMed] [Google Scholar]
- 30.Batool S, Nawaz MS, Kamal MA. (2013).In silico analysis of the amido phosphoribosyltransferase inhibition by PY873, PY899 and a derivative of isophthalic acid. Invest New Drugs, 31, 1355–63 [DOI] [PubMed] [Google Scholar]
- 31.Smith JL, Zaluzec EJ, Wery JP, Niu L, Switzer RL, Zalkin H, Satow Y. (1994).Structure of the allosteric regulatory enzyme of purine biosynthesis. Science, 264, 1427–33 [DOI] [PubMed] [Google Scholar]
- 32.Wong JY, Bernlohr DA, Turnbough CL, Switzer RL. (1981).Purification and properties of glutamine phosphoribosylpyrophosphate amidotransferase from Bacillus subtilis. Biochemistry, 20, 5669–74 [DOI] [PubMed] [Google Scholar]
- 33.Zhou G, Broyles SS, Dixon JE, Zalkin H. (1992).Avian glutamine phosphoribosylpyrophosphate amidotransferase propeptide processing and activity are dependent upon essential cysteine residues. J Biol Chem, 267, 7936–42 [PubMed] [Google Scholar]
- 34.Mena NP, Bulteau AL, Salazar J, Hirsch EC, Nunez MT. (2011).Effect of mitochondrial complex I inhibition on Fe-S cluster protein activity. Biochem Biophys Res Commun, 409, 241–6 [DOI] [PubMed] [Google Scholar]
- 35.Sharma N, Ahalawat N, Sandhu P, Strauss E, Mondal J, Anand R. (2020).Role of allosteric switches and adaptor domains in long-distance cross-talk and transient tunnel formation. Sci Adv, 6, eaay7919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li SX, Tong YP, Xie XC, Wang QH, Zhou HN, Han Y, Zhang ZY, Gao W, Li SG, Zhang XC, Bi RC. (2007).Octameric structure of the human bifunctional enzyme PAICS in purine biosynthesis. J Mol Biol, 366, 1603–14 [DOI] [PubMed] [Google Scholar]
- 37.Skerlova J, Unterlass J, Gottmann M, Marttila P, Homan E, Helleday T, Jemth AS, Stenmark P. (2020).Crystal structures of human PAICS reveal substrate and product binding of an emerging cancer target. J Biol Chem [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ray SP, Deaton MK, Capodagli GC, Calkins LA, Sawle L, Ghosh K, Patterson D, Pegan SD. (2012).Structural and biochemical characterization of human adenylosuccinate lyase (ADSL) and the R303C ADSL deficiency-associated mutation. Biochemistry, 51, 6701–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wall M, Shim JH, Benkovic SJ. (2000).Human AICAR transformylase: role of the 4-carboxamide of AICAR in binding and catalysis. Biochemistry, 39, 11303–11 [DOI] [PubMed] [Google Scholar]
- 40.Greasley SE, Horton P, Ramcharan J, Beardsley GP, Benkovic SJ, Wilson IA. (2001).Crystal structure of a bifunctional transformylase and cyclohydrolase enzyme in purine biosynthesis. Nat Struct Biol, 8, 402–6 [DOI] [PubMed] [Google Scholar]
- 41.Vergis JM, Bulock KG, Fleming KG, Beardsley GP. (2001).Human 5-aminoimidazole-4-carboxamide ribonucleotide transformylase/inosine 5'-monophosphate cyclohydrolase. A bifunctional protein requiring dimerization for transformylase activity but not for cyclohydrolase activity. J Biol Chem, 276, 7727–33 [DOI] [PubMed] [Google Scholar]
- 42.Bulock KG, Beardsley GP, Anderson KS. (2002).The kinetic mechanism of the human bifunctional enzyme ATIC (5-amino-4-imidazolecarboxamide ribonucleotide transformylase/inosine 5'-monophosphate cyclohydrolase). A surprising lack of substrate channeling. J Biol Chem, 277, 22168–74 [DOI] [PubMed] [Google Scholar]
- 43.Hartman SC, Buchanan JM. (1959).Nucleic acids, purines, pyrimidines (nucleotide synthesis). Annu Rev Biochem, 28, 365–410 [DOI] [PubMed] [Google Scholar]
- 44.Miller BG, Wolfenden R. (2002).Catalytic proficiency: the unusual case of OMP decarboxylase. Annu Rev Biochem, 71, 847–85 [DOI] [PubMed] [Google Scholar]
- 45.Schmitt DL, An S. (2017).Spatial Organization of Metabolic Enzyme Complexes in Cells. Biochemistry, 56, 3184–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sweetlove LJ, Fernie AR. (2018).The role of dynamic enzyme assemblies and substrate channelling in metabolic regulation. Nat Commun, 9, 2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kastritis PL, Gavin AC. (2018).Enzymatic complexes across scales. Essays Biochem, 62, 501–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lynch EM, Kollman JM, Webb BA. (2020).Filament formation by metabolic enzymes-A new twist on regulation. Curr Opin Cell Biol, 66, 28–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Raushel FM, Thoden JB, Holden HM. (2003).Enzymes with molecular tunnels. Acc Chem Res, 36, 539–48 [DOI] [PubMed] [Google Scholar]
- 50.Srere PA. (1985).The metabolon. Trends Biochem Sci, 10, 109–10 [Google Scholar]
- 51.Ovadi J, Saks V. (2004).On the origin of intracellular compartmentation and organized metabolic systems. Mol Cell Biochem, 256-257, 5–12 [DOI] [PubMed] [Google Scholar]
- 52.Srere PA. (1987).Complexes of sequential metabolic enzymes. Annu Rev Biochem, 56, 89–124 [DOI] [PubMed] [Google Scholar]
- 53.Gassmann MG, Stanzel A, Werner S. (1999).Growth factor-regulated expression of enzymes involved in nucleotide biosynthesis: a novel mechanism of growth factor action. Oncogene, 18, 6667–76 [DOI] [PubMed] [Google Scholar]
- 54.Mueller EJ, Meyer E, Rudolph J, Davisson VJ, Stubbe J. (1994).N5-carboxyaminoimidazole ribonucleotide: evidence for a new intermediate and two new enzymatic activities in the de novo purine biosynthetic pathway of Escherichia coli. Biochemistry, 33, 2269–78 [DOI] [PubMed] [Google Scholar]
- 55.Rudolph J, Stubbe J. (1995).Investigation of the mechanism of phosphoribosylamine transfer from glutamine phosphoribosylpyrophosphate amidotransferase to glycinamide ribonucleotide synthetase. Biochemistry, 34, 2241–50 [DOI] [PubMed] [Google Scholar]
- 56.Mazzarino RC, Baresova V, Zikánová M, Duval N, Wilkinson TG, Patterson D, Vacano GN. (2020).The CRISPR-Cas9 crGART HeLa transcriptome: A novel cell model of de novo purine synthesis deficiency. bioRxiv, 2020.06.23.167924 [Google Scholar]
- 57.Mazzarino RC, Baresova V, Zikanova M, Duval N, Wilkinson TG 2nd, Patterson D, Vacano GN. (2019).The CRISPR-Cas9 crADSL HeLa transcriptome: A first step in establishing a model for ADSL deficiency and SAICAR accumulation. Mol Genet Metab Rep, 21, 100512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mazzarino RC, Baresova V, Zikánová M, Duval N, Wilkinson TG, Patterson D, Vacano GN. (2020).The CRISPR-Cas9 crATIC HeLa transcriptome: Characterization of a novel cellular model for ATIC deficiency and ZMP accumulation. bioRxiv [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Keller KE, Tan IS, Lee YS. (2012).SAICAR stimulates pyruvate kinase isoform M2 and promotes cancer cell survival in glucose-limited conditions. Science, 338, 1069–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Keller KE, Doctor ZM, Dwyer ZW, Lee YS. (2014).SAICAR induces protein kinase activity of PKM2 that is necessary for sustained proliferative signaling of cancer cells. Mol Cell, 53, 700–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yan M, Chakravarthy S, Tokuda JM, Pollack L, Bowman GD, Lee YS. (2016).Succinyl-5-aminoimidazole-4-carboxamide-1-ribose 5'-Phosphate (SAICAR) Activates Pyruvate Kinase Isoform M2 (PKM2) in Its Dimeric Form. Biochemistry, 55, 4731–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zahra K, Dey T, Ashish, Mishra SP, Pandey U. (2020).Pyruvate Kinase M2 and Cancer: The Role of PKM2 in Promoting Tumorigenesis. Front Oncol, 10, 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Asby DJ, Cuda F, Beyaert M, Houghton FD, Cagampang FR, Tavassoli A. (2015).AMPK Activation via Modulation of De Novo Purine Biosynthesis with an Inhibitor of ATIC Homodimerization. Chem Biol, 22, 838–48 [DOI] [PubMed] [Google Scholar]
- 64.Liu X, Chhipa RR, Pooya S, Wortman M, Yachyshin S, Chow LM, Kumar A, Zhou X, Sun Y, Quinn B, McPherson C, Warnick RE, Kendler A, Giri S, Poels J, Norga K, Viollet B, Grabowski GA, Dasgupta B. (2014).Discrete mechanisms of mTOR and cell cycle regulation by AMPK agonists independent of AMPK. Proc Natl Acad Sci U S A, 111, E435–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Racanelli AC, Rothbart SB, Heyer CL, Moran RG. (2009).Therapeutics by cytotoxic metabolite accumulation: pemetrexed causes ZMP accumulation, AMPK activation, and mammalian target of rapamycin inhibition. Cancer Res, 69, 5467–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.An S, Kumar R, Sheets ED, Benkovic SJ. (2008).Reversible compartmentalization of de novo purine biosynthetic complexes in living cells. Science, 320, 103–6 [DOI] [PubMed] [Google Scholar]
- 67.Baresova V, Krijt M, Skopova V, Souckova O, Kmoch S, Zikanova M. (2016).CRISPR-Cas9 induced mutations along de novo purine synthesis in HeLa cells result in accumulation of individual enzyme substrates and affect purinosome formation. Mol Genet Metab, 119, 270–77 [DOI] [PubMed] [Google Scholar]
- 68.Baresova V, Skopova V, Sikora J, Patterson D, Sovova J, Zikanova M, Kmoch S. (2012).Mutations of ATIC and ADSL affect purinosome assembly in cultured skin fibroblasts from patients with AICA-ribosiduria and ADSL deficiency. Hum Mol Genet, 21, 1534–43 [DOI] [PubMed] [Google Scholar]
- 69.Baresova V, Skopova V, Souckova O, Krijt M, Kmoch S, Zikanova M. (2018).Study of purinosome assembly in cell-based model systems with de novo purine synthesis and salvage pathway deficiencies. PLoS One, 13, e0201432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chan CY, Zhao H, Pugh RJ, Pedley AM, French J, Jones SA, Zhuang X, Jinnah H, Huang TJ, Benkovic SJ. (2015).Purinosome formation as a function of the cell cycle. Proc Natl Acad Sci U S A, 112, 1368–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhao H, Chiaro CR, Zhang L, Smith PB, Chan CY, Pedley AM, Pugh RJ, French JB, Patterson AD, Benkovic SJ. (2015).Quantitative analysis of purine nucleotides indicates that purinosomes increase de novo purine biosynthesis. J Biol Chem, 290, 6705–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Doigneaux C, Pedley AM, Mistry IN, Papayova M, Benkovic SJ, Tavassoli A. (2020).Hypoxia drives the assembly of the multienzyme purinosome complex. J Biol Chem, 295, 9551–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Williamson J, Petralia RS, Wang YX, Mattson MP, Yao PJ. (2017).Purine Biosynthesis Enzymes in Hippocampal Neurons. Neuromolecular Med, 19, 518–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mangold CA, Yao PJ, Du M, Freeman WM, Benkovic SJ, Szpara ML. (2018).Expression of the purine biosynthetic enzyme phosphoribosyl formylglycinamidine synthase in neurons. J Neurochem, 144, 723–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yamada S, Sato A, Sakakibara SI. (2020).Nwd1 Regulates Neuronal Differentiation and Migration through Purinosome Formation in the Developing Cerebral Cortex. iScience, 23, 101058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schmitt DL, Sundaram A, Jeon M, Luu BT, An S. (2018).Spatial alterations of De Novo purine biosynthetic enzymes by Akt-independent PDK1 signaling pathways. PLoS One, 13, e0195989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.French JB, Zhao H, An S, Niessen S, Deng Y, Cravatt BF, Benkovic SJ. (2013).Hsp70/Hsp90 chaperone machinery is involved in the assembly of the purinosome. Proc Natl Acad Sci U S A, 110, 2528–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chan CY, Pedley AM, Kim D, Xia C, Zhuang X, Benkovic SJ. (2018).Microtubule-directed transport of purine metabolons drives their cytosolic transit to mitochondria. Proc Natl Acad Sci U S A, 115, 13009–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fu R, Sutcliffe D, Zhao H, Huang X, Schretlen DJ, Benkovic S, Jinnah HA. (2015).Clinical severity in Lesch-Nyhan disease: the role of residual enzyme and compensatory pathways. Mol Genet Metab, 114, 55–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhao A, Tsechansky M, Ellington AD, Marcotte EM. (2014).Revisiting and revising the purinosome. Mol Biosyst, 10, 369–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhao A, Tsechansky M, Swaminathan J, Cook L, Ellington AD, Marcotte EM. (2013).Transiently transfected purine biosynthetic enzymes form stress bodies. PLoS One, 8, e56203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Deng Y, Gam J, French JB, Zhao H, An S, Benkovic SJ. (2012).Mapping protein-protein proximity in the purinosome. J Biol Chem, 287, 36201–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kyoung M, Russell SJ, Kohnhorst CL, Esemoto NN, An S. (2015).Dynamic architecture of the purinosome involved in human de novo purine biosynthesis. Biochemistry, 54, 870–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wan C, Borgeson B, Phanse S, Tu F, Drew K, Clark G, Xiong X, Kagan O, Kwan J, Bezginov A, Chessman K, Pal S, Cromar G, Papoulas O, Ni Z, Boutz DR, Stoilova S, Havugimana PC, Guo X, Malty RH, Sarov M, Greenblatt J, Babu M, Derry WB, Tillier ER, Wallingford JB, Parkinson J, Marcotte EM, Emili A. (2015).Panorama of ancient metazoan macromolecular complexes. Nature, 525, 339–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pareek V, Tian H, Winograd N, Benkovic SJ. (2020).Metabolomics and mass spectrometry imaging reveal channeled de novo purine synthesis in cells. Science, 368, 283–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pelet A, Skopova V, Steuerwald U, Baresova V, Zarhrate M, Plaza JM, Hnizda A, Krijt M, Souckova O, Wibrand F, Andorsdottir G, Joensen F, Sedlak D, Bleyer AJ, Kmoch S, Lyonnet S, Zikanova M. (2019).PAICS deficiency, a new defect of de novo purine synthesis resulting in multiple congenital anomalies and fatal outcome. Hum Mol Genet, 28, 3805–14 [DOI] [PubMed] [Google Scholar]
- 87.Spurr IB, Birts CN, Cuda F, Benkovic SJ, Blaydes JP, Tavassoli A. (2012).Targeting tumour proliferation with a small-molecule inhibitor of AICAR transformylase homodimerization. Chembiochem, 13, 1628–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tavassoli A, Benkovic SJ. (2005).Genetically selected cyclic-peptide inhibitors of AICAR transformylase homodimerization. Angew Chem Int Ed Engl, 44, 2760–3 [DOI] [PubMed] [Google Scholar]
- 89.Pedley AM, Karras GI, Zhang X, Lindquist S, Benkovic SJ. (2018).Role of HSP90 in the Regulation of de Novo Purine Biosynthesis. Biochemistry, 57, 3217–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.French JB, Jones SA, Deng H, Pedley AM, Kim D, Chan CY, Hu H, Pugh RJ, Zhao H, Zhang Y, Huang TJ, Fang Y, Zhuang X, Benkovic SJ. (2016).Spatial colocalization and functional link of purinosomes with mitochondria. Science, 351, 733–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.An S, Deng Y, Tomsho JW, Kyoung M, Benkovic SJ. (2010).Microtubule-assisted mechanism for functional metabolic macromolecular complex formation. Proc Natl Acad Sci U S A, 107, 12872–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Schmitt DL, Cheng YJ, Park J, An S. (2016).Sequestration-Mediated Downregulation of de Novo Purine Biosynthesis by AMPK. ACS Chem Biol, 11, 1917–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mathews CK. (1993).The cell-bag of enzymes or network of channels? J Bacteriol, 175, 6377–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Prouteau M, Loewith R. (2018).Regulation of Cellular Metabolism through Phase Separation of Enzymes. Biomolecules, 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Strom AR, Brangwynne CP. (2019).The liquid nucleome - phase transitions in the nucleus at a glance. J Cell Sci, 132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Banani SF, Lee HO, Hyman AA, Rosen MK. (2017).Biomolecular condensates: organizers of cellular biochemistry. Nat Rev Mol Cell Biol, 18, 285–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bracha D, Walls MT, Brangwynne CP. (2019).Probing and engineering liquid-phase organelles. Nat Biotechnol, 37, 1435–45 [DOI] [PubMed] [Google Scholar]
- 98.Doigneaux C, Pedley AM, Mistry IN, Papayova M, Benkovic SJ, Tavassoli A. (2020).Hypoxia Drives the Assembly of the Multi-Enzyme Purinosome Complex. J Biol Chem, 295, 9551–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Soding J, Zwicker D, Sohrabi-Jahromi S, Boehning M, Kirschbaum J. (2020).Mechanisms for Active Regulation of Biomolecular Condensates. Trends Cell Biol, 30, 4–14 [DOI] [PubMed] [Google Scholar]
- 100.Mateju D, Franzmann TM, Patel A, Kopach A, Boczek EE, Maharana S, Lee HO, Carra S, Hyman AA, Alberti S. (2017).An aberrant phase transition of stress granules triggered by misfolded protein and prevented by chaperone function. EMBO J, 36, 1669–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rai AK, Chen JX, Selbach M, Pelkmans L. (2018).Kinase-controlled phase transition of membraneless organelles in mitosis. Nature, 559, 211–16 [DOI] [PubMed] [Google Scholar]
- 102.Wippich F, Bodenmiller B, Trajkovska MG, Wanka S, Aebersold R, Pelkmans L. (2013).Dual specificity kinase DYRK3 couples stress granule condensation/dissolution to mTORC1 signaling. Cell, 152, 791–805 [DOI] [PubMed] [Google Scholar]
- 103.Alberti S, Gladfelter A, Mittag T. (2019).Considerations and Challenges in Studying Liquid-Liquid Phase Separation and Biomolecular Condensates. Cell, 176, 419–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Pedley AM, Boylan JB, Wolstenholme CH, Liu Y, Kennedy EL, Chan CY, An S, Kyoung M, Zhang X, Benkovic SJ. (to be submitted).Purine enzyme assemblies display liquid-like properties regulated by molecular chaperones.
- 105.Labuschagne CF, van den Broek NJ, Mackay GM, Vousden KH, Maddocks OD. (2014).Serine, but not glycine, supports one-carbon metabolism and proliferation of cancer cells. Cell Rep, 7, 1248–58 [DOI] [PubMed] [Google Scholar]
- 106.Ali ES, Sahu U, Villa E, O'Hara BP, Gao P, Beaudet C, Wood AW, Asara JM, Ben-Sahra I. (2020).ERK2 Phosphorylates PFAS to Mediate Posttranslational Control of De Novo Purine Synthesis. Mol Cell, 78, 1178–91 e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wang W, Fridman A, Blackledge W, Connelly S, Wilson IA, Pilz RB, Boss GR. (2009).The phosphatidylinositol 3-kinase/akt cassette regulates purine nucleotide synthesis. J Biol Chem, 284, 3521–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Saha A, Connelly S, Jiang J, Zhuang S, Amador DT, Phan T, Pilz RB, Boss GR. (2014).Akt phosphorylation and regulation of transketolase is a nodal point for amino acid control of purine synthesis. Mol Cell, 55, 264–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Liu C, Knudsen GM, Pedley AM, He J, Johnson JL, Yaron TM, Cantley LC, Benkovic SJ. (2019).Mapping Post-Translational Modifications of de Novo Purine Biosynthetic Enzymes: Implications for Pathway Regulation. J Proteome Res, 18, 2078–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.An S, Kyoung M, Allen JJ, Shokat KM, Benkovic SJ. (2010).Dynamic regulation of a metabolic multi-enzyme complex by protein kinase CK2. J Biol Chem, 285, 11093–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Verrier F, An S, Ferrie AM, Sun H, Kyoung M, Deng H, Fang Y, Benkovic SJ. (2011).GPCRs regulate the assembly of a multienzyme complex for purine biosynthesis. Nat Chem Biol, 7, 909–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ben-Sahra I, Hoxhaj G, Ricoult SJH, Asara JM, Manning BD. (2016).mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science, 351, 728–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Pedley AM, Benkovic SJ. (2018).Detecting Purinosome Metabolon Formation with Fluorescence Microscopy. Methods Mol Biol, 1764, 279–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie 1. Intracellular Transport of Purinosomes along Microtubules. Two purinosomes (FGAMS-EGFP, green) are shown to move along microtubules (silicon rhodamine tubulin, gray) and merge into a larger purinosome at 260 seconds. Scale bar = 1 μm. Movie was generated from time-lapse images reported in Chan et al., 2018 (Chan et al., 2018).