

Abstract
The mechanisms through which tau and amyloid-beta (Aβ) accumulate in the brain of Alzheimer’s disease patients may differ but both are related to neuronal networks. We examined such mechanisms on neuroimaging in 58 participants with atypical Alzheimer’s disease (posterior cortical atrophy or logopenic progressive aphasia). Participants underwent Aβ-PET, longitudinal tau-PET, structural MRI and resting-state functional MRI, which was analyzed with graph theory. Regions with high levels of Aβ were more likely to be functional hubs, with a high number of functional connections important for resilience to cascading network failures. Regions with high levels of tau were more likely to have low clustering coefficients and degrees, suggesting a lack of trophic support or vulnerability to local network failures. Regions strongly functionally connected to the disease epicenters were more likely to have higher levels of tau and, less strongly, of Aβ. The regional rate of tau accumulation was associated with tau levels in functionally connected regions, in support of tau accumulation in a functional network. This study elucidates the relations of tau and Aβ to functional connectivity metrics in atypical Alzheimer’s disease, strengthening the hypothesis that the spread of the 2 proteins is driven by different biological mechanisms related to functional networks.
Keywords: Alzheimer’s disease, amyloid, functional connectivity, tau, trans-neuronal spread
Introduction
Amyloid β (Aβ) extracellular senile plaques and intracellular aggregations of hyperphosphorylated tau protein known as neurofibrillary tangles are key pathological hallmarks of Alzheimer’s disease (AD) (Braak and Braak 1991). Amyloid and tau are present in the typical amnestic variant of the disease as well as in atypical variants like logopenic progressive aphasia (LPA) and posterior cortical atrophy (PCA), with topographical patterns differing across variants, particularly for tau (Ossenkoppele et al. 2016; Jones et al. 2017; Tetzloff et al. 2018; Sintini et al. 2020). Large-scale brain networks, such as the default mode network, are also affected in AD, and again differences exist across disease variants, with the involvement of syndrome-specific networks like the language network for LPA and the higher visual network for PCA (Seeley et al. 2009; Lehmann et al. 2013, 2015; Whitwell et al. 2015).
The notion that, in AD, neuronal networks are intertwined with neurodegeneration and pathologic proteins has been vastly discussed in the literature (Brettschneider et al. 2015). From an imaging perspective, existing multimodal studies combine resting-state functional MRI (fMRI) to measure task-free functional connectivity across brain regions, structural MRI for atrophy and Aβ- and tau-PET (Zhou et al. 2012; Jones et al. 2017; Mutlu et al. 2017; Cope et al. 2018; Hoenig et al. 2018; Ossenkoppele et al. 2019; Pereira et al. 2019; Franzmeier et al. 2020; Vogel et al. 2020). Having different spatiotemporal patterns of deposition, Aβ and tau do not relate to functional connectivity in the same way (Jones et al. 2017). This was also shown in a recent animal model, demonstrating that the former induces neuronal hyperactivity while the latter suppresses neuronal activity, and the 2 together synergize to impair neuronal networks (Busche et al. 2019). According to the cascading network failure (CNF) hypothesis (Jones et al. 2016), large-scale brain networks are implicated in the pathophysiology of tau deposition in AD and amyloid is a mediator in this relationship, with tau deposition patterns closely resembling those of functional networks (Jones et al. 2017). The CNF model hypothesizes a tau-associated local network failure followed by a global compensatory phenomenon associated with Aβ: after global functional hubs saturate their ability to offer resilience to local network failures, tau accumulation within those failing networks rapidly accelerates (Jones et al. 2016; Jones et al. 2017). Functional hubs are areas of the cortex that contain a large number of functional connections that are not unimodal, but instead integrate information from diverse sources (Mesulam 1998; Sporns et al. 2007; Buckner et al. 2009). This is why the CNF model predicts that functional ‘hubness’ will associate with Aβ deposition irrespective of the clinical phenotype, but tau accumulation will vary by clinical phenotype within specific functional networks related to the cognitive function impaired. Graph-theoretical analysis of resting-state fMRI signal allows a quantification of the topographical properties of the regions that compose the brain networks involved in AD (Bullmore and Sporns 2009), which can subsequently be used to model the spatial associations between functional connectivity and pathological protein burden. Three main alternatives to the CNF model exist on these associations that can be investigated with graph-theoretical metrics: 1) functional network failure via molecular spread (i.e., pathologic proteins spread through functional networks in a prion-like manner), 2) metabolic susceptibility (i.e., functional hubs, like the precuneus and posterior cingulate cortex, are more susceptible to pathologic proteins because of high metabolic demand), and 3) the trophic failure hypothesis (i.e., deteriorated functional networks weaken inter-regional trophic support and pathologic proteins spread more easily within regions lacking trophic factors) (Zhou et al. 2012). On the other hand, the CNF model predicts that all graph-theoretical metrics will be related to measures of AD pathology, but these associations will be different for Aβ (i.e., functional ‘hubness’) and tau (i.e., specific functional network connections) and may vary by disease stage. It is worth mentioning that these hypotheses on mechanisms of protein spreading are not mutually exclusive and different mechanisms could be simultaneously playing a role in the disease course. Imaging and experimental studies have shown that neural activity stimulates Aβ production and therefore Aβ deposition maps onto functional hubs (Cirrito et al. 2005; Buckner et al. 2009; Bero et al. 2011; Mutlu et al. 2017; Busche et al. 2019), in agreement with the cascading failure model. On the contrary, findings on the association between tau and functional connectivity metrics are more complex. It has been suggested that, in AD, tau spreads trans-neuronally (Clavaguera et al. 2009; de Calignon et al. 2012; Liu et al. 2012; Ahmed et al. 2014), possibly from disease-specific epicenters, since the topological patterns of tau deposition in the brain closely resemble the functional networks obtained using such epicenters as seeds in resting-state fMRI analyses (Hoenig et al. 2018; Pereira et al. 2019; Ossenkoppele et al. 2019). Disease epicenters may be intended as preferential locations of pathology accumulation and sources of pathology spreading but it is unknown whether they represent the very genesis point of the disease. A positive correlation between regional tau pathology and the number and strength of regional functional connections has also been reported in a study that employed the graph-theoretical approach (Cope et al. 2018). A recent longitudinal study has suggested that the accumulation of tau pathology occurs through functional networks, rather than spatial proximity (Franzmeier et al. 2020), which is a concept also supported by observations in animal models (Clavaguera et al. 2009; Liu et al. 2012; de Calignon et al. 2012; Ahmed et al. 2014).
The majority of existing studies are cross-sectional and focused on typical amnestic AD, while little is known about associations between pathologic protein deposition and resting-state functional connectivity in the atypical presentations of the disease. This is a key research question, since the mechanisms of protein spreading through various brain networks can potentially explain the heterogeneity in the neurodegeneration patterns of AD variants. As we and others have reported, cross-sectional tau maps onto cognitive dysfunctions and discriminates well between AD variants (Ossenkoppele et al. 2016; Tetzloff et al. 2018; Sintini et al. 2020), while longitudinal accumulation partially converges to common areas (Sintini et al. 2019). How these patterns of tau deposition relate to the graph-theoretical properties of the brain regions that they cover has not yet been studied in atypical AD. Atypical AD also provides an ideal cohort to assess these relationships since tau appears to be a strong driver of neurodegeneration (Sintini et al. 2018), with less influence of other proteins, such as the TAR DNA binding protein of 43 kDa (Sahoo et al. 2018), which plays an important role in typical AD, particularly in older patients (Josephs et al. 2014). In addition, the CNF model also makes testable predictions based on phenotype in that amyloid will have the same relationship to functional ‘hubness’ across phenotypes, but tau will vary its relationship to match the functional network that is impaired in an individual roughly captured by clinical phenotype.
We aimed to address these questions in a cohort of 58 participants with biomarker-supported diagnoses of PCA or LPA, using cross-sectional resting-state fMRI, Aβ-PET and longitudinal tau-PET images. We hypothesized that regional Aβ, baseline tau and longitudinal rates of change in tau will relate differently to graph-theoretical and functional connectivity metrics.
Materials and Methods
Participants
Fifty-eight atypical AD participants (31 meeting clinical criteria for PCA; Crutch et al. 2012 and 27 meeting clinical criteria for LPA; Gorno-Tempini et al. 2011) were included in the study. Participants were recruited by the Neurodegenerative Research Group (NRG; PI’s Josephs and Whitwell) between 2016 and 2019 and underwent structural MRI and resting-state fMRI, [11C]Pittsburgh Compound B PET for Aβ, and [18F]flortaucipir PET for tau. Thirty-four participants (15 PCA and 19 LPA) also underwent 1-year follow-up structural MRI and [18F]flortaucipir PET. The Aβ PET scans were analyzed to determine Aβ positivity as previously described (Jack et al. 2017) and all participants were determined to be Aβ(+). Participants were excluded from the study if they had a stroke or tumor that could explain their symptoms, if they had poor vision (20/400), if MRI was contraindicated (e.g., metal in head, cardiac pace maker), if there were conditions that may confound brain imaging studies (e.g., structural abnormalities, including subdural hematoma or intracranial neoplasm), or if they met specific criteria for another neurodegenerative disorder. All participants underwent a clinical and neuropsychological evaluation (Tetzloff et al. 2018), including: Montreal Cognitive Assessment Battery (MoCA) to assess general cognitive function; Clinical Dementia Rating (CDR) scale to assess functional impairment; Boston Naming Test (BNT) to assess naming; Boston Diagnostic Aphasia Examination (BDAE) repetition to assess sentence repetition; Wechsler Memory Scale-III (WMS-III) Visual reproduction test to assess visual memory; Visual Object and Space Perception Battery (VOPS) incomplete letters test to assess visual perception. Apolipoprotein E (APOE) genotyping was performed on 44 participants. Demographics and clinical scores were compared between PCA and LPA with t-test or Fisher’s exact test. Twelve cognitively unimpaired (CU) individuals were also recruited by the NRG and included in the study as a control group. The median age of the CU individuals was 59 (inter-quartile range: 57, 64); 8 were female; the median MoCA score was 27 (inter-quartile range: 25, 28). The study was approved by the Mayo Clinic IRB, and all participants provided written informed consent to participate in this study.
Image Acquisition
All PET scans were acquired using PET/CT scanners (GE Healthcare, Milwaukee, Wisconsin) operating in 3D mode. For tau-PET, an intravenous bolus injection of approximately 370 MBq (range 333–407 MBq) of [18F]flortaucipir was administered, followed by a 20-min PET acquisition performed 80 min after injection. For Aβ-PET, participants were injected with [11C]Pittsburgh Compound B (PiB) of approximately 628 MBq (range 385–723 MBq) and, after a 40–60-min uptake period, a 20-min PiB scan was obtained. Both PiB and flortaucipir PET scans consisted of four 5-min dynamic frames following a low dose CT transmission scan. Standard corrections were applied. Emission data were reconstructed into a 256 × 256 matrix with a 30-cm field of view (in-plane pixel size = 1.0 mm). Flortaucipir PET scans were performed at baseline and at 1-year follow-up. All participants also underwent a 3 T head MRI protocol at both time-points that included a magnetization prepared rapid gradient echo (MPRAGE) sequence (TR/TE/TI, 2300/3/900 ms; flip angle 8°, 26-cm field of view; 256 × 256 in-plane matrix with a phase field of view of 0.94, and slice thickness of 1.2 mm; Jack et al. 2008) and resting-state gradient echo-planar imaging (TR/TE = 3000/30 ms, 90° flip angle, slice thickness 3.3 mm, in-plane resolution 3.3 mm, and 160 volumes). Participants were instructed to keep their eyes open during the resting-state fMRI scanning. The MRI scans were performed on one of 2 GE scanners (GE Healthcare, Milwaukee, Wisconsin) with identical protocols. The MRI scans were performed a median of 1 day from the PET scans at both baseline and follow-up.
Image Processing: Structural MRI and PET
Each PET image was rigidly registered to its corresponding MPRAGE using SPM12 (Wellcome Trust Centre for Neuroimaging). Standard uptake value ratios (SUVR) were calculated normalizing each Aβ- and tau-PET image to the cerebellar crus gray matter. Using ANTs (Avants et al. 2008), the Brainnetome atlas (http://www.brainnetome.org/), which has been validated for both structural and functional images (Fan et al. 2016; Brown et al. 2019), was propagated to the native MPRAGE space and median Aβ and tau SUVR were calculated in each region-of-interest (ROI), in the gray and white matter segmentations binarized at 0.5. The Brainnetome atlas has 246 cortical and subcortical ROIs; however, in our analyses, we included only the 210 cortical ROIs in the frontal lobe (superior, middle and inferior frontal gyri, orbital gyrus, precentral gyrus, paracentral lobule), insular lobe, limbic lobe (cingulate gyrus), temporal lobe (superior, middle and inferior temporal gyri, fusiform gyrus, parahippocampal gyrus, posterior superior temporal sulcus), parietal lobe (superior and inferior parietal lobule, precuneus, postcentral gyrus), and occipital lobe (medio-ventral and lateral occipital cortex). Subcortical ROIs were excluded from the main analyses due to the issue of off-target binding of the tau-PET tracer; additionally, atypical AD pathology affects primarily the cortex. Masking atlas ROIs based on the segmentation avoids outlying voxels that are mostly non-tissue, and it thus reduces the effects of partial volume. Tissue probabilities were determined for each MPRAGE using Unified Segmentation (Ashburner and Friston 2005) in SPM12, with the Mayo Clinic Adult Lifespan Template (MCALT) tissue priors and settings (Schwarz et al. 2017). Annualized rates of change in tau SUVR were calculated as the difference in regional SUVR between follow-up and baseline, divided by the scan interval in years.
Image Processing: Functional MRI
All functional images were preprocessed using CONN functional connectivity toolbox (www.nitrc.org/projects/conn). The preprocessing included discarding the first 10 volumes to obtain steady-state magnetization, slice time correction, re-alignment and unwarp (i.e., subject motion estimation and correction), outliers detection (i.e., excessive head motion), segmentation and direct normalization to MNI template space, smoothing using spatial convolution with a Gaussian kernel of 6-mm full width half-maximum, nuisance regression (white matter and CSF signal; head-motion parameters from the re-alignment step and their first derivatives), bandpass filtering in the 0.009–0.08-Hz frequency. The fMRI data sets with more than 3 mm of absolute translational movement or 3° of absolute rotational movement were excluded from the analyses. From the original data (60 participants and 13 CU), 3 outliers (2 participants and one CU individual) were detected and excluded. No significant differences were observed across groups (PCA, LPA, CU) on the mean 6 head motion parameters (x = 0.12, 0.11, 0.10 mm, P = 0.78; y = 0.25, 0.14, 0.11 mm, P = 0.07; z = 0.30, 0.35, 0.30 mm, P = 0.75; pitch = 0.4°, 0.3°, 0.3°, P = 0.60; roll = 0.2°, 0.2°, 0.2°, P = 0.63; yaw = 0.3°, 0.4°, 0.2°, P = 0.31). After preprocessing, the functional images were parcellated with the Brainnetome atlas, as the PET images. The mean blood-oxygen-level-dependent (BOLD) time series within each ROI of the atlas was extracted, and the Pearson’s R correlation coefficients were calculated across all ROI pairs. The association matrices were generated by transforming the R coefficients with the Fisher’s R-to-Z transformation. Functional connectivity between each pair of ROIs was therefore quantified by their Z value. For each participant, a non-directional unweighted adjacency matrix was computed by thresholding the association matrix, interpreting each ROI as a node, and each thresholded functional connection as an edge. We computed the adjacency matrices using a relative network density threshold of 0.05, meaning that in each participant a fixed percentile (i.e., 5%) of edges was included. This threshold was selected because it jointly maximized the difference between global efficiency of our networks relative to lattice graphs as well as their local efficiency relative to random graphs, leading to brain networks with small-world properties. This optimization was performed with the CONN toolbox. A graph-theoretical analysis was then performed to investigate the topological properties of each ROI within the graph. The graph-theoretical metrics that were assessed were the degree, which is the number of functional connections of each node and provides a proxy for functional ‘hubness’, and the clustering coefficient, which characterizes the level of connections among all nodes within a node neighboring sub-graph and provides a proxy for trophic support. All the results presented here were obtained using the 0.05 threshold when computing the graph-theoretical metrics; however, to test their robustness, we repeated the main analyses with different threshold that led to more sparse (0.02) and less sparse networks (0.10 and 0.15) and these results are reported as supplemental.
Statistical Analyses
The group-average Fisher’s R-to-Z transformed association matrices of the CU individuals and PCA and LPA participants were compared by a pairwise subtraction. PCA and LPA functional connectivity matrices were also compared to the matrices derived from fMRIs of 50 young healthy individuals (age 21–35) from the Human Connectome Project (HCP) (http://www.humanconnectomeproject.org/), using t-tests with FWE multiple comparisons correction. HCP images were pre-processed using the same pipeline applied to the atypical AD participants. To describe the spatial patterns of tau and Aβ deposition, the group-average PiB and flortaucipir SUVR voxel-based maps for PCA and LPA were created. To test the validity of the 3 protein spreading hypotheses (functional ‘hubness’, trophic failure and trans-neuronal spread), we computed Pearson’s R correlation coefficients on syndrome-specific group-average data, similarly to previous studies (Cope et al. 2018; Franzmeier et al. 2020). The first variables in the correlations were group-average regional values of: 1) cross-sectional PiB SUVR, 2) cross-sectional flortaucipir SUVR, and 3) rates of change in flortaucipir SUVR. The second variables were group-average regional values of: 1) degree, for the functional ‘hubness’ hypothesis; 2) clustering coefficients, for the trophic failure hypothesis; and 3) functional connectivity to a syndrome- and protein-specific epicenter, for the trans-neuronal spread hypothesis. Epicenters were defined as the ROIs with the highest SUVR within the group at baseline (Mutlu et al. 2017; Ossenkoppele et al. 2019). As an alternative approach, similarly to previous studies (Zhou et al. 2012; Brown et al. 2019), we identified as epicenters those ROIs whose functional connectivity to the other regions was best correlated to regional protein SUVR and these results are reported as supplemental. The epicenter analyses were repeated using the first 5 ROIs with the highest SUVR or that led to highest correlations, to investigate their stability. Correlations were computed across all the cortical ROIs. Correlations including subcortical ROIs (amygdala, hippocampus, basal ganglia, thalamus) are reported as supplemental. The correlation analyses between protein SUVR and graph-theory metrics were repeated using group-average degrees and clustering coefficients derived from HCP young healthy individuals, in order to better understand the role of temporal disease factors. Linear regression on graph-theory metrics and protein SUVR were also computed within each lobe and each network. For the network-level correlations, which are reported as supplemental, PET and graph-theory data were obtained using the 400 cortical ROIs of the Schaefer atlas, organized in 7 functional networks (visual, somatomotor, default mode, control, dorsal attention, salience, limbic) (Schaefer et al. 2018). The rates of change in tau SUVR were also correlated to the tau-weighted functional connectivity metric proposed in a recent study (Franzmeier et al. 2020) to test the trans-neuronal spread hypothesis. Tau-weighted functional connectivity is calculated, for each ROI, as the mean of functional connectivity values (Z values) between the given ROI and all the other ROIs, divided by the baseline flortaucipir SUVR of all the other ROIs. Unlike the study that proposed this method (Franzmeier et al. 2020), we avoided dividing by the Euclidean distances between the ROIs (i.e., tau- and distance-weighted functional connectivity) in order to model the hypothesis that tau spreads through functional connectivity, rather than proximity. Given that brain biological regional quantities suffer from autocorrelation, we compared all the group-level correlations P values with: 1) the exact P values obtained using 1000 null-model functional connectivity matrices, i.e., shuffling mean LPA and PCA functional connectivity matrices while preserving weight- and degree-distribution using functions of the Brain Connectivity Toolbox (https://sites.google.com/site/bctnet/); 2) the exact P values derived from 1000 surrogate brain maps with autocorrelated spatial heterogeneity generated with BrainSMASH (Burt et al. 2020), i.e., shuffling SUVR maps while maintaining spatial autocorrelation in the data. We also computed the exact P values derived from 1000 spatially naïve surrogate brain maps generated with BrainSMASH, i.e., randomly shuffling SUVR maps. Within-subject correlations were also computed as well as between-subject correlations across the average cortex values of SUVR and clustering coefficient. The between-subject correlation was not computed for the degree, since, by definition, the average degree is constant across participants (i.e., 5% of functional connections are taken into account to build each graph). The group-average correlations involving the degree were repeated using a different measurement of the total functional connectivity of each node. To obtain the group-level measure of this ‘total flow’ or ‘global connectivity’ (Zhou et al. 2012; Mutlu et al. 2017), the t-scores from group-level one-sample t-tests were summed for each node, thresholding at P < 0.05 after FWE correction (for baseline values) and at P < 0.001 (for participants that had a follow-up scan). We further examined the process of tau accumulation using a mixed-effect model that predicted the rates of change in tau from baseline tau and Aβ levels, tau-weighted functional connectivity and the degree across all cortical ROIs of the 34 atypical AD participants with follow-up images. All the variables were normalized, and 2 interaction terms were also included to model the interaction between baseline tau and Aβ levels and between degree and tau-weighted functional connectivity. For comparison purposes, the mixed-effect model was also developed using tau- and distance-weighted functional connectivity (Franzmeier et al. 2020). The mixed-effect model was developed in R version 3.6.0 (http://www.r-project.org/), while all the other analyses were performed in Matlab 2018a (The Mathworks, Inc.). In the mixed-effect model, the T statistics were calculated with the R package lme4, while the P values were calculated with the R package arm.
Results
Participants
Demographics and clinical scores of the 2 syndromes are reported in Table 1. LPA participants performed significantly worse than PCA in BNT (P = 0.04) and BDAE repetition (P = 0.006) and significantly better in VOSP letters (P < 0.001).
Table 1.
Demographics and clinical scores
| PCA (N = 31) | LPA (N = 27) | P | |
|---|---|---|---|
| Female sex, N (%) | 21 (68%) | 17 (63%) | 0.78 |
| Age at baseline (years) | 62 (59, 68) | 67 (61, 72) | 0.10 |
| Disease duration (years) | 4 (3, 5) | 2.5 (2, 3) | 0.17 |
| Left handedness, N (%) | 4 (13%) | 4 (15%) | 1.0 |
| Global PiB SUVR | 2.45 (2.20, 2.60) | 2.42 (2.19, 2.90) | 0.28 |
| APOE ɛ4 frequency, N (%) | 11 (48%)* | 8 (38%)** | 0.56 |
| MoCA (/30) | 16 (12, 21) | 19 (16, 22) | 0.86 |
| CDR Dementia Staging Instrument (/18) | 3 (2, 6) | 1.5 (1, 3) | 0.07 |
| BNT | 12 (10,14) | 11 (6, 13) | 0.04 |
| BDAE repetition | 9 (6, 10) | 7 (6, 8) | 0.006 |
| WMS-III VR% retention MOANS | 8 (5, 10) | 9 (7, 12) | 0.06 |
| VOSP letters | 13 (5, 16) | 19 (18, 20) | <0.001 |
Notes: Data are shown as median (inter-quartile range), or N (%). PiB SUVR, Pittsburgh Compound B standardized uptake value ratio; APOE, Apolipoprotein E; MoCA, Montreal Cognitive Assessment Battery; CDR, Clinical Dementia Rating (sum of boxes); BNT, Boston Naming Test; BDAE, Boston Diagnostic Aphasia Examination; WMS-III VR% retention MOANS, Wechsler Memory Scale-III Visual Reproduction % Mayo Older American Normative scale; VOSP, Visual Object and Space Perception battery. P values were obtained with t-test for continuous variables and Fisher’s exact test for categorical variables. Significant P values are bolded
*Data available for 23 participants
**Data available for 21 participants
Regional Functional Connectivity
Atypical AD participants showed a general decline in the strength of within- and between-lobe functional connections relative to the older CU individuals (Fig. 1A,B). Similarly, relative to HCP young healthy individuals, PCA and LPA participants showed marked diffuse reductions in functional connectivity, particularly in the occipital and limbic lobes for PCA and in the temporal lobe for LPA (Supplementary Fig. 1). Both phenotypes also showed regions of increased functional connectivity compared to both HCP individuals (Supplementary Fig. 1) and older CU individuals (Fig. 1B), particularly in the frontal and parietal lobes. Differences between the PCA and LPA groups were more subtle, with PCA having weaker connections in the occipital lobe (in blue) and LPA having weaker connections in the frontal lobe (in red) (Fig. 1B, ‘PCA minus LPA’). Mean functional connectivity for each group (PCA, LPA, CU) are reported as supplemental (Supplementary Fig. 2). Mean connectivity was lower for participants relative to CU individuals, particularly for PCA (P = 0.004) and less strikingly for LPA (P = 0.06); no differences were found between PCA and LPA (P = 0.30).
Figure 1.
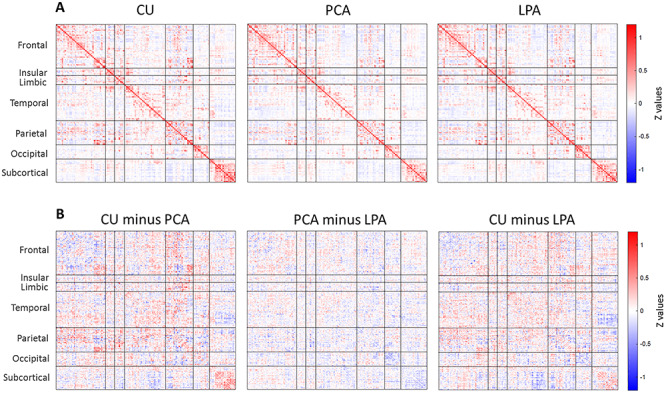
Group-average Fisher’s Z transformed functional connectivity matrix (i.e., association matrix) for CU, PCA and LPA (A). Pairwise subtractions of the matrices (B).
Baseline Associations between Tau, Aβ and Connectivity
All correlations reported here were obtained with a graph-theory threshold of 0.05 while correlations obtained using 0.02, 0.10 and 0.15 are reported in Supplementary Table 1. Figure 2A shows the average voxel-based maps of baseline PiB and flortaucipir SUVR in PCA and LPA for reference. PiB SUVR was positively associated to the degree (PCA: R = 0.51, P < 0.001; LPA: R = 0.38, P < 0.001), in support of the functional ‘hubness’ hypothesis for Aβ. An analogous positive association was found when using the degree calculated from the functional connectivity matrices of the young healthy HCP individuals (Supplementary Table 2). PiB SUVR was also positively associated to the clustering coefficient in PCA (R = 0.30, P < 0.001). The correlation between PiB SUVR and degree in LPA was not significant when compared to either the null-models of functional connectivity or the surrogate autocorrelated brain SUVR maps, while, on the contrary, the same correlation in PCA survived both autocorrelation tests (Table 2). Flortaucipir SUVR was negatively associated to the degree (PCA: R = −0.16, P = 0.02; LPA: R = −0.20, P = 0.004) and the clustering coefficient (PCA: R = −0.15, P = 0.03; LPA: R = −0.34, P < 0.001), in support of the trophic failure or vulnerability to failure hypothesis for tau. However, among these correlations, only the one between flortaucipir SUVR and clustering coefficient in LPA survived the autocorrelation tests (Table 2). A negative correlation between tau and clustering coefficient in LPA was also present when using HCP functional connectivity, while, on the contrary, the negative correlation between tau and degree was not present in either phenotype (Supplementary Table 2). The positive association between Aβ and degree and the negative one between tau and clustering coefficient were consistent across graph-theory thresholds for both phenotypes (Supplementary Table 1). The functional ‘hubness’ hypothesis for Aβ was strengthened by the correlation of PiB SUVR with the ‘total flow’ (PCA: R = 0.35, P < 0.001; LPA: R = 0.31, P < 0.001), while the same measure had a negative association to flortaucipir SUVR (PCA: R = −0.41, P < 0.001; LPA: R = −0.29, P < 0.001) (Supplementary Fig. 3). The within-subject correlations performed across all ROIs of each participant revealed extensive inter-subject variability but confirmed the significant differences (P < 0.001) between tau and Aβ associations to degree and clustering coefficient, with Aβ having more positive associations to these metrics and tau more negative associations (Fig. 2C). For tau, part of the variability in the within-subject correlations was explained by the characteristics of the patients: patients that were younger, with a more aggressive disease and more extensive cortical tau pathology, had less negative correlations between tau and degree or tau and clustering coefficient, potentially due to saturation effect (Supplementary Fig. 4). These trends were not observed for the within-subject amyloid correlations, potentially due to amyloid being extremely widespread. At the whole-brain level, a negative correlation between the mean cortex SUVR and the mean clustering coefficient was present for tau (R = −0.27, P = 0.04), while no significant relationship was found for Aβ (R = 0.22, P = 0.09) (Fig. 2D). Differences in these relationships existed across lobes and networks (Table 3 and Supplementary Table 3). The positive association between Aβ and degree was strongest in the limbic lobe and in the limbic network for both variants. The negative association between tau and clustering coefficient was strongest in the occipital and limbic network for PCA and in the parietal lobe and somatomotor network for LPA. PCA had positive associations between tau and clustering coefficient in the limbic, insular and temporal lobe, which led to a low R2 calculated across all the cortical ROIs (Table 3).
Figure 2.
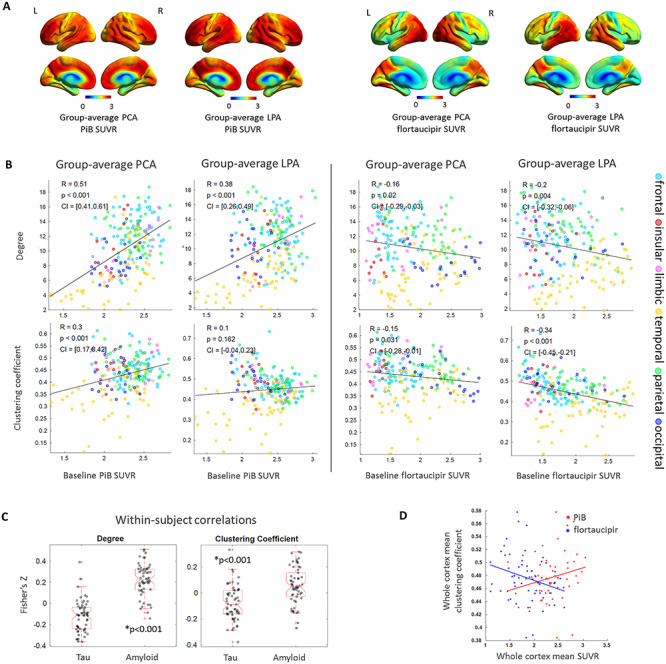
Group-average PiB and flortaucipir SUVR for PCA and LPA (A). Group-average ROI-level associations of PiB SUVR and flortaucipir SUVR with degree and clustering coefficient (B). Within-subject ROI-level Fisher’s Z transformed correlations between SUVR, degrees and clustering coefficients (C). Whole-cortex clustering coefficient versus whole-cortex PiB and flortaucipir SUVR (D).
Table 2.
P values of the correlation between variable 1 and variable 2 obtained from: traditional Pearson’s correlations between variable 1 and variable 2, without taking into account autocorrelation; exact P values derived from the comparison with the null distribution Pearson’s R coefficients calculated from variable 1 and 1000 null-model FC matrices, i.e., shuffling mean LPA and PCA functional connectivity matrices while preserving weight- and degree-distribution; exact P values derived from the comparison with the null-distribution Pearson’s R coefficients calculated from variable 2 and 1000 surrogate brain maps with autocorrelated spatial heterogeneity generated with BrainSMASH, i.e., shuffling SUVR maps while maintaining spatial autocorrelation in the data; exact P values derived from the comparison with the null-distribution Pearson’s R coefficients calculated from variable 2 and 1000 spatially naïve surrogate brain maps generated with BrainSMASH, i.e., randomly shuffling SUVR maps
| Variable 1 | Variable 2 | Phenotype | Pearson’s R, P value | Exact P value from null-model FC matrices | Exact P value from surrogate autocorrelated SUVR maps | Exact P value from surrogate spatially naïve SUVR maps |
|---|---|---|---|---|---|---|
| Aβ | Degree | LPA | R = 0.38, P < 0.001 | P = 0.14 | P = 0.118 | P < 0.001 |
| PCA | R = 0.51, P < 0.001 | P < 0.001 | P = 0.018 | P < 0.001 | ||
| Clustering Coefficient | LPA | ns | — | — | — | |
| PCA | R = 0.30, P < 0.001 | P < 0.001 | P = 0.131 | P < 0.001 | ||
| epicenter FC | LPA | R = 0.18, P = 0.009 | P = 0.01 | P = 0.138 | P = 0.004 | |
| PCA | R = 0.30, P < 0.001 | P < 0.001 | P = 0.034 | P < 0.001 | ||
| Tau | Degree | LPA | R = −0.20, P = 0.004 | P = 0.367 | P = 0.434 | P = 0.004 |
| PCA | R = −0.16, P = 0.02 | P = 1 | P = 0.466 | P = 0.023 | ||
| Clustering Coefficient | LPA | R = −0.34, P < 0.001 | P < 0.001 | P = 0.045 | P < 0.001 | |
| PCA | R = −0.15, P = 0.03 | P = 0.188 | P = 0.423 | P = 0.028 | ||
| epicenter FC | LPA | R = 0.58, P < 0.001 | P < 0.001 | P < 0.001 | P < 0.001 | |
| PCA | R = 0.71, P < 0.001 | P < 0.001 | P < 0.001 | P < 0.001 | ||
| Tau rates of change | Degree | LPA | R = 0.23, P < 0.001 | P = 0.175 | P = 0.242 | P < 0.001 |
| PCA | ns | — | — | — | ||
| Clustering Coefficient | LPA | ns | — | — | — | |
| PCA | ns | — | — | — | ||
| Tau weighted FC | LPA | R = 0.30, P < 0.001 | P < 0.001 | P = 0.063 | P < 0.001 | |
| PCA | R = 0.24, P < 0.001 | P = 1 | P = 0.227 | P = 0.001 | ||
| epicenter FC | LPA | R = 0.16, P = 0.023 | P = 0.011 | P = 0.225 | P = 0.017 | |
| PCA | R = −0.17, P = 0.017 | P = 0.020 | P = 0.45 | P = 0.018 |
Notes: P values less than 0.05 are bolded. ns, not significant; FC, functional connectivity
Table 3.
Lobe-level linear regression on Aβ and degree and on tau and clustering coefficient (P < 0.05)
| Amyloid and degree | Tau and clustering coefficient | |||
|---|---|---|---|---|
| PCA | LPA | PCA | LPA | |
| All cortical ROIs | R 2 = 0.27 | R 2 = 0.14 | R 2 = 0.02(−) | R 2 = 0.11(−) |
| Frontal | ns | ns | R 2 = 0.10(−) | R 2 = 0.21(−) |
| Insular | R 2 = 0.50 | ns | R 2 = 0.37 | ns |
| Limbic | R 2 = 0.75 | R 2 = 0.55 | R 2 = 0.43 | ns |
| Temporal | R 2 = 0.32 | R 2 = 0.33 | R 2 = 0.11 | ns |
| Parietal | ns | ns | R 2 = 0.24(−) | R 2 = 0.47(−) |
| Occipital | ns | ns | R 2 = 0.49(−) | R 2 = 0.23(−) |
Notes: ns, not significant. R-squared are reported. The (−) denotes a negative relationship
The disease epicenters were located in the right precuneus (PCA) and left precuneus (LPA) for Aβ and in the left lateral occipital (PCA) and left posterior superior temporal sulcus (LPA) for tau (Fig. 3A). The regional burden of both Aβ and tau was proportional to the strength of the regional functional connectivity to the disease epicenters. However, the proportionality was considerably stronger in tau (PCA: R = 0.71, P < 0.001; LPA: R = 0.58, P < 0.001) than in Aβ (PCA: R = 0.30, P < 0.001; LPA: R = 0.18, P = 0.009), pointing to the validity of functional network failure and the trans-neuronal spread hypothesis for tau more than Aβ. All these correlations survived the comparison with null-models of functional connectivity (Table 2). These correlations exhibited similar trends when correcting for the Euclidean distance of each ROI to the epicenter (Fig. 3B). This same trend was confirmed by the within-subject correlations, which were statistically higher for tau than Aβ when using subject-specific epicenters, but not when using group-level epicenters (Fig. 3C). Supplementary Table 4 shows the correlations obtained using as epicenters the 5 ROIs with highest PiB and flortaucipir SUVR: while regions with similarly high levels of tau led to similarly high correlations, the same was not true for Aβ. Supplementary Table 5 shows the results obtained using an alternative definition of epicenters, i.e., those ROIs whose functional connectivity was most strongly correlated to the other regions’ SUVR. For tau, such epicenters were located in the expected ‘signature’ regions, with high tau burden (occipital for PCA and left temporal for LPA), but the same was not true for Aβ. The combined findings from Figure 3, Supplementary Tables 4 and 5 support the notion that tau spreads within the functional networks of regions with high tau levels. Including the subcortical ROIs did not alter the results obtained for Aβ and tau in the cortical regions (Supplementary Fig. 5).
Figure 3.
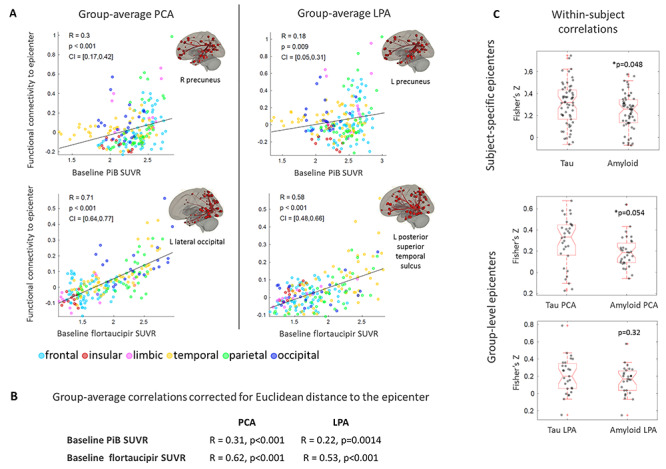
Group-average ROI-level correlations between PiB and flortaucipir SUVR and functional connectivity to the disease epicenter (A). Partial correlations between the same quantities, regressing out the Euclidean distance to the epicenter (B). Within-subject ROI-level Fisher’s Z transformed correlations between PiB and flortaucipir SUVR and functional connectivity to the subject-specific epicenter (i.e., ROI with the highest SUVR for the subject) and to the group-level epicenter (i.e., ROI with highest group-average SUVR) (C).
Associations between Connectivity and Longitudinal Tau Accumulation
The rate of change in flortaucipir SUVR was averaged across the 15 PCA and 19 LPA participants that had follow-up data and the voxel-based maps revealed high rates of accumulation in the bilateral frontal regions (PCA, LPA), lateral temporal regions (PCA, LPA), right medial occipital regions (PCA), and right temporo-parietal regions (LPA) (Fig. 4A). In both phenotypes, the tau-weighted functional connectivity was the metric most strongly correlated to the regional rates of change in tau (PCA: R = 0.24, P < 0.001, LPA: R = 0.30, P < 0.001) (Fig. 4B). In LPA, ROIs that experienced higher rates of tau accumulation were likely to have higher degree (R = 0.23, P < 0.001) (Fig. 4B) and higher ‘total flow’ (R = 0.21, P = 0.003) (Supplementary Fig. 3). These relationships were not present in PCA. The rate of change in tau was not correlated to the clustering coefficient in either phenotype (Fig. 4B). Among these correlations on the rates of change in tau, only the one with tau-weighted functional connectivity in LPA survived the comparison with null-models of functional connectivity (Table 2). In LPA, the rate of change in tau and the functional connectivity to the disease epicenter (i.e., ROI with highest baseline flortaucipir SUVR) were moderately positively correlated (R = 0.16, P = 0.02); however, interestingly, in PCA they were negatively correlated (R = −0.17, P = 0.017) (Fig. 4B). These correlations between tau rates of change and functional connectivity to the epicenters were significant when compared to the null models of functional connectivity (Table 2). The ROIs whose functional connectivity led to the highest correlation to the rates of change in tau throughout the cortex were located in the inferior temporal and frontal lobes for PCA and in the frontal and parietal lobes for LPA (Supplementary Table 5). The within-subject correlations of the rates of tau accumulation with functional connectivity metrics did not show statistically significant patterns (Fig. 4C). This could be due to the annual rates of change in tau SUVR being too subtle to show a clear within-subject pattern, as in the group-average analyses. However, the mixed-effect model that was fit on all the PCA and LPA participants with follow-up data reinforced the findings of the group-average correlations, revealing a positive effect of the tau-weighted functional connectivity (P = 0.002) and of the degree (P = 0.03) on the rates of tau accumulation (Table 3). The interaction term between the degree and tau-weighted functional connectivity was significant but did not invalidate the main effects (Supplementary Fig. 6). The mixed-effect model also showed that the baseline level of tau had a mild negative effect on the rates of tau accumulation (P = 0.06). Note that clustering coefficient was not included in the model variables because it was not correlated with tau rates of change in either phenotype at the group-level (Fig. 4). The tau- and distance-weighted functional connectivity led to a slightly stronger relationship to tau rates of changes (T = 4.9, P < 0.001) compared to tau-weighted functional connectivity (T = 3.0, P = 0.002, Table 4).
Figure 4.
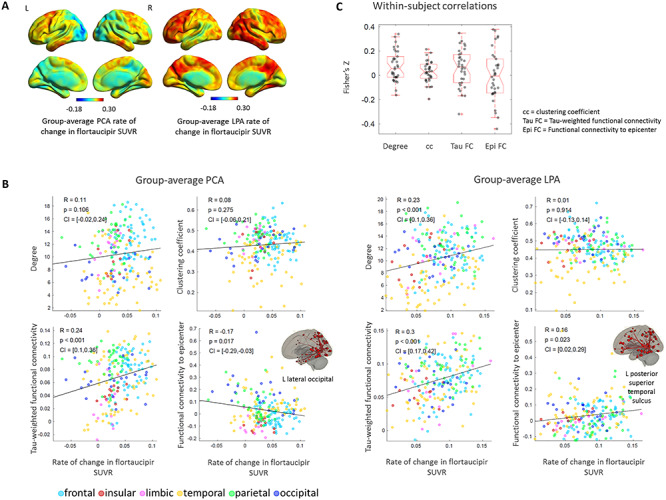
Group-average rate of change in flortaucipir SUVR in PCA and LPA (A). Group-average ROI-level associations between rate of change in flortaucipir SUVR and: degree, clustering coefficient, tau-weighted functional connectivity and functional connectivity to the disease epicenter (B). Within-subject ROI-level Fisher’s Z transformed correlations between the same metrics (C).
Table 4.
Mixed-effect model to evaluate the effect of baseline tau, amyloid, degree and tau-weighted functional connectivity (FC) on the rates of tau accumulation across all the 210 cortical regions of the 34 atypical AD participants with follow-up tau-PET images
| Rate of change in tau ~ tau + amyloid + tau-weighted FC + degree + tau: amyloid + degree: tau-weighted FC + (1 + tau + amyloid | id) + (1 | region) | |||
|---|---|---|---|
| T statistic | P value | Interpretation | |
| Tau | −1.9 | 0.06 | Inversely proportional to tau change (ns) |
| Amyloid | 1.4 | 0.18 | Proportional to tau change (ns) |
| Tau-weighted FC | 3.0 | 0.002 | Proportional to tau change |
| Degree | 2.3 | 0.03 | Proportional to tau change |
| Tau: amyloid | −1.6 | 0.11 | Interaction term (ns) |
| Degree: tau-weighted FC | −2.4 | 0.01 | Interaction term |
Notes: P values less than 0.05 and the corresponding T statistic are bolded. ns, not significant; FC, functional connectivity
Discussion
This study employed multimodal neuroimaging to describe associations between resting-state functional connectivity and the 2 hallmarks of Alzheimer’s disease pathology: tau and Aβ. In 2 variants of atypical AD, i.e., posterior cortical atrophy and logopenic progressive aphasia, we have shown that associations with tau and Aβ differ, supporting the hypothesis that the propagation mechanisms of these 2 proteins also differ. Even though our analyses cannot prove any causal relationship, they point to factors related to functional ‘hubness’ being key for Aβ deposition and region-to-region functional connectivity being key to the mechanism through which tau propagates. Additionally, tau was higher in the regions with low clustering coefficient, suggesting that tau propagation might be facilitated by the lack of regional trophic support or poor resilience to failure. Together, these findings are entirely consistent with the CNF model of AD pathophysiology (Jones et al. 2016, 2017), but only partially consistent with a pure neuron-to-neuron spreading model. The CNF model does not preclude nor necessitate neuron-to-neuron spreading, but neuron-to-neuron spreading alone is not able to account for all of the findings in this study. Therefore, such spreading models represent an incomplete mechanistic account of the relationship between molecular pathology and functional connectivity in AD.
Amyloid-β was primarily correlated with 2 quantities derived from resting-state fMRI: the degree and the functional connectivity to the disease epicenter, defined as the region with highest Aβ burden. These 2 measures may not be independent of each other, since, for both PCA and LPA, the Aβ epicenter was located in the precuneus, which is a functional hub and is connected to other high degree hub areas. These findings are in agreement with a previous study which included healthy and AD participants (Mutlu et al. 2017), but they differ in the order of importance of the correlations. In our analyses, for both PCA and LPA, the positive association to the degree was much stronger than the one to the functional connectivity to the epicenter, suggesting the functional ‘hubness’ hypothesis as the most likely for Aβ (Buckner et al. 2009). However, this difference could be explained by the Aβ burden being much more severe in our cohort of patients than in a cohort involving participants that were healthy or at the initial stage of the disease (Mutlu et al. 2017). Additionally, it must be noted that it remains unclear if the disease originates in one or more epicenters and how to unequivocally define them: our analyses provide some evidence in support of trans-neuronal spread or network accumulation hypotheses for Aβ but do not rule out other mechanisms (Ossenkoppele et al. 2019). In both phenotypes, functional connectivity to regions that did not display the highest group-average PiB SUVR, like inferior parietal and middle frontal regions, was a strong predictor of Aβ in the functionally connected regions, challenging the hypothesis that Aβ propagates through the brain from a single epicenter. Lastly, regions with high amyloid burden, such as frontal and parietal regions, displayed increased functional connectivity in the atypical AD groups relative to controls, consistently with the CNF hypothesis. Interestingly, the positive associations of group-average PiB SUVR with degree and clustering coefficient were weaker or absent using functional connectivity data from young healthy individuals, particularly in PCA, hinting at a link between amyloid and increased functional connectivity.
Our results on regional clustering coefficient and degree in relation to tau support the validity of the trophic failure hypothesis, as we found that regions with higher tau burden were more likely to have low clustering coefficient and low degree. However, such correlations were modest, with some of them not surviving the autocorrelation tests, suggesting that other mechanisms might play more determining roles in the propagation of tau. Our results do not confirm the findings of a recent imaging study, which reported a positive association between regional tau burden and both degree and clustering coefficient in AD participants (Cope et al. 2018), in agreement with experimental work suggesting that tau could increase neural activity (Devos et al. 2013; Pooler et al. 2013).These discrepancies might be due to differences in disease stage of the participants, since a decrease in trophic support and a disruption of functional connections might be downstream events of tau accumulation (Cope et al. 2018). Another inconsistency between our 2 studies is that we are investigating atypical AD cohorts, which show more extensive patterns of cortical tau deposition than typical AD (van der Flier et al. 2011; Whitwell et al. 2018; Whitwell et al. 2019). The negative relationship we found between regional tau and both degree and ‘total flow’ (i.e., a measure of the total functional connectivity of a region, computed from the association matrix) resembles the findings of a previous study on atrophy (Mutlu et al. 2017). This is not surprising as atrophy patterns resemble those of tau in AD (Sintini et al. 2018; Iaccarino et al. 2018). A sensible explanation for this negative correlation could be that regions with high levels of tau in PCA and LPA, e.g., occipital and lateral temporal, do not belong to functional hubs but have more specialized functional connections and are more vulnerable to failure with limited options for compensation. Another potential explanation is that, since tau is thought to disrupt functional connectivity, as a result, the regions that have more tau have also less functional connections. This explanation is backed up by the observations we made on reduced functional connectivity in PCA and LPA compared to CU individuals and by the absence of a negative association between tau in atypical AD and degree of connectivity in young healthy individuals. The fact that some findings are significant in only one of the 2 phenotypes suggests that the regions involved in the phenotype may play a primary role, with the different expression of pathological proteins being driven not necessarily by network properties, but by intrinsic molecular properties. Lastly, another potential explanation for the negative association between tau and degree/clustering coefficient is that, if the regions with high tau burden were functional hubs, AD would be a much faster disease, assuming that tau spreads trans-neuronally.
While we did not find a positive relationship between degree and baseline levels of tau, we found it with longitudinal rates of tau accumulation. This could suggest that the mere number of functional connections in a region (i.e., degree) is not a primary mechanism for tau deposition, as opposed to the strength of the region’s functional connection to the syndrome-specific failing network, but, as the disease evolves over time, regions that have more functional connections are also more exposed to tau accumulation, as tau transfers between regions trans-synaptically. This is consistent with the fact that the regions that display more tau are not the ones that experience the higher rates of accumulation (Sintini et al. 2019). In other words, the spatial disconnection between the regions with higher tau burden at baseline and the regions with higher rates of tau accumulation translates into different associations of these quantities to functional connectivity measures. Regions functionally connected to the disease epicenters showed level of tau pathology highly correlated to the strength of this connection: such correlations were relatively high even within-subject and statistically higher for tau than for Aβ. Additionally, the correlations between tau and functional connectivity to the epicenters were the only ones that, in both phenotypes, survived both autocorrelation tests, i.e., null models of functional connectivity and surrogate autocorrelated SUVR maps. This aspect crucially points to a trans-neuronal spread mechanism and/or accumulation in failing networks for tau. Particularly, the notion that more than one region may serve as disease epicenter supports the CFN hypothesis. Unlike baseline levels of tau, the longitudinal rates of tau accumulation were only moderately predicted by the strength of the functional connectivity to the disease epicenter in LPA and negatively correlated to it in PCA, suggesting again that other factors may play a role in the accumulation of tau in a region, as the disease progresses. For example, the mechanisms of tau accumulation might adapt as the initial sites reach a plateau. It is likely that PCA participants were at a more advanced disease stage and, therefore, the regions more strongly functionally connected to the disease epicenter had already a high tau burden and thus experienced lower rates of accumulation. While the rate of change in tau accumulation was not correlated to the connectivity to the sites with highest tau SUVR, i.e., left temporal regions for LPA and occipital regions for PCA, it was associated to functional connectivity to other regions, i.e., frontoparietal regions for LPA and frontotemporal for PCA. These findings support the hypothesis that, during the course of the disease, the initial tau epicenter or seeding region leads to other epicenters that cascade systematically through the cortex (Vogel et al. 2020). The notion of epicenters that evolve throughout the course of the disease is entirely compatible with the predictions of the cascading network failure hypothesis. We cannot exclude the fact that our results may be dependent upon the window of the disease we are studying, since it is likely that earlier in the disease process rates of tau accumulation would have been greater in regions closer to the epicenter. It also must be noted that PCA participants were at a slightly more advanced stage of the disease compared to LPA, although the disease durations between the 2 variants was not significantly different, and they had more disrupted functional connections relative to CU individuals. This aspect might explain the differences we found on the relationships between connectivity and tau accumulation between PCA and LPA. On tau accumulation, our analyses are consistent with a recently published study on typical AD (Franzmeier et al. 2020), confirming that the best predictor of the rate at which a region accumulates tau is the level of tau in all the regions functionally connected to it and the strength of such connections. The fact that including Euclidean distance in the tau-weighted functional connectivity only moderately improved the predictions points to functional connection rather than proximity being a key factor in tau propagation. The dependence of tau rates on functional connectivity in a network cannot differentiate a neuron-to-neuron spreading mechanism from an accumulation of tau within a failing network. Therefore, both mechanisms may explain this phenomenon, but neuron-to-neuron spreading is unable to explain the findings related to functional ‘hubness’ and clustering coefficient varying by protein and phenotype as predicted by the CNF model.
Healthy functional brain networks are often described as small-world graphs, because they are characterized by a high level of clustering and low shortest-path lengths between the nodes. Alzheimer’s disease can alter such small-world properties (Stam et al. 2007; Supekar et al. 2008; Sanz-Arigita et al. 2010). In the current study, we did not investigate how the disease-related changes in resting-state functional connectivity in atypical AD might affect Aβ and tau propagation, as it was outside of our primary scope. Future work will need to address this aspect, including a larger population of cognitively unimpaired individuals.
Our study elucidated differences in the relations of tau and Aβ to the resting-state functional connectivity metrics in PCA and LPA. The relationships between proteins and connectivity were consistent across these 2 atypical AD syndromes as would be expected since they have the same underlying pathology, although regional patterns of reduced connectivity and tau uptake differed. A major strength of our study is that functional connectivity measurements were derived from the same patient population as Aβ and tau PET measurement, and not from healthy controls. Several correlations that were significant at P < 0.05 did not survive one or both autocorrelation tests. This certainly raises questions on whether such correlations were simply a product of the brain topological properties and it emphasizes the importance of the comparison with null-models in future functional connectivity studies. We believe that the correlations that were not significant when compared to the ones obtained with null-models of functional connectivity matrices or with surrogate autocorrelated brain maps should not be dismissed as meaningless but warrant more investigations with larger patients populations, e.g., the negative association between baseline tau levels and degree of functional connectivity. The main strength of this study is that it combines Aβ and longitudinal tau data with resting-state functional MRI data in a large cohort of PCA (31 participants) and LPA (27 participants). A limitation is that, in our main analyses, we employed group-average data, assuming uniform effects within the disease groups. However, this assumption is in part justified by having patient populations that are relatively large with unique and characteristic clinical phenotypes. We performed within-subject correlations between Aβ and tau SUVR and graph-theory metrics, but the high variability in the results suggests that more advanced analyses or novel MRI and PET acquisition protocols might be needed in order to evaluate such relationships at the patient level. A limitation in our workflow is the use of ROI-based raw SUVR change to measure the longitudinal rate of tau accumulation. We adopted this approach consistently with previous studies (Jack et al. 2018; Harrison et al. 2018; Sintini et al. 2019; Franzmeier et al. 2020), but it must be noted that the ROI-based difference in SUVR between 2 time points cannot distinguish between a steady accumulation (i.e., the voxels inside the ROI increase their level of tau) and a sequential spread (i.e., some of voxels inside the ROI that at baseline did not have tau, do have tau at follow-up) (Jones et al. 2017). It also must be noted that the calculation of longitudinal changes in SUVR could be influenced by biological changes in the cerebellar crus gray matter, which served as the reference region. Modeling functional ‘hubness’ and trophic support with graph-theoretical metrics (i.e., degree and clustering coefficient) is an approach that relies on reasonable assumptions and has been employed in several studies, but it has not been validated experimentally. Specifically, clustering coefficient might be problematic as a model of trophic support because fMRI measurements include long-range functional connections. For this reason, final evidence on spreading mechanisms of tau and Aβ will come only from human clinical trials. Nevertheless, our findings shed light on potential mechanisms of protein accumulation in atypical AD. Understanding these mechanisms will be important in the development of future treatment and interventions in AD.
Supplementary Material
Contributor Information
Irene Sintini, Department of Radiology, Mayo Clinic, Rochester, MN 55905, USA.
Jonathan Graff-Radford, Department of Neurology, Mayo Clinic, Rochester, MN 55905, USA.
David T Jones, Department of Radiology, Mayo Clinic, Rochester, MN 55905, USA; Department of Neurology, Mayo Clinic, Rochester, MN 55905, USA.
Hugo Botha, Department of Neurology, Mayo Clinic, Rochester, MN 55905, USA.
Peter R Martin, Department of Health Science Research, Mayo Clinic, Rochester, MN 55905, USA.
Mary M Machulda, Department of Psychiatry and Psychology, Mayo Clinic, Rochester, MN 55905, USA.
Christopher G Schwarz, Department of Radiology, Mayo Clinic, Rochester, MN 55905, USA.
Matthew L Senjem, Department of Radiology, Mayo Clinic, Rochester, MN 55905, USA; Department of Information Technology, Mayo Clinic, Rochester, MN 55905, USA.
Jeffrey L Gunter, Department of Radiology, Mayo Clinic, Rochester, MN 55905, USA.
Clifford R Jack, Jr, Department of Radiology, Mayo Clinic, Rochester, MN 55905, USA.
Val J Lowe, Department of Radiology, Mayo Clinic, Rochester, MN 55905, USA.
Keith A Josephs, Department of Neurology, Mayo Clinic, Rochester, MN 55905, USA.
Jennifer L Whitwell, Department of Radiology, Mayo Clinic, Rochester, MN 55905, USA.
Notes
We would like to greatly thank AVID Radiopharmaceuticals, Inc., for their support in supplying the AV-1451 precursor, chemistry production advice and oversight, and FDA regulatory cross-filing permission and documentation needed for this work.
Conflict of Interest: J.L.W., K.A.J., C.G.S., M.M.M., D.T.J. receive research support from the NIH. M.L.S. owns stock in Align Technology, Inc., Gilead Sciences, Inc., Globus Medical Inc., Inovio Biomedical Corp., Johnson & Johnson, LHC Group, Inc., Medtronic, Inc., Mesa Laboratories, Natus Medical Incorporated, Oncothyreon, Inc., Parexel International Corporation, Varex Imaging Corporation. V.J.L. serves as a consultant for Bayer Schering Pharma, Philips Molecular Imaging, Piramal Imaging and GE Healthcare and receives research support from GE Healthcare, Siemens Molecular Imaging, AVID Radiopharmaceuticals, the NIH (NIA, NCI), and the MN Partnership for Biotechnology and Medical Genomics. C.R.J. serves on a scientific advisory board for Eli Lilly & Company, as a speaker for Eisai and on an independent data safety monitoring board for Roche but he receives no personal compensation from any commercial entity; receives research support from the NIH, and the Alexander Family Alzheimer’s Disease Research Professorship of the Mayo Clinic.
Funding
National Institutes of Health (R01-AG50603, R01-NS89757).
References
- Ahmed Z, Cooper J, Murray TK, Garn K, McNaughton E, Clarke H, Parhizkar S, Ward MA, Cavallini A, Jackson S, et al. 2014. A novel in vivo model of tau propagation with rapid and progressive neurofibrillary tangle pathology: the pattern of spread is determined by connectivity, not proximity. Acta Neuropathol. 127:667–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner J, Friston KJ. 2005. Unified segmentation. NeuroImage. 26:839–851. [DOI] [PubMed] [Google Scholar]
- Avants BB, Epstein CL, Grossman M, Gee JC. 2008. Symmetric diffeomorphic image registration with cross-correlation: evaluating automated labeling of elderly and neurodegenerative brain. Med Image Anal. 12:26–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bero AW, Yan P, Roh JH, Cirrito JR, Stewart FR, Raichle ME, Lee JM, Holtzman DM. 2011. Neuronal activity regulates the regional vulnerability to amyloid-beta deposition. Nat Neurosci. 14:750–U353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E. 1991. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82:239–259. [DOI] [PubMed] [Google Scholar]
- Brettschneider J, Del Tredici K, Lee VMY, Trojanowski JQ. 2015. Spreading of pathology in neurodegenerative diseases: a focus on human studies. Nat Rev Neurosci. 16:109–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JA, Deng J, Neuhaus J, Sible IJ, Sias AC, Lee SE, Kornak J, Marx GA, Karydas AM, Spina S, et al. 2019. Patient-tailored, connectivity-based forecasts of spreading brain atrophy. Neuron. 104:856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckner RL, Sepulcre J, Talukdar T, Krienen FM, Liu HS, Hedden T, Andrews-Hanna JR, Sperling RA, Johnson KA. 2009. Cortical hubs revealed by intrinsic functional connectivity: mapping, assessment of stability, and relation to Alzheimer's disease. J Neurosci. 29:1860–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullmore ET, Sporns O. 2009. Complex brain networks: graph theoretical analysis of structural and functional systems. Nat Rev Neurosci. 10:186–198. [DOI] [PubMed] [Google Scholar]
- Burt JB, Helmer M, Shinn M, Anticevic A, Murray JD. 2020. Generative modeling of brain maps with spatial autocorrelation. Neuroimage. 220:117038. doi: 10.1016/j.neuroimage.2020.117038. Epub 2020 Jun 22. PMID: 32585343. [DOI] [PubMed] [Google Scholar]
- Busche MA, Wegmann S, Dujardin S, Commins C, Schiantarelli J, Klickstein N, Kamath TV, Carlson GA, Nelken I, Hyman BT. 2019. Tau impairs neural circuits, dominating amyloid-β effects, in Alzheimer models in vivo. Nature neuroscience. 22(1):57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC, Schoepp DD, Paul SM, Mennerick S, Holtzman DM. 2005. Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron. 48:913–922. [DOI] [PubMed] [Google Scholar]
- Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, Fraser G, Stalder AK, Beibel M, Staufenbiel M, et al. 2009. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol. 11:909–U325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cope TE, Rittman T, Borchert RJ, Jones PS, Vatansever D, Allinson K, Passamonti L, Rodriguez PV, Bevan-Jones WR, O'Brien JT, et al. 2018. Tau burden and the functional connectome in Alzheimer's disease and progressive supranuclear palsy. Brain. 141:550–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crutch SJ, Lehmann M, Schott JM, Rabinovici GD, Rossor MN, Fox NC. 2012. Posterior cortical atrophy. Lancet Neurol. 11:170–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Calignon A, Polydoro M, Suarez-Calvet M, William C, Adamowicz DH, Kopeikina KJ, Pitstick R, Sahara N, Ashe KH, Carlson GA, et al. 2012. Propagation of tau pathology in a model of early Alzheimer's disease. Neuron. 73:685–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devos SL, Goncharoff DK, Chen G, Kebodeaux CS, Yamada K, Stewart FR, Schuler DR, Maloney SE, Wozniak DF, Rigo F, et al. 2013. Antisense reduction of tau in adult mice protects against seizures. J Neurosci. 33:12887–12897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan LZ, Li H, Zhuo JJ, Zhang Y, Wang JJ, Chen LF, Yang ZY, Chu CY, Xie SM, Laird AR, et al. 2016. The human Brainnetome atlas: a new brain atlas based on connectional architecture. Cereb Cortex. 26:3508–3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzmeier N, Neitzel J, Rubinski A, Smith R, Strandberg O, Ossenkoppele R, Hansson O, Ewers M, Adni . 2020. Functional brain architecture is associated with the rate of tau accumulation in Alzheimer's disease. Nat Commun. 11:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorno-Tempini ML, Hillis AE, Weintraub S, Kertesz A, Mendez M, Cappa SF, Ogar JM, Rohrer JD, Black S, Boeve BF, et al. 2011. Classification of primary progressive aphasia and its variants. Neurology. 76:1006–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison TM, La Joie R, Maass A, Baker SL, Swinnerton K, Fenton L, Mellinger TJ, Edwards L, Pham J, Miller BL. 2018. Longitudinal tau accumulation and atrophy in aging and Alzheimer's disease. Ann Neurol. 85:229–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoenig MC, Bischof GN, Seemiller J, Hammes J, Kukolja J, Onur OA, Jessen F, Fliessbach K, Neumaier B, Fink GR, et al. 2018. Networks of tau distribution in Alzheimer's disease. Brain. 141:568–581. [DOI] [PubMed] [Google Scholar]
- Iaccarino L, Tammewar G, Ayakta N, Baker SL, Bejanin A, Boxer AL, Gorno-Tempini ML, Janabi M, Kramer JH, Lazaris A, et al. 2018. Local and distant relationships between amyloid, tau and neurodegeneration in Alzheimer's disease. Neuroimage Clin. 17:452–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack Jr CR, Lowe VJ, Senjem ML, Weigand SD, Kemp BJ, Shiung MM, Knopman DS, Boeve BF, Klunk WE, Mathis CA, et al. 2008. 11C PiB and structural MRI provide complementary information in imaging of Alzheimer's disease and amnestic mild cognitive impairment. Brain. 131:665–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Wiste HJ, Schwarz CG, Lowe VJ, Senjem ML, Vemuri P, Weigand SD, Therneau TM, Knopman DS, Gunter JL, et al. 2018. Longitudinal tau PET in ageing and Alzheimer's disease. Brain. 141:1517–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Wiste HJ, Weigand SD, Therneau TM, Lowe VJ, Knopman DS, Gunter JL, Senjem ML, Jones DT, Kantarci K, et al. 2017. Defining imaging biomarker cut points for brain aging and Alzheimer's disease. Alzheimers Dement. 13:205–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DT, Graff-Radford J, Lowe VJ, Wiste HJ, Gunter JL, Senjem ML, Botha H, Kantarci K, Boeve BF, Knopman DS, et al. 2017. Tau, amyloid, and cascading network failure across the Alzheimer's disease spectrum. Cortex. 97:143–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DT, Knopman DS, Gunter JL, Graff-Radford J, Vemuri P, Boeve BF, Petersen RC, Weiner MW, Jack CR, Alzheimer's Dis Neuroimaging Initi . 2016. Cascading network failure across the Alzheimer's disease spectrum. Brain. 139:547–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josephs KA, Whitwell JL, Weigand SD, Murray ME, Tosakulwong N, Liesinger AM, Petrucelli L, Senjem ML, Knopman DS, Boeve BF, et al. 2014. TDP-43 is a key player in the clinical features associated with Alzheimer's disease. Acta Neuropathol. 127:811–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann M, Madison C, Ghosh PM, Miller ZA, Greicius MD, Kramer JH, Coppola G, Miller BL, Jagust WJ, Gorno-Tempini ML, et al. 2015. Loss of functional connectivity is greater outside the default mode network in nonfamilial early-onset Alzheimer's disease variants. Neurobiol Aging. 36:2678–2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann M, Madison CM, Ghosh PM, Seeley WW, Mormino E, Greicius MD, Gorno-Tempini ML, Kramer JH, Miller BL, Jagust WJ, et al. 2013. Intrinsic connectivity networks in healthy subjects explain clinical variability in Alzheimer's disease. Proc Natl Acad Sci USA. 110:11606–11611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Drouet V, Wu JW, Witter MP, Small SA, Clelland C, Duff K. 2012. Trans-synaptic spread of tau pathology in vivo. PLoS One. 7:e31302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesulam MM 1998. From sensation to cognition. Brain. 121:1013–1052. [DOI] [PubMed] [Google Scholar]
- Mutlu J, Landeau B, Gaubert M, de La Sayette V, Desgranges B, Chetelat G. 2017. Distinct influence of specific versus global connectivity on the different Alzheimer's disease biomarkers. Brain. 140:3317–3328. [DOI] [PubMed] [Google Scholar]
- Ossenkoppele R, Iaccarino L, Schonhaut DR, Brown JA, La Joie R, O'Neil JP, Janabi M, Baker SL, Kramer JH, Gorno-Tempini ML, et al. 2019. Tau covariance patterns in Alzheimer's disease patients match intrinsic connectivity networks in the healthy brain. Neuroimage Clin. 23:101848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossenkoppele R, Schonhaut DR, Scholl M, Lockhart SN, Ayakta N, Baker SL, O'Neil JP, Janabi M, Lazaris A, Cantwell A, et al. 2016. Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer's disease. Brain. 139:1551–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira JB, Ossenkoppele R, Palmqvist S, Strandberg TO, Smith R, Westman E, Hansson O. 2019. Amyloid and tau accumulate across distinct spatial networks and are differentially associated with brain connectivity. Elife. 8:e50830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pooler AM, Phillips EC, Lau DHW, Noble W, Hanger DP. 2013. Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep. 14:389–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahoo A, Bejanin A, Murray ME, Tosakulwong N, Weigand SD, Serie AM, Senjem ML, Machulda MM, Parisi JE, Boeve BE, et al. 2018. TDP-43 and Alzheimer's disease pathologic subtype in non-amnestic Alzheimer's disease dementia. J Alzheimer's Dis. 64:1227–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz-Arigita EJ, Schoonheim MM, Damoiseaux JS, Rombouts SARB, Maris E, Barkhof F, Scheltens P, Stam CJ. 2010. Loss of 'Small-World' networks in Alzheimer's disease: graph analysis of fMRI resting-state functional connectivity. PLoS One. 5:e13788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer A, Kong R, Gordon EM, Laumann TO, Zuo XN, Holmes AJ, Eickhoff SB, Yeo BTT. 2018. Local-global parcellation of the human cerebral cortex from intrinsic functional connectivity MRI. Cereb Cortex. 28:3095–3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz CG, Gunter JL, Ward CP, Vemuri P, Senjem ML, Wiste HJ, Petersen RC, Knopman DS, Jack CR. 2017. The MAYO CLINIC ADULT LIFE span template: better quantification across the LIFE span. Alzheimers Dement. 13:P93–P94. [Google Scholar]
- Seeley WW, Crawford RK, Zhou J, Miller BL, Greicius MD. 2009. Neurodegenerative diseases target large-scale human brain networks. Neuron. 62:42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sintini I, Graff-Radford J, Senjem ML, Schwarz CG, Machulda MM, Martin PR, Jones DT, Boeve BF, Knopman DS, Kantarci K, et al. 2020. Longitudinal neuroimaging biomarkers differ across Alzheimer’s disease phenotypes. Brain. 143:2281–2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sintini I, Martin PR, Graff-Radford J, Senjem ML, Schwarz CG, Machulda MM, Spychalla AJ, Drubach DA, Knopman DS, Petersen RC. 2019. Longitudinal tau-PET uptake and atrophy in atypical Alzheimer's disease. NeuroImage Clin. 23:101823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sintini I, Schwarz CG, Martin PR, Graff-Radford J, Machulda MM, Senjem ML, Reid RI, Spychalla AJ, Drubach DA, Lowe VJ. 2018. Regional multimodal relationships between tau, hypometabolism, atrophy, and fractional anisotropy in atypical Alzheimer's disease. Hum Brain Mapp. 40:1618–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sporns O, Honey CJ, Kotter R. 2007. Identification and classification of hubs in brain networks. PLoS One. 2:e1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stam CJ, Jones BF, Nolte G, Breakspear M, Scheltens P. 2007. Small-world networks and functional connectivity in Alzheimer's disease. Cereb Cortex. 17:92–99. [DOI] [PubMed] [Google Scholar]
- Supekar K, Menon V, Rubin D, Musen M, Greicius MD. 2008. Network analysis of intrinsic functional brain connectivity in Alzheimer's disease. PLoS Comput Biol. 4:e1000100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetzloff KA, Graff-Radford J, Martin PR, Tosakulwong N, Machulda MM, Duffy JR, Clark HM, Senjem ML, Schwarz CG, Spychalla AJ, et al. 2018. Regional distribution, asymmetry, and clinical correlates of tau uptake on [18F]AV-1451 PET in atypical Alzheimer's disease. J Alzheimers Dis. 62:1713–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Flier WM, Pijnenburg YAL, Fox NC, Scheltens P. 2011. Early-onset versus late-onset Alzheimer's disease: the case of the missing APOE ɛ4 allele. Lancet Neurol. 10:280–288. [DOI] [PubMed] [Google Scholar]
- Vogel JW, Iturria-Medina Y, Strandberg OT, Smith R, Levitis E, Evans AC, Hansson O. 2020. Spread of pathological tau proteins through communicating neurons in human Alzheimer's disease. Nat Commun. 11:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitwell JL, Graff-Radford J, Tosakulwong N, Weigand SD, Machulda M, Senjem ML, Schwarz CG, Spychalla AJ, Jones DT, Drubach DA, et al. 2018. [F-18]AV-1451 clustering of entorhinal and cortical uptake in Alzheimer's disease. Ann Neurol. 83:248–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitwell JL, Jones DT, Duffy JR, Strand EA, Machulda MM, Przybelski SA, Vemuri P, Gregg BE, Gunter JL, Senjem ML, et al. 2015. Working memory and language network dysfunctions in logopenic aphasia: a task-free fMRI comparison with Alzheimer's dementia. Neurobiol Aging. 36:1245–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitwell JL, Martin P, Graff-Radford J, Machulda MM, Senjem ML, Schwarz CG, Weigand SD, Spychalla AJ, Drubach DA, Jack CR, et al. 2019. The role of age on tau PET uptake and gray matter atrophy in atypical Alzheimer's disease. Alzheimers Dement. 15:675–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Gennatas ED, Kramer JH, Miller BL, Seeley WW. 2012. Predicting regional neurodegeneration from the healthy brain functional connectome. Neuron. 73:1216–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.