

There is a Blood Commentary on this article in this issue.
Key Points
CRISPR-Cas9 CISH deletion enhances the metabolic fitness and antitumor activity of armored IL-15–secreting CB-derived CAR-NK cells.
Abstract
Immune checkpoint therapy has resulted in remarkable improvements in the outcome for certain cancers. To broaden the clinical impact of checkpoint targeting, we devised a strategy that couples targeting of the cytokine-inducible Src homology 2–containing (CIS) protein, a key negative regulator of interleukin 15 (IL-15) signaling, with fourth-generation “armored” chimeric antigen receptor (CAR) engineering of cord blood–derived natural killer (NK) cells. This combined strategy boosted NK cell effector function through enhancing the Akt/mTORC1 axis and c-MYC signaling, resulting in increased aerobic glycolysis. When tested in a lymphoma mouse model, this combined approach improved NK cell antitumor activity more than either alteration alone, eradicating lymphoma xenografts without signs of any measurable toxicity. We conclude that targeting a cytokine checkpoint further enhances the antitumor activity of IL-15–secreting armored CAR-NK cells by promoting their metabolic fitness and antitumor activity. This combined approach represents a promising milestone in the development of the next generation of NK cells for cancer immunotherapy.
Visual Abstract

Introduction
Natural killer (NK) cells mediate potent cytotoxicity against tumor cells1 and are attractive candidates for the next-generation cancer immunotherapies.2 Moreover, their ready availability from various sources, such as umbilical cord blood (CB), boosts their potential as a third-party product for widespread clinical scalability.3,4 A recent advance in the development of NK-cell–based immunotherapy is the demonstration that chimeric antigen receptor (CAR) engineering can enhance their effector function.5-7 We have shown that CB-NK cells transduced with a fourth-generation vector encoding anti-CD19 CAR and interleukin-15 (IL-15) induce greater in vivo expansion and longer-term persistence than nontransduced (NT) NK cells.6 While our preclinical study using an aggressive model of NK-resistant Raji lymphoma confirmed that this approach can prolong survival of mice,6 it was not curative, leading us to question whether the antitumor activity of IL-15–secreting CAR-NK cells could be further enhanced by inhibiting key cytokine-related immune checkpoints.
The suppressor-of-cytokine signaling (SOCS) family of proteins play important roles in NK cell biology by attenuating JAK-STAT–mediated cytokine signaling and NK cell cytotoxicity against cancer.8,9 One of its members, the cytokine-inducible Src homology 2–containing protein (CIS), is encoded by the CISH gene. CIS contains a central Src homology 2 that interacts with phosphorylated tyrosine motifs in target proteins such as those belonging to the JAK-STAT signaling pathway and a C-terminal 40-amino-acid motif known as the SOCS box that ubiquitinates the target proteins and directs them for proteosomal degradation.10,11 CIS is induced by cytokines such as IL-2 and IL-1512,13 and is an important intracellular checkpoint in NK cells.10 Given that our CAR19-specific CB-derived NK cells are designed to secrete IL-15, we hypothesized that CIS would be a logical checkpoint to target to enhance their antitumor potency. Here, we show that a combined strategy of IL-15 CAR engineering and CISH knockout (KO) in CB-derived NK cells significantly improved tumor control. This gain of effector function is attributed to enhanced IL-15 signaling secondary to CISH KO, with consequent activation of the Akt/mTORC1/c-MYC pathway and increased NK cell glycolysis in response to tumor. Thus, we demonstrate that deleting a critical cytokine checkpoint in IL-15–secreting CAR-NK cells improves their metabolic “fitness,” permitting greater in vivo persistence and cytotoxic function. Our data support the use of a 2-step strategy that combines engineering CAR-NK cells to secrete IL-15 with cytokine checkpoint gene editing to further enhance their therapeutic potential in the clinic.
Materials and methods
Retrovirus transfection and transduction
The retroviral vector encoding iC9.CAR19.CD28-ζ-2A-IL-15 was kindly provided by Gianpietro Dotti (University of North Carolina).14,15 CAR19.CD28-ζ (without IL-15) was used as a control.
CRISPR-Cas9 gene editing of CISH
CISH KO was performed using ribonucleoprotein (RNP) complex, in both NT and CAR-NK cells (for details, see supplemental Methods, available on the Blood Web site). To assess KO efficiency, we used polymerase chain reaction (PCR) gel electrophoresis, western blot, and Sanger sequencing. Details on the protocols are included in supplemental Methods.
NK cell functional and cytotoxicity assays
Cytokine production, degranulation, chromium release assay, Incucyte real-time assay, and annexin V/DRAQ7 viability assays were used as previously described.6 Details of these assays are provided in the supplemental Methods.
Mass cytometry and antibody conjugation
A panel comprising 37 metal-tagged antibodies was used for the in-depth characterization of NK cells16 (supplemental Table 1 and supplemental Methods).
Seahorse assays
The extracellular acidification rate (ECAR) and oxygen consumption rate were measured using the Agilent Seahorse XFe96 Analyzer (Agilent) per the manufacturer’s instructions. NT-NK cells (control or CISH KO) and CAR-transduced NK cells (control or CISH KO) were assayed alone or purified after 2 hours of coculture with Raji. Where indicated, NK cells were preincubated with rapamycin (100 ng/mL, Miltenyi Biotec) for 4 hours prior to the Seahorse assay.
Xenogeneic lymphoma models
We used an aggressive NK-resistant Raji NOD/SCID IL-2Rγnull (NSG) xenograft model as previously described.6 Mice (10-12 weeks old; The Jackson Laboratory, Bar Harbor, ME) were irradiated with 300 cGy at day −1 and engrafted with FFluc-Raji cells. Where indicated, NT (control or CISH KO), CAR19/IL-15 CB-NK (control or CISH KO), or CAR19 (no IL-15) CB-NK (control or CISH KO) cells were injected through the tail vein. Mice were subjected to weekly bioluminescence imaging (Xenogen IVIS-200 Imaging System).17 Trafficking, persistence and expansion of NK cells were measured by flow cytometry. The antibodies used for flow cytometry staining are described in detail in the supplemental Methods.
Off-target identification
The genome-wide, unbiased identification of double-stranded breaks enabled by sequencing (GUIDE-seq) method was employed for unbiased discovery of off-target editing events.18 Details of this technology and target enrichment via rhAmpSeq19 are included in supplemental Methods.
Additional detailed methods are provided in supplemental Data.
Results
Phenotypic and molecular signaling alterations associated with CISH deletion
The central hypothesis of this study predicts that knocking out the CISH gene in CAR-NK cells will enhance their effector function against tumor cells, much in the way that targeting PD-1 improves outcomes by removing a critical immune checkpoint in T cells.20,21 Our approach for combined retroviral transduction with the iC9/CAR19/IL-15 construct and RNP-mediated gene editing of CISH is shown in Figure 1A-B. On day 7, we nucleofected CAR-NK cells with Cas9 alone (Cas9 mock) or Cas9 preloaded with gRNA targeting CISH exon 4. The iC9/CAR19/IL-15 transduction efficiency and cell viability on day 7 were >90% and remained stable over time in both control and gene-edited cells (supplemental Figure 1A-B). The efficiency of CISH KO was high (81% to 98%) in both the nontransduced (NT) control and CAR-expressing NK cells by PCR (Figure 1B) and western blot (Figure 1C) and remained stable over time (supplemental Figure 1C-D). This on-target efficiency was confirmed by Sanger sequencing (Figure 1D). Importantly, CISH expression in NK cells transduced with either iC9/CAR19/IL-15 or NT (control) and cultured with IL-2 and K562 feeder cells expressing membrane-bound IL-21, 4-1BB ligand, and CD48 (referred to as uAPC) increased over time (Figure 1E). This indicates that CAR-modified and NT-NK cells are subject to the same counter-regulatory circuits leading to modulation of CIS levels.
Figure 1.
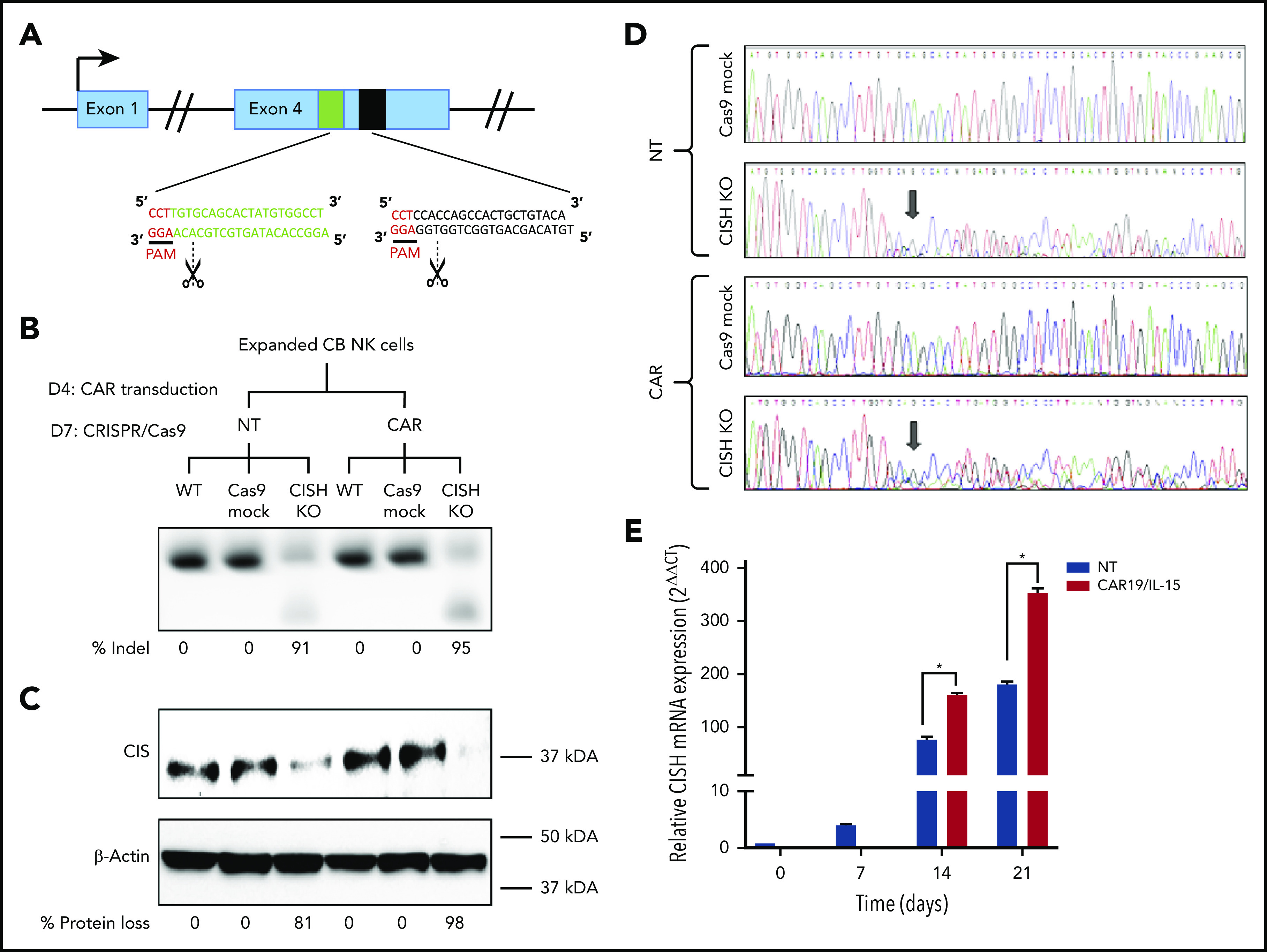
CRISPR-Cas9–mediated deletion of CISH in iC9/CAR19/IL-15 NK cells. (A) Schematic representation of CRISPR-Cas9–mediated CISH KO using 2 guide RNAs (gRNA) targeting exon 4 of the CISH gene. PAM, protospacer-adjacent motif. (B-C) CB-NK cells were expanded with K562 based feeder cells and IL-2 and then either left NT or transduced with a retroviral vector expressing iC9/CAR19/IL-15 construct on day 4 (D4) of expansion. On day 7 (D7) of expansion, NT and iC9/CAR19/IL-15–expressing CB-NK cells were nucleofected with Cas9 alone (Cas9 mock), Cas9 preloaded with gRNA targeting CISH exon 4 (CISH KO), or nonnucleofected (wild-type [WT]). The CISH KO efficiency was determined by PCR (B) and western blot analysis (C). (D) Sanger sequencing results showing multiple peaks reflecting nonhomologous end-joining (NHEJ) events in NT or iC9/CAR19/IL-15 (CAR) NK cells that underwent CISH KO compared with single peaks in control (Cas9 mock). Arrows indicate the base pair position where the gene editing started. (E) Bar graphs showing the relative mRNA expression levels of CISH determined on days 0, 7, 14, and 21 of expansion in NT (blue) and iC9/CAR19/IL-15 (red) NK cells by reverse transcription polymerase chain reaction (RT-PCR) (n = 3). Note that on days 0 and 7 only data for NT-NK cells are included since the CAR transduction step is performed on day 4 of expansion. 18 S ribosomal RNA (18S) was used as the internal reference gene. Bars represent mean values with standard deviation, *P ≤ .05.
Next, to gain insight into the phenotypic changes that accompany CISH KO in CAR-NK cells, we used cytometry by time of flight and a panel of 37 antibodies against inhibitory and activating receptors, as well as differentiation, homing, and activation markers (supplemental Table 1). CISH KO resulted in increased expression of markers of activation and cytotoxicity (Figure 2A), including granzyme B, perforin, TRAIL, and CD3ζ; transcription factors such as eomesodermin (eomes) and T-bet; adaptor molecules such as DAP12; and activating co-receptors/proliferation markers such as DNAM, CD25, and Ki67. A similar profile of upregulated markers was identified after CISH KO in NT-NK cells (supplemental Figure 2A). Using viSNE, a t-distributed stochastic neighbor-embedding (t-SNE) algorithm, we observed marked phenotypic differences between control and CISH KO iC9/CAR19/IL-15 NK cells, with predominantly proliferative and cytotoxic features (eg, increased expression of CD25, Ki67, CD3ζ, perforin, and granzyme B) in CISH KO iC9/CAR19/IL-15 NK cells (Figure 2B). Despite higher expression of proliferation markers on CISH KO iC9/CAR19/IL-15 NK cells on phenotyping, the proliferative capacity of NK cells during in vitro expansion did not differ significantly following CISH KO (supplemental Figure 3A). This is likely related to the use of uAPCs that can optimally expand both engineered and nonengineered NK cells in vitro.5,17 Moreover, despite the activation phenotype associated with CISH KO, we did not observe any evidence of activation-induced cell death in NT CISH KO or CAR19/IL-15 CISH KO cells following stimulation with tumor targets (supplemental Figure 3B).
Figure 2.
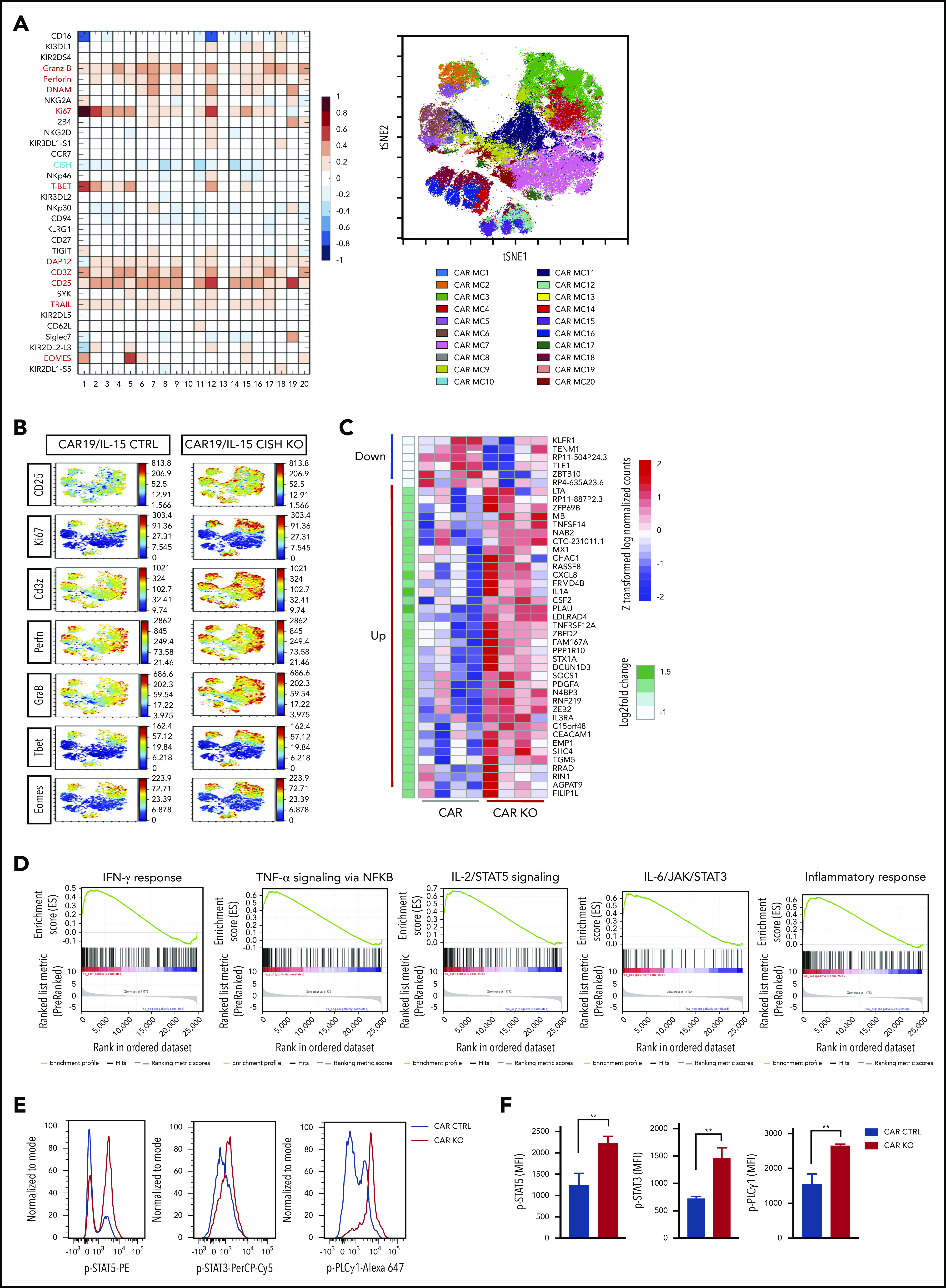
Phenotype and molecular signature of iC9/CAR19/IL-15 CISH KO NK cells. (A) Comparative heatmap of mass cytometry data showing the expression of NK cell surface markers, transcription factors, and cytotoxicity markers in iC9/CAR19/IL-15 CISH KO compared with iC9/CAR19/IL-15 control NK cells. Each column represents a separate cluster identified by FlowSOM analysis, and each row reflects the expression of a certain marker for each annotation. Color scale shows the expression level for each marker, with red representing higher expression and blue lower expression in iC9/CAR19/IL-15 CISH KO NK cells. The t-SNE map generated from FlowSOM analysis in the right panel shows the 20 NK cell metaclusters (MCs) represented in the mass cytometry heatmap in the left panel. (B) Individual t-SNE maps show the expression of selected NK cell markers for iC9/CAR19/IL-15 CISH KO compared with iC9/CAR19/IL-15 control NK cells. Color scale indicates signal intensity, ranging from low (blue) to high (red) after arcsine transformation. (C) Global gene expression analysis by RNA sequencing. Heatmap displays the genes that were differentially expressed in purified iC9/CAR19/IL-15 CISH KO vs iC9/CAR19/IL-15 control NK cells (n = 4). Color scale shows the expression level of each marker, with red representing higher expression and blue lower expression in iC9/CAR19/IL-15 control (CAR) or iC9/CAR19/IL-15 CISH KO (CAR KO) NK cells (q < 0.1 and absolute log2foldchange > 0.8). (D) GSEA showing enrichment in IFN-γ response, TNF-α signaling via NF-κB, IL-2/STAT5 signaling, IL-6/JAK/STAT3 signaling and inflammatory response in iC9/CAR19/IL-15 CISH KO compared with iC9/CAR19/IL-15 control NK cells. (E) Representative histogram showing enhanced phosphorylation of STAT5 (p-STAT5), STAT3 (p-STAT3) and phospholipase C γ 1 (p-PLCγ1) in iC9/CAR19/IL-15 CISH KO vs iC9/CAR19/IL-15 control NK cells after coculture with Raji cells for 30 minutes. Blue histograms represent CAR control, and red histograms represent CAR CISH KO. (F) Bar graphs showing mean fluorescence intensity (MFI) of p-STAT5, p-STAT3, and p-PLCγ1 in iC9/CAR19/IL-15 CISH KO vs iC9/CAR19/IL-15 control NK cells (n = 3). Blue bars represent CAR control and red bars represent CAR CISH KO. Bars represent mean values with standard deviation. **P ≤ .01.
To assess the consequences of CISH KO on the transcriptomic and signaling pathway responses of NK cells, we performed RNA-sequencing studies of CISH KO NT and iC9/CAR19/IL-15 NK cells vs their unmodified controls. CISH KO led to upregulation of genes related to inflammatory and immune responses (eg, tumor necrosis factor [TNF] and interferon [IFN] signaling such as TNFRSF12A, MX1, and T-bet regulation such as ZEB2) as well as cytokine signaling (eg, IL1A, IL3RA, and CXCL8) (Figure 2C; supplemental Figure 2B) in both NT and iC9/CAR19/IL-15 NK cells. Gene set enrichment analysis (GSEA) supported enrichment of pathways involved in TNF-α, IFN-γ, IL-2/STAT5, and IL-6/JAK/STAT3 signaling, as well as in those related to inflammatory immune responses (Figure 2D; supplemental Figure 2C). We confirmed our findings at the protein level by showing enhanced phosphorylation of STAT5, STAT3, and phospholipase C γ 1 (PLCγ1) in CISH KO iC9/CAR19/Il-15 NK cells (Figure 2E-F). It is worthwhile to note that CIS and other SOCS proteins have been reported to downregulate CD3ζ22 and PLCγ123 in T cells, and our data support a similar effect in NK cells after CISH KO for both CD3ζ (Figure 2A-B) and p-PLCγ1 (Figure 2E-F). Considered together, these phenotypic and molecular signaling results support the hypothesis that targeting CIS in CAR-NK cells removes an important immune checkpoint.
CISH ablation enhances the antitumor activity of iC9/CAR19/Il-15 NK cells
When tested against CD19+ Raji lymphoma cells, our CISH KO NT or iC9/CAR19/IL-15 NK cells produced more IFN-γ and TNF-α, and displayed greater degranulation (CD107a) and cytotoxicity against their targets than did their respective NT and iC9/CAR19/IL-15 NK controls (Figure 3A-D). Furthermore, CISH KO iC9/CAR19/IL-15 NK cells killed Ramos lymphoma cell line and primary chronic lymphocytic leukemia targets cells (n = 3) more efficiently than controls (supplemental Figure 4A-B). This outcome was reinforced by the effect of CISH KO on the formation of the immunologic synapse (IS) between NT or iC9/CAR19/IL-15 NK cells and tumor cells. Indeed, polarization of perforin-centroid defined microtubule-organizing center (MTOC) was augmented by CISH deletion as reflected by a shortened MTOC-to-IS distance compared with controls (Figure 3E-F), a finding that typically correlates with increased effector cell function and cytotoxicity.24 While we did not observe an upregulation in the expression of adhesion molecules (eg, LFA-1, CD11b, or CD2), CISH deletion resulted in an increase in the CAR mean fluorescence intensity (supplemental Figure 5). Upregulation of CAR expression following CISH KO is likely related to the increased activation state of the iC9/CAR19/IL-15 NK cells and supports prior reports that cell activation can increase expression of SFG retroviral vector–encoded transgenes.14,25
Figure 3.
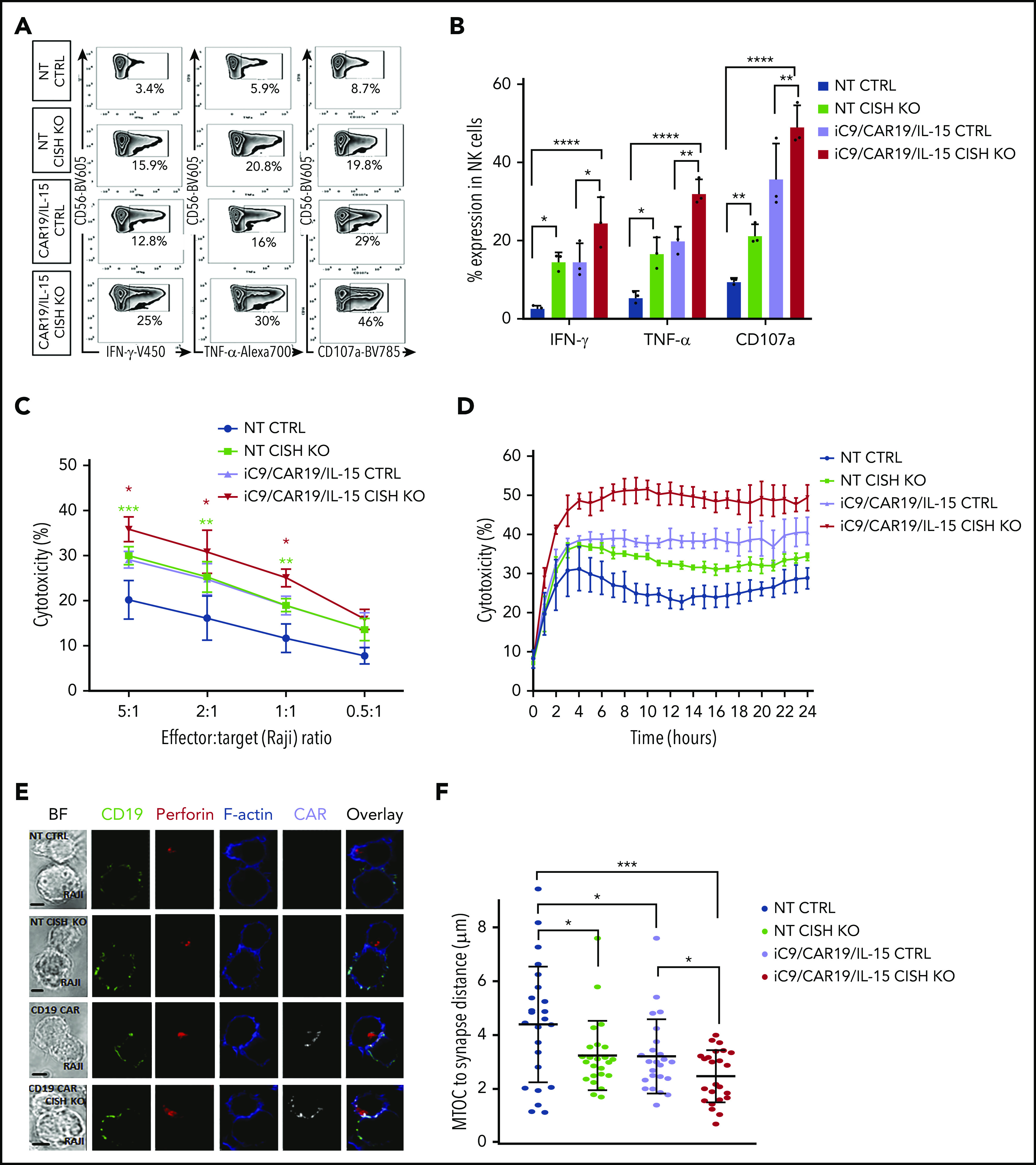
CISH deletion improves function and cytotoxicity of NT and iC9/CAR19/IL-15 NK cells. (A) Representative FACS plots of cytokine production (IFN-γ and TNF-α) and CD107a degranulation by NT control, NT CISH KO, iC9/CAR19/IL-15 control, or iC9/CAR19/IL-15 CISH KO NK cells after coculture with Raji target cells for 6 hours. Inset values indicate the frequency of IFN-γ–, TNF-α–, and CD107a-positive cells from each group. (B) Bar plots summarize the flow cytometry data on cytokine production (IFN-γ and TNF-α) and CD107a degranulation by NT control (blue bars), NT CISH KO (green bars), iC9/CAR19/IL-15 control (purple bars) or iC9/CAR19/IL-15 CISH KO NK cells (red bars) after coculture with Raji target cells for 6 hours (n = 3). Statistical significance is indicated as *P ≤ .05, **P ≤ .01, and ****P ≤ .0001; bars represent mean values with standard deviation. (C) Cytotoxicity of NT control, NT CISH KO, iC9/CAR19/IL-15 control, or iC9/CAR19/IL-15 CISH KO NK cells against Raji targets at different effector-to-target (E:T) ratios, as measured by 51Cr-release assay (n = 3). The bars represent mean values with standard deviation. The red asterisks represent the statistical significance between iC9/CAR19/IL-15 CISH KO vs iC9/CAR19/IL-15 control NK cells (*P ≤ .05). The green asterisks represent the statistical significance between NT CISH KO vs NT control NK cells (***P ≤ .001; **P ≤ .01). (D) Cytotoxicity of NT control, NT CISH KO, iC9/CAR19/IL-15 control, or iC9/CAR19/IL-15 CISH KO NK cells against Raji targets over 24 hours at 1:1 E:T ratio as measured by Incucyte live-imaging cell killing assay (n = 3). Bars represent mean values with standard deviation. At 10 hours, NT CISH KO vs NT control (P = .04), iC9/CAR19/IL-15 CISH KO vs iC9/CAR19/IL-15 control (P = .007). (E) Confocal microscopy showing representative synapse images of NT control, NT CISH KO, iC9/CAR19/IL-15 control or iC9/CAR19/IL-15 CISH KO NK cells conjugated with Raji cells. Images show conjugates in bright field (BF) or stained with anti-CD19 (green), anti-perforin (red), phalloidin-F-actin (blue), and anti-CAR (gray), and an overlay of fluorescence channels are also shown. (F) NT control, NT CISH KO, iC9/CAR19/IL-15 control, or iC9/CAR19/IL-15 CISH KO NK cells were assessed for their ability to polarize lytic granules to Raji cells as measured by distance from the MTOC to the IS. Results from 3 independent donors are shown. Each data point represents a single IS. Statistical significance is indicated as *P ≤ .05 and ***P ≤ .001.
Robust metabolic changes associated with CIS checkpoint elimination
Although blocking the cytokine checkpoint CIS seemed like a logical approach, we remained concerned over the possible impact of increased IL-15 signaling on prominent metabolic pathways in NK cells. In one negative scenario, long-term exposure to IL-15 could suppress rather than boost metabolic rates, leading to NK cell exhaustion.26 In another scenario, it might produce undue systemic toxicity.27,28 We therefore evaluated the effects of CISH KO, with or without iC9/CAR19/IL-15 transduction, on 2 major regulators of NK cell metabolism: mTORC1, which controls pathways responsible for proliferation and cytotoxicity,29,30 and MYC, which upregulates glucose transporters and glycolytic enzymes that promote glycolysis.31 Indeed, GSEA revealed an enrichment of genes involved in phosphatidylinositol 3-kinase (PI3K)/Akt/mTOR, mTORC1, c-MYC, and glycolysis (Figure 4A).
Figure 4.
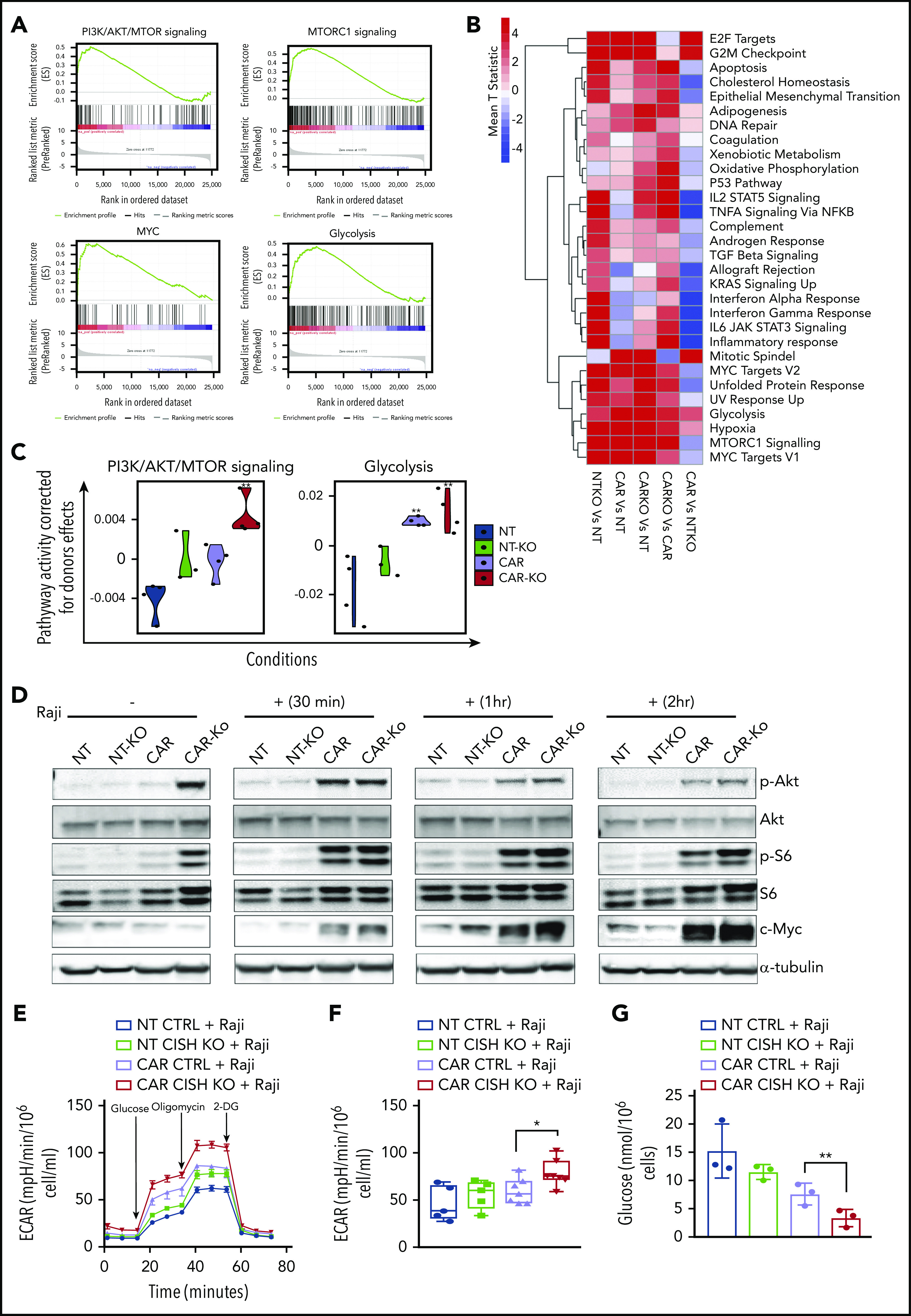
Metabolic changes associated with iC9/CAR19/IL-15 CISH KO NK cells. (A) GSEA plots showing enrichment in PI3K/Akt/MTOR, mTORC1, MYC, and glycolysis pathways in iC9/CAR19/IL-15 CISH KO NK cells compared with iC9/CAR19/IL-15 control NK cells. (B) Comparative mean T statistic heat map of RNA sequencing data showing the expression of metabolic pathways in NT control (NT), NT CISH KO (NTKO), iC9/CAR19/IL-15 control (CAR), or iC9/CAR19/IL-15 CISH KO (CARKO) NK cells that are significantly different (q < 0.01) in ≥1 of the 5 comparisons. Each column represents a separate comparison, and each row reflects the expression of a certain hallmark pathway for each annotation. Color scale indicates signal intensity, ranging from lower (blue) to higher (red) expression. (C) Violin plots showing PI3K/Akt/mTORC1 and glycolysis signaling in NT (blue), NT-KO (green), CAR (purple), or CAR-KO (red) NK cells after correction for donor effect. Pathway activity of samples is regressed against donor and the residual is the corrected pathway activity. P values reported are computed relative to NT using the linear regression approach discussed in Materials and methods. **P ≤ .01. (D) NT control (NT), NT CISH KO (NT-KO), iC9/CAR19/IL-15 control (CAR), or iC9/CAR19/IL-15 CISH KO (CAR-KO) NK cells were cultured without (−) or with (+) Raji cells for 30 minutes, 1 hour, or 2 hours; NK cells were then purified, and the protein expression levels of p-Akt, Akt, p-S6, S6, c-MYC, and α-tubulin in NK cells were determined by western blot analysis. Representative blots from 2 independent experiments are shown. (E) A series of ECARs was calculated for NT control (blue lines), NT CISH KO (green lines), iC9/CAR19/IL-15 control (purple lines), or iC9/CAR19/IL-15 CISH KO (red lines) NK cells cocultured with Raji targets for 2 hours and subsequently purified and treated with 2 g/L d-glucose, 1 μM oligomycin, and 100 mM 2-deoxyglucose (2-DG). A representative graph from 5 independent experiments is shown. (F) Box plots summarize the ECAR data by NT control (blue box), NT CISH KO (green box), iC9/CAR19/IL-15 control (purple box), or iC9/CAR19/IL-15 CISH KO (red box) NK cells cocultured with Raji (n = 5). Statistical significance is indicated as *P ≤ .05; bars represent mean values with standard deviation. (G) Bar graph summarizes the glucose concentration in the supernatant of the different NK cell conditions cocultured with Raji for 2 hours: NT control (blue), NT CISH KO (green), iC9/CAR19/IL-15 control (purple), or iC9/CAR19/IL-15 CISH KO (red) NK cells (n = 3). Bars represent mean values with standard deviation (**P ≤ .01).
The above results raise a pivotal question: are the metabolic gene expression patterns seen with CISH KO distinct, or do they overlap with those seen with unmodified or CAR19/IL-15 NK cells? The heatmap in Figure 4B displays the various hallmark pathways and how they change (upregulated or downregulated) among the various comparisons. In general, compared with NT-NK cells, each engineering strategy (CISH KO or CAR transduction) alone or in combination led to enrichment of metabolic pathways (Figure 4B). We observed some overlap between the pathways upregulated by CISH ablation or iC9/CAR19/IL-15 transduction alone; however, the combination of both approaches clearly led to more robust metabolic changes (Figure 4B). Of note, certain pathways were specifically activated after CISH KO and were not induced with CAR transduction alone; these included pathways related to cytokine signaling and inflammatory response (Figure 4B).
We next focused on the metabolic pathways that are functionally relevant for NK cell antitumor activity; overall, our linear regression model showed an additive effect of CISH ablation and CAR transduction, with the largest increase in PI3K/Akt/mTOR and glycolysis pathways being achieved with a combination of the 2 strategies, suggesting that both genetic manipulations are needed to achieve optimal NK cell effector functions (Figure 4C). This interpretation was confirmed by a greater phosphorylation of Akt and ribosomal protein S6 (S6), a downstream target of mTORC1, and increased expression of c-MYC, an important mediator of glycolysis, in particular in CISH KO CAR19/IL-15 NK cells in response to Raji targets (Figure 4D; supplemental Figure 6). Notably, p-S6 and p-Akt were upregulated in CISH KO CAR19/IL-15 NK cells even in the absence of Raji, although this was not associated with increased tonic signaling (supplemental Figure 7). It is likely by removing the CIS brake, the endogenously secreted IL-15 by CAR19/IL-15 NK cells can trigger the mTORC1/Akt pathway with a lower threshold of activity. In contrast c-MYC, which is the precursor of glycolysis, was only upregulated in response to Raji tumor. To pursue the functional implications of these findings, we first blocked the mTORC1 pathway by treating the CAR19/IL-15 NK cells (±CISH KO) with rapamycin and observed a decrease in their cytotoxicity against Raji lymphoma compared with untreated cells (supplemental Figure 8A), which asserts the importance of the mTORC1 pathway as part of the mechanism by which CISH KO enhances the cytotoxicity of CAR19/IL-15 NK cells. We then used Seahorse assays to measure the glycolytic response of NK cells to tumor targets and showed that in response to Raji cells, either CISH KO alone or CAR transduction alone in NK cells could increase glycolysis, as measured by ECAR, although the best result (and the only statistically significant one) was achieved by combining CISH KO and CAR19/IL-15 transduction (Figure 4E-F). Consistent with these findings, CAR-NK cells with CISH deletion showed the greatest glucose consumption, when cocultured with Raji, compared with controls (Figure 4G). The addition of rapamycin completely abrogated any glycolytic advantage of CISH KO in CAR19/Il-15 NK cells or NT-NK cells (supplemental Figure 8B-C), further supporting the importance of the mTORC1 pathway as an upstream regulator of glycolysis in this setting. Importantly, in the absence of Raji targets, there was no difference in glycolysis among the different NK conditions (supplemental Figure 8D-E) which parallels the c-MYC expression profile. CISH KO iC9/CAR19/IL-15 NK cells also had higher oxygen consumption rate compared with control iC9/CAR19/IL-15 NK cells (supplemental Figure 9A) and induced an increase in mitochondria numbers and the mitochondrial/nuclear volume ratio as assessed by confocal microscopy (supplemental Figure 9B-C). These data suggest that CISH KO can also enhance the metabolism of CAR-NK cells by increasing mitochondrial activity.
Finally, we propose a model (visual abstract), in which CISH KO in CAR-NK cells enhances IL-15 signaling by releasing a checkpoint brake, in turn leading to increased activation of the Akt/mTORC1 axis and in the presence of tumor ensuing c-MYC activation, culminating in greater glycolytic capacity of CAR-NK cells and a resultant increase in their ability to respond to tumor targets.
CIS checkpoint disruption combined with iC9/CAR19/IL-15 transduction improves tumor control in a Raji lymphoma model
Using an aggressive Raji lymphoma mouse model (Figure 5A), we next investigated whether adoptive transfer of CISH KO NK cells in the absence of CAR transduction could boost the control of disease in tumor-bearing mice. First, mice received a single IV infusion of NK cells (10 × 106/mouse) that were either unmodified (NT control) or electroporated with Cas9 alone (Cas9 control) or had CISH KO. The tumor burden, monitored by bioluminescence imaging (BLI), increased in all animals through day 28 of the study, with no significant differences in survival noted between the CISH KO group and controls (supplemental Figure 10). We next investigated the in vivo antitumor activity of NK cells modified with both CISH KO and iC9/CAR19/IL-15 transduction. Mice received iC9/CAR19/IL-15 or CISH KO iC9/CAR19/IL-15 NK cells, and tumor growth was followed by weekly BLI imaging. Survival was monitored and all surviving animals were sacrificed at day 35 and their organs examined for evidence of lymphoma. While animals treated with iC9/CAR19/IL-15 NK cells had evidence of tumor in their organs and succumbed to lymphoma, those treated with CISH KO iC9/CAR19/IL-15 were tumor free and did not have evidence of lymphoma in their spleen, liver, or bone marrow (Figure 5B-D; supplemental Figure 11A-B). Since dual-engineered cells had shown greater potency than either modification alone, even at low E:T ratios in vitro, we hypothesized that it would also be more effective at controlling Raji lymphoma cells at lower infusion doses. Indeed, when as few as 3 × 106 CISH KO iC9/CAR19/IL-15 NK cells were administered, they significantly improved survival (P = .003) and the control of Raji lymphoma compared with control NK cells, although the mice eventually succumbed to tumor by day 46 (Figure 5E-G). The higher dose (10 × 106) of the CISH KO iC9/CAR19/IL-15 NK cells eradicated lymphoma in mice, as demonstrated by BLI and pathologic examination, and led to significantly prolonged survival times (Figure 5E-H). This result was associated with improved NK cell persistence (up to 7 weeks after infusion) in mice that received the CISH KO iC9/CAR19/IL-15 cells compared with iC9/CAR19/IL-15 NK controls (Figure 5I; supplemental Figure 11C).
Figure 5.
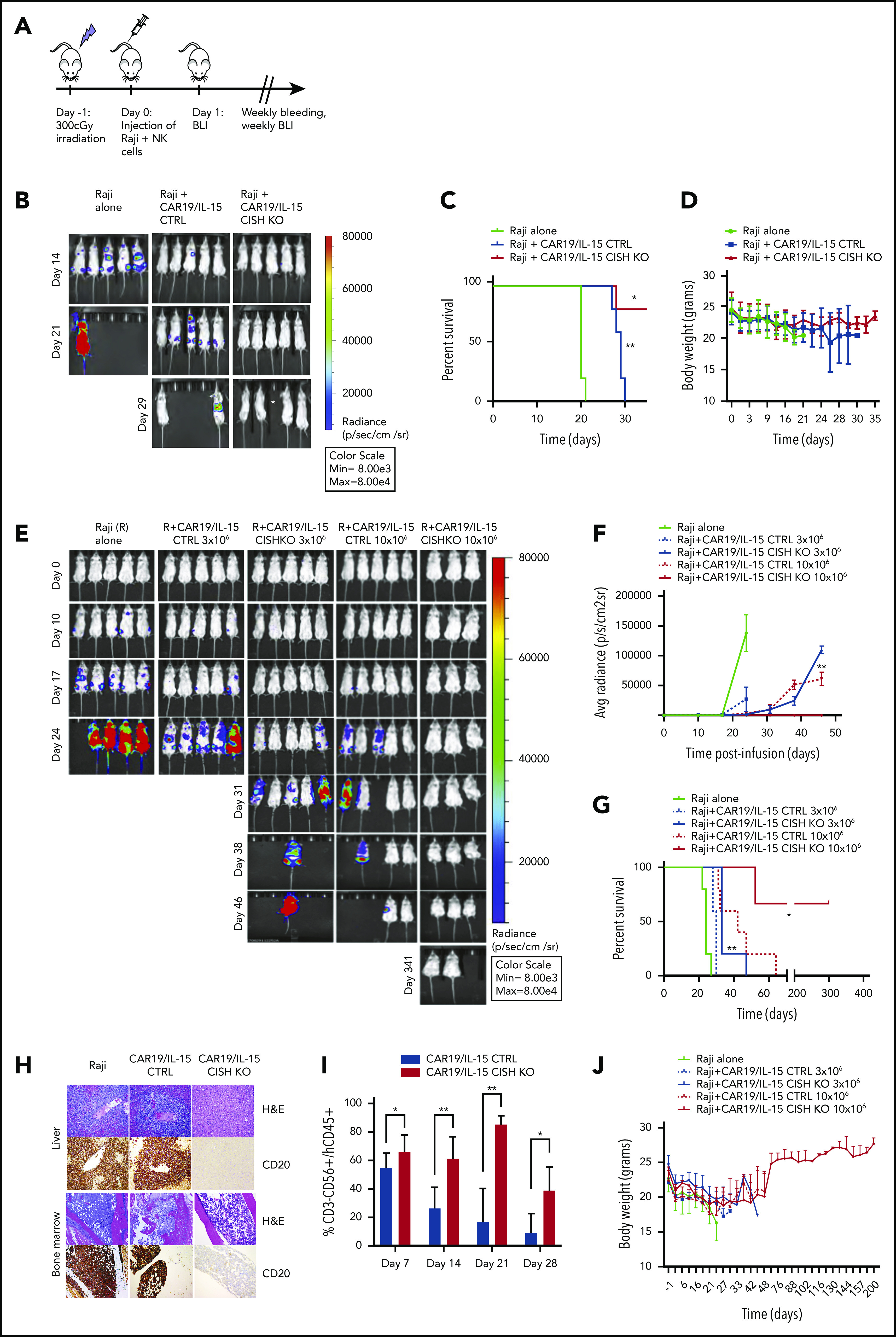
CISH KO iC9/CAR19/IL-15 NK cells improve tumor control and survival in a Raji lymphoma mouse model at low infusion doses. (A) Schematic diagram representing the timeline of the in vivo experiments. (B) BLI imaging of an independent mouse experiment where mice received either Raji alone or Raji plus 10 × 106 CAR19/IL-15 control or Raji plus 10 × 106 CAR19/IL-15 CISH KO (n = 5 mice per group). *This mouse died accidentally during bleeding. (C) Survival curve for the 3 groups of mice described in panel B (Raji alone, green; Raji plus 10 × 106 CAR19/IL-15 control, blue; Raji plus 10 × 106 CAR19/IL-15 CISH KO, red). Statistical significance is represented by *P ≤ .05 for the comparison of the red and blue curves and **P ≤ .01 for the comparison of the blue and gray curves. (D) Graph representing the body weights of mice over time in the 3 different groups described in panel B. Bars represent mean values with standard deviation. (E) BLI data from 5 groups of NSG mice treated with Raji alone (n = 5), Raji plus 1 dose of 3 × 106 of iC9/CAR19/IL-15 control NK cells (n = 5) or iC9/CAR19/IL-15 CISH KO NK cells (n = 5), or Raji plus 1 dose of 10 × 106 of iC9/CAR19/IL-15 control NK cells (n = 5) or iC9/CAR19/IL-15 CISH KO NK cells (n = 3). (F-G) The average radiance (F) and survival curves (G) are shown for the 5 groups of mice described in panel E. Statistical significance is represented by *P ≤ .05 or **P ≤ .01. (H) Photomicrographs of hematoxylin and eosin (H&E) and immunohistochemical CD20 staining of liver (top) and bone marrow (BM) (bottom) from mice engrafted with Raji B cell lymphoma either untreated or treated with iC9/CAR19/IL-15 CISH KO or iC9/CAR19/IL-15 CTLR NK cells (10 × 106 dose level). Representative images show absence of neoplastic B cells in liver and bone marrow of a mouse treated with iC9/CAR19/IL-15 CISH KO NK cells in comparison with similar treatment with iC9/CAR19/IL-15 NK cells retaining CISH expression. Images were taken at 10× (liver) and 5× (bone marrow) using a Leica DFC 495 camera. (I) Bar graph showing the percentage of NK cells (CD3−CD56+CD45+) present in peripheral blood from mice treated with iC9/CAR19/IL-15 control vs iC9/CAR19/IL-15 CISH KO NK cells at days 7, 14, 21, and 28. Bars represent mean values with standard deviation. Statistical significance is represented by *P ≤ .05 or **P ≤ .01. (J) Graph showing body weights of NSG mice groups described in panel E over time.
CISH KO was not associated with signs of increased toxicity in mice, including organ damage or increase in systemic inflammatory cytokines or increased weight loss compared with control groups (Figure 5D,H,J; supplemental Figure 12). After the initial weight loss observed in all groups after irradiation, mice treated with CISH KO iC9/CAR19/IL-15 cells recovered their weight to baseline over an extended period of follow-up (Figure 5J). Moreover, CAR-NK cells were not detectable in the organs of animals at autopsy, indicating that CISH KO does not induce uncontrolled expansion and persistence of CAR-NK cells. We conclude that silencing CISH in the context of CAR19/IL-15 transduction in NK cells can secure robust control of tumor cells in vivo without appreciable toxicity.
The presence of IL-15 in the CAR construct is necessary for CISH KO–mediated improvement in NK cell function
Since our CAR construct expresses both CAR19 and IL-15, it was important to understand whether the improvement in NK cell function following CISH KO is dependent on the presence of IL-15 in the CAR construct. To address this question, we transduced NK cells with a vector expressing CAR19 without the IL-15 transgene (referred to as CAR19 NK cells) and analyzed their CISH expression after culture with IL-2 and uAPC. CAR19 NK cells showed a time-dependent increase in CISH expression to the same extent as observed with NT control, but to a lower magnitude than in iC9/CAR19/IL-15 NK cells (supplemental Figure 13A). The efficiency of CISH KO in CAR19 NK cells was similar to what was achieved in CAR19/IL-15 NK cells (supplemental Figure 13B) and improved their cytotoxicity against Raji in vitro (supplemental Figure 13C) similar to NT-NK cells following CISH KO. However, when tested in our Raji lymphoma mouse model, CISH KO failed to improve the antitumor activity of CAR19 NK cells in vivo (supplemental Figure 13D-F), pointing to the essential role of IL-15 in mediating the effect of CISH KO on CAR-NK cell antitumor activity.
Safety evaluation of CISH KO iC9/CAR19/IL-15 NK cells for relapsed/refractory B-cell malignancy
To investigate the possibility that enhanced IL-15 signaling may result in autonomous dysregulated growth of CISH KO CB-NK cells, we cultured NT (control or CISH KO) or iC9/CAR19/IL-15 CB-NK cells (control or CISH KO) in media in the absence of exogenous IL-2 or feeder cells for 45 days. Cultured CISH KO NT or iC9/CAR.19/IL-15 CB-NK cells did not show signs of abnormal growth over 6 weeks (supplemental Figure 14A), after which the cells stopped expanding. Moreover, we did not observe an increase in the expression of genes related to chromosomal instability32 such as aurora kinases or genes associated with DNA damage following CISH KO (supplemental Figure 14B).
Even though CISH KO CAR-NK cells lacked evidence of serious toxicity in our in vivo model, we planned additional in vitro and vivo experiments to ascertain that CISH KO CAR-NK cells could be swiftly eliminated in the event of toxicity in early phase clinical testing. Thus, we relied on the presence of iC9 as a suicide gene in our vector to confirm that CISH KO CAR-NK cells could be induced to undergo apoptosis in the presence of a small-molecule dimerizer, AP1903. The addition of as little as 10 nM of AP1903 to cultures of CAR-NK cells induced their apoptosis within 4 hours, and CISH KO did not affect the action of the dimerizer (supplemental Figure 15A). The suicide gene was also effective at eliminating the CAR-NK cells in vivo (supplemental Figure 15B-C). Mice engrafted with Raji tumor received either control or CISH KO CAR-NK cells (n = 5 mice per group) followed by treatment with the dimerizer on days 7 and 9 after NK cell infusion. The animals were then sacrificed on day 12. Administration of the small-molecule dimerizer resulted in a striking reduction of the transduced cells (both control and CISH KO) in the blood and tissues (liver, spleen, and bone marrow) in all treated mice (supplemental Figure 15B-C).
Identifying possible off-target editing events mediated by the CRISPR-Cas9 RNP complexes is crucial before the approach described here can be moved to the clinic. Thus, we used Guide-seq and rhAmpSeq technologies (Integrated DNA Technologies [IDT]) to assess genome-wide off-target effects for our CISH gRNAs. Guide-seq experiments were performed using HEK293 cells that constitutively express Streptococcus pyogenes Cas9 nuclease paired with highly modified synthetic gRNAs18 to identify off-target sites with the highest potential to be edited for each CRISPR RNA (crRNA) (Figure 6A-B). Potential Cas9 off-target cleavage sites were then quantified in NK cells electroporated with RNP complexes targeting the CISH locus using rhAmpSeq technology. Cells treated with wild-type S pyogenes. Cas9 protein had a low frequency of off-target editing events with either crRNA1, crRNA2, or the combination of both crRNAs (Figure 6C-D). The use of a high-fidelity Cas9 protein (Alt-R HiFi Cas9 v3; IDT)33 further reduced the off-target events to < 0.5% (Figure 6C-D). These data support the translation of this approach to the clinic.
Figure 6.
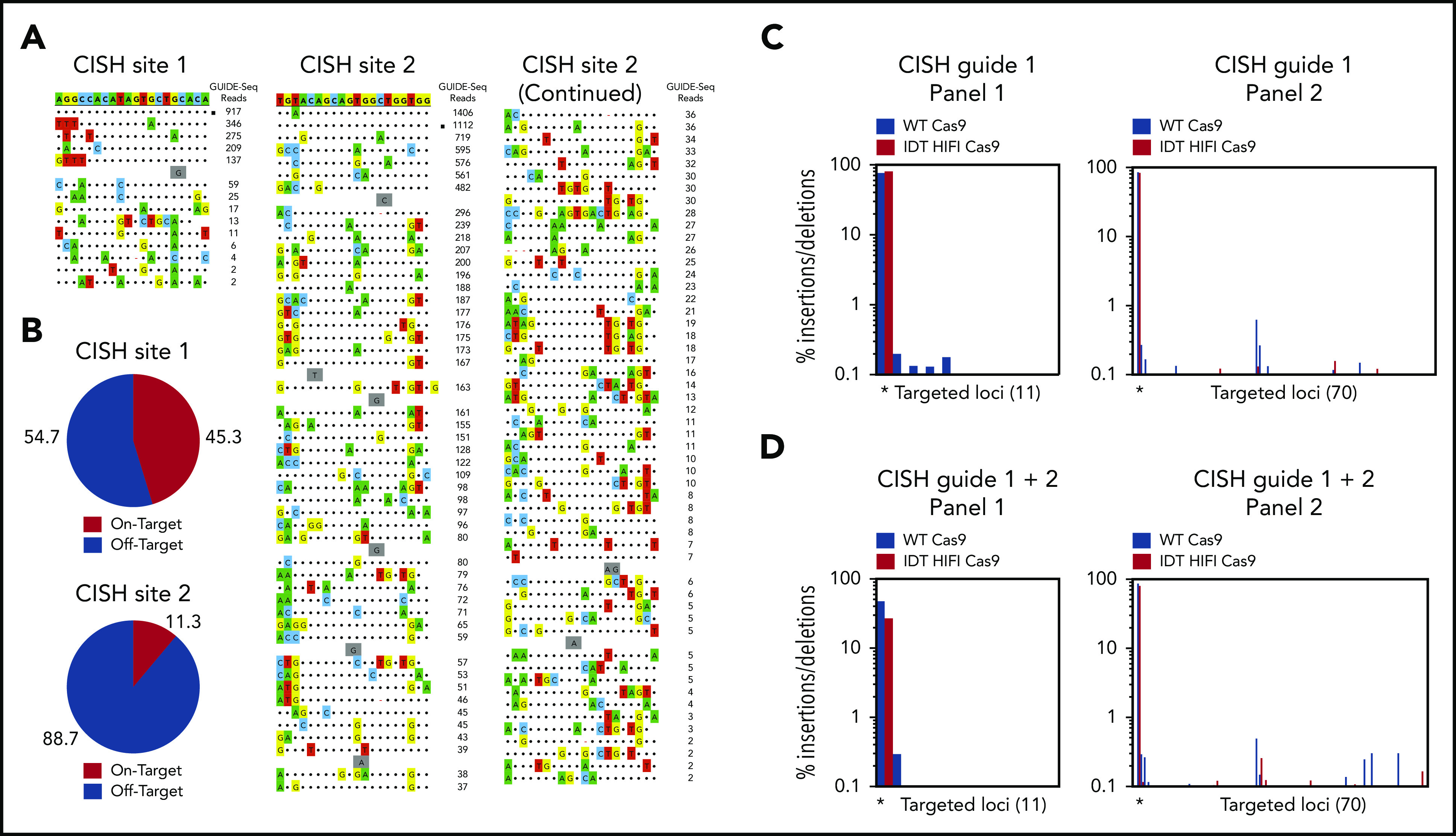
Identification of Cas9 off-target sites by GUIDE-seq and quantification of potential Cas9 off-target cleavage sites using rhAmpSeq technology. (A) Sequences of off-target sites identified by GUIDE-seq for 2 guides targeting the CISH locus. The guide sequence is listed on top with off-target sites shown below. The on-target site is identified with a black square. Mismatches to the guide are shown and highlighted in color with insertions shown in gray. The number of GUIDE-seq sequencing reads are shown to the right of each site. 10 µM Alt-R crRNA XT complexed to Alt-R transactivating CRISPR RNA was delivered into HEK293 cells that constitutively express Cas9 nuclease by nucleofection. (B) Pie charts indicate the fractional percentage of the total unique, CRISPR-Cas9–specific read counts that are on-target (red) and off-target (blue). Total editing at the on- and off-target sites identified by GUIDE-Seq was measured using rhAmpSeq, a multiplexed targeted enrichment approach for next-generation sequencing. For each of the 2 CISH targeting guides, amplicons were designed around each Cas9 cleavage site with reads >1% of the on-target GUIDE-seq reads. RNP complexes formed with either WT Cas9 (blue) or Alt-R HiFi Cas9 (red) were delivered via electroporation into expanded NK cells. (C) Insertion/deletion formation at each targeted loci for CISH guide 1 (panel 1, 11-plex) and CISH guide 2 (panel 2, 70-plex) when a single RNP complex was delivered. The on-target locus is indicated with a black asterisk underneath the first 2 bars of each graph. (D) Insertion/deletion formation at each targeted loci when CISH guide 1 and CISH guide 2 were codelivered. The on-target locus is indicated with a black asterisk underneath the first 2 bars of each graph.
Discussion
NK cell differentiation, effector function and survival, defined as ‘fitness’, are coupled to metabolic reprogramming processes. However, signals and checkpoints that regulate NK cell fitness and function in the tumor microenvironment are not well defined. Our study focuses on the impact of targeting CIS, a negative regulator of cytokine signaling10,34 in CAR-NK cells engineered to constitutively express IL-15.
Having demonstrated the antitumor activity of armored CAR-NK cells preclinically and in the clinic,5,6 we exploited the potential mechanistic synergy that could be achieved by deleting a cytokine checkpoint in IL-15–secreting CAR-NK cells. Here we report that combining IL-15-secreting CAR-NK cells with CISH deletion enhances antitumor activity more than either strategy alone. Using a xenograft mouse model of NK-resistant Raji lymphoma, we showed that CISH KO CAR19/IL-15 CB-NK cells persist twice as long as control CAR19/IL-15 NK cells. We attribute this gain of function to IL-15–driven Akt/mTORC1 and MYC signaling secondary to removal of the CIS checkpoint, with a consequent increase in glycolytic activity. These data are consistent with a recent report showing that deleting CISH in iPSC NK cells improves their metabolism and antitumor activity.34 Our findings have clinical relevance because they establish, to our knowledge for the first time, proof-of-principle that targeting a critical cytokine checkpoint in cytokine-secreting armored CAR-NK cells promotes their fitness and endows them with superior antitumor function.
This increased antitumor activity was not associated with higher toxicity in the animals. While the NSG mouse model has limitations for the study of cytokine release syndrome,35 the absence of overt toxicity such as rapid weight loss and early deaths support the safety of our approach. A legitimate concern with CISH KO is that in the absence of a cytokine checkpoint, IL-15 may drive malignant transformation of CAR-NK cells. However, our extensive studies using transcriptomic analysis, autonomous growth experiments, and in vivo studies did not show any evidence of long-term toxicity or malignant transformation of CISH KO CAR19/IL-15 NK cells.
Previous reports have shown that CISH deletion in NK cells is beneficial only in murine tumor models where IL-15 or IL2 was present in the tumor microenvironment or administered exogenously.10,36 In our study in xenografted mice, there was no appreciable increase in the activity of NT NK cells or CAR19 NK cells (without IL-15 transgene) after CISH KO; the only improvement was noted in mice treated with CISH KO CAR19/IL-15 NK cells. This suggests that the robust antitumor activity of CISH KO CAR-NK cell depends upon the availability of IL-15 in the tumor microenvironment. Thus, incorporation of the IL-15 gene in the CAR construct, which avoids the toxicities seen with systemic IL-15,27,28 likely played an important role in the enhanced antitumor activity of CISH KO iC9/CAR19/IL-15 NK cells in our tumor model. Given the absence or limited concentration of IL-15 in the microenvironment of some cancers, expressing this cytokine in the CAR construct is essential to achieving an optimal therapeutic effect from CISH KO.
Our results suggest that removal of the CIS checkpoint may increase the metabolic fitness of armored CAR-NK cells by enhancing the activity of downstream metabolic pathways. In the model we propose (visual abstract), CISH KO releases the brake on IL-15 signaling, which in turn enhances Akt/mTORC1 activity,30 leading to upregulation of c-MYC and therefore glycolysis, specifically in response to tumor targets. We have previously shown that upon activation, CAR19/IL-15 NK cells secrete more IL-15.6 We now propose that upon activation by the tumor, the higher levels of IL-15 in the microenvironment leads to increased IL-15/Akt/mTORC1 and c-MYC signaling in CISH KO CAR19/IL-15 NK cells. We would emphasize that in our studies, c-MYC expression and the resultant shift in metabolism toward aerobic glycolysis31 in CISH KO iC9/CAR19/IL-15 CB-derived NK cells were only apparent when the NK cells were cultured with tumor targets. This observation is important, as an exaggerated increase in glycolysis in ex vivo–expanded T cells has been reported to severely impair the ability of CD8+ T cells to persist long-term and form memory cells in vivo.37 Hence, careful tuning of NK cell metabolism to ensure a timely increase in aerobic glycolysis in response to tumor stimulation would be desirable.
In summary, this is the first report of a genetic engineering strategy combining armored CAR engineering and CISH deletion in CB-derived NK cells. When tested in a preclinical tumor model, this cellular product eliminated CD19+ lymphoma cells without signs of serious toxicity. Our findings support the merging of armored CAR engineering and cytokine checkpoint gene editing to enhance the therapeutic potential of NK cells in the clinic.
Supplementary Material
The online version of this article contains a data supplement.
Acknowledgments
Supported in part by the generous philanthropic support to The University of Texas MD Anderson Cancer Center Moonshot program, by grants from the Cancer Prevention and Research Institute of Texas (RP160693) and the National Institutes of Health, National Cancer Institute (1 R01 CA211044-01 and PO1 5P01CA148600-03) and by Cancer Center Support (CORE) grant CA016672 (to the Flow Cytometry and Cellular Imaging Facility and the A sequencing core facility at Anderson Cancer Center). The authors acknowledge the support of a Deutsche Knochen Mark Spenderdatei Mechtild Harf research grant and a Society for Immunotherapy of Cancer-Amgen Cancer Immunotherapy in Hematologic Malignancies fellowship award (M.D.).
Footnotes
All requests for raw data and materials will be reviewed by MD Anderson Cancer Center to verify if the request is subject to any intellectual property or confidentiality obligations. Any data and materials that can be shared by the corresponding author will be released freely or via a Material Transfer Agreement if deemed necessary.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: M.D., R.B., and P.P.B. performed experiments and interpreted and analyzed data; N.B. performed western blot and seahorse assay experiments and interpreted and analyzed data; E.G., N.U., A.K.N.C., J.L., G.O., E.E., M. Kaplan, V.N., M.B., L.B., X.W., and X.R.J. assisted with experiments; V.M., Y.X., Y.S., K.C., and J.W. assisted with RNA-sequencing analysis; D. Mak assisted with mass cytometry sample processing; S.K. and N.P. assisted with mass cytometry analysis; M.F., A.R., and N.V. performed and commented on confocal microscopy for mitochondrial studies; M.S.S., G.R.R., R.T., M.S.M., G.K., H.L., and M.A.B. performed off-target effect experiments and analysis and commented on the manuscript; N.W.F. performed pathologic examination and staining of mice tissues; M. Konopleva, N.V., E.J.S., R.E.C., K.C., E.L., S. Ang, S. Acharya, R.J.S., S.L., and L.S. commented on the manuscript; D. Marin, M.M., L.N.K., N.I., M.S., L.L., H.S., F.L.W.I.L., P.P.B., and Y.L. provided advice on experiments and commented on the manuscript; K.R. and M.D. designed and directed the study; and K.R., M.D., and L.M.-F. wrote the manuscript.
Conflict-of-interest disclosure: K.R., M.D., R.B., P.P.B., E.L., E.J.S, R.E.C., D.M., and The University of Texas MD Anderson Cancer Center (MDACC) have an institutional financial conflict of interest with Takeda Pharmaceutical for the licensing of the technology related to the research reported here. MDACC has implemented an Institutional Conflict of Interest Management and Monitoring Plan to manage and monitor the conflict of interest with respect to MDACC’s conduct of any other ongoing or future research related to this relationship. M.A.B., M.S.S., G.R.R., M.S.M., G.K., and R.T. are employed by IDT, which manufactures reagents similar to some described in the paper; M.A.B., M.S.M., R.T., and G.R.R. own equity in DHR, the parent company of IDT. H.L. is a consultant for IDT. The remaining authors declare no competing financial interests.
Correspondence: Katayoun Rezvani, Department of Stem Cell Transplantation and Cellular Therapy, The University of Texas MD Anderson Cancer Center, Houston, TX, 77030-4009; e-mail: krezvani@mdanderson.org.
REFERENCES
- 1.Morvan MG, Lanier LL. NK cells and cancer: you can teach innate cells new tricks. Nat Rev Cancer. 2016;16(1):7-19. [DOI] [PubMed] [Google Scholar]
- 2.Daher M, Rezvani K. Next generation natural killer cells for cancer immunotherapy: the promise of genetic engineering. Curr Opin Immunol. 2018;51:146-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mehta RS, Shpall EJ, Rezvani K. Cord blood as a source of natural killer cells. Front Med (Lausanne). 2016;2:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sarvaria A, Jawdat D, Madrigal JA, Saudemont A. Umbilical cord blood natural killer cells, their characteristics, and potential clinical applications. Front Immunol. 2017;8:329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu E, Marin D, Banerjee P, et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N Engl J Med. 2020;382(6):545-553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu E, Tong Y, Dotti G, et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia. 2018;32(2):520-531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pfefferle A, Huntington ND. You have got a fast CAR: chimeric antigen receptor NK cells in cancer therapy. Cancers (Basel). 2020;12(3):E706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krebs DL, Hilton DJ. SOCS proteins: negative regulators of cytokine signaling. Stem Cells. 2001;19(5):378-387. [DOI] [PubMed] [Google Scholar]
- 9.Linossi EM, Babon JJ, Hilton DJ, Nicholson SE. Suppression of cytokine signaling: the SOCS perspective. Cytokine Growth Factor Rev. 2013;24(3):241-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Delconte RB, Kolesnik TB, Dagley LF, et al. CIS is a potent checkpoint in NK cell-mediated tumor immunity. Nat Immunol. 2016;17(7):816-824. [DOI] [PubMed] [Google Scholar]
- 11.Zhang JG, Farley A, Nicholson SE, et al. The conserved SOCS box motif in suppressors of cytokine signaling binds to elongins B and C and may couple bound proteins to proteasomal degradation. Proc Natl Acad Sci USA. 1999;96(5):2071-2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yoshimura A, Nishinakamura H, Matsumura Y, Hanada T. Negative regulation of cytokine signaling and immune responses by SOCS proteins. Arthritis Res Ther. 2005;7(3):100-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Inagaki-Ohara K, Hanada T, Yoshimura A. Negative regulation of cytokine signaling and inflammatory diseases. Curr Opin Pharmacol. 2003;3(4):435-442. [DOI] [PubMed] [Google Scholar]
- 14.Hoyos V, Savoldo B, Quintarelli C, et al. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia. 2010;24(6):1160-1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vera J, Savoldo B, Vigouroux S, et al. T lymphocytes redirected against the kappa light chain of human immunoglobulin efficiently kill mature B lymphocyte-derived malignant cells. Blood. 2006;108(12):3890-3897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li L, Chen H, Marin D, et al. A novel immature natural killer cell subpopulation predicts relapse after cord blood transplantation. Blood Adv. 2019;3(23):4117-4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shah N, Martin-Antonio B, Yang H, et al. Antigen presenting cell-mediated expansion of human umbilical cord blood yields log-scale expansion of natural killer cells with anti-myeloma activity. PLoS One. 2013;8(10):e76781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsai SQ, Zheng Z, Nguyen NT, et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat Biotechnol. 2015;33(2):187-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dobosy JR, Rose SD, Beltz KR, et al. RNase H-dependent PCR (rhPCR): improved specificity and single nucleotide polymorphism detection using blocked cleavable primers. BMC Biotechnol. 2011;11(1):80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cherkassky L, Morello A, Villena-Vargas J, et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest. 2016;126(8):3130-3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chong EA, Melenhorst JJ, Lacey SF, et al. PD-1 blockade modulates chimeric antigen receptor (CAR)-modified T cells: refueling the CAR. Blood. 2017;129(8):1039-1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matsuda T, Yamamoto T, Kishi H, Yoshimura A, Muraguchi A. SOCS-1 can suppress CD3zeta- and Syk-mediated NF-AT activation in a non-lymphoid cell line. FEBS Lett. 2000;472(2-3):235-240. [DOI] [PubMed] [Google Scholar]
- 23.Palmer DC, Guittard GC, Franco Z, et al. Cish actively silences TCR signaling in CD8+ T cells to maintain tumor tolerance. J Exp Med. 2015;212(12):2095-2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mukherjee M, Mace EM, Carisey AF, Ahmed N, Orange JS. Quantitative imaging approaches to study the CAR immunological synapse. Mol Ther. 2017;25(8):1757-1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krenciute G, Prinzing BL, Yi Z, et al. Transgenic expression of IL15 improves antiglioma activity of IL13Rα2-CAR T cells but results in antigen loss variants. Cancer Immunol Res. 2017;5(7):571-581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Felices M, Lenvik AJ, McElmurry R, et al. Continuous treatment with IL-15 exhausts human NK cells via a metabolic defect. JCI Insight. 2018;3(3):96219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Conlon KC, Lugli E, Welles HC, et al. Redistribution, hyperproliferation, activation of natural killer cells and CD8 T cells, and cytokine production during first-in-human clinical trial of recombinant human interleukin-15 in patients with cancer. J Clin Oncol. 2015;33(1):74-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Waldmann TA, Lugli E, Roederer M, et al. Safety (toxicity), pharmacokinetics, immunogenicity, and impact on elements of the normal immune system of recombinant human IL-15 in rhesus macaques. Blood. 2011;117(18):4787-4795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Donnelly RP, Loftus RM, Keating SE, et al. mTORC1-dependent metabolic reprogramming is a prerequisite for NK cell effector function. J Immunol. 2014;193(9):4477-4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marçais A, Cherfils-Vicini J, Viant C, et al. The metabolic checkpoint kinase mTOR is essential for IL-15 signaling during the development and activation of NK cells. Nat Immunol. 2014;15(8):749-757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Donnelly RP, Finlay DK. Glucose, glycolysis and lymphocyte responses. Mol Immunol. 2015;68(2 2 Pt C):513-519. [DOI] [PubMed] [Google Scholar]
- 32.Carter SL, Eklund AC, Kohane IS, Harris LN, Szallasi Z. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat Genet. 2006;38(9):1043-1048. [DOI] [PubMed] [Google Scholar]
- 33.Vakulskas CA, Dever DP, Rettig GR, et al. A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nat Med. 2018;24(8):1216-1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu H, Blum RH, Bernareggi D, et al. Metabolic reprograming via deletion of CISH in human iPSC-derived NK cells promotes in vivo persistence and enhances anti-tumor activity. Cell Stem Cell. 2020;27(2):224-237.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Giavridis T, van der Stegen SJC, Eyquem J, Hamieh M, Piersigilli A, Sadelain M. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat Med. 2018;24(6):731-738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Putz EM, Guillerey C, Kos K, et al. Targeting cytokine signaling checkpoint CIS activates NK cells to protect from tumor initiation and metastasis. OncoImmunology. 2017;6(2):e1267892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sukumar M, Liu J, Ji Y, et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J Clin Invest. 2013;123(10):4479-4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.