

Abstract
Clinical exome sequencing is a powerful approach to overcome the wide clinical and genetic heterogeneity of mucopolysaccharidosis. These data could be useful for prenatal diagnosis of MPS VII, genetic counseling, and preimplantation genetic testing.
Keywords: clinical exome sequencing, glycosaminoglycans, Mucopolysaccharidosis VII, prenatal diagnosis, Sly syndrome, β‐glucuronidase
Clinical exome sequencing is a powerful approach to overcome the wide clinical and genetic heterogeneity of mucopolysaccharidosis. These data could be useful for prenatal diagnosis of MPS VII, genetic counseling, and preimplantation genetic testing.

1. INTRODUCTION
The aim of this study was to describe a new variant in GUSB gene related to severe recurrent fetal hydrops. A consanguineous couple with previous fetal losing by fetal hydrops (FH). At second gestation, generalized edema was presented. Fetal sample was analyzed by next‐generation sequencing using CES (clinical exome sequencing). We identified in homozygous form the variant NM_000181.3:c.107G > T; p.Arg36Leu; Chr7:g.65982077C > A in the GUSB gene. No activity of β‐glucuronidase was detected in fetal cells. The pregnancy ended after stillbirth at 30 weeks and 3 days. We describe a new pathogenic variant in GUSB gene related to mucopolysaccharidosis VII associated with severe hydrops fetalis. These data could be useful for early prenatal diagnosis of MPS VII, genetic counseling, and preimplantation genetic testing.
Fetal hydrops (FH) is an uncommon complication of pregnancy but it is often lethal. FH is an abnormal collection of fluid in visceral cavities and soft fetal tissues. Cardiovascular diseases, chromosomal disorders, infections, metabolic disorders, tumors, and others can lead to FH. It has classically been divided into immune and nonimmune hydrops. 1 The features are detected by ultrasound. It is defined as the presence of 2 or plus abnormal fluid collections such as ascites, pleural effusions, pericardial effusion, or generalized skin edema (defined as nuchal fold > 6 mm). 2 Nonimmune fetal hydrops (NIFH) refers specifically to cases not caused by red cell alloimmunization. 3 The prevalence of NIFH ranges from 1/1700 to 1/3000 pregnancies 4 , 5 and represents almost 90% of cases of hydrops. 6
Once more frequent etiologies of NIHF are ruled out, inborn errors of metabolism (IEM) should be considered. Mucopolysaccharidosis type VII (MPS VII, MIM #253220) is among the most common lysosomal storage disease diagnosed in NIFH. 7
MPS VII also known as Sly syndrome is a rare disorder caused by mutations in GUSB gene (MIM* 611 499) inherited in autosomal‐recessive pattern. The GUSB gene is located on chromosome 7q21.11 and encodes beta‐glucuronidase (EC 3.2.1.31). 8 The enzyme deficiency results in accumulation of glycosaminoglycans (GAG) heparan sulfate, dermatan sulfate, chondroitin‐4‐sulfate, and chondroitin‐6‐sulfate in lysosomes of several tissues. 9
Patients with MPS VII have a broad range of clinical signs and symptoms including coarse facies, skeletal dysplasia, joint contractures, short stature, hernias, hepatosplenomegaly, neurological impairment, cardiac involvement, pulmonary insufficiency, hearing impairment, recurrent upper respiratory infections, middle ear infections, and corneal clouding. The clinical spectrum ranges from a severe form with lethal fetal hydrops to attenuated forms with survival into adulthood despite physical, behavioral disturbances such as hyperactivity and cognitive impairment, but it can be possible to a milder phenotype with later onset, fewer clinical manifestations, and normal or near‐normal intelligence instead. 10 , 11 , 12
Usually, the diagnosis happens in postnatal period after clinical suspicion and using biochemical methods to identifying glycosaminoglycan deficiency or excess in biological fluids such as urine, blood, and cerebrospinal fluid (CSF). In prenatal period, biochemical diagnosis is possible analyzing enzymatic activity in amniotic fluid. The enzyme assays are considered the gold standard for diagnosis and can be used with GAG analysis for prognosis and treatment efficacy monitoring. 13
The diagnosis of MPS is currently based on the quantification of GAG, measurement of enzyme activities, and identification of genetic variants. A correct genetic diagnosis allows specific therapeutic actions and also permits phenotype prediction, carrier identification, genetic counseling, and prenatal diagnosis. New technologies, such as next‐generation sequencing (NGS), are becoming more accessible and relatively affordable for the MPS diagnostic routine. 14
2. CASE
A 33‐year‐old Moroccan pregnant woman was derived to our fetal medicine unit due to increased nuchal translucency of 3.3 mm at 12 weeks and 6 days of gestation. It was her second gestation. She had a previous pregnancy evaluated also in our fetal medicine unit with a female fetus that presented with nonimmune fetal hydrops at 20 weeks (Figure 1). We performed a chorionic villus sampling (CVS) that demonstrated a female karyotype and chromosomal genomic hybridization (CGH) array are both normal. TORCH screening and maternal serum indirect Coombs test were also negative. The pregnancy ended after stillbirth at 28 weeks’ gestation.
Figure 1.
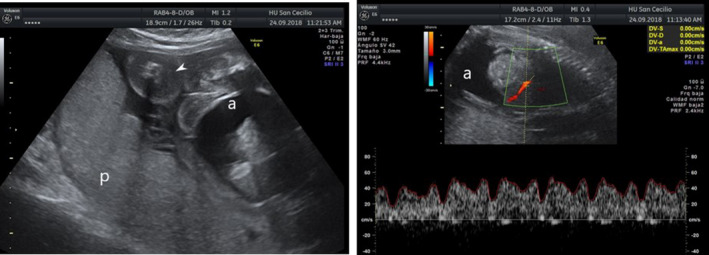
First pregnancy with nonimmune fetal hydrops at 20 weeks: placentomegaly (p), ascites (a), skin edema (white arrow)
In her actual pregnancy, the only risk factors were a BMI of 31.2 and consanguinity (first cousins). In this occasion, we investigated about family history in‐depth, and we found a high consanguinity grade (Figure 2). The mother of the patient and the father of the husband are brothers. The husband's sister was married to a cousin of both and had three sons: a healthy daughter, a daughter with severe developmental delay, and a son with macrocephaly and growth restriction. Two other cousins in the family were married between them, and they had lost two fetuses at 4 months of gestation. These fetal losses had not been studied in Morocco.
Figure 2.
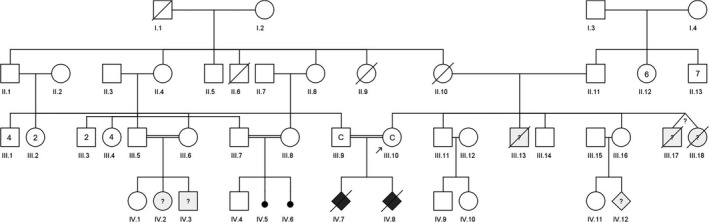
Pedigree chart of patient´s family. Phenotype description: III.10 and III.9 asymptomatic consanguineous couples that were carrier of GUSB variant in heterozygous way (C), IV.2 severe developmental delay, IV.3 macrocephaly, IV.7. female fetus with generalized fetal hydrops that it was not studied (MPSVII case suspected), IV.8 male fetus carrier of GUSB variant in homozygous way (MPSVII case confirmed), and IV.12 unknown
During this second pregnancy, the ultrasound examination performed first in our unit at 16 weeks and 5 days of gestation (Aplio i700; Canon Medical Systems, Japan) revealed a nuchal fold of 7.0 mm, generalized edema without other morphological abnormalities or signs of fetal anemia, normal Doppler, normal placenta, and amniotic fluid (Figure 3).
Figure 3.
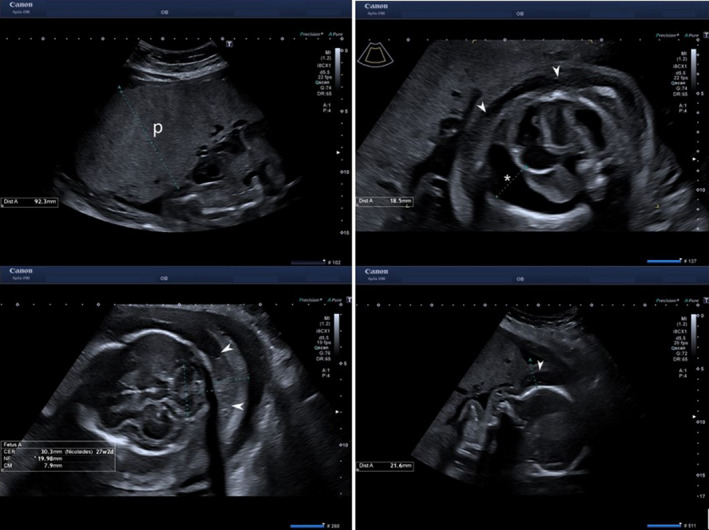
Second pregnancy with progressive fetal hydrops at 27 weeks: placentomegaly (p), skin edema (white arrow), and hydrothorax (white asterisk)
Amniocentesis sampling demonstrated a normal male karyotype and CGH array without alterations. Maternal serum TORCH and indirect Coombs test were also negative.
We offered to the patients a clinical exome sequencing targeted to fetal hydrops phenotype disorders.
Amniotic fluid (AF) (20 mL) and umbilical cord blood (2 mL) were obtained using the percutaneous ultrasound‐guided standard procedure. AF was collected in sterile tubes without additives. We also obtained blood samples from the parents by venipuncture using a standardized procedure. All blood samples were collected in tubes with EDTA K3.
For genetic studies, we performed the next procedure: DNA extraction and quantification from the collected samples, preparation of a library of fragments of the genome, selection of the regions under study using the SureSelectXT Human All Exon V5 capture method (Agilent Technologies), clonal amplification and sequencing of the selected regions on the Illumina HiSeq platform following the paired‐end strategy and bioinformatic study of the DNA sequence obtained by comparison with the reference nucleotide sequence (GRCh38). This analysis considers variants those alterations with a number of readings > 10x and a ratio of the number of variant reads to total number of reads > 0.2. The regions analyzed include exons and adjacent intronic zones (± 8 bp) of the genes under study.
We selected a list of genes described in the OMIM and HGMD databases as genes associated with phenotype. We analyzed the variants described in HGMD, variants with deleterious effect (frameshift, stop codon, nonsense, essential splicing, etc), de novo variants, and homozygous variants (due to the history of inbreeding) present in the genes detailed in the supplementary table. We confirm by Sanger sequencing the pathogenic or likely pathogenic variants identified.
A trio‐based CES targeted to phenotype was requested after informed consent, explaining to the parents its meaning, limitations, and time necessary for results.
Three variants of unknown clinical significance were identified: one in GUSB gene and two in RYR1 gene. The presence of the following variants was identified in genes whose phenotypes associated could be compatible, a priori, with the clinical features described in the patient:
NM_000181.3: c.107G > T; p.Arg36Leu; Chr7: g.65982077C > A, in homozygous form, in the GUSB gene.
NM_000540.2: c.4980C > T; p.Arg1660 =; Chr19: g.38485635C > T, in homozygous form, in the RYR1 gene.
NM_000540.2: c.14270G > A; p.Arg4757His; Chr19: g.38578015G > A, in homozygous form, in the RYR1 gene.
The genetic study was reported by the laboratory as nonconcluding. Genetic assessment of the results was carried out. We refuse variants of RYR1 gene as explanatory of the findings because its clinical description in OMIM 8 database was not related to hydrops. We proposed to the parents that we to check our suspicion for a probable mucopolysaccharidosis associated with the GUSB gene variant by an additional biochemical study. The parents accepted our proposal to obtain a new sampling for studies at 27 weeks and 5 days pregnancy.
The fetal enzyme assay was carried out in amniocytes and leukocytes obtained from AF and blood of umbilical cord, respectively. Beta‐glucuronidase activity was determined in these samples as previously described 15 with the fluorogenic substrate 4‐methylumbelliferyl‐b–D‐glucuronide (Sigma‐Aldrich‐Merck).
No activity of β‐glucuronidase was detected in two different fetal cell types (amniocytes and leukocytes) by two different methods, respectively. The value for beta‐glucuronidase enzymatic activity assay performed in leukocytes isolated from umbilical cord blood was 0 nmol/ (h*mg prot) (reference values: 147 ‐ 1148). The beta‐glucuronidase activity was undetectable with this method. The result for beta‐glucuronidase enzymatic activity assay performed in amniocytes cultured was 0.5 nmol/ (h*mg prot) (reference values: 58,0 ‐ 195, 0). Those results indicate that the fetus is affected by MPS VII.
Both parents were heterozygous carrier for the GUSB gene variant identified previously in fetal sample (NM_000181.3: c.107G > T; p.R36L). These data confirm the consanguineous inheritance transmission pattern.
The pregnancy ended up in spontaneous preterm delivery at 30 weeks and 3 days of pregnancy, with a newborn of 2750 gr that died in the first week of life. The couple was referred for human reproductive assisted techniques in our regional reference center.
The novel identified variant in GUSB gene was submitted to ClinVar (accession VCV000694267) with likely pathogenic (Class 4) classification by ACMG criteria.
3. DISCUSSION
The β‐glucuronidase is a lysosomal homotetramer(15) and comprised of 651 amino acid residues (78 kD)(16). The GUSB monomer is comprised of three distinct structural domains, the first domain is a jelly roll shape, which is highly distorted and forms a barrel‐like structure along with two b‐hairpin insertions. The hairpin loop of the jelly roll motif was considered to be an essential part of GUSB for lysosomal targeting (17). Therefore, it is also called a lysosomal targeting motif. The second domain is structurally similar to the immunoglobulin constant domains. The third is ana/bor TIM barrel domain motif, which possesses the active site of the enzyme and a characteristic feature of glycosyl hydrolase enzymes. The active site of each monomer is present at the interface of oligomer, and the tetramer complex has four active sites.
We used CES to identify a mutation (NM_000181.3:c.107G > T; p.R36L; Chr7:g.65982077C > A) in GUSB gene in a fetus affected by hydrops fetalis.
Subsequently, we speculated that the variant of GUSB gene is critical for the proper function of the gene and might had led to an absence of the β‐glucuronidase or reduce activity of the enzyme. The results of the biochemical studies confirmed our suspicion. The variant NM_000181.3:c.107G > T (p.R36L) in GUSB gene is a missense mutation. It is a base transversion of guanine for thymine in the position 107 of the gene coding sequence, presumably it causes a change of arginine (R) for leucine (L) in the codon 36 of the protein. This variant has not been described previously in the literature revised. The variant is not found in GnomAD exome databases and in GnomAD genome databases either. The computational prediction tools unanimously support a deleterious effect on the gene. To date, 54 disease‐causing mutations have been reported in GUSB gene (https://www.ncbi.nlm.nih.gov/clinvar. Web page date access 21/06/2020). The variant p.R36L is located very close to Glyco_hydro_2_N domain of the β‐glucuronidase. We suspect that this circumstance could cause altered function of the enzyme, but more studies are necessary to stablish this hypothesis.
Prenatal biochemical diagnosis of mucopolysaccharidosis type VII has long been possible. 4 , 16 , 17 , 18 Although enzyme activity assay is considered the gold standard for the diagnosis of MPS disorders, molecular genetic testing is recommended and, whenever possible, diagnostic conclusions should be made taking the clinical, biochemical, and molecular genetic results into consideration. 14
New technologies, such as next‐generation sequencing (NGS), are becoming more accessible and relatively affordable for the MPS diagnostic routine. This technology was revealed as a powerful approach to overcome the wide clinical and genetic heterogeneity of MPS, allowing the simultaneous screening of several MPS‐related genes with shorter turn‐around times for the final report.
For those individuals at a high risk of having a child affected by an MPS VII condition, it is possible to offer a range of reproductive options including both preconception testing (preimplantation genetic diagnosis [PGD]) and prenatal testing (free fetal DNA testing, chorionic villus sampling [CVS], or amniocentesis). It is essential that those at risk of having a child affected by an MPS VII disease are counseling in relation to their reproductive options. 19
A better understanding of the relationship between genotype and phenotype will allow progress in the diagnosis and treatment of this disease.
CONFLICT OF INTEREST
There are no conflicts of interest to disclose. There is no funding or financial support to declare for this study. The manuscript complies with the ethical approval requirements. The data that support the findings of this study are available from the corresponding author upon reasonable request.
AUTHOR CONTRIBUTION
Poyatos‐Andújar, A: conceived and wrote the manuscript. García‐Linares, S.: performed genetic analysis. Carretero, P., Ocon, O, and Fresneda, M.D: contributed clinical data. Gort, L.: performed biochemicals analysis. Molina, FS.: reviewed the manuscript.
ACKNOWLEDGMENTS
Published with written consent of the patient.
Poyatos‐Andújar AM, García‐Linares S, Carretero P, et al. Prenatal mucopolysaccharidosis VII: A novel pathogenic variant identified in GUSB gene. Clin Case Rep.2021;9:790–795. 10.1002/ccr3.3644
REFERENCES
- 1. Parks WT. A pathologist’s approach to nonimmune hydrops. J Fetal Med. 2015;2(3):143‐149. [Google Scholar]
- 2. Molina FS, Avgidou K, Kagan KO, Poggi S, Nicolaides KH. Cystic hygromas, nuchal edema, and nuchal translucency at 11–14 weeks of gestation. Obstet Gynecol. 2006;107(3):678‐683. [DOI] [PubMed] [Google Scholar]
- 3. Norton ME, Chauhan SP, Dashe JS. Society for maternal‐fetal medicine (SMFM) clinical guideline #7: Nonimmune hydrops fetalis. Am J Obstet Gynecol. 2015;212(2):127‐139. [DOI] [PubMed] [Google Scholar]
- 4. Machin GA. Hydrops revisited: Literature review of 1, 414 cases published in the 1980s. Am J Med Genet. 1980s;34(3):366‐390. [DOI] [PubMed] [Google Scholar]
- 5. Heinonen S, Ryynänen M, Kirkinen P. Etiology and outcome of second trimester non‐immunologic fetal hydrops. Acta Obstet Gynecol Scand. 2000;79(1):15‐18. [PubMed] [Google Scholar]
- 6. Santolaya J, Alley D, Jaffe R, Warsof SL. Antenatal classification of hydrops fetalis. Obstet Gynecol. 1992;79(2):256‐259. [PubMed] [Google Scholar]
- 7. Al‐Kouatly HB, Felder L, Makhamreh MM, Kass SL, Vora NL, Berghella V, Berger S, Wenger DA, Luzi P. Lysosomal storage disease spectrum in nonimmune hydrops fetalis: a retrospective case control study. Prenatal Diagnosis. 2020;40(6):738–745. 10.1002/pd.5678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. McKusick‐Nathans Institute of Genetic Medicine, Johns Hopkins University (Baltimore M. Online Mendelian Inheritance in Man, OMIM. https://www.omim.org/entry/611499. Accessed May 20, 2020.
- 9. Sly WS, Quinton BA, McAlister WH, Rimoin DL. Beta glucuronidase deficiency: Report of clinical, radiologic, and biochemical features of a new mucopolysaccharidosis. J Pediatr. 1973;82(2):249‐257. [DOI] [PubMed] [Google Scholar]
- 10. Montaño AM, Lock‐Hock N, Steiner RD, et al. Clinical course of sly syndrome (mucopolysaccharidosis type VII). J Med Genet. 2016;53(6):403‐418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zielonka M, Garbade SF, Kölker S, Hoffmann GF, Ries M. Quantitative clinical characteristics of 53 patients with MPS VII: A cross‐sectional analysis. Genet Med. 2017;19(9):983‐988. [DOI] [PubMed] [Google Scholar]
- 12. Morrison A, Oussoren E, Friedel T, Cruz J, Yilmaz N. Pathway to diagnosis and burden of illness in mucopolysaccharidosis type VII – a European caregiver survey. Orphanet J Rare Dis. 2019;14(1):254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stapleton M, Arunkumar N, Kubaski F, Mason RW, Tadao O, Tomatsu S. Clinical presentation and diagnosis of mucopolysaccharidoses. Mol Genet Metab. 2018;125(1–2):4‐17. [DOI] [PubMed] [Google Scholar]
- 14. Kubaski F, de Oliveira PF, Michelin‐Tirelli K, et al. Diagnosis of Mucopolysaccharidoses. Diagnostics. 2020;10(3):172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Beaudet AL, DiFerrante NM, Ferry GD, Nichols BL, Mullins CE. Variation in the phenotypic expression of β‐glucuronidase deficiency. J Pediatr. 1975;86(3):388‐394. [DOI] [PubMed] [Google Scholar]
- 16. Poenaru L, Castelnau L, Mossman J, Boué J, Dreyfus JC. Prenatal diagnosis of a heterozygote for mucopolysaccharidosis type VII (β‐glucuronidase deficiency). Prenat Diagn. 1982;2(4):251‐256. [DOI] [PubMed] [Google Scholar]
- 17. Den Hollander NS, Kleijer WJ, Schoonderwaldt EM, Los FJ, Wladimiroff JW, Niermeijer MF. In‐utero diagnosis of mucopolysaccharidosis type VII in a fetus with an enlarged nuchal translucency. Ultrasound Obstet Gynecol. 2000;16(1):87‐90. [DOI] [PubMed] [Google Scholar]
- 18. Saxonhouse MA, Behnke M, Williams JL, Richards D, Weiss MD. Mucopolysaccharidosis type VII presenting with isolated neonatal ascites. J Perinatol. 2003;23(1):73‐75. [DOI] [PubMed] [Google Scholar]
- 19. Wilson A, Lavery C, Stewart F, et al. Mucopolysaccharidosis and Adulthood: Genetics, Inheritance, and Reproductive Options. J Child Sci. 2018;8(1):e138‐e143. [Google Scholar]