

Abstract
Acquired hemophilia should be evaluated in pediatric patients with bleeding and isolated prolonged aPTT. Immunosuppressive treatment should be initiated even in minor bleedings. Bypassing agents like rFVIIa can be used in children with success.
Keywords: Acquired hemophilia A, factor VIII deficiency, pediatric population, recombinant factor VII activated
Acquired hemophilia should be evaluated in pediatric patients with bleeding and isolated prolonged aPTT. Immunosuppressive treatment should be initiated even in minor bleedings. Bypassing agents like rFVIIa can be used in children with success.
1. INTRODUCTION
Despite articles regarding acquired hemophilia A in adult population, there is lack of experience in management of pediatric patients. We report a case of AHA in a 7‐year‐old girl, receiving recombinant activated factor VII and prednisone. Our experience shows that these drugs can be used effectively in children with AHA.
Acquired hemophilia A (AHA) is a rare, potentially life‐threatening hemorrhagic disorder caused by the development of autoantibodies directed in most of the cases against functional epitopes of factor VIII. The incidence is estimated to be 0.2‐1 cases per 1 million people per year 1 ; it shows two peaks in general population, one in females in the peri‐ and postpartum period (usually in primiparous women within 3 months from delivery) and another one in elderly patients. In this population, acquired hemophilia is related to underlying conditions such as malignancies, autoimmune disorders, dermatologic conditions, or drugs in half of patients, while in the rest, the cause remains idiopathic. 2
Clinical features differ widely from the ones present in “classical” congenital hemophilia. It is known that 80% of patients develop deep soft tissues bleedings involving skin, muscles, and mucous membranes, gastrointestinal and urogenital tract. Severe retroperitoneal and brain hemorrhages are described and potentially life‐threatening. Hemarthroses, on the other hand, though described, are uncommon. 1 , 2
AHA in children is extremely rare with limited number of cases described in the literature, and very little is known about its epidemiology, diagnosis, clinical course, and treatment. Franchini et al described 42 cases of pediatric acquired hemophilia, of whom 37 cases reported a “de‐novo” autoantibody formation, while in 5 cases, a transplacental inhibitor transmission was detected in neonates. An underlying autoimmune disorder was found in six cases, while the majority of children, instead, had no underlying conditions and no family history of bleeding, with the presence of a recent history of upper airways infections or antibiotics use, especially penicillin or penicillin‐like antibiotics. In particular, two cases occurred in children with a recent history of streptococcal pharyngitis treated with penicillin. Twelve cases remained idiopathic. 1
Disease diagnosis represents a clinical challenge. It must be considered every time a child with no personal or family history of bleeding develops an isolated prolongation of the activated partial thromboplastin time (aPTT). The cause of aPTT prolongation could be due to a number of causes such as coagulation factor deficiency (FVIII, IX, XI, XII) or the presence of coagulation factor inhibitors. The importance of mixing test to determine the correction of aPTT at time 0 and 2 hours after the incubation of patient's plasma and normal plasma in a 1:1 ratio at 37°C could be of aid in differential diagnosis. Subsequent finding of extremely low or absent factor VIII levels together with the presence of its inhibitor remains necessary in order to confirm the diagnosis. The inhibitor levels are measured in Bethesda Units (BU) by using Bethesda or modified Nijmegen assay (increased sensitivity) and classifying patients with low (<5 BU/mL) and high (>5 BU/mL) titer. Enzyme‐linked immunosorbent assay (ELISA) detection of anti–factor VIII autoantibodies is also available and more easily interpreted compared to Bethesda and modified Nijmegen assay in order to test both the antibody isotype and concentration. 3
As for the therapeutic goals they are divided in the arrest of the acute bleeding and the eradication of the autoantibody, obtained by administering immunosuppressant therapy and should be promptly started. 2 , 4 First‐line hemostatic treatment choices are represented by bypassing agents: recombinant activated factor VII (rFVIIa; NovoSeven®), activated prothrombin concentrate complex (APCC; FEIBA®), and replacement therapy with recently available recombinant porcine factor VIII (rpFVIII, Obizur®), prior to antiporcine titer evaluation, due to their extremely high efficacy (around 90%). Treatment efficiency and dosage modification are assessed by clinical examination, hemoglobin monitoring, and repeated imaging. 5 , 6 , 7 However, the use of rpFVIII is not indicated for the pediatric population at the moment, according to European Medical Agency (EMA). Lastly, invasive procedures should be avoided, if not urgently needed, and the number of venous punctures should be limited, to prevent ulterior major bleeding.
On the other hand, inhibitor eradication in adult population is recommended for all patients and based on immunosuppressive therapy (IST) with steroids (prednisone 1 mg/kg/d PO for 4‐6 weeks) alone or in combination with cyclophosphamide (1.5‐2 mg/kg/day PO for 4‐6 weeks). 3 , 8 The choice of single or combination therapy is based on both the patient's performance/comorbidity status and FVIII activity (<1%) and inhibitor titer levels (>20 BU). 3 , 5 Although not approved for AHA patients, given its proven efficacy in case reports, rituximab, anti‐CD20 monoclonal antibody, was already part of second‐line treatment, at the standard dosage of 375 mg/mq IV, weekly for a total of 4 infusions. 9 According to the latest recommendations, its use is suggested in first‐line in patients with poor prognostic markers or in those with corticosteroid contraindication. 8
Given the limited experience in pediatric population, the management of children with AHA is extrapolated from adult data. However, the outcome seems to be more favorable than in adults, since inhibitors usually resolve more quickly, probably because most cases are secondary to treatable conditions, in contrast with adult population. 1 Furthermore, probably due to low comorbidity burden, tolerance to immunosuppressive treatment is improved compared to the elderly population with less frequent infection episodes. Therefore, single agent therapy with steroids could be preferred compared to combination therapy with cytostatic cyclophosphamide.
Here we report a case of a 7‐year‐old female patient affected with AHA, and successfully treated with prednisone and rFVIIa therapy.
2. CASE REPORT
A 7‐year‐old girl presented with recurrent self‐limiting epistaxis in the absence of personal or family history of bleeding. Anamnesis revealed that the mother and male sibling were beta‐thalassemia carriers.
In complete welfare, our patient started was admitted for the first time at the pediatric emergency department and after otolaryngologist evaluation was diagnosed with bleeding varicose vessel in her nasal septum and oral tranexamic acid was prescribed. After one week, due to frequent hemorrhagic episodes and asthenia onset, she was admitted once again at the emergency department. Her blood examinations revealed microcytic anemia (hemoglobin 7.9 g/dL, mean corpuscular volume 59.4 fl), normal platelet count (320 × 109/L), and an isolated prolonged aPTT (116 seconds). Iron oral supplement was started, and the patient was discharged.
A few days later, she was finally admitted to our Hematology‐Oncology Pediatric Department. Blood tests showed increasing hemoglobin levels and reticulocytosis, confirming the iron deficiency and initial response to oral therapy. On the other hand, aPTT prolongation (116 seconds) was confirmed, and did not improve after incubation with mixing test in a 1:1 relation with plasma from a healthy control. Clotting factors concentration revealed almost complete FVIII deficiency (0.7%). Other factors and lupus anticoagulant count were normal. Immunoglobulins, autoimmunity screening, antimycoplasma antibodies, and thyroid function all tested negative. Antistreptolysin O titer tested positive (1002 UI/mL, reference <250 UI/mL), but her throat swab culture resulted negative for group A Streptococcal infection. While waiting for these laboratory results, we decided to wait and see, since the patient was asymptomatic, apart from previous minor bleeding, and her hematologic assessment seemed to show initial response to iron therapy.
A week later, the girl was admitted again to our department complaining of intense headache and mental confusion, and presenting at general examination with a vast hematoma, undetectable few days before, extending from her left hip to the knee with walking impairment (Figure 1). We ran an emergency MRI and doppler ultrasound in order to exclude compartment syndrome. Ultrasonography showed patency of the deep venous system, while MRI confirmed the presence of a large inflammatory component of high intensity on STIR, hyperintense in both T2 and FAT SAT with slight peripheral edema, compatible with a muscular hematoma (Figure 2). Her blood tests revealed severe anemia (hemoglobin 5 gr/dL), heart rate was accelerated, and so two emergency red blood cell transfusions were executed. Coagulation assessment revealed 0% of factor VIII activity levels, with the aPTT confirmed at 117 seconds. FVIII Inhibitor assay was positive with high titer (8.9 BU). Clinical, anamnestic, and laboratory diagnostic criteria confirmed acquired hemophilia.
Figure 1.
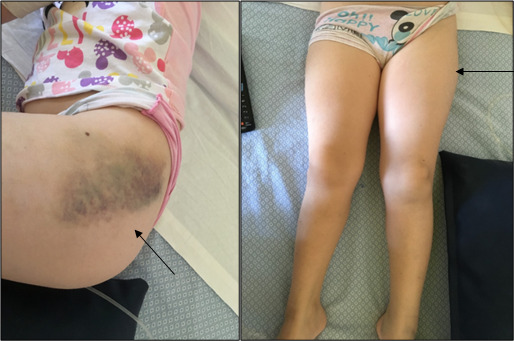
Areas of demarcation showing the left thigh hematoma and the left leg being swollen (arrows)
Figure 2.
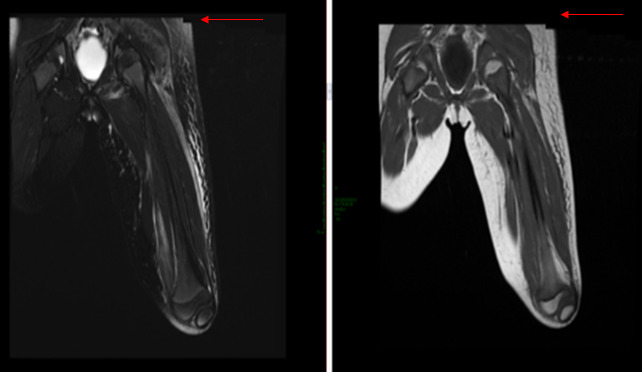
Magnetic resonance imaging showing notable soft tissue edema/inflammation in the left thigh, hyperintense in T2 and FAT SAT with slight peripheral edema, compatible with a muscular hematoma (arrows)
We started intravenous administration of rFVIIa, the only available bypassing agent, at the dose of 90 µg/kg every 2‐3 hours and oral prednisone (1 mg/kg/day). After initial clinical and laboratoristic improvement, with hematoma resolution and increase of hemoglobin level, the time of administration of rFVIIa was gradually prolonged until complete suspension in six days.
During the two weeks of hospitalization, she achieved a complete response, did not develop other spontaneous bleedings and achieved hemostatic control documented by progressively decreasing aPTT count and undetectable inhibitor's titer at a second measure.
She was discharged on oral prednisone, tapered until suspension in eight weeks.
After 6 months of follow‐up, the patient is symptom‐free at the moment, with normal levels of both aPTT and FVIII activity, and the absence of autoantibodies.
3. DISCUSSION
Very little is known about epidemiology, diagnosis, pathogenetic association, clinical course, and treatment of AHA in pediatric population, because of its extremely rare incidence.
The diagnosis is often delayed because of lack of recognition, especially in asymptomatic patients or with minor bleeding episodes. Given the prolonged aPTT, differential diagnosis should include clotting factor deficiencies (factor VIII, IX, XI, von Willebrand), congenital or due to autoantibodies, and presence of lupus anticoagulants. As mentioned mixing test can be of aid in order to distinguish factor deficiency from autoantibodies in base of aPTT correction at time 0 and after 2 hours of incubation. In AHA patients, aPTT is immediately corrected or improved at time 0 and again prolonged after two hours of incubation, revealing strong suspicion of clotting factor inhibitors which are time‐ and temperature‐dependent. Lupus anticoagulant presence is also characterized by prolonged aPTT, but prothrombotic activity, not the bleeding tendency, together with lack of correction both at time 0 and after 2 hours of mixing test, exclude AHA as differential diagnosis. 9 In our case report, the patient presents with recurrent minor bleeding episode, in the absence of personal and family history of bleeding, together with prolonged aPTT, not corrected after 2 hours of incubation in mixing test. The diagnosis should have been more promptly, although the recurrent minor epistaxis was probably a confounding factor and was undervalued due to lack of familiarity with the disease by the attending physicians.
Factor VIII activity and inhibitor titers do not always relate with clinical severity and inhibitor potency, probably due to its nonlinear complex inactivation pattern underestimating true inhibitor potency. 2 , 10 , 11 However, given that high bleed‐related mortality and that initial bleeding tendency were not predictive of later major bleeding, it is suggested that the therapy should be started immediately following diagnosis. 9 Furthermore, in GTH‐AH 01/2010 study and EACH2 registry presence of sever deficiency of FVIII (<1%) or high inhibitor titer (>20 BU) were the most important prognostic factors in order to achieve lower response rates and longer time to response. 3 , 5 Also the presence of anti‐FVIII IgA autoantibodies in ELISAs was predictors of relapse. 12 However, the data utilized are from adult population and on the basis of limited pediatric experience causes are frequently reversible, with possibility of spontaneous resolution. After the late diagnosis of AHA, the girl presented with minor bleeding episodes, that did not correlate with severe FVIII deficiency and high inhibitor titer. The patient was discharged and observed, but as mentioned previously, the subsequent bleedings were major, and did not relate with first clinical onset. Therefore, even though the clinical course of pediatric patients seems less aggressive in the literature, in the absence of major bleeding events, we suggest prompt start of IST with steroids alone, to be associated with hemostatic agents in case of worsening of patient's conditions.
Management of AHA classically involves control of acute bleeding episodes, treatment of underlying illness, and use of immunosuppressive medications with an excellent outcome when diagnosed timely. 13 As for the prevention and control of bleeding, two types of therapy are used in first‐line in adults: replacement therapy with rpFVIII and bypassing agents with ACPP and rFVIIa. Treatment efficiency and dosage modification are assessed by clinical examination, hemoglobin monitoring, and repeated imaging. Laboratory testing is not of aid in directly monitoring the drug's clinical effect, apart from rpFVIII in which factor VIII activity can guide successive administrations. 6 Given the high rates of efficiency in achieving response in short periods of therapy, without significant difference between them, in case of availability of all three of them, the choice is made based on the level of antiporcine FVIII antibody titers. In case of the absence of antiporcine inhibitors, rpFVIII should be first choice, while in patients with high titers of antibody, bypassing agents should be considered. As for the safety profiles, thromboembolic and cardiovascular events were reported in less than 5% of patients treated with rFVIIa and APCC, while rpFVIIIa had no allergic reactions, thrombocytopenia or thrombotic events compared to plasma derived porcine FVIII. 5 , 6 , 7 However, as mentioned before, rpFVIII is not indicated for pediatric and adolescent population at the moment. Other hemostatic approaches include human FVIII replacement and desmopressin were used in AHA patients with low inhibitor titers and higher FVIII activity with significantly lower efficacy, so its use is limited in the lack of availability of first‐line agents at the medical center. 5 Tranexamic acid can be concomitantly used with other hemostatic agents, especially in mucocutaneous bleedings in favorable risk‐benefit assessment, although with caution due to higher risk of thromboembolic events. Urological bleeding represents the only contraindication to tranexamic acid. 2 , 8
When the patient presented with severe anemia, intense headache and confusion in order to achieve hemostasis we decided to start intravenous administration of rFVIIa, the only available hemostatic agent in that moment, at the dosage of 90μg/kg every 3 hours. It was gradually suspended in six days on the basis of resolution of bleeding symptoms confirmed with rise of Hb level and repeated imaging. The use of rFVIIa is approved for all age groups and it has been used successfully in clinical practice for more than two decades following its approval in 1995, showing a consistently favorable safety profile in all licensed indications. 14 rFVIIa is indicated for both the treatment of bleeds and prevention of bleeding in patients with acquired hemophilia undergoing surgery or invasive procedures. 15
Inhibitor eradication is essential in order to improve patient's status and prevent future both clinical (bleeding) and biochemical (low FVIII activity with inhibitor reappearance) relapse. IST achieves remission in about 60%‐80% of patients according to previous studies. 5 , 6 , 7 The possible choices in first‐line are steroids alone or in association with cytostatic agent cyclophosphamide or rituximab. In elderly frail patients with important comorbidities, steroids alone are of aid without increasing the risk of infection. 6 Prognostic factors were mentioned previously, and according to the latest recommendations compared to the previous ones, the combination therapy should be used in patients with extremely low FVIII activity and high inhibitor titer. 8 As for the children with AHA, cyclophosphamide should be avoided if possible, given its cytostatic activity. On the other hand, if steroids alone do not provide satisfactory response, addition of rituximab could improve response rates of inhibitor eradication, as described in a pediatric patient. 16
In our case association to hemostatic treatment, we used oral prednisone with clinical resolution after seven days. After complete remission was achieved, the dosage was tapered until suspension in eight weeks. Remission is defined by factor VIII’s activity higher than 50% and factor VIII inhibitor levels being undetectable. 4
Like for diagnosis and management, follow‐up in pediatric patients is similar to those of adult patients. Outcome in children is more favorable than in adults and inhibitors tend to disappear quickly, probably because most cases are secondary to treatable conditions like infections or antibiotic drug use. It is believed that a regular follow‐up including physical examination, full blood count, coagulation assessment, and FVIII inhibitor assay is necessary monthly for first six months, every 2‐3 months until first year, and every 6 months for at least 1‐2 years. 2 , 10 , 17 On a six‐month follow‐up, our patient remained symptom‐free off immunosuppressant therapy and did not experience other episodes of bleeding; her aPTT is normal, and FVIII inhibitors are absent.
4. CONCLUSION
Although extremely rare in children, acquired hemophilia must be considered in differential diagnosis in front of previously healthy children with bleeding symptoms and an isolated prolonged aPTT. Early detection is vital for improving disease outcome. Our case report showed a good clinical response to the treatment with rFVIIa and prednisone, although IST therapy might have been started earlier even in the presence of minor bleeding. This case adds data to the very limited evidence currently available on treatment for AHA in pediatric patients. More research is warranted, given the severity of the disease.
5. ETHICS STATEMENT
Informed consent was obtained from the patient regarding the report of her clinical scenario data in an anonymous way.
CONFLICT OF INTEREST
The authors stated that they had no interests that might be perceived as posing a conflict or bias.
AUTHOR CONTRIBUTIONS
All authors have made substantial contributions to all of the following: VC: obtained patient consent, collected initial data, and critically revised the manuscript. UM: interpreted data and wrote the first and final draft of the manuscript. DN: involved in the acquisition of data, analysis, and interpretation of data. GG: critically revised the article for important intellectual content and approved the final version for submission.
ACKNOWLEDGMENTS
The authors acknowledge Alessandra Di Lelio and Antonio Nicolucci (CORESEARCH—Center for Outcomes Research and Clinical Epidemiology, Pescara, Italy) for their support in medical writing and editorial assistance. Published with written consent of the patient.
Giuffrida G, Markovic U, Parisi M, Nicolosi D, Calafiore V. Acquired hemophilia in a 7‐year‐old girl successfully treated with recombinant FVIIA and steroids: A case report. Clin Case Rep.2021;9:638–643. 10.1002/ccr3.3588
Funding informationThe editorial assistance was provided by CORESEARCH SRL through a Novo Nordisk S.p.A. unconditional grant. The authors of the publication are fully responsible for the contents and conclusions. Novo Nordisk S.p.A. did not influence and has not been involved in the data interpretation presented in the manuscript
DATA AVAILABILITY STATEMENT
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
REFERENCES
- 1. Franchini M, Zaffanello M, Lippi G. Acquired hemophilia in pediatrics: A systematic review. Pediatr Blood Cancer. 2010;55(4):606‐611. [DOI] [PubMed] [Google Scholar]
- 2. Kruse‐Jarres R, Kempton CL, Baudo F, et al. Acquired hemophilia A: Updated review of evidence and treatment guidance. Am J Hematol. 2017;92(7):695‐705. [DOI] [PubMed] [Google Scholar]
- 3. Tiede A, Klamroth R, Scharf RE, et al. Prognostic factors for remission of and survival in acquired hemophilia A (AHA): Results from the GTH‐AH 01/2010 study. Blood. 2015;125(7):1091‐1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Collins PW. Management of acquired haemophilia A. J Thromb Haemost. 2011;9:226‐235. [DOI] [PubMed] [Google Scholar]
- 5. Baudo F, Collins P, Huth‐Kühne A, et al. Management of bleeding in acquired hemophilia A: Results from the European Acquired Haemophilia (EACH2) registry. Blood. 2012;120(1):39‐46. [DOI] [PubMed] [Google Scholar]
- 6. Kruse‐Jarres R, St‐Louis J, Greist A, et al. Efficacy and safety of OBI‐1, an antihaemophilic factor VIII (recombinant), porcine sequence, in subjects with acquired haemophilia A. Haemophilia. 2015. [DOI] [PubMed] [Google Scholar]
- 7. Tiede A, Giangrande P, Teitel J, et al. Clinical evaluation of bleeds and response to haemostatic treatment in patients with acquired haemophilia: A global expert consensus statement. Haemophilia. 2019;25(6):969‐978. [DOI] [PubMed] [Google Scholar]
- 8. Tiede A, Collins P, Knoebl P, et al. International recommendations on the diagnosis and treatment of acquired hemophilia A. Haematologica. 2020. 105(2020): 10.3324/haematol.2019.230771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Huth‐Kühne A, Baudo F, Collins P, et al. International recommendations on the diagnosis and treatment of patients with Acquired hemophilia a. Haematologica. 2009;94(4):566‐575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Franchini M, Vaglio S, Marano G, et al. Acquired hemophilia A: a review of recent data and new therapeutic options. Hematology. 2017;22(9):514‐520. [DOI] [PubMed] [Google Scholar]
- 11. Cugno M, Gualtierotti R, Tedeschi A, Meroni PL. Autoantibodies to coagulation factors: From pathophysiology to diagnosis and therapy. Autoimmun Rev. 2014;13(1):40‐48. [DOI] [PubMed] [Google Scholar]
- 12. Tiede A, Hofbauer CJ, Werwitzke S, et al. Anti‐factor VIII IgA as a potential marker of poor prognosis in acquired hemophilia A: Results from the GTH‐AH 01/2010 study. Blood. 2016;127(19):2289‐2297. [DOI] [PubMed] [Google Scholar]
- 13. Singh N, Singh Lubana S, Dabrowski L, Acquired Hemophilia A: A Potentially Fatal Bleeding Disorder. Cureus. 2019;12(6): e8744 https://doi/org/10.7759/cureus.8744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Neufeld EJ, Négrier C, Arkhammar P, et al. Safety update on the use of recombinant activated factor VII in approved indications. Blood Rev. 2015;29:S34‐S41. [DOI] [PubMed] [Google Scholar]
- 15. Tiede A, Worster A. Lessons from a systematic literature review of the effectiveness of recombinant factor VIIa in acquired haemophilia. Ann Hematol. 2018;97(10):1889‐1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fletcher M, Crombet O, Morales‐Arias J. Successful treatment of acquired hemophilia a with rituximab and steroids in a 5‐year‐old girl. J Pediatr Hematol Oncol. 2014;36(2):e103‐e104. [DOI] [PubMed] [Google Scholar]
- 17. Franchini M, Mannucci PM. Acquired haemophilia A: A 2013 update. Thromb Haemost. 2013;110(12):1114‐1120. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.