

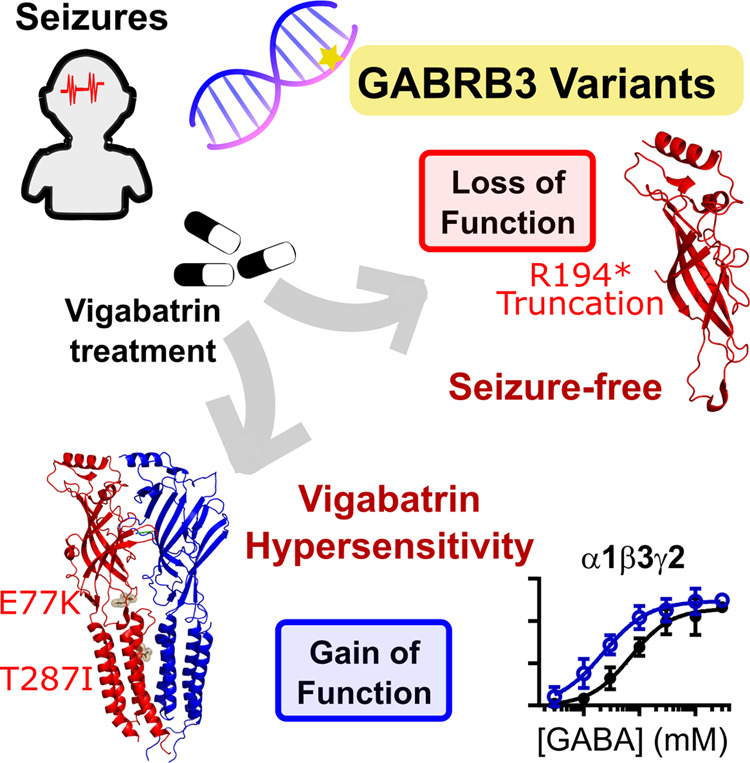
Abstract
Variants in the GABRB3 gene encoding the β3-subunit of the γ-aminobutyric acid type A ( receptor are associated with various developmental and epileptic encephalopathies. Typically, these variants cause a loss-of-function molecular phenotype whereby γ-aminobutyric acid has reduced inhibitory effectiveness leading to seizures. Drugs that potentiate inhibitory GABAergic activity, such as nitrazepam, phenobarbital or vigabatrin, are expected to compensate for this and thereby reduce seizure frequency. However, vigabatrin, a drug that inhibits γ-aminobutyric acid transaminase to increase tonic γ-aminobutyric acid currents, has mixed success in treating seizures in patients with GABRB3 variants: some patients experience seizure cessation, but there is hypersensitivity in some patients associated with hypotonia, sedation and respiratory suppression. A GABRB3 variant that responds well to vigabatrin involves a truncation variant (p.Arg194*) resulting in a clear loss-of-function. We hypothesized that patients with a hypersensitive response to vigabatrin may exhibit a different γ-aminobutyric acid A receptor phenotype. To test this hypothesis, we evaluated the phenotype of de novo variants in GABRB3 (p.Glu77Lys and p.Thr287Ile) associated with patients who are clinically hypersensitive to vigabatrin. We introduced the GABRB3 p.Glu77Lys and p.Thr287Ile variants into a concatenated synaptic and extrasynaptic γ-aminobutyric acid A receptor construct, to resemble the γ-aminobutyric acid A receptor expression by a patient heterozygous for the GABRB3 variant. The mRNA of these constructs was injected into Xenopus oocytes and activation properties of each receptor measured by two-electrode voltage clamp electrophysiology. Results showed an atypical gain-of-function molecular phenotype in the GABRB3 p.Glu77Lys and p.Thr287Ile variants characterized by increased potency of γ-aminobutyric acid A without change to the estimated maximum open channel probability, deactivation kinetics or absolute currents. Modelling of the activation properties of the receptors indicated that either variant caused increased chloride flux in response to low concentrations of γ-aminobutyric acid that mediate tonic currents. We therefore propose that the hypersensitivity reaction to vigabatrin is a result of GABRB3 variants that exacerbate GABAergic tonic currents and caution is required when prescribing vigabatrin. In contrast, drug strategies increasing tonic currents in loss-of-function variants are likely to be a safe and effective therapy. This study demonstrates that functional genomics can explain beneficial and adverse anti-epileptic drug effects, and propose that vigabatrin should be considered in patients with clear loss-of-function GABRB3 variants.
Keywords: developmental and epileptic encephalopathies, GABAA receptor, gain-of-function variants, vigabatrin, benzodiazepines
This study elucidated the mechanism behind the markedly different response to vigabatrin in patients with genetic epilepsies associated with the same gene. Vigabatrin compensated for the truncation of a β3 γ-aminobutyric acid receptor subunit, but likely exacerbates gain-of-function variants with patients experiencing hypersensitivity associated with hypotonia, sedation and respiratory suppression.
Graphical Abstract
Graphical Abstract.
Introduction
Developmental and epileptic encephalopathies (DEEs) are a group of severe childhood epilepsies often associated with co-morbidities including developmental delay (DD), intellectual disability, movement disorders and autistic features (French, 2006). Variants in the GABRB3 gene that code for β3 γ-aminobutyric acid type A (GABAA) receptor subunits are associated with DEE (Johannesen et al., 2016; Møller et al., 2016; Absalom et al., 2019; Hernandez et al., 2019; Maljevic et al., 2019).
GABAA receptors are ligand-gated ion channels that regulate neurotransmission via the inhibitory neurotransmitter, GABA. Nineteen genes code for subunits that make up the various GABAA receptor subtypes. These subunits mix and match to form distinct receptor subtypes (Johannesen et al., 2016; Liao et al., 2019) that express at different cellular and brain regions to regulate neurophysiological responses such as movement, learning and memory.
The β3 subunit of the GABAA receptor is widely expressed across many regions of the brain (Fritschy et al., 1992; Persohn et al., 1992; Wisden and Seeburg, 1992; Pirker et al., 2000) often combining with various α subunits (such as α1) and γ2 subunits to form pentameric synaptic GABAA receptors with a 2α:2β:γ stoichiometry (Fig. 1A). These receptors respond to high levels of synaptically released GABA to mediate a phasic response. In addition, the β3 subunit is found at extrasynaptic sites, combining with either α5 or δ subunits resulting in extrasynaptic receptors that respond to comparatively low levels of GABA to mediate a tonic GABAergic response.
Figure 1.

Structural location of GABAA receptor variants. (A) Pentameric structure from the extracellular side of the membrane of the α1β3γ2 synaptic GABAA receptor (Masiulis et al., 2019). α1 subunits are in blue, β3 in red and γ2 in green. (B) Schematic of a single β3 GABAA receptor showing the location of the β3E77, β3R194 and β3T287 residues (red) in the extracellular and the four transmembrane domains M1–M4 (black). (C) Also shown is the structure of the interface between β3 and α1 subunits from side-on showing the truncation of the β3R194* subunit, the structure displaying the β3E77 residue (black sticks and spheres) in the coupling region connecting the extracellular domain and the transmembrane domains, and the β3T287 residue (black sticks and spheres) at the top of the pore lining M2 region. (D) The sequence alignment for selected subunits is shown below in the loop 2 and M2 regions that contain the β3E77K and β3T287I variants, respectively (red).
As GABAA receptors work to maintain normal brain function, genetic variants in individual genes such as the GABRB3 can lead to neurophysiological dysfunction including seizure disorders such as febrile seizures, generalized epilepsy with febrile seizures+, myoclonic atonic epilepsy, West syndrome, Dravet syndrome and other severe unclassified DEEs (Johannesen et al., 2016; Møller et al., 2016; Absalom et al., 2019; Hernandez et al., 2019; Maljevic et al., 2019). An analysis of the known variants in GABAA receptors proposed that most variants lead to loss-of-function molecular phenotypes through impaired transcription or translation, misfolding and degradation, endoplasmic reticulum retention of truncated or functional receptors or impairments in receptor activation (Hernandez and Macdonald, 2019). However, increased GABAergic activity can also lead to seizure phenotypes, such as in loss-of-function SLC6A1 variants (Mattison et al., 2018), and loss-of-function GABAA receptor molecular phenotypes cannot simply be assumed. Nevertheless, with the prevalence of loss-of-function GABAA receptor variants, drugs that enhance GABAergic neurotransmission could be expected to help reduce seizures in these patients, yet many patients with DEE are either refractory to current treatments or they worsen symptoms or cause a severe adverse reaction.
Vigabatrin is an irreversible GABA transaminase inhibitor used as adjunctive therapy for focal seizures and monotherapy for infantile spasms. It is thought to alleviate seizures by increasing tonic, or persistent, GABAergic inhibitory currents (Wu et al., 2001, 2003). Therefore, it may be anticipated that vigabatrin would be useful in patients with GABRB3 variants that have a reduction in the number or a functional impairment of β3-containing GABAA receptors.
However, recent reports described two patients presenting with GABRB3 variants that responded adversely to vigabatrin. When administered vigabatrin, these patients responded with reduced seizures but suffered severe adverse effects, such as decreased alertness, extreme drowsiness, hypotonia, sedation and respiratory difficulties, that reversed upon treatment cessation. Next-generation sequencing identified the GABRB3 variants p.Glu77Lys and p.Thr287Ile (Papandreou et al., 2016; Kothur et al., 2018). Our hypothesis is that the adverse responses to vigabatrin are a consequence of the molecular phenotype of the individual GABRB3 variants.
In this study, we sought to determine whether the molecular phenotype of the variants could explain the patients hypersensitivity to vigabatrin. Our results demonstrated that patients who responded adversely to vigabatrin had atypical gain-of-function molecular phenotypes when compared to patients that responded well to vigabatrin who had typical loss-of-function phenotype. We propose that functionally analysing variants of patients is essential to avoid adverse effects when prescribing drug treatments.
Materials and methods
This study was performed in accordance with ethical principles for medical research outlined in the Declaration of Helsinki. All genetic studies were performed with informed consent of the patients or their responsible relatives and were approved by the local ethical committees. All procedures using Xenopus laevis frogs and harvesting of oocytes were approved by the animal ethics committee of the University of Sydney (animal ethics committee No. 2016/970) in accordance with the National Health and Medical Research Council of Australia code for the care and use of animals. Frogs were housed in custom-built tanks with a plastic shelter, with three frogs to a tank. Water was reticulated and water quality checked weekly. Frogs were fed twice weekly with Wardley’s reptile sticks (Wardley, USA) and Irradiated Adult Xenopus Diet (Xenopus Express, Florida, USA).
Patients
We report the electrophysiological effects of variants previously reported and the detailed clinical case of two previously reported and one novel GABRB3 gene variant (maternally inherited with the affected relative included) (Papandreou et al., 2016; Kothur et al., 2018). We chose to analyse patients on the following criteria: one variant identified as loss-of-function (novel variant p. Arg194*) associated with a favourable response to vigabatrin; and two variants (p.Glu77Lys and p.Thr287Ile) associated with the adverse reaction of hypersensitivity to vigabatrin (Papandreou et al., 2016; Kothur et al., 2018).
Electrophysiological analyses were performed for two GABRB3 variants and compared to the wild-type (WT) to correlate their functional response to their pharmacoresponse to vigabatrin and nitrazepam where used.
Molecular biology
The human concatenated γ2-β3-α1-β3-α1 receptor construct has previously been described (Absalom et al., 2019; Liao et al., 2019). The γ2-β3-α5-β3-α5 subunit was created by subcloning α5 subunits with flanking linker sequences into the γ2-β3-α1-β3-α1 construct. Briefly, vectors containing DNA encoding the α5 subunits with linker sequences and unique restriction sites were purchased from Genscript (Singapore). The α5 subunits were cut and β3 subunits were ligated with the vector containing the γ2 subunit to create concatenated receptors with linker sequences in the order of γ2-(AGS)5-β3-(AGS)5LGS(AGS)-α5-AGT(AGS)5-β3-(AGS)4ATGAGS-α5.To create β3E77K and β3T287I sets of concatenated receptors, human cDNA for monomeric β3 GABAA receptor subunits were mutated using QuikChange II Site Directed Mutagenesis Kit (Agilent Technologies, Mulgrave, Australia) then subcloned into the γ2-β3-α1-β3-α1 or The γ2-β3-α5-β3-α5 construct using unique restriction sites. Fidelity of all coding sequences were verified by double stranded sequencing. DNA gel electrophoresis ensured incorporation of the five subunits. cRNA was produced from linearized cDNA using the mMessage mMachine T7 Transcription kit (Thermo Fisher, Scoresby, Australia).
Xenopus surgery and oocyte preparation
In brief, a section of ovarian lobe from X. laevis was surgically removed under anaesthesia induced by tricaine, cut and digested with 35 mg of collagenase-A in 15 ml OR2 (in mM: NaCl, 82.5; HEPES, 5; MgCl2, 2 and KCl, 2; pH 7.4) at 18°C for ∼1 h until oocytes were fully detached. Oocytes were then injected with 2 ng of cRNA per cell encoding a concatenated receptor and incubated for 2–4 days at 18°C in ND96 solution (in mM: NaCl, 96; KCl, 2; MgCl2, 1; CaCl2, 1.8; HEPES, 5; pH 7.4) supplemented with 2.5 mM pyruvate, 0.5 mM theophylline and 50 µg/ml gentamicin.
Two-electrode voltage-clamp recording
Cell currents were recorded using the two-electrode voltage-clamp method as previously described (Absalom et al., 2019). Briefly, cells were impaled with microelectrodes filled with 3 M KCl then voltage clamped at −60 mV. Currents were recorded using a GeneClamp 500B (Axon Instrument, Foster City, USA) or OC-725C amplifier Clamp (Warner Instrument Corp., Hamden, USA) and digitized with a Powerlab 8/36 and LabChart version 8.03 (ADInstruments, Sydney, Australia).
For concentration–response curves and estimated open probability (Est POmax), a 3 mM concentration of GABA was applied as a reference three times and peak currents were normalized to the mean current of the last two GABA applications. When estimating POmax, after three consecutive applications of reference 3 mM GABA solution, 10 mM GABA, 1 µM diazepam and 3 µM etomidate was co-applied at γ2-β3-α1-β3-α1 and 10 mM GABA and 10 µM etomidate was co-applied at γ2-β3-α5-β3-α5 receptors. A washout period of 10–12 min was performed between GABA applications to prevent effects from desensitization. Experiments were performed over a minimum of two different batches of oocytes.
Receptor desensitization assay
To measure current decay rates, dead volume was reduced and single concentrations of GABA were applied for 120 s. Traces were fitted to an exponential decay in GraphPad Prism (v8).
Data analysis and statistics
Concentration–response curves were fitted using GraphPad Prism 8 to a monophasic Hill equation of the form:
where Imax is the maximum current, EC50 is the concentration eliciting half-maximum response, [A] is the ligand concentration and nH is the Hill slope. Individual oocytes where a complete concentration–response curve was taken are recorded as a single n. Responses were normalized to the fitted maximum response of individual curves. The EC50 is from the fitting of Hill equations to all data, while the logEC50, Imax and nH values are the mean and error derived from fitting curves to individual experiments.
For nitrazepam concentration–response curves, the per cent modulation was derived by the equation:
and fitted to the Hill equation for parameters of the curves.
The Estimated POmax for individual experiments was derived by the equation:
To determine the decay rates, individual traces from the peak current were fitted to the equation:
where I is the current at time t, I0 is the current at time point 0, Plateau is the current at steady-state desensitization and k is the decay constant.
The modelled response was determined by assuming an equal expression ratio (1:1:1:1) of WT, γ-β3•-α-β3-α, γ-β3-α-β3•-α and γ-β3•-α-β3•-α receptors and given by the equation:
,where the Est Po(wt), nH(wt) and EC50wt are the Est Pomax, Hill co-efficient and EC50 of the wild-type receptor, Est Po(1), nH(1) and EC50(1) are the respective parameters of the γ-β3•-α-β3-α receptor, Est Po(2), nH(2) and EC50(2) are respective parameters of the γ-β3-α-β3•-α receptor Est Po (3), nH(3) and EC50(3) are the respective of the γ-β3•-α-β3•-α receptors.
The change in response was determined by subtracting the WT concentration–response curve. The GABA concentration that vigabatrin induces was increased by 30-fold to compensate for the higher EC50 values determined using concatenated receptors expressed in oocytes compared to free subunits expressed in recombinant mammalian cells (Mortensen et al., 2011).
For statistical analysis, Est POmax values and parameters derived from concentration–response curves were compared with a one-way ANOVA with Tukey’s post hoc test. Significance values of P < 0.05, P < 0.01 and P < 0.001 are shown in the results section. For desensitization, the decay rates were plotted against log10 GABA concentrations and fitted to a linear curve. An F-test was performed to determine if the relationship between the decay constants and GABA concentration for each variant and the WT could be described by a single curve. If not, a Mann–Whitney test was then performed to determine if the data were significantly different between the WT and variant receptors. Power calculations were performed for comparisons between maximum currents, logEC50s and Est Pomax with a one-way ANOVA with an estimated effect size of 0.5, power of 0.85 and a significance level of 0.05 for a minimum number of experiments of 9.
Blinding and randomization
Experiments were not blinded or randomized, but performed on a semi-automated recording apparatus that enabled four experiments to be performed at any one time.
Data availability
Raw data are available on request.
Results
Patient that responds to vigabatrin without hypersensitivity
The vigabatrin-responsive patient chosen represented a typical loss-of-function GABRB3 variants that was prescribed vigabatrin as monotherapy. This variant is a novel case of a maternally-inherited truncation variant p. Arg194* (cases 1 and 2). This variant leads to an aberrant protein sequence of the affected β3 subunit, and the resultant truncated protein cannot express the critical extracellular and pore-forming transmembrane motifs. Hence, these patients would only have functional β3 subunits from the unaffected allele.
Clinical details of patient and affected mother that responds to vigabatrin without hypersensitivity
Cases 1 and 2 (p.Arg194*) are a 29 month old girl and her affected mother. The girl presented with infantile spasms at the age of 7 months. EEG at onset showed hypsarrhythmia, which later evolved into multifocal discharges. Introduction of vigabatrin was tolerated without side effect but was not sufficient to achieve seizure control and prednisolone was added with good effect. In the following period she was seizure free on monotherapy with 130 mg/kg vigabatrin, which was later switched to valproic acid. At last reported follow-up at 21 months of age she had global DD and a right-sided hemiplegia. She could sit unsupported in a kyphotic position and was able to say a few words. The mother of the patient presented with atonic and generalized tonic clonic seizures at the age of two years. She was treated with valproic acid with good effect and has been seizure free since the age of 18 years. She has never tried vigabatrin. She has mild learning difficulties.
Molecular phenotype of vigabatrin-responsive patients without hypersensitivity
The β3R194* truncation variant can be safely predicted to have a loss-of-function molecular phenotype. The β3R194* variant introduces a stop codon within the extracellular domain leading to a truncation of the protein upstream of the transmembrane domain (Fig. 1B and C). This truncated subunit would not form receptor subunits as no transmembrane regions or pore region would be translated.
Vigabatrin-hypersensitive patients
The vigabatrin-hypersensitive patients were chosen for their clinical response to vigabatrin but had exacerbated hypotonia sedation and/or respiratory difficulties that reverted upon cessation of therapy. These included a patient (case 3) with de novo GABRB3 variant p.Glu77Lys (Kothur et al., 2018), that was previously reported as part of 105 patients investigated using a gene panel where the detailed case was not previously reported, and a previously reported de novo GABRB3 variant p.Thr287Ile (Papandreou et al., 2016) with an updated case report (case 4). The p.Glu77Lys and p.Thr287Ile variants were located at the conserved extracellular loop 2 and M2 domains (Fig. 1B–D), respectively that play key roles in channel activation.
Clinical details of vigabatrin-hypersensitive patients
Case 3 (p.Glu77Lys) is a 4-year-old girl who presented with spasms since the age of 5 months in the form of sudden jerking of arms and legs occurring in clusters of 5–10 spasms, with 4–5 clusters per day (Table 1). Her early development prior to spasms was normal, although there was loss of social smile and reduced cooing for 1 month before spasm onset. On examination her head circumference was at 90th centile, weight was 90th centile and length was 75th centile. She was hypotonic with poor neck control, delayed visual maturation and responsiveness. Her EEG showed high amplitude spike and wave and polyspike and wave in the posterior region, and MRI brain showed the prominence of CSF spaces and mild ventriculomegaly. Her CSF studies including neurotransmitters were normal. She was treated with corticosteroid according to UKISS protocol (10 mg prednisolone four times per day) but continued to have spasms up to 60 spasms/day despite treatment with high dose steroids, plus nitrazepam, sodium valproate, zonisamide and pyridoxine. Due to failure to respond, vigabatrin was introduced, but was associated with extreme drowsiness and exacerbation of hypotonia on only 70 mg/kg/day (800 mg BD), and was therefore weaned off over the next few weeks. She was subsequently stabilized and spasms ceased on a combination of nitrazepam and the ketogenic diet. At 18 months of age, she became seizure free except for infrequent myoclonic seizures. Her ongoing EEG showed frequent slowing over the bilateral frontal region but no epileptiform discharges were noted. Following the cessation of spasms, she showed significant improvement in gross motor development. However, she continued to have persistent language delay with limited social interaction suggestive of autism spectrum disorder.
Table 1.
Clinical phenotypes
| Case 1 | Case 2 | Case 3 (Kothur et al., 2018) | Case 4 (Papandreou et al., 2016) | |
|---|---|---|---|---|
| Current age (year), sex | 2, female | 29, female | 4, female | 3.2, male |
| Detection method | Multigene epilepsy panel, Sanger confirmation | Sanger | Multigene epilepsy panel, Sanger confirmation | Multigene epilepsy panel, Sanger confirmation |
| Gene | GABRB3 | GABRB3 | GABRB3 | GABRB3 |
| Nucleotide change | c.580C>T | c.580C>T | c.229G > A | c.860C>T |
| Protein change | p.Arg194* | p.Arg194* | p.Glu77Lys | p.Thr287Ile |
| Type of variant | Stop | Stop | Missense | Missense |
| Inheritance | Maternal | N/A | De novo | De novo |
| Age of onset epilepsy | 7 months | 2 years | 5 months | 3 months |
| Epilepsy syndrome | Infantile spasms | Atonic seizures | Infantile spasms | Unclassified DEE |
| Seizure type | Infantile spasms | Atonic seizures | Infantile spasms | Tonic, myoclonic and focal motor seizures |
| Development | DD | Learning difficulties | Hypotonia, delayed visual maturation | Hypotonia, severe global DD |
| Other features | Right sided hemiplegia, plagiocephaly | Autistic features | Microcephaly, mild dysmorphism | |
| Interictal EEG findings | Hypsarrhythmia | NA | High amplitude Spike and Wave and poly-spike | High amplitude multifocal discharges |
| MRI findings | Porencephalic cyst left ACM area with secondary Wallerian degeneration tractus corticospinalis brainstem left: prenatal ischemic insult left ACM area; almost no myelinization left hemisphere (7 months) | NA | Prominence of CSF spaces and mild ventriculomegaly (5 months) | Normal (14 months) |
| Medication tried | Prednisolone, vigabatrin | Valproic acid | Steroids, vigabatrin, valproate, zonisamide, pyridoxine, ketogenic diet, nitrazepam | Vigabatrin, carbamazepine, topiramate, sodium valproate |
| Reaction to vigabatrin | Seizure reduction, no side effects | NA | Severe drowsiness and hypotonia | Severe hypotonia, sedation, respiratory difficulties |
| Ongoing treatment | Valproic acid | None | Nil | Ketogenic diet, levetiracetam |
| Seizure outcomes | Seizure free by 10 months | Seizure free from 18 years | Infrequent myoclonic seizures, cessation of spasms | Ongoing focal motor, tonic, myoclonic seizures |
| Developmental outcome | Global DD, speech delay | Severe autism and language delay (few words at 5 years) and hyperactivity | Severe global DD |
Did not include GABA peak as this is filtered in routine reporting.
Case 4 (p.Thr287Ile) is a male infant who presented with neonatal hypotonia, then focal tonic seizures from 3 months of age. EEG showed multifocal discharges but not hypsarrhythmia. Introduction of vigabatrin resulted in seizure cessation but caused severe exacerbation of hypotonia, sedation and respiratory difficulties necessitating cessation of vigabatrin. Unfortunately seizures recurred from 5 months of age, and were refractory to carbamazepine, levetiracetam, topiramate, sodium valproate and the ketogenic diet. At last reported follow-up at 3 years and 2 months, he continues to have refractory epilepsy with 10–20 seizures per day of multiple seizure types with behavioural arrest, focal motor, myoclonic jerks and brief tonic seizures. There is global DD with paucity of movements yet anti-gravity movements, some pyramidal signs with brisk reflexes and upgoing plantars, and some vocalizations. There is subtle dysmorphic features and microcephaly (0.4th centile).
Assessing the molecular phenotype of GABRB3 missense variants
Assessing the function of GABAA variants is complicated by the fact that variants are typically dominant and synaptic receptors form with a 2α:2β:1γ stoichiometry. As patients have one normal and one mutated allele, a mixture of receptors with WT and variant subunits will form. For patients carrying a variant in the β3-subunit (β3E77K and β3T287I), two simple receptor populations express with all WT subunits or with two β3 variant subunits. These receptors can easily be expressed in vitro with free γ2 and α1 subunits, but would be predicted to each only comprise of 25% of the receptor population within the patient (Fig. 2A). The two other receptors expressed contain one WT β3 and one variant β3 subunit. For a random distribution of subunits, these receptors will comprise ∼50% of the receptor population and cannot easily be evaluated by the heterologous expression of free subunits (Fig. 2A).
Figure 2.

Design and expression levels of synaptic receptor constructs. (A) Diagram depicting the mixture of receptors formed on the cell surface from a de novo variant. Assuming equivalent expression and random assembly, four different receptors will be expressed at equal ratios. (B) Concatenated DNA constructs used to determine the functional effect of each variant at all expressed receptors. Five subunits were linked in the order γ2-β3-α1-β3-α1 such that cRNA injected into Xenopus oocytes resulted in pentameric receptors of the same orientation as synaptic receptors. Three separate constructs were created for the β3E77K and β3T287I variants where the variant was introduced into the 2nd subunit (red), the 4th subunit (purple) or both the 2nd and 4th subunits (blue) of the construct. This replicated the effect of the variant in vivo, where a WT receptor, two receptors containing a single copy of the variant and a receptor containing two copies of the variant are expressed. (C) Representative trace from two-electrode voltage recordings of a response to 3 mM GABA (black bars) of Xenopus oocytes injected with the WT concatenated receptor construct. (D) Bar graph of maximum currents elicited by 3 mM GABA at receptors (black) and at receptors containing β3E77K or β3T287I variants with one copy of the variant in the 2nd subunit (red), one copy in the 4th subunit (purple) and two copies in the 2nd and 4th subunit (blue). Dots represent individual experiments and bars represent mean ± SD.
We recently developed an approach to determine the function of each of these subunit combinations (Absalom et al., 2019) by engineering all five GABAA receptor subunits into the native receptor arrangement using linkers with unique restriction sites (γ2-β3-α1-β3-α1) (Fig. 2A and B). This approach enables us to generate a library of receptors containing one or two copies of the mutation by a ‘cut and paste’ approach and then express them individually, recapitulating the full molecular phenotype of the variant. We analyse the function of all possible receptors by performing two-electrode voltage-clamp electrophysiology on concatenated WT, one copy or two copies of the β3E77K and β3T287I mutations expressed in Xenopus oocytes.
β3E77K and β3T287I variants do not alter maximal GABA-evoked current amplitudes
Using this approach we developed single and double mutated receptor constructs from the original WT construct (γ2-β3-α1-β3-α1) for each variant. This totalled six unique receptor constructs and mRNA was synthesized for each construct and injected into Xenopus oocytes. All three receptor constructs for each variants expressed robustly with I3mM_GABA-evoked currents in the 0.9–2.2 µA range (Fig. 2C). No significant difference in the maximum currents elicited by 3 mM GABA were observed between all six receptor constructs [one-way ANOVA, F(6,111) = 2.062, P = 0.0633; Table 2]. While not significantly different, the γ2-β3E77K-α1-β3E77K-α1 receptor had approximately a 2-fold reduction in maximum current that may arguably be a result of a reduced maximum open probability, reduced cell surface expression or impaired protein folding. However, as the single subunit β3E77K variant receptors exhibited similar absolute currents to the WT, it is unlikely that such a minor change in a 25% receptor subpopulation has much overall effect in a patient.
Table 2.
Functional parameters of receptors containing one, or two, copies of each variant
| β3 Variants | Construct | Activation properties |
||
|---|---|---|---|---|
| I3mM GABA ± SEM (nA) | EC50 (μM)−logEC50 ± SEM | Est Po | ||
| WTa | γ2-β3-α1-β3-α1 | 2095 ± 126 (13)b | 69 (14) 4.11 ± 0.06 |
0.94 ± 0.03 (10) |
| γ2-β3-α5-β3-α5 | 808 ± 90 (20) | 108 (10) 3.93 ± 0.05 |
0.90 ± 0.01 (10) | |
| β3E77K | γ2-β3E77K-α1-β3-α1 | 1731 ± 241 (21) | 20 (10) 4.62** ± 0.07 |
0.98 ± 0.04 (10) |
| γ2-β3-α1-β3E77K-α1 | 1764 ± 245 (13) | 27 (10) 4.63** ± 0.06 |
1.02 ± 0.04 (10) | |
| γ2-β3E77K-α1-β3E77K-α1 | 946 ± 140 (16) | 21 (10) 4.69** ± 0.06 |
0.94 ± 0.04 (8) | |
| γ2-β3E77K-α5-β3E77K-α5 | 465 ± 66 (20) | 49 (10) 4.31*** ± 0.04 |
0.95 ± 0.05 (10) | |
| β3T287I | γ2-β3T287I-α1-β3-α1 | 1838 ± 310 (21) | 43 (10) 4.36 ± 0.08 |
0.97 ± 0.02 (10) |
| γ2-β3-α1-β3T287I-α1 | 1936 ± 316 (20) | 23 (10) 4.62** ± 0.12 |
0.93 ± 0.01 (11) | |
| γ2-β3T287I-α1-β3T287I-α1 | 2181 ± 223 (14) | 22 (10) 4.65** ± 0.06 |
1.00 ± 0.02 (10) | |
| γ2-β3T287I-α5-β3T287I-α5 | 712 ± 13 (20) | 45 (10) 4.30*** ± 0.08 |
1.01* ± 0.03 (10) | |
WT values except rate constants are also reported in Absalom et al. (2019).
Number of individual oocytes (n) are in parentheses.
P < 0.05.
P < 0.01.
P < 0.001, one-way ANOVA, Dunnett’s t-test c.f. WT.
β3E77K and β3T287I variants increases the potency of GABA
GABAA receptors are activated by GABA in a concentration-dependent manner, where a reduction in GABA potency has commonly been attributed to loss-of-function GABRB3 variants (Janve et al., 2016; Møller et al., 2017). The β3E77 residue is located in loop 2, part of the ‘coupling domain’ incorporating loops two, seven and nine, the pre-M1 helix and the M2 and M3 loop, that alters conformation during channel activation (Fig. 3A) (Kash et al., 2004; Laverty et al., 2019). Therefore, to determine if the GABA-activation properties of synaptic receptors were altered by the β3E77K variant, we estimated the maximal open probability and constructed concentration–response curves of receptors to GABA.
Figure 3.

Gain-of-function molecular phenotype of β3E77K variant. (A) The location of the β3E77K variant in a schematic of the β3 subunit and in the protein structure displaying a close-up of the coupling interface between the extracellular and transmembrane regions. (B) Bar graphs representing individual experiments (dots) and the mean ± SD for the estimated open probability of WT (black) and β3E77K constructs containing one copy of the variant in the 2nd subunit (red), one copy in the 4th subunit (purple) and two copies in the 2nd and 4th subunit (blue). The estimated open probability was calculated by comparing the response at 3 mM GABA to the response at 10 mM GABA, 3 μM etomidate and 1 μM diazepam. (C) Representative traces of currents recorded from Xenopus oocytes injected with WT (black) and β3E77K constructs containing one copy of the variant in the 2nd subunit (red), one copy in the 4th subunit (purple) and two copies in the 2nd and 4th subunit (blue). GABA concentrations were applied as indicated by the black bars to construct the concentration–response curves. Scale bars indicate 200 nA and 30 s, except for the blue bar that represents 50 nA and 30 s. (D) Concentration–response curves of WT and (i) concatenated constructs containing the β3E77K variant in the 2nd (red) or 4th subunit and (ii) the concatenated construct containing a β3E77K variant in the 2nd and 4th subunits. Symbols represent mean ± SD and data were fitted to the Hill equation.
The potency of GABA at the WT γ-β3-α1-β3-α1 receptor construct was 69 μM and the maximum estimated open probability 0.94 (Fig. 3B–D). With observed values in the 0.93-1.0 range, none of the receptors containing the β3E77K variant altered the maximum estimated open probability [one-way ANOVA, F(6,62) = 1.17, P = 0.34; Table 2]. In contrast, there was a significant difference in the GABA potency at the three different receptors [one-way ANOVA, F(6,67) = 6.208 P < 0.0001 logEC50].
Receptors with one copy of the β3E77K variant significantly increased the GABA potency with EC50’s ranging between 20-34 µM for γ-β3E77K-α1-β3-α1 and γ-β3-α1-β3E77K-α1 (P < 0.01 Dunnett’s post hoc test; logEC50 c.f. WT; Table 2). Receptors with two copies of the β3E77K variant also displayed a significant increase in the GABA potency compared to WT, reducing the EC50 to 21 μM (Table 2).
The β3T287 residue is located in the M2 region that lines the channel pore, lying one turn of the helix above a key leucine conserved across ligand-gated ion channels that forms a narrow diameter within the pore (Phulera et al., 2018; Laverty et al., 2019) (Fig. 4A). The M2 helix undergoes a tilt when the receptor transitions to an open state, and as such we estimated the maximal open probability and constructed concentration–response curves of β3T287I concatenated receptors to GABA.
Figure 4.

Gain-of-function molecular phenotype of β3T287I variant. (A) The location of the β3T287I variant in a schematic of the β3 subunit and in the protein structure displaying a close-up of the M2 domains of the β3 (red) and α1 (blue) subunits. (B) Bar graphs representing individual experiments (dots) and the mean ± SD for the estimated open probability of WT (black) and β3T287I constructs containing one copy of the variant in the 2nd subunit (red), one copy in the 4th subunit (purple) and two copies in the 2nd and 4th subunit (blue). The estimated open probability was calculated by comparing the response at 3 mM GABA to the response at 10 mM GABA, 3 μM etomidate and 1 μM diazepam. (C) Representative traces of currents recorded from Xenopus oocytes injected with WT (black) and β3T287I constructs containing one copy of the variant in the 2nd subunit (red), one copy in the 4th subunit (purple) and two copies in the 2nd and 4th subunit (blue). GABA concentrations were applied as indicated by the black bars to construct the concentration–response curves. Scale bars indicate 200 nA and 30 s, except for the purple bar that represents 2 000 nA and 30 s. (D) Concentration–response curves of WT and (i) concatenated constructs containing the β3T287I variant in the 2nd (red) or 4th subunit and (ii) the concatenated construct containing a β3T287I variant in the 2nd and 4th subunits. Symbols represent mean ± SD and data were fitted to the Hill equation.
None of the receptors containing β3T287I variants altered the Est Pomax with values ranging between 0.93 and 1.0 [one-way ANOVA, F(6,62) = 1.17, P = 0.34; Fig. 4B; Table 2]. Receptors with one copy of the variant either significantly increased the GABA potency, reducing the EC50 to 23 µM for γ-β3-α1-β3T287I-α1, P < 0.01 Dunnett’s post hoc test; logEC50 c.f. WT or the increase was not significant (EC50 = 43 µM for γ-β3T287I-α1-β3-α1, P > 0.05; Dunnett’s post hoc test; Fig. 4C and D; Table 2). Receptors with two copies of the β3T287I variants displayed significant increase in the GABA potency compared to WT, reducing the EC50 to 22 μM (P < 0.01, Dunnett’s post hoc test; Fig. 4C and D, Table 2).
Thus, the common molecular phenotype for the vigabatrin-hypersensitive variants that we identified in receptors containing the β3E77K or the β3T287I variant is an increase in the GABA potency, and therefore an increase in the activation of the receptors at lower GABA concentrations. This contrasts from previous findings that variants in β3 subunits typically result in loss-of-function receptors. However, variants may have other characteristics that would lead to loss in inhibitory GABAergic transmission and thus we next investigated desensitization profiles that have been reported to have changed in other GABAA receptor subunit variants (Butler et al., 2018).
β3E77K and β3T287I variants do not alter macroscopic desensitization characteristics of the receptors
Upon activation by GABA, the channel pore of the GABAA receptor opens, then shifts to an equilibrium that includes a desensitized state that is unable to conduct ions. In the case of GABAA receptors, chloride passes from the outside to the inside of the cell to hyperpolarize the cell, acting as a ‘brake’ for neuronal firing. If the receptor is highly desensitized the required chloride is unable to pass through the receptor and neuronal firing continues. Conversely, if the receptor does not desensitize then chloride will continue to permeate for extended times and neuronal firing could be excessively reduced. Variants in GABAA receptor subunits have previously been identified that increase the desensitization properties of the receptor (Shen et al., 2017; Butler et al., 2018) leading to an overall loss-of-function profile.
To quantify the macroscopic desensitization, we applied a single concentration of GABA, fitted the current decay to an exponential curve and determined the rate constant at different concentrations of GABA (Fig. 5A–C). As the potency of receptors with two copies of the variant was altered the most, we performed these experiments only at receptors containing two copies of the variants where any change in desensitization would be the largest. For WT receptors, the rate constants at concentrations of 0.1, 0.3, 1 and 3 mM GABA were 11 ± 2 ms, 22 ± 2 ms, 28 ± 2 ms and 31 ± 2 ms, for the β3E77K were 30 ± 4 ms, 42 ± 3 ms, 56 ± 5 ms and 47 ± 6 ms and for the β3T287I receptors were 20 ± 3 ms, 30 ± 6 ms, 28 ± 6 ms and 36 ± 6 ms (n = 6).
Figure 5.
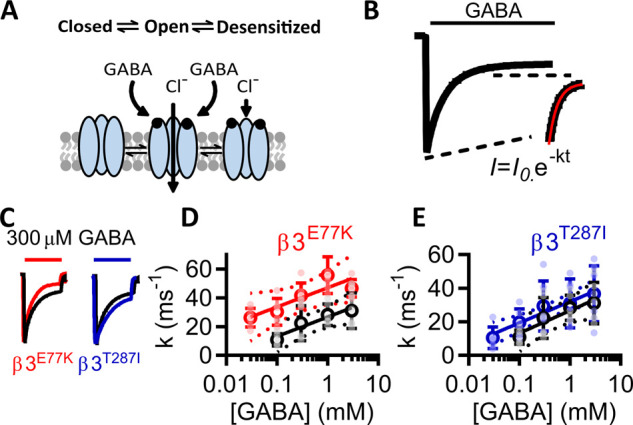
Desensitization properties of β3E77K and β3T287I variants. (A) Schematic displaying desensitization of the receptor, whereby subsequent to GABA binding, GABAA receptors shift between open and closed desensitizing states. (B) To measure the rate of desensitization, the recording apparatus was configured to remove dead volume and GABA was applied for 120 s, and the deactivation rates in the presence of GABA were fitted to an exponential decay function, with the deactivation constant determined for different GABA concentrations against WT and double mutant receptors. (C) Comparison of the WT and double mutant β3E77K and β3T287I receptors. Traces normalized to the peak current are shown for responses to 300 µM GABA at WT (black), β3E77K (red) and β3T287I receptors (blue) receptors, with the scale bar indicating 50 s. (D) and (E) Deactivation constants were plotted against the log of GABA concentrations and fitted to a linear function (95% confidence interval in dotted line) for the WT (black) and (D) β3E77K (red) and (E) β3T287I receptors (blue) receptors.
To compare the rate constants between receptors, we plotted decay constants as a function of the log10 GABA concentration. Empirically, the data fitted well to a linear curve (Fig. 5D and E). An F-test demonstrated that the WT and β3T287I linear functions could be described by the same curve [F(2,45) = 2.34, P = 0.0108], but the WT and β3E77K linear functions could be described by different curves [F-test, F(2,51) = 28.09, P < 0.001]. A further Mann–Whitney test demonstrated that the WT and β3E77K data were not significantly different (P = 0.111). This demonstrates that the variants do not lead to a loss-of-function molecular phenotypes through alterations in the desensitization profiles.
Taken together, these data demonstrate that both the β3E77K and β3T287I variants result in a significant increase in the sensitivity of the receptor to GABA without increased desensitization, consistent with a gain-of-function molecular phenotype.
Response of β3E77K and β3T287I variants to nitrazepam
Benzodiazepines, including nitrazepam, are positive allosteric modulators of GABAA receptors that predominately affect phasic currents, usually through prolonging inhibitory postsynaptic currents (Otis and Mody, 1992). As nitrazepam reduced seizures without adverse effects for the patient with the β3E77K variant, we evaluated the sensitivity of receptors to nitrazepam and compared the potency and efficacy of nitrazepam modulation of GABA (EC10)-elicited currents (1 µM) at WT and variant receptors.
We constructed concentration–response curves to nitrazepam at WT receptors and receptors containing two copies of the β3E77K or β3T287I variant. The WT receptor had an EC50 of 122 μM (−logEC50 = 6.96 ± 0.03) to nitrazepam and a maximum modulation (Emax) of 186 ± 4.6%. (n = 6) (Fig. 6A). Neither the β3E77K and β3T287I variant significantly altered the potency of nitrazepam with EC50’s of 99 and 89 nM, respectively [−logEC50 = 7.04 ± 0.03 and 7.05 ± 0.02, n = 5, one-way ANOVA, F(3,17) = 3.258 P > 0.05]. The efficacy of nitrazepam was significantly reduced to 122% at β3E77K [one-way ANOVA, F(3,16) = 61.9, P < 0.0001] (Emax = 122 ± 3%, n = 5, Dunnett’s post hoc test, P < 0.0001) but not at β3T287I receptors (Emax = 174 ± 4.7%, n = 5, Dunnett’s post hoc test, P > 0.05). These data suggest that nitrazepam is less efficacious at modulating receptors containing a β3E77K variant, likely exerting greater effects in the patient via WT receptor subtypes.
Figure 6.

Vigabatrin and GABAA receptor variants at receptors mediating tonic currents. (A) Concentration–response curves of the positive modulation of EC10 GABA currents by nitrazepam at WT (black), β3E77K (red) and β3T287I (blue) receptors The maximum modulation was significantly reduced at β3E77K receptors compared to WT (P < 0.01, one-way ANOVA with Dunnett’s post hoc test). (B) Bar graph of maximum currents elicited by 3 mM GABA at WT γ2-β3-α5-β3-α5 receptors (black) and receptors containing two copies of the β3E77K (red) or β3T287I (blue). Dots represent individual experiments and bars represent mean ± SD. (C) Bar graphs representing individual experiments (dots) and the mean ± SD for the estimated open probability of WT γ2-β3-α5-β3-α5 (black) β3E77K (red) or β3T287I (blue) receptors. The estimated open probability was calculated by comparing the response of 3 mM GABA was compared to the response to 10 mM GABA and 10 μM etomidate. (D) Concentration–response curves of WT γ2-β3-α5-β3-α5 (black) β3E77K (red) or β3T287I (blue) receptors to GABA. Symbols represent mean ± SD and data were fitted to the Hill equation. (E) Depiction of an inhibitory synapse describing the mechanism of action of vigabatrin. GABA transaminase catalyses the breakdown of GABA into succinic semi-aldehyde, which is inhibited by vigabatrin. This reverses the transport gradient of GAT-1, leading to non-vesicular release of GABA into the synapse to increases the tonic current of the post-synaptic neuron. In two patients with hypersensitivity to vigabatrin, a variant was identified in the β3 subunit of the GABAA receptor that increased the response of the receptor to vigabatrin, while the patients with a truncation of the GABRB3 gene that causes a loss-of-function variant, responded to vigabatrin without hypersensitivity. (F) Modelled change in response compared to the WT of β3E77K (red) and β3T287I receptors (blue) compared to the WT, showing the predicted difference in current levels at different GABA concentrations. The x-axis indicates no change. The shaded region is the expected [GABA]0 concentration elicited by vigabatrin. (G) Structure of the α1β3γ2 GABAA receptor (Masiulis et al., 2019) showing the location of the β3E77 residue within loop 2 (black sticks and spheres) and the β3S76 residue that is homologous to the A52 residue of the GlyR that is associated with hyperekplexia (Plested et al., 2007). The β3T287 residue is shown in a side and overhead view of the M2 region (black sticks and spheres). Also shown is the central β3L284 and β3T288 residue that is also associated with DEE (Hernandez et al., 2017).
β3E77K and β3T287I variants increase GABA potency of extrasynaptic α5-containing receptors
β3 subunits are incorporated into a variety of GABAA receptors, including extrasynaptic α5β3γ receptors that mediate tonic currents. To determine how the β3E77K and β3T287I variants affected these receptors, we created a γ2-β3-α5-β3-α5 WT, γ2-β3E77K-α5-β3E77K-α5 and γ2-β3T287I-α5-β3T287I-α5 constructs. mRNA was synthesized for each construct and injected into Xenopus oocytes. All three receptor constructs expressed robustly with I3mM_GABA-evoked currents in the 0.5–0.8 µA range (Fig. 6B). No significant difference in the maximum currents elicited by 3 mM GABA were observed for the variants [one-way ANOVA, F(2,59) = 3.004, P = 0.0571; Table 2], although similar to the α1 constructs the γ2-β3E77K-α5-β3E77K-α5 receptor had the lowest absolute currents.
The potency of GABA at the WT γ-β3-α5-β3-α5 receptor construct was 108 μM and an Est POmax value of 0.90 (Fig. 6C and D). The Est POmax values were in a range of 0.90–1.01 and no significant difference was observed [one-way ANOVA, F(2,27) = 2.867, P = 0.07; Table 2]. However, both variants significantly reduced the GABA potency by ∼2-fold compared to WT [one-way ANOVA, F(2,27) = 15.37 P < 0.0001 logEC50, P < 0.0001]. Taken together, these data demonstrate that both the β3E77K and β3T287I variants result in a significant increase in the sensitivity of the receptor to GABA without increased desensitization, consistent with a gain-of-function molecular phenotype. This is the same functional change occurring in α5β3γ2 extrasynaptic receptor as is occurring when β3E77K and β3T287I variants are incorporated into the α1β3γ2 synaptic receptor.
Why would gain of function variants lead to vigabatrin hypersensitivity?
Vigabatrin is an irreversible inhibitor of GABA transaminase that blocks intracellular GABA degradation at the presynaptic terminal. The increased intracellular GABA concentrations reverse the direction of GABA transport, leading to non-vesicular release of GABA into the extracellular space (Wu et al., 2001, 2003) (Fig. 6E). In hippocampal cultures, 4 days of vigabatrin exposure caused an average increase in neuronal inhibition of 9 nS, with an implied [GABA]0 of 0.5 μM. This results in large increases in tonic currents of the postsynaptic cell with little to no decrease in phasic inhibitory currents (Wu et al., 2003).
The β3R194* reduces the chloride flux at all GABA concentrations and, due to the reduction in functional receptors, the tonic current induced by vigabatrin likely would be lower than the WT. In contrast, the population of receptors expressed by both the β3E77K and β3T287I increased the chloride flux at lower GABA concentrations. The expression of concatenated GABAA receptors in oocytes leads to a 30-fold higher GABA EC50 than in recombinant mammalian cells, where the EC50 ∼ 2 μM (Mortensen et al., 2011). Therefore, at GABA concentrations in the oocytes equivalent to the [GABA]0 (∼15 μM), we record the highest increases in current elicited at receptors containing the β3E77K and β3T287I variants compared to WT, with ∼20% increase in currents. Vigabatrin, through the reversal of GABA transport in the presynaptic neuron, would therefore likely lead to considerably increased tonic inhibition in these variants compared to the WT receptor. Benzodiazepines, that do not elicit phasic currents from synaptic receptors, are unlikely to lead to the same adverse effect.
Discussion
GABAA receptors are the primary mediators of inhibitory neurotransmission in the developing and adult brain. Variants in GABAA receptors that alter the function of the receptor can modify neuronal excitability, leading to neurodevelopment disorders including epilepsy. Pathogenic variants in GABRB3 are commonly identified in individuals with mild to severe forms of epilepsy (Janve et al., 2016; Møller et al., 2017), signifying the importance of this gene in brain development and function. Located within an imprinting region that spans the 15q11-13 chromosome, variants in the GABRB3 gene are often clustered around central functional domains such as the extracellular GABA binding site, transmembrane helixes supporting or lining the pore (M1 and M2) or loop regions that couple ligand binding to channel gating (Niturad et al., 2017). We, and others, have shown that variants in GABAA receptors often result in loss of receptor function predominantly manifesting as mutations that reduce receptor function by impairing channel gating or as a result of reduced subunit expression due to impaired transcription, impaired translation, misfolding and degradation, truncation, endoplasmic reticulum retention (Janve et al., 2016; Møller et al., 2017; Absalom et al., 2019; Hernandez and Macdonald, 2019; Hernandez et al., 2019).
Both the β3E77 and β3T287 residues are located in key activation regions of the receptor within loop 2 and the pore-lining transmembrane domain (M2) of the subunit, respectively. Loop 2 alters conformation during receptor activation, whereby the M2 region tilts to convert the channel from closed to open state (Laverty et al., 2019; Masiulis et al., 2019). Variants in these structural regions have been identified in GABAA receptor subunits, and other ligand-gated ion channels, that lead to genetic disorders with both loss- and gain-of-function variants being reported for the different ion channels (Fig. 6E) (Plested et al., 2007). The β3T287I variant at the 13′ position faces the adjacent M2 domain of the neighbouring subunit, as opposed to the ion pore itself (Fig. 6E), and with a homologous variant identified in the GABRB1 gene (p.Thr287Ile), albeit with no functional data (Lien et al., 2016), the location appears to be a hotspot for spontaneous mutations. Alternatively, an evaluation of the β2 glutamate to lysine variant homologous to β3E77K showed no change in either the maximum absolute currents of GABA or concentration–response curve (Kash et al., 2004). For variants in these regions, it appears essential to perform functional genomics to ascertain the molecular phenotype of the variant.
The structural location of the variants may also underlie the differences in the sensitivities to nitrazepam. The β3E77 residue within loop 2 couples to the M2-M3 loop to mediate transitions earlier in the activation process than the M2 region where the β3T287 residue is located. The difference between nitrazepam efficacy in these two variants is most likely due to β3E77K altering the equilibrium between pre-activated conformational states of the receptor that benzodiazepines also alter (Gielen et al., 2012). In contrast, β3T287I, within the M2 region, is more likely to alter the equilibrium between conformational states later in the activation process, perhaps even between closed and open states, leading to little to no change in the efficacy of nitrazepam.
In this study, we describe four individuals with epilepsy and DD, three of which were prescribed and had different reactions to the anti-epileptic drug vigabatrin. One patient had a truncation within the GABRB3 gene that introduces a stop codon in the DNA coding sequence upstream of the transmembrane regions (β3R194* truncation). This truncation would abolish the functional expression of the β3 subunit, a classical loss-of-function variant. Interestingly this patient had worsening seizures in response to oxcarbazepine, a sodium channel blocker. Oxcarbazepine inhibits GABA release from the presynaptic terminal initiated via presynaptic NaV1.1 channels. As this patient has a typical loss-of-function GABRB3 variant, the addition of oxcarbazepine would further reduce the GABA-mediated inhibitory currents that are already lower than normal. This adverse response could have been avoided if the genetic information was known prior to treatment. In contrast this patient responded well to vigabatrin with reduced seizure frequency, becoming seizure free at 12 months as vigabatrin enhances GABA-mediated inhibitory currents.
In this patient, vigabatrin would be expected to increase the levels of tonic inhibition through the non-vesicular release of GABA mediated via the reversal of GABA transport induced by the blockade of GABA transaminase. GABAA receptors at the synapse are now persistently activated by the increased GABA concentrations, whereby these receptors will be continually activated leading to excess tonic currents. As a means to alleviate seizures that are ultimately caused by reduced GABAA receptor expression, increased tonic inhibition is already known to be a key compensatory mechanism responsible for the relatively mild phenotype of mice with homozygous α1 GABAA receptor deletions. Apart from a slight handling tremor, these mice have no pronounced phenotype, and analysis of tonic currents at cerebral granular cells identified ∼60% increases in tonic current, identifying a plausible compensatory mechanism (Ortinski et al., 2006). Utilizing vigabatrin to increase the tonic current in patients with GABRB3 truncation variants would thus represent a rational approach to therapy.
The two patients carrying the heterozygous missense GABRB3 variants (p.Glu77Lys and p.Thr287Ile) initially showed a promising reduction in seizures to vigabatrin, but subsequently deteriorated with decreased alertness, extreme drowsiness, hypotonia, sedation and respiratory difficulties. These patients exhibited an atypical gain-of-function molecular phenotype as evidenced by increased GABA sensitivity, no alterations to receptor desensitization characteristics, and little change to the Est open probability or maximum GABA currents. The enhanced sensitivity to GABA at β3E77K and β3T287I containing receptors is likely to underlie the hypersensitive clinical reaction to vigabatrin. The increase in non-vesicular GABA release that is converted to a tonic current will be exacerbated at receptors containing variants that increase the potency of GABA. Furthermore, when the β3E77K or β3T287I variants are incorporated into α5β3γ2 receptors that normally mediate tonic currents, the resulting receptors also display increased GABA sensitivity. Under vigabatrin treatment, any compensation mechanisms appear unable to prevent the hypersensitive reactions in these two patients with GABRB3 variants that result in gain-of-function molecular phenotypes. Despite good efficacy for treating infantile spasms, including our case 1, ∼5% of patients exhibit symptoms of vigabatrin hypersensitivity manifesting as significant drowsiness, hypotonia and respiratory difficulties (Lux et al., 2005), and similarly to what was observed with our patients, the adverse response was reversible (Lux et al., 2005; Hernàndez Vega et al., 2014).
Benzodiazepines, however, did not exacerbate symptoms or lead to severe adverse reactions, despite having a mechanism of action that increases GABAergic inhibitory currents. Our patient that carried the β3E77K (case 3) variant had a significant reduction in seizures over a 6-month period when treated with nitrazepam combined with a ketogenic diet with no overt adverse response. When nitrazepam was analysed, it had reduced efficacy at this variant but maintains normal efficacy at WT. Furthermore, benzodiazepines act differently to vigabatrin. They are positive allosteric modulators of synaptic GABAA receptors that selectively bind to α-γ2 subunit interfaces, mainly prolonging phasic currents rather than increasing tonic currents (Sigel and Ernst, 2018). These results indicate that functional assessment of direct acting GABAA receptor antiepileptics is important in understanding the mechanism for how patients with distinct GABAA variants may respond to certain drugs or drug classes.
Gain-of-function variants in GABRB3 gene have never been previously reported, although a report of gain-of-function variants causing spontaneous GABA ion channel opening and increased GABA potency of recombinant receptors containing the GABRB1 (p.Leu285Arg) variant located within the M2 domain were associated with alcohol dependence in mice (Anstee et al., 2013). Furthermore enhanced GABA potency has been reported for a variant in the GABRA5 variant (p.Val294Leu), three in GABRA3 and one in GABRA1. However unlike the β3E77K and β3T287I variants, these variants apparently either resulted in greater receptor desensitization (Butler et al., 2018; Steudle et al., 2020) or exhibited significantly reduced current amplitudes (Niturad et al., 2017), indicating a possible net loss of GABAergic neurotransmission. The peak concentration of GABA in the synaptic cleft will reach ∼1 mM, a saturating GABA concentration. However, this concentration only lasts for less than a millisecond before the GABA is taken up by presynaptic transporters whereby the binding occupancy of GABAA receptors are not always saturated (Nusser et al., 1997). It would therefore be predicted that in the patients there will be either an increase in the peak amplitude of IPSCs, or a prolonged response of IPSCs, depending on the cell type and synaptic properties in different regions of the brain. Regardless, we would expect a generalized increase in inhibitory GABAergic neurotransmission within these patients.
The idea that increased GABAergic function causes DEE may seem counterintuitive, but extracellular GABA per se can have a pro-convulsive action on the epileptic network through disinhibition and alterations in neuronal development (Pavlov and Walker, 2013). Epilepsy-causing loss-of-function variants in the gene encoding the GABA uptake transporter SLC6A1 result in an accumulation of GABA in the synapses that leads to over activation of synaptic and extrasynaptic GABAA receptors (Mattison et al., 2018). Furthermore, the GABRB3 variants described here (p.Glu77Lys and p.Thr287Ile) are de novo and manifest during embryonic development. During development, GABA is excitatory and depolarizing, controlling cell proliferation, growth, migration and differentiation via extrasynaptic GABAergic signalling. Thus increased GABA potency at extrasynaptic receptors will affect development and the formation of normal neuronal networks. As the GABRB3 gene has been associated with epilepsy, development, motor, learning and memory (Tanaka et al., 2012) and autism (Delahanty et al., 2011) and given its location throughout the brain, the gain-of-function molecular phenotype is likely contributing to the clinical presentations of seizures, head lag, autism and DD.
Treatments for patients with gain-of-function mutations must differ to those with loss-of-function mutations since further enhancing ‘hyperactive’ GABAA receptors could potentially worsen symptoms. Indeed a patient carrying the GABRA5 variant that had a gain-of-function phenotype experienced increased seizure frequency while on phenobarbital, and extreme sedation while on clonazepam, albeit this could also be the natural course of the patient’s epilepsy (Butler et al., 2018). Evidently, caution must be taken when interpreting why a patient with a specific variant responds differently to one drug than another. Even without background genetic effects, the pharmacodynamics will be complex where both variant and WT receptors will be affected by the drug.
In conclusion, this is the first study that describes GABAA receptor variants that exhibit a gain-of-function molecular phenotype and may be a hitherto under-acknowledged cause of DEEs. The gain-of-function molecular phenotype can explain the hypersensitive reaction to vigabatrin seen in our patients and caution is required when prescribing vigabatrin to patients with GABAA receptor variants. We propose that different molecular phenotypes will require different drug treatments targeted at the functional effect of the epilepsy-causing variant, paving the way for a precision medicine approach to the treatment of some forms of DEE.
Acknowledgements
We thank the families and patients that contributed to this study.
Funding
N.L.A. was supported by the Lambert Initiative for Cannabinoid Therapeutics, a philanthropically funded research centre at the University of Sydney. N.L.A., P.K.A., V.W.Y.L. and M.C. were supported by the Australian National Health and Medical Research Council (APP1185122 and APP1124567). Both V.W.Y.L. and P.K.A. were supported by the Australian Research Council of Australia (LP160100560). M.T.B. was supported by the Australian National Health and Medical Research Council (APP1092046). R.S.M. was supported by the Lundbeck Foundation (R324-2019-1083). I.S.M., M.C. and M.T.B. receive grants and salary support from the Lambert Initiative for Cannabinoid Therapeutics. Exome sequencing and analysis support for one of the three patient was provided by Broad Institute of MIT and Harvard Center for Mendelian Genomics funded by the National Institutes of Health grant UM1HG008900. A.M. and M.K. are supported by the National Institute for Health Research Great Ormond Street Hospital Biomedical Research Centre (NIHR GOSH BRC). The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health.
Competing interests
M.C. receives funding from the Lambert Initiative for Cannabinoid Therapeutics, a philanthropically funded research centre at the University of Sydney M.C., N.A., K.K., P.A. received funding from the National Health and Medical Research Council of Australia, Ideas grant 1185122. ISM receives grants and salary support from National Health and Medical Research Council of Australia and from Lambert Initiative for Cannabinoid Therapeutics and has patents to WO2018107216A1, WO2017004674A1 and WO2011038451A1 issued and licensed; and patents to AU2017904438, AU2017904072 and AU2018901971 pending. None of the patents relate to the work presented in this manuscript. I.S.M. and M.T.B. also act as consultants for Kinoxis Therapeutics. A.M. receives grants and salary support from the NIHR GOSH BRC and Rosetrees Trust and received an honorarium from Biocodex. All other authors have nothing to disclose.
Glossary
- DEEs =
developmental and epileptic encephalopathies
- Est Po =
estimated receptor open probability
- GABA =
γ-aminobutyric acid
- WT =
wild-type
Contributor Information
Nathan L Absalom, Faculty of Medicine and Health, School of Pharmacy, Brain and Mind Centre, The University of Sydney, Sydney, New South Wales 2006, Australia.
Vivian W Y Liao, Faculty of Medicine and Health, School of Pharmacy, Brain and Mind Centre, The University of Sydney, Sydney, New South Wales 2006, Australia.
Kavitha Kothur, Kids Neuroscience Centre at The Children’s Hospital at Westmead, Westmead, New South Wales 2145, Australia.
Dinesh C Indurthi, Faculty of Medicine and Health, School of Pharmacy, Brain and Mind Centre, The University of Sydney, Sydney, New South Wales 2006, Australia.
Bruce Bennetts, Department of Molecular Genetics, The Children’s Hospital at Westmead, Westmead, New South Wales 2145, Australia; Discipline of Paediatrics and Adolescent Health, The Children's Hospital at Westmead Clinical School, The University of Sydney, 2145, Australia.
Christopher Troedson, T.Y. Nelson Department of Neurology and Neurosurgery, The Children's Hospital at Westmead, Westmead, New South Wales 2145, Australia.
Shekeeb S Mohammad, Kids Neuroscience Centre at The Children’s Hospital at Westmead, Westmead, New South Wales 2145, Australia.
Sachin Gupta, T.Y. Nelson Department of Neurology and Neurosurgery, The Children's Hospital at Westmead, Westmead, New South Wales 2145, Australia.
Iain S McGregor, Faculty of Science, Lambert Initiative for Cannabinoid Therapeutics, Brain and Mind Centre, The University of Sydney, Sydney, New South Wales 2006, Australia.
Michael T Bowen, Faculty of Science, Lambert Initiative for Cannabinoid Therapeutics, Brain and Mind Centre, The University of Sydney, Sydney, New South Wales 2006, Australia.
Damien Lederer, Institute of Pathology and Genetics, Center for Human Genetics, Gosselies 6041, Belgium.
Sandrine Mary, Institute of Pathology and Genetics, Center for Human Genetics, Gosselies 6041, Belgium.
Liesbeth De Waele, Department of Development and Regeneration, KULeuven, Leuven 3000, Belgium.
Katrien Jansen, Department of Development and Regeneration, KULeuven, Leuven 3000, Belgium.
Deepak Gill, Kids Neuroscience Centre at The Children’s Hospital at Westmead, Westmead, New South Wales 2145, Australia.
Manju A Kurian, Molecular Neurosciences, UCL Great Ormond Street Institute of Child Health, London WC1E 6BT, UK; Department of Neurology, Great Ormond Street Hospital for Children, London WC1N 3JH, UK.
Amy McTague, Molecular Neurosciences, UCL Great Ormond Street Institute of Child Health, London WC1E 6BT, UK; Department of Neurology, Great Ormond Street Hospital for Children, London WC1N 3JH, UK.
Rikke S Møller, Department of Epilepsy Genetics and Personalized Medicine, Danish Epilepsy Centre, Dianalund 4293, Denmark; Department of Regional Health Research, University of Southern Denmark, Odense 5230, Denmark.
Philip K Ahring, Faculty of Medicine and Health, School of Pharmacy, Brain and Mind Centre, The University of Sydney, Sydney, New South Wales 2006, Australia.
Russell C Dale, Kids Neuroscience Centre at The Children’s Hospital at Westmead, Westmead, New South Wales 2145, Australia.
Mary Chebib, Faculty of Medicine and Health, School of Pharmacy, Brain and Mind Centre, The University of Sydney, Sydney, New South Wales 2006, Australia.
References
- Absalom NL, Ahring PK, Liao VW, Balle T, Jiang T, Anderson LL, et al. Functional genomics of epilepsy-associated mutations in the GABAA receptor subunits reveal that one mutation impairs function and two are catastrophic. J Biol Chem 2019; 294: 6157–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anstee QM, Knapp S, Maguire EP, Hosie AM, Thomas P, Mortensen M, et al. Mutations in the Gabrb1 gene promote alcohol consumption through increased tonic inhibition. Nat Commun 2013; 4: 2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler KM, Moody OA, Schuler E, Coryell J, Alexander JJ, Jenkins A, et al. De novo variants in GABRA2 and GABRA5 alter receptor function and contribute to early-onset epilepsy. Brain 2018; 141: 2392–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiron C, Dulac O. The pharmacologic treatment of Dravet syndrome. Epilepsia 2011; 52: 72–5. [DOI] [PubMed] [Google Scholar]
- Delahanty RJ, Kang JQ, Brune CW, Kistner EO, Courchesne E, Cox NJ, et al. Maternal transmission of a rare GABRB3 signal peptide variant is associated with autism. Mol Psychiatry 2011; 16: 86–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French JA. Refractory epilepsy: one size does not fit all. Epilepsy Curr 2006; 6: 177–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritschy JM, Benke D, Mertens S, Oertel WH, Bachi T, Möhler H. Five subtypes of type A gamma-aminobutyric acid receptors identified in neurons by double and triple immunofluorescence staining with subunit-specific antibodies. Proc Natl Acad Sci U S A 1992; 89: 6726–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gataullina S, Bienvenu T, Nabbout R, Huberfeld G, Dulac O. Gene mutations in paediatric epilepsies cause NMDA-pathy, and phasic and tonic GABA-pathy. Dev Med Child Neurol 2019; 61: 891–8. [DOI] [PubMed] [Google Scholar]
- Gielen MC, Lumb MJ, Smart TG. Benzodiazepines modulate GABAA receptors by regulating the preactivation step after GABA binding. J Neurosci 2012; 32: 5707–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez CC, Macdonald RL. A structural look at GABAA receptor mutations linked to epilepsy syndromes. Brain Res 2019; 1714: 234–47. [DOI] [PubMed] [Google Scholar]
- Hernandez CC, XiangWei W, Hu N, Shen D, Shen W, Lagrange AH, et al. Altered inhibitory synapses in de novo GABRA5 and GABRA1 mutations associated with early onset epileptic encephalopathies. Brain 2019; 142: 1938–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez CC, Zhang Y, Hu N, Shen D, Shen W, Liu X, et al. gabaa receptor coupling junction and pore GABRB3 mutations are linked to early-onset epileptic encephalopathy. Sci Rep 2017; 7: 15903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández Vega Y, Kaliakatsos M, U-King-Im JM, Lascelles K, Lim M. Reversible vigabatrin-induced life-threatening encephalopathy. JAMA Neurol 2014; 71: 108–9. [DOI] [PubMed] [Google Scholar]
- Janve VS, Hernandez CC, Verdier KM, Hu N, Macdonald RL. Epileptic encephalopathy de novo GABRB mutations impair γ-aminobutyric acid type A receptor function. Ann Neurol 2016; 79: 806–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannesen K, Marini C, Pfeffer S, Møller RS, Dorn T, Niturad CE, et al. Phenotypic spectrum of GABRA1: from generalized epilepsies to severe epileptic encephalopathies. Neurology 2016; 87: 1140–51. [DOI] [PubMed] [Google Scholar]
- Kash TL, Dizon MJ, Trudell JR, Harrison NL. Charged residues in the beta2 subunit involved in GABAA receptor activation. J Biol Chem 2004; 279: 4887–93. [DOI] [PubMed] [Google Scholar]
- Kothur K, Holman K, Farnsworth E, Ho G, Lorentzos M, Troedson C, et al. Diagnostic yield of targeted massively parallel sequencing in children with epileptic encephalopathy. Seizure 2018; 59: 132–40. [DOI] [PubMed] [Google Scholar]
- Kuenzle C, Steinlin M, Wohlrab G, Boltshauser E, Schmitt B. Adverse effects of vigabatrin in Angelman syndrome. Epilepsia 1998; 39: 1213–5. [DOI] [PubMed] [Google Scholar]
- Laverty D, Desai R, Uchański T, Masiulis S, Stec WJ, Malinauskas T, et al. Cryo-EM structure of the human α1β3γ2 GABAA receptor in a lipid bilayer. Nature 2019; 565: 516–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao VWY, Chua HC, Kowal NM, Chebib M, Balle T, Ahring PK. Concatenated gamma-aminobutyric acid type A receptors revisited: finding order in chaos. J Gen Physiol 2019; 151: 798–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lien E, Våtevik AK, Østern R, Haukanes BI, Houge G. A second patient with a de novo GABRB1 mutation and epileptic encephalopathy. Ann Neurol 2016; 80: 311–2. [DOI] [PubMed] [Google Scholar]
- Lux AL, Edwards SW, Hancock E, Johnson AL, Kennedy CR, Newton RW, et al. The United Kingdom Infantile Spasms Study (UKISS) comparing hormone treatment with vigabatrin on developmental and epilepsy outcomes to age 14 months: a multicentre randomised trial. Lancet Neurol 2005; 4: 712–7. [DOI] [PubMed] [Google Scholar]
- Maljevic S, Møller RS, Reid CA, Pérez-Palma E, Lal D, May P, et al. Spectrum of GABAA receptor variants in epilepsy. Curr Opin Neurol 2019; 32: 183–90. [DOI] [PubMed] [Google Scholar]
- Masiulis S, Desai R, Uchański T, Serna Martin I, Laverty D, Karia D, et al. GABAA receptor signalling mechanisms revealed by structural pharmacology. Nature 2019; 565: 454–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattison KA, Butler KM, Inglis GAS, Dayan O, Boussidan H, Bhambhani V, et al. SLC6A1 variants identified in epilepsy patients reduce γ-aminobutyric acid transport. Epilepsia 2018; 59: e135–41. [DOI] [PubMed] [Google Scholar]
- Møller RS, Larsen LH, Johannesen KM, Talvik I, Talvik T, Vaher U, et al. Gene panel testing in epileptic encephalopathies and familial epilepsies. Mol Syndromol 2016; 7: 210–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Møller RS, Wuttke TV, Helbig I, Marini C, Johannesen KM, Brilstra EH, et al. Mutations in GABRB3: from febrile seizures to epileptic encephalopathies. Neurology 2017; 88: 483–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortensen M, Patel B, Smart TG. GABA potency at GABA(A) receptors found in synaptic and extrasynaptic zones. Front Cell Neurosci 2011; 6: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasrallah FA, Balcar VJ, Rae CD. Activity-dependent gamma-aminobutyric acid release controls brain cortical tissue slice metabolism. J Neurosci Res 2011; 89: 1935–45. [DOI] [PubMed] [Google Scholar]
- Niturad CE, Lev D, Kalscheuer VM, Charzewska A, Schubert J, Lerman-Sagie T, et al. Rare GABRA3 variants are associated with epileptic seizures, encephalopathy and dysmorphic features. Brain 2017; 140: 2879–94. [DOI] [PubMed] [Google Scholar]
- Nusser Z, Cull-Candy S, Farrant M. Differences in synaptic GABA(A) receptor number underlie variation in GABA mini amplitude. Neuron 1997; 19: 697–709. [DOI] [PubMed] [Google Scholar]
- Ortinski PI, Turner JR, Barberis A, Motamedi G, Yasuda RP, Wolfe BB, et al. Deletion of the GABA(A) receptor alpha1 subunit increases tonic GABA(A) receptor current: a role for GABA uptake transporters. J Neurosci 2006; 26: 9323–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otis TS, Mody I. Modulation of decay kinetics and frequency of GABAA receptor-mediated spontaneous inhibitory postsynaptic currents in hippocampal neurons. Neuroscience 1992; 49: 13–32. [DOI] [PubMed] [Google Scholar]
- Papandreou A, McTague A, Trump N, Ambegaonkar G, Ngoh A, Meyer E, et al. GABRB3 mutations: a new and emerging cause of early infantile epileptic encephalopathy. Dev Med Child Neurol 2016; 58: 416–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlov I, Walker MC. Tonic GABA(A) receptor-mediated signalling in temporal lobe epilepsy. Neuropharmacology 2013; 69: 55–61. [DOI] [PubMed] [Google Scholar]
- Persohn E, Malherbe P, Richards JG. Comparative molecular neuroanatomy of cloned GABAA receptor subunits in the rat CNS. J Comp Neurol 1992; 326: 193–216. [DOI] [PubMed] [Google Scholar]
- Phulera S, Zhu H, Yu J, Claxton DP, Yoder N, Yoshioka C, et al. Cryo-EM structure of the benzodiazepine-sensitive α1β1γ2S tri-heteromeric GABAA receptor in complex with GABA. elife 2018; 7: pii:e39383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirker S, Schwarzer C, Wieselthaler A, Sieghart W, Sperk G. GABA(A) receptors: immunocytochemical distribution of 13 subunits in the adult rat brain. Neuroscience 2000; 101: 815–50. [DOI] [PubMed] [Google Scholar]
- Plested AJ, Groot-Kormelink PJ, Colquhoun D, Sivilotti LG. Single-channel study of the spasmodic mutation alpha1A52S in recombinant rat glycine receptors. J Physiol 2007; 581: 51–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen D, Hernandez CC, Shen W, Hu N, Poduri A, Shiedley B, et al. De novo GABRG2 mutations associated with epileptic encephalopathies. Brain 2017; 140: 49–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigel E, Ernst M. The benzodiazepine binding sites of GABAA receptors. Trends Pharmacol Sci 2018; 39: 659–71. [DOI] [PubMed] [Google Scholar]
- Steudle F, Rehman S, Bampali K, Simeone X, Rona Z, Hauser E, et al. A novel de novo variant of GABRA1 causes increased sensitivity for GABA in vitro. Sci Rep 2020; 10: 2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M, DeLorey TM, Delgado-Escueta A, Olsen RW. GABRB3, epilepsy, and neurodevelopment. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, editors. Jasper's basic mechanisms of the epilepsies. 4th edn. Bethesda, MD: National center for biotechnology information (US; ); 2012. [PubMed] [Google Scholar]
- Wirrell EC, Laux L, Donner E, Jette N, Knupp K, Meskis MA, et al. Optimizing the diagnosis and management of Dravet syndrome: recommendations from a North American Consensus Panel. Pediatr Neurol 2017; 68: 18–34. [DOI] [PubMed] [Google Scholar]
- Wisden W, Seeburg PH. GABAA receptor channels: from subunits to functional entities. Curr Opin Neurobiol 1992; 2: 263–9. [DOI] [PubMed] [Google Scholar]
- Wu Y, Wang W, Richerson GB. GABA transaminase inhibition induces spontaneous and enhances depolarization-evoked GABA efflux via reversal of the GABA transporter. J Neurosci 2001; 21: 2630–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Wang W, Richerson GB. Vigabatrin induces tonic inhibition via GABA transporter reversal without increasing vesicular GABA release. J Neurophysiol 2003; 89: 2021–34. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Raw data are available on request.