

Abstract
Purpose: Cystinuria is a genetic disorder with both autosomal recessive and incompletely dominant inheritance. The disorder disrupts cystine and other dibasic amino acid transport in proximal tubules of the kidney, resulting in recurrent kidney stone formation. Currently, there are no consensus guidelines on evaluation and management of this disease. This article represents the consensus of the author panel and will provide clinicians with a stepwise framework for evaluation and clinical management of patients with cystinuria based on evidence in the existing literature.
Materials and Methods: A search of MEDLINE®/PubMed® and Cochrane databases was performed using the following key words: “cystine nephrolithiasis,” “cystinuria,” “penicillamine, cystine,” and “tiopronin, cystine.” In total, as of May 2018, these searches yielded 2335 articles, which were then evaluated for their relevance to the topic of evaluation and management of cystinuria. Evidence was evaluated by the Grading of Recommendations Assessment, Development, and Evaluation (GRADE) system.
Results: Twenty-five articles on the topic of cystinuria or cystine nephrolithiasis were deemed suitable for inclusion in this study. The literature supports a logical evaluation process and step-wise treatment approach beginning with conservative measures: fluid intake and dietary modification. If stone formation recurs, proceed to pharmacotherapeutic options by first alkalinizing the urine and then using cystine-binding thiol drugs.
Conclusions: The proposed clinical pathways provide a framework for efficient evaluation and treatment of patients with cystinuria, which should improve overall outcomes of this rare, but highly recurrent, form of nephrolithiasis.
Keywords: cystinuria, calculi, genetic, nephrolithiasis, urolithiasis
Introduction
Cystinuria was first described in 1810 by William Hyde Wollaston, but erroneously called cystic oxide. Sir Archibald Garrod then correctly described it in the early 1900s. It is a genetic disorder that results from mutations in the genes SLC3A1 and SLC7A9, which code for the two subunits of a transporter and mediate nearly complete reabsorption of cystine and other dibasic amino acids (ornithine, lysine, and arginine) in the renal proximal tubule and intestine. The disorder may be inherited in an autosomal recessive manner or as an autosomal dominant condition with variable expressivity.1
Defects in reabsorption of cystine in the renal proximal tubular cells lead to excessive cystine in urine, prompting cystine precipitation in the distal renal tubule. Affected patients have a high propensity for cystine stone formation due to the relatively low solubility of cystine in urine, which may result in hundreds of stones per year as well as development of chronic kidney disease (CKD) in severe cases.2 Crystallization occurs when concentrations of cystine exceed 250 mg/L of urine.
Apart from formation of cystine stones, there are no other clinical manifestations secondary to the excessive loss of the three amino acids in urine: ornithine, lysine, and arginine. Cystine stones comprise about 1% of all stones in adults and about 6% to 10% of stones in children. The estimated incidence of cystinuria is 1/7000 live births in the United States, although this estimate varies by geographic location and ethnicity.3 The average age at first presentation for cystinuria and kidney stones is 12 to 13 years. Recurrent episodes of nephrolithiasis may increase subsequent development of hypertension, CKD, and end-stage kidney disease.2,4
Because cystinuria is a rare disease, many physicians, including nephrologists and urologists, may have limited experience caring for patients with this disorder. To address this gap, the International Cystinuria Foundation (www.cystinuria.org) recruited a panel of recognized experts who sought to perform a systematic review with the purpose of summarizing the current clinical approaches to management of cystinuria in adults and children.
Materials and Methods
We performed a systematic review in accordance with the PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analyses) statement. A search of MEDLINE®/PubMed® and Cochrane databases was performed using the following key words: “cystine nephrolithiasis,” “cystinuria,” “penicillamine, cystine,” and “tiopronin, cystine.” In total, as of May 2018, these searches yielded 2335 articles, which were then evaluated for their relevance to the topic of evaluation and management of cystinuria according to PRISMA guidelines. A total of 2110 articles were excluded from screening (n = 125 articles were left); 100 potential articles were excluded from eligibility after reviewing abstracts (n = 25 articles were left). The remaining 25 full-text articles were assessed and no further articles removed, leaving 25 eligible articles meeting criteria for inclusion in the recommendations for treatment of patients with cystine nephrolithiasis (Fig. 1). Evidence was evaluated by the Grading of Recommendations Assessment, Development, and Evaluation (GRADE) system. Case reports and case series were excluded. In addition to the above 25 articles, 14 extra basic science articles and noncystinuria review articles in the field of stone prevention were also used as background for this article (giving a total of 39 references).5,6
FIG. 1.
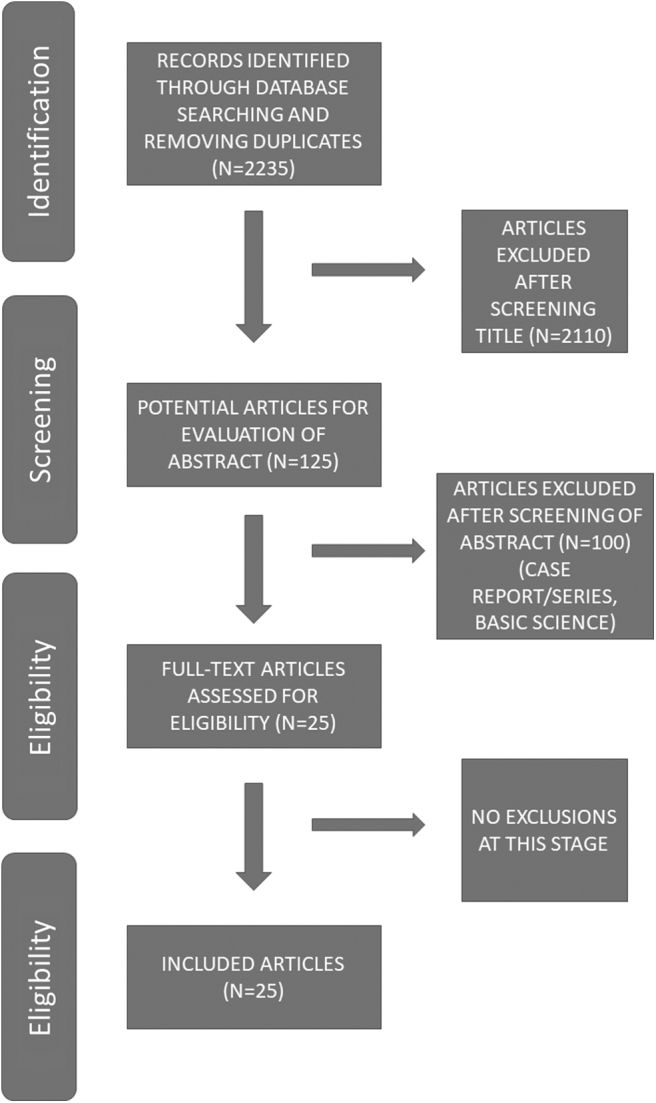
Article search methodology.
Quality of the evidence was assessed using the GRADE system: high = A, moderate = B, low = C, and very low = D. When sufficient evidence was lacking for definitive conclusions, expert opinions were formulated based upon group consensus.
Diagnosis
History
Clinical principle
Clinicians should obtain a detailed medical, dietary, and kidney stone history on a patient newly diagnosed with nephrolithiasis. Cystinuria should be considered in all patients if stone composition has never been confirmed and/or if a urine cystine screen has never been performed. Suspicion should be particularly high in children if a patient presents with one or more large stones or recurrent stones without a clinical diagnosis.
A detailed history includes the following: age of onset of stone disease; frequency of stone passage; stone number and size; stone-related procedure history; verification of cystine stone composition; family history of cystinuria or other stone disease; comorbid conditions that may affect the care or medical therapy of a patient with cystinuria (i.e., CKD); and lifestyle factors that may increase the risk of stone disease (i.e., activities or occupations that may expose the patient to prolonged periods of dehydration or that may prohibit adequate intake of fluids). Dietary factors that increase the risk of stone formation for patients with cystinuria should also be elicited as in other stone formers, including low fluid intake and excessive dietary intake of sodium or nondairy animal protein.7
Diagnosis
A diagnosis is confirmed if any of the following exist:
(a) Stone composition determined by X-ray crystallography or infrared spectroscopy.
(b) Microscopy of spot urine demonstrating hexagonal crystals in the sediment.
(c) Screening with the cyanide–nitroprusside qualitative test may be performed. When urine cystine excretion is greater than 75 mg/L, this spot test will turn the urine purple. Quantitative testing is then recommended, such as (1) 24-hour urine cystine measurement or (2) random spot urinary cystine, ornithine, arginine, and lysine excretion normalized by creatinine excretion.
Clinical recommendation
Because the dietary and pharmacologic treatment of patients with cystinuria is unique among stone formers, confirmation of stone composition either by review of outside medical records or stone analysis is essential as patients with cystinuria may rarely form noncystine stones. If these data are not obtainable, a quantitative 24-hour urinary cystine level should be obtained; patients with heterozygous or homozygous mutations in the two genes known to cause cystinuria (SLC3A1 and SLC7A9) will have a 24-hour cystine excretion greater than 80 mg/day. Microscopy using a spot urine sample demonstrating hexagonal crystals is pathognomonic of cystinuria8; however, quantitative 24-hour urinary testing should then be performed (Standard; Evidence Strength: Expert opinion).
Evaluation
Clinicians should perform a comprehensive metabolic evaluation, including serum studies and 24-hour urine stone risk factors, in all patients with newly diagnosed cystine nephrolithiasis or in recurrent cystine stone formers who have not previously undergone metabolic evaluation. Twenty-four hour urine cystine levels, urinary cystine supersaturation, and capacity can be measured, but the optimal method of monitoring urine chemistry is uncertain (Standard; Evidence Strength: Expert opinion). Serum studies include serum electrolytes, bicarbonate, blood urea nitrogen, calcium, urate, and creatinine. One or two 24-hour urine samples should be collected while consuming a self-selected diet that can serve as a baseline to follow-up on the effects of a dietary or pharmacologic intervention. Pertinent 24-hour stone risk factors include the following: total volume, urine pH, cystine, sodium, citrate, urate, creatinine (validates complete collection), and possibly urine urea nitrogen. Of note, patients with cystinuria may demonstrate urinary risk factors for formation of noncystine stones as well.
Radiology
All patients should have undergone radiologic imaging of kidneys at the time of cystinuria diagnosis to assess the existing stone burden. Cystine stones are not very radiopaque and therefore plain films may fail to detect stones. The imaging at baseline could consist of an ultrasound or computed tomography without contrast. Besides establishing the magnitude of the initial stone burden, this study will serve as a baseline for later imaging (Standard; Evidence Strength: Expert opinion).
Genetic testing
At present, there is no clear indication for genetic testing since the phenotype of cystinuria does not appear to vary based upon the genotype, whether caused by two SLC3A1 and SLC7A9 mutations or one of each. Therapy also does not depend on the underlying genotype. However, as more is learned about the pathobiology associated with various genotypes and as the cost of genetic testing reduces, it is possible that genetic testing will be performed more frequently in the future.
Treatment
Daily fluid intake
Clinicians should recommend a daily fluid intake to maintain a minimum urine output of 3.0 L/day/1.73 m2 spread throughout the day and evening to decrease urine cystine concentration and cystine crystallization. Twenty-four hour urine cystine excretion can be used to modify this recommendation (Standard, Evidence Strength: Grade C). A single retrospective study demonstrated that a daily urine volume of 3.1 L may be associated with lower rates of cystine stone recurrence than a daily urine volume of 2.4 L.9 If 24-hour urine data are available, clinicians may use daily cystine excretion as a guide to modify this recommendation since urine cystine concentrations of >250 to 300 mg/L are associated with higher supersaturation and risk of stone formation.9,10
To decrease urine cystine concentration to <250 mg/L (∼1 mmol/L), oral fluid intake should be 2.5 to 4 L/day for adults, as tolerated, and scaled down appropriately for children to maintain the same relationship per 1.73 m2 body surface area as in adults. Desired urine output is at least >750 mL/24 hours in infants, >1000 mL/24 hours in young children up to 5 years of age, >1500 mL/24 hours up to 10 years of age, >2000 mL/24 hours in older children and adolescents, and >3000 mL/24 hours in older adolescents and adults.
Dietary sodium intake
Clinicians should recommend that dietary sodium intake be restricted to <2500 mg/day (Standard, Evidence Strength: Grade C). Dietary sodium restriction has been shown to decrease urinary cystine excretion. Several studies have shown that cystine excretion increases linearly with urinary sodium excretion. Thus, urinary cystine decreases significantly on a low-sodium diet. These studies have demonstrated a benefit of sodium restriction (dietary <2500 mg/day) for adults and children not treated with cystine-binding thiol drugs (CBTDs), as well as in adult patients using CBTDs.11–14 However, no study has been performed to demonstrate that sodium restriction actually prevents cystine stone formation.
Dietary animal protein intake
Clinicians should recommend low intake of nondairy animal protein for adults and for patients to consider protein intake from nonanimal sources (Standard, Evidence Strength: Grade C). Foods of animal origin are rich in cystine and methionine, which is metabolized to cystine. Studies have shown that a high-protein diet with a large percentage of animal protein (70%) was associated with greater cystine excretion than a lower-protein diet comprising equal amounts of animal and plant protein.15 A potential criticism of this study is that the experimental dietary parameters were extreme—under standard real-world conditions, compliance with these diets may be challenging.
In addition, restriction of animal protein intake is associated with reduced net acid load requiring less urinary acid excretion. Potential benefits include a higher urine pH, greater cystine solubility, and reduction of the dose of alkali needed to achieve an increased urine pH (see below). In adults, these may be more easily accomplished on a diet restricted to <8 ounces of dietary animal protein per day. However, there have been no randomized controlled studies examining the effects of dietary protein restriction or vegetarian diets on prevention of cystine stone formation. In general, excessive dietary protein restriction is not recommended for children due to the requirement of protein intake for optimal growth; however, children should continue to achieve the recommended dietary allowance for protein in their diets.15
Urinary alkalinization therapy
Urinary alkalinization therapy should be prescribed to patients with recurrent cystine stones to achieve a urine pH goal of 7.0 to 7.5 (Standard, Evidence Strength: Grade C). Cystine solubility is pH dependent and optimal urine pH in these patients is 7.0 to 7.5.10,16,17 Higher urine pH also improves the efficacy of CBTDs in vitro.18 Potassium citrate or potassium bicarbonate is generally recommended as a first-line therapy for urinary alkalinization in patients with cystinuria due to its effect of raising the urinary pH without increasing cystine excretion, unlike sodium citrate.19 It should also be noted that overalkalinization (>7.5) has the potential to induce the formation of calcium phosphate stones; therefore, the composition of new stones should be determined in patients taking an alkali rather than assuming it to be cystine. Sodium bicarbonate is sometimes associated with fewer gastrointestinal adverse effects than potassium citrate. Alkalinization with sodium bicarbonate or sodium citrate should be considered a second-line therapy since increased urinary sodium increases urinary cystine excretion.11–14,19 The recommended target urine pH is 7.0 to 7.5.
Pharmacotherapy with CBTDs
Clinicians should offer CBTDs to patients with frequent, recurrent cystine stones who have failed therapy with fluids, diet, and alkalinization. The use of a capacity assay (Litholink, Chicago, IL; www.litholink.com) is recommended given the unreliability of cystine measurements once CBTDs are introduced—with the goal of treatment being positive capacity ideally >50 mg/L (Standard, Evidence Strength: Grade C). The two most commonly used and studied CBTDs are alpha-mercaptopropionyl glycine (tiopronin) and D-penicillamine. This class of drugs, each containing a thiol group, accomplishes disulfide exchange with cystine. The result is the formation of a soluble drug–cystine complex. Both medications reduce free urinary cystine levels and decrease the risk of recurrence of cystine stones in retrospective nonrandomized studies.20–24 However, these drugs do not reduce cystine excretion.19,25 Few studies have directly compared D-penicillamine and tiopronin, but the single largest study demonstrated equal effectiveness of the two drugs. Of note, tiopronin had fewer adverse effects.24 Citing this study, the 2014 American Urological Association (AUA) guideline for the medical management of kidney stones recommended tiopronin as first-line pharmacotherapy for patients with cystinuria for whom alkalinization and high fluid intake were not sufficient to reduce further stone formation.3 Oral vitamin B6 (50 mg daily) should be given to prevent possible B6 (pyridoxine) deficiency that can result from penicillamine chelation of pyridoxine.
In July 2019, FDA approved a new preparation of tiopronin called Thiola EC, for enteric coating. Our experience with this preparation is quite limited at the time of this publication. The tablets reduce pill burden as they weigh 300 mg, instead of the original 100-mg tablet. The enteric coating leads to a more sustained release that may have a clinical benefit, which has not yet been demonstrated. In addition, this preparation does not require careful spacing with respect to meals, unlike the original Thiola preparation, and does prohibit administration of the drug during pregnancy.
Tiopronin dosing for adults usually begins at 600 to 900 mg daily, divided three times a day. In children, the FDA-approved tiopronin dosing should begin at 15 mg/kg/day and is only indicated for pediatric patients weighing 20 kg or more. d-penicillamine is dosed at 500 to 1500 mg/day, divided into 2 or 3 doses. For clinicians who are unable to obtain cystine concentration, a reasonable therapy target goal is 1 mg tiopronin for every 1 mg of cystine excretion per 24 hours.
Many clinicians who treat cystinuria suggest that ideal dosing should reduce cystine concentration to <250 to 350 mg/L. However, optimal dosing is not as straightforward. The measurement of cystine concentration is confounded by the presence of thiol-binding drugs, which render the measurement of cystine levels inaccurate. Specifically, assays cannot distinguish cystine from the soluble cysteine–thiol drug complex. For example, with colorimetric assays, cystine concentration cannot be reliably measured and may detect free cystine or the cysteine–thiol drug complex.7 A high-performance liquid chromatographic method may be able to distinguish the two, but often sample preparation will disrupt the cysteine–thiol drug complex and will impact measurement and reported results.26 All of these parameters may impact the report of cystine excretion and cystine supersaturation. An in vitro-based study suggests that liquid chromatography–tandem mass spectrometry can differentiate cystine from the cysteine–tiopronin complex, although this is not clinically available.27 Hence, it has been difficult to determine the best values to interpret in follow-up 24-hour urine samples in patients with cystinuria currently treated with thiol drugs.
A proprietary solid-phase assay is fortunately available to directly and accurately measure cystine supersaturation in the presence or absence of CBTDs.28 Available through Litholink (www.litholink.com), the assay coins the measurement as capacity. A known amount of solid cystine is added to patient urine and incubated. The amount of solid phase is then measured. In an undersaturated urine sample, the solid phase recovered is less than originally added since some of it dissolves in undersaturated urine. This is reported as a positive capacity. In a saturated urine sample, the solid phase recovered is greater than originally added since supersaturated urine adds mass to the original solid cystine. This is reported as a negative capacity.
In a small study of seven patients, patients served as their own controls and were tested both off CBTDs and then on CBTDs, and capacity improved while on CBTDs.29 A more recent and larger prospective cohort study supports the clinical utility of a capacity tool among a mixed population of CBTDs and conservative treatments. They sought to correlate capacity with stone formation activity. They showed that capacity ranged from −841 to +307 and, more importantly, demonstrated an inverse relationship of capacity with stone activity: stone quiescence vs stone activity, mean ± standard deviation 48 ± 107 vs −38 ± 163 mg/L, P < 0.001, respectively.30 Therefore, a capacity of >50 mg/L has been commonly accepted as a target threshold for clinicians.
Pharmacotherapy with captopril, an angiotensin-converting enzyme (ACE) inhibitor containing a thiol group, has not been reproducibly demonstrated to decrease cystine stone formation in adults. At a maximum dose of 150 mg/day, urinary excretion of cystine does not achieve a sufficient number of millimoles of thiol to have an appreciable effect on cystine solubility. Therefore, captopril is not recommended for the routine treatment of adult patients with cystinuria.31–33 Nevertheless, captopril (0.3 to 1.5 mg/kg/day) may be an appropriate therapeutic option to treat hypertension in cystinuric patients or in children who cannot use oral tiopronin due to its capsule size.
Follow-Up Care
-
(1)
Obtain a single 24-hour urine sample within 1 to 2 months of initiation of dietary modification or pharmacotherapy. After initial follow-up, periodic 24-hour urine studies should be performed to measure changes produced by the prescribed dietary or pharmacologic therapy, with a goal of urinary cystine concentration <250 mg/L and pH >7.0. The frequency of testing may be reduced to semiannual or annual tests when metabolic activity has stabilized (Standard; Evidence Strength: Expert opinion). Several observational studies have demonstrated that among patients with calcium-based stones, urinary compositional changes are associated with a decrease in recurrent stone episodes; we extrapolate these observations to patients with cystinuria.34–37 Moreover, previous studies suggest that close follow-up of patients with cystinuria is essential to encourage compliance with medical therapy.38 Patient compliance, as well as the ongoing evaluation of other noncystine urinary stone risk factors, should also be evaluated as part of this follow-up regimen.
-
(2)
For patients prescribed CBTDs, a comprehensive metabolic panel and spot measurement of the urine protein/creatinine ratio should be obtained at baseline (within 3–6 months of initiating treatment and every 6–12 months yearly thereafter). Serum B6 levels should be monitored annually for penicillamine use (Standard; Evidence Strength: Expert opinion). Side effects of penicillamine can be monitored by routine laboratory testing for anemia, neutropenia, thrombocytopenia, liver function, and urine albumin excretion and for tiopronin, urine albumin excretion.24 Monitoring of complete blood counts and liver function tests are no longer required according to the FDA-approved prescribing information for Thiola EC. The most common histologic finding causing proteinuria is membranous nephropathy. Other potential side effects, which should be screened by a review of systems, include gastrointestinal symptoms (taste disturbance, nausea, emesis, diarrhea, and abdominal pain), dermatological symptoms, hyperelasticity (rash and urticaria), photosensitivity, hair loss, and hypersensitivity reactions. Anecdotal evidence suggests that adverse events can be rechallenged after their resolution at the clinician's discretion. Patients with cystinuria are at risk for CKD, therefore serum creatinine should be measured at least yearly. CBTDs should be discontinued if they are associated with significant proteinuria.
-
(3)
Clinicians should obtain periodic imaging with ultrasonography or low-dose CT to assess stone recurrence, stone burden, and stone growth (Standard; Evidence Strength: Expert opinion). Low-radiation noncontrast CT is nevertheless the gold standard imaging modality for acute urolithiasis and has the highest sensitivity and specificity for stone disease. Renal ultrasound may suffice for screening purposes and avoids exposure to ionizing radiation, although sensitivity is poor for smaller stones. Recently, digital tomosynthesis has been proposed as another option to noncontrast CT with lower cost and decreased ionizing radiation.39 Imaging frequency can be tailored to patients' symptoms or frequency of stone events. For patients who are stable without symptoms or recurrent stone formation, a minimum 12-month imaging interval is recommended. For all stone formers, the AUA guideline recommends a 1-year imaging interval for asymptomatic patients, but frequency can be decreased over time provided there is no stone recurrence.34
-
(4)
Siblings of people with cystinuria should undergo screening for cystinuria (Standard; Evidence Strength: Expert opinion). At present, routine genotyping of people with cystinuria is not recommended. Screening of siblings of a newly identified patient with cystinuria can be performed with a single 24-hour urine collection to measure urine cystine excretion or with a random urine screen for ratio of amino acids to urine creatinine. Finding asymptomatic cystinuria >80 mg/24 hours should lead to performance of a renal ultrasound.
Future directions
Recently, the possibility of development of new drugs for the treatment of cystinuria has been reported. A series of diamines, which mimic cystine and interfere with its crystallization, have shown efficacy in vitro and in a knockout mouse model.40–42 The effect of these molecules is attributed to steric hindrance, where side chains prevent additional molecules of cystine from being added to crystals. No studies of these drugs in humans with cystinuria have been performed as of yet.
Another promising compound is alpha-lipoic acid, which has a significant inhibitory effect on cystine stone formation in vitro and in the knockout mouse model.43 Currently, alpha-lipoic acid is being tested in people with cystinuria in a trial.44
Ultimately, cystinuria could be amenable to correction by genetic therapy. Targeting a functional cystine transporter to the proximal tubule is particularly complex. A recent proposal, not yet tested in an animal model, takes advantage of the ability of certain aminoglycosides to bridge nonsense mutations. These drugs are concentrated in the nephron and could allow a more functional cystine transporter to be translated in the appropriate location.45
Conclusions
Based on the cited literature, clinical principles, and expert recommendations, a stepwise algorithm for evaluation and medical management of patients with cystinuria and urolithiasis is provided (Fig. 2). Briefly, the steps are as follows: a high index of suspicion is necessary in patients who present early in life, along with confirmatory stone analysis and 24-hour urinary analysis. Following a cystine diagnosis, lifestyle and dietary recommendations (maintaining 3 L of urine output/day and moderate dietary sodium and animal protein) are suggested. If conservative measures fail to reduce the urinary cystine concentration to <250 mg/L, the next step is initiating alkali therapy with potassium citrate two or three times per day to achieve a 24-hour urinary pH of 7.0 to 7.5.
FIG. 2.

Recommended clinical algorithm.
If these measures fail to reduce the urinary cystine concentration to <250 mg/L or cystine stones recur, initiation of therapy with CBTDs should be considered. Urine studies should be repeated and thiol doses titrated as necessary to create positive capacity (Litholink) ideally >50 mg/L. Recurrent stone formation may also indicate that higher doses are appropriate. Last, lifelong follow-up with both serial 24-hour urine analysis and routine imaging is necessary to prevent stone recurrence.
Acknowledgments
The authors would like to thank Gayle Sweeney of the International Cystinuria Foundation for her support of this initiative. Additionally, the authors would like to extend their gratitude to their editor, Jessica Bogdany, who spent countless hours managing the editing of the manuscript. The authors sincerely hope the collaborative work will move the needle of care for those suffering from cystinuria.
Abbreviations Used
- AUA
American Urological Association
- CBTDs
cystine-binding thiol drugs
- CKD
chronic kidney disease
- CT
computed tomography
- GRADE
Grading of Recommendations Assessment, Development, and Evaluation
- PRISMA
Preferred Reporting Items for Systematic Reviews and Meta-Analyses
Author Disclosure Statement
B.H.E. is a consultant for Allena Pharmaceuticals and Retrophin and is a patent holder and owner of Dr. Arnie's, Inc. D.S.G. is a consultant and has received honoraria from Retrophin and is a patent holder and owner of Dr. Arnie's, Inc. M.A.B. provides medical advisory to Retrophin. G.C.C. is a consultant for Allena Pharmaceuticals, Shire, RenalGuard Solutions, and Akuous. He has also received grant funding from SHOEBOX Audiometry and personal fees from UpToDate. G.M.P. is a consultant for Retrophin. J.C.L. has received grant funding from Retrophin. A.L.Z. is a consultant for Retrophin. R.L.S. is a consultant and speaker for Boston Scientific; a speaker for Cook Medical and Karl Storz; and a speaker and advisory board member for Retrophin. He is also the Chief Medical Officer for Kalera Medical, Inc., and Pacific Litho San Diego, LLC. All other authors declare that no competing financial interests exist.
Funding Information
No funding was received for this article.
References
- 1. Fattah H, Hambaroush Y, Goldfarb DS. Cystine nephrolithiasis. Transl Androl Urol 2014;3:228–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Prot-Bertoye C, Lebbah S, Daudon M, et al. CKD and its risk factors among patients with cystinuria. Clin J Am Soc Nephrol 2015;10:842–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rogers A, Kalakish S, Desai RA, Assimos DG. Management of cystinuria. Urol Clin North Am 2007;34:347–362 [DOI] [PubMed] [Google Scholar]
- 4. Edvardsson VO, Goldfarb DS, Lieske JC, et al. Hereditary causes of kidney stones and chronic kidney disease. Pediatr Nephrol 2013;28:1923–1942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shekelle PG, Woolf SH, Eccles M, et al. Developing clinical guidelines. West J Med 1999;170:348–351 [PMC free article] [PubMed] [Google Scholar]
- 6. Lavergne V, Nolin TD, Hoffman RS, et al. The EXTRIP (EXtracorporeal TReatments In Poisoning) workgroup: Guideline methodology. Clin Toxicol (Phila) 2012;50:403–413 [DOI] [PubMed] [Google Scholar]
- 7. Goldfarb DS, Coe FL, Asplin JR. Urinary cystine excretion and capacity in patients with cystinuria. Kidney Int 2006;69:1041–1047 [DOI] [PubMed] [Google Scholar]
- 8. Lewis HB The occurrence of cystinuria in healthy young men and women. Ann Intern Med 1932;6:183–192 [Google Scholar]
- 9. Barbey F, Joly D, Rieu P, et al. Medical treatment of cystinuria: Critical reappraisal of long-term results. J Urol 2000;163:1419–1423 [DOI] [PubMed] [Google Scholar]
- 10. Nakagawa Y, Asplin JR, Goldfarb DS, et al. Clinical use of cystine supersaturation measurements. J Urol 2000;164:1481–1485 [PubMed] [Google Scholar]
- 11. Jaeger P, Portmann L, Saunders A, et al. Anticystinuric effects of glutamine and of dietary sodium restriction. N Engl J Med 1986;315:1120–1123 [DOI] [PubMed] [Google Scholar]
- 12. Lindell A, Denneberg T, Edholm E, et al. The effect of sodium intake on cystinuria with and without tiopronin treatment. Nephron 1995;71:407–415 [DOI] [PubMed] [Google Scholar]
- 13. Norman RW, Manette WA. Dietary restriction of sodium as a means of reducing urinary cystine. J Urol 1990;143:1193–1195 [DOI] [PubMed] [Google Scholar]
- 14. Rodriguez LM, Santos F, Malaga S, et al. Effect of a low sodium diet on urinary elimination of cystine in cystinuric children. Nephron 1995;71:416–418 [DOI] [PubMed] [Google Scholar]
- 15. Rodman JS, Blackburn P, Williams JJ, et al. The effect of dietary protein on cystine excretion in patients with cystinuria. Clin Nephrol 1984;22:273–278 [PubMed] [Google Scholar]
- 16. Dent CE, Senior B. Studies on the treatment of cystinuria. Br J Urol 1955;27:317–332 [DOI] [PubMed] [Google Scholar]
- 17. Pak CY, Fuller CJ. Assessment of cystine solubility in urine and of heterogeneous nucleation. J Urol 1983;129:1066–1070 [DOI] [PubMed] [Google Scholar]
- 18. Asplin DM, Asplin JR. The interaction of thiol drugs and urine pH in the treatment of cystinuria. J Urol 2013;189:2147–2151 [DOI] [PubMed] [Google Scholar]
- 19. Fjellstedt E, Denneberg T, Jeppsson JO, et al. A comparison of the effects of potassium citrate and sodium bicarbonate in the alkalinization of urine in homozygous cystinuria. Urol Res 2001;29:295–302 [DOI] [PubMed] [Google Scholar]
- 20. Remien A, Kallistratos G, Burchardt P. Treatment of cystinuria with thiola (alpha-mercaptopropionyl glycine). Eur Urol 1975;1:227–228 [PubMed] [Google Scholar]
- 21. Halperin EC, Thier SO, Rosenberg LE. The use of D-penicillamine in cystinuria: Efficacy and untoward reactions. Yale J Biol Med 1981;54:439–446 [PMC free article] [PubMed] [Google Scholar]
- 22. Lindell A, Denneberg T, Hellgren E, et al. Clinical course and cystine stone formation during tiopronin treatment. Urol Res 1995;23:111–117 [DOI] [PubMed] [Google Scholar]
- 23. Pareek G, Steele TH, Nakada SY. Urological intervention in patients with cystinuria is decreased with medical compliance. J Urol 2005;174:2250–2252, discussion 2252. [DOI] [PubMed] [Google Scholar]
- 24. Pak CY, Fuller C, Sakhaee K, et al. Management of cystine nephrolithiasis with alpha-mercaptopropionylglycine. J Urol 1986;136:1003–1008 [DOI] [PubMed] [Google Scholar]
- 25. Johansen K, Gammelgard PA, Jorgensen FS. Treatment of cystinuria with alpha-mercaptopropionylglycine. Scand J Urol Nephrol 1980;14:189–192 [DOI] [PubMed] [Google Scholar]
- 26. Campanella L, Crescentini G, Avino P. Simultaneous determination of cysteine, cystine and 18 other amino acids in various matrices by high-performance liquid chromatography. J Chromatogr 1999;833:137–145 [DOI] [PubMed] [Google Scholar]
- 27. Giesen CD, Chirackal RS, Brady C, et al. Interference of tiopronin with urine cystine measurement is assay-dependent. Abstract presented at: Kidney Week with American Society of Nephrology, New Orleans, LA, November2, 2017 [Google Scholar]
- 28. Coe FL, Clark C, Parks JH, et al. Solid phase assay of urine cystine supersaturation in the presence of cystine binding drugs. J Urol 2001;16:688–693 [PubMed] [Google Scholar]
- 29. Dolin DJ, Asplin JR, Grasso M, et al. Effect of CBTD on urinary cystine capacity in patients with cystinuria. J Endourol 2005;19:429–432 [DOI] [PubMed] [Google Scholar]
- 30. Friedlander JI, Antonelli JA, Canvasser N, et al. Do urinary cystine parameters predict clinical stone activity? J Urol 2018;199:495–499 [DOI] [PubMed] [Google Scholar]
- 31. Cohen TD, Streem SB, Hall P. Clinical effect of captopril on the formation and growth of cystine calculi. J Urol 1995;154:164–166 [PubMed] [Google Scholar]
- 32. Coulthard M, Richardson J, Fleetwood A. Captopril is not clinically useful in reducing the cystine load in cystinuria or cystinosis. Pediatr Nephrol 1991;5:98. [DOI] [PubMed] [Google Scholar]
- 33. Sloand JA, Izzo JL Jr. Captopril reduces urinary cystine excretion in cystinuria. Arch Intern Med 1987;147:1409–1412 [PubMed] [Google Scholar]
- 34. Pearle MS, Goldfarb DS, Assimos DG, et al. Medical management of kidney stones: AUA guideline. J Urol 2014;192:316–324 [DOI] [PubMed] [Google Scholar]
- 35. Robinson MR, Leitao VA, Haleblian GE, et al. Impact of long-term potassium citrate therapy on urinary profiles and recurrent stone formation. J Urol 2009;181:1145–1150 [DOI] [PubMed] [Google Scholar]
- 36. Pak CY, Heller HJ, Pearle MS, et al. Prevention of stone formation and bone loss in absorptive hypercalciuria by combined dietary and pharmacological interventions. J Urol 2003;169:465–469 [DOI] [PubMed] [Google Scholar]
- 37. Borghi L, Schianchi T, Meschi T, et al. Comparison of two diets for the prevention of recurrent stones in idiopathic hypercalciuria. N Engl J Med 2002;346:77–84 [DOI] [PubMed] [Google Scholar]
- 38. Pietrow PK, Auge BK, Weizer AZ, et al. Durability of the medical management of cystinuria. J Urol 2003;169:68–70 [DOI] [PubMed] [Google Scholar]
- 39. Cabrera FJ, Kaplan AG, Youssef RF, et al. Digital tomosynthesis: A viable alternative to noncontrast computed tomography for the follow-up of nephrolithiasis? J Endourol 2016;30:366–370 [DOI] [PubMed] [Google Scholar]
- 40. Rimer JD, An Z, Zhu Z, et al. Crystal growth inhibitors for the prevention of L-cystine kidney stones through molecular design. Science 2010;330:337–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yang Y, Albanyan H, Lee S, et al. Design, synthesis, and evaluation of l-cystine diamides as l-cystine crystallization inhibitors for cystinuria. Bioorg Med Chem Lett 2018;28:1303–1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hu L, Yang Y, Aloysius H, et al. l-Cystine diamides as l-cystine crystallization inhibitors for cystinuria. J Med Chem 2016;59:7293–7298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zee T, Bose N, Zee J, et al. α-Lipoic acid treatment prevents cystine urolithiasis in a mouse model of cystinuria. Nat Med 2017;23:288–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. ClinicalTrials.gov. Lipoic acid supplement for cystine stone (ALA). Last updated April 17, 2019. https://clinicaltrials.gov/ct2/show/NCT02910531 (accessed January13, 2020)
- 45. Tokhmafshan F, Dickinson K, Akpa MM, et al. A no-nonsense approach to hereditary kidney disease. Pediatr Nephrol 2019. DOI: 10.1007/s00467-019-04394-5 [DOI] [PubMed] [Google Scholar]