

Abstract
The material-phase diffusion coefficient (Dm) and material/air partition coefficient (Kma) are the key parameters controlling the emissions of semivolatile organic compounds (SVOCs) from source materials. In indoor environments, air temperature is subject to change and can significantly affect the emission rates of SVOCs from building materials and consumer products. In this study, the emissions of organophosphate ester flame retardants (OPEFRs) from customized polyisocyanurate foam materials were measured in 44-mL microchambers at 23, 35, and 55 °C. The values of Dm and Kma at different temperatures were determined. The results showed that the increase of temperature can significantly enhance the emissions of OPEFRs from the foam materials, and the emissions of OPEFRs were found to transfer from SVOC-type to volatile organic compound (VOC)-type with the increase of temperature. A correlation for OPEFRs between the steady-state emission rate and temperature and correlations between Dm, Kma, and temperature were obtained. The research results shed light on the effect of temperature on the mechanisms governing emissions of SVOCs.
Keywords: Organophosphate ester flame retardants (OPEFRs), Semivolatile organic compounds (SVOCs), Chamber testing, Emission mechanism, Temperature influence
1. Introduction
Organophosphate flame retardants (OPFRs) have been increasingly produced and are extensively used in a wide range of products since the phase-out of brominated flame retardants (Greaves and Letcher, 2017; Van der Veen and de Boer, 2012). OPFRs can be found in building materials and consumer products such as polyvinyl chloride (PVC) flooring, plastics, textile coatings, furniture, and electrical and electronic products (Van der Veen and de Boer, 2012; Wei et al., 2015; Wensing et al., 2005; Stapleton et al., 2009) and belong to the family of semivolatile organic compounds (SVOCs), the vapor pressures of which range from 10−14 to 10−4 atm at 25 °C (Weschler and Nazaroff, 2008). They are usually used as additives that are not chemically bonded to the materials. As a result, slow emissions are likely to occur from the original source to the environment at room temperature (Wei et al., 2015; Stapleton et al., 2009; Hartmann et al., 2004). OPFRs are ubiquitous in indoor environments and have been found in indoor air (Hartmann et al., 2004; Bradman et al., 2014; Staaf and Ostman, 2005), house dust (Bradman et al., 2014; Van den Eede et al., 2011; Bergh et al., 2011; Brommer et al., 2012), etc. Humans can be exposed to OPFRs via inhalation, dermal absorption, and ingestion (Wei et al., 2015; US ATSDR, 2012). Exposure to OPFRs can result in various health effects, such as damage to the reproductive system, adverse neurodevelopmental effects, endocrine disruption, and carcinogenicity (Van der Veen and de Boer, 2012; Bradman et al., 2014; US ATSDR, 2012).
Understanding the emission mechanisms of SVOCs from various sources is important to characterize their fate and transport in the indoor environment and further develop strategies to limit human exposure to these compounds. However, although an increasing number of studies are available on the emissions of SVOCs, such as phthalates (Xu and Little, 2006; Xu et al., 2012; Liu et al., 2013; Liang and Xu, 2014a; 2014b; Cao et al., 2016; Mao et al., 2018; Eichler et al., 2018), relatively few studies have targeted measurements of OPFRs emission parameters (Wei et al., 2015; Kemmlein et al., 2003; Ni et al., 2007; Liang et al., 2018a). Collectively, these studies have revealed that the emissions of SVOCs from building materials and consumer products are controlled by material-phase concentration (C0, μg/m3), material-phase diffusion coefficient (Dm, m2/h), material/air partition coefficient (Kma, dimensionless), mass transfer coefficients (hm, m/h), and surface/air partition coefficient (Ksa, m).
Temperature is likely to significantly influence the emission of SVOCs in indoor environments. Indoor air temperature is subject to increase due to various factors, such as outdoor temperature increase and failures of heating, ventilation, and air-conditioning (HVAC) systems. These factors can greatly enhance the emission rates of SVOCs from indoor sources. In addition to indoor environments, people also spend a considerable amount of time in automobiles, where air temperature can go up to 76 °C (McLaren et al., 2005). These high temperatures can result in extremely high emission rates from materials in automobiles, such as cable insulation, car seats, and interior trim and potential high exposure levels. Recent studies (Besis et al., 2017; Christia et al., 2018) have found high levels of brominated flame retardants (BFRs) and OPFRs in interior car dust. Hoffman et al. (2017) suggested that temperature fluctuations in automobiles (e.g., from sitting in the sun) can result in seasonal differences of exposure to OPFRs used in automobiles.
Previous studies have been focused on the influence of temperature on the emission of phthalates and flame retardants in indoor environments (Kemmlein et al., 2003; Ni et al., 2007; Clausen et al., 2012; Fujii et al., 2003; Ekelund et al., 2008, 2010; Liang and Xu, 2014a). These studies showed that the emissions of phthalates from PVC products were considerably enhanced as a result of temperature increase. In studying the emission of phthalates and phthalate alternatives from PVC flooring and mattress covers, Liang and Xu (2014a) proposed an equation describing the relationship between the ratio of material-phase concentration (C0) to gas-phase concentration in equilibrium with the material (y0) and temperature. However, whether this relationship can be applied to other types of SVOCs remains unclear. Kemmlein et al. (2003) studied the emissions of organophosphate and brominated flame retardants from consumer products and building materials in emission test chambers and cells. They found that the emission rate of OPFRs increased significantly with the increase of temperature. However, the mechanisms of temperature influence on OPFRs remain unclear in this study. Ni et al. (2007) measured the emissions of OPFRs using a passive flux sampler. Their measurement results showed that the emission rate of tris(1-chloro-2-propyl) phosphate (TCIPP) from wallpaper samples increased 4.5-fold when the temperature increased from 25 to 60 °C. A linear relationship was found between the TCIPP emission rate and the reciprocal of temperature. Although this relationship is very useful to predict emission rates for OPFR emission tests, it lacks a rational basis and cannot represent the fundamental mechanism of temperature influence on OPFR emissions. To date, no systematic investigations have been conducted to characterize the effect of temperature on the mechanisms governing emissions of OPFRs, which is a prerequisite to reduce the uncertainty and increase the applicability of existing emission models and data.
The aim of this study was to investigate the influence of temperature on the emission of organophosphate ester flame retardants (OPEFRs), a subclass of OPFRs from building materials. The specific objectives included (1) to conduct a series of controlled tests in stainless steel microchambers to characterize OPEFR emissions from polyisocyanurate (PIR) foam at three different temperatures; (2) to determine Dm and Kma for OPEFRs at different temperatures; (3) to investigate the nature of OPEFRs emission rates, Dm, and Kma as a function of temperature and provide correlation relationships. We chose the PIR foam for this research because PIR foams are extensively used as rigid thermal insulation.
2. Methods and materials
2.1. Chemicals
Certified tris(2-chloroethyl) phosphate (TCEP, CAS# 115-96-8), TCIPP (CAS# 13674-84-5), and tris(1,3-dichloro-2-propyl) phosphate (TDCIPP, CAS# 13674-87-8) calibration standards were purchased from AccuStandard Inc. (New Haven, CT, USA). An isotopically-labeled compound, tributyl phosphate-D27 (99.5% purity, Cambridge Isotope Laboratories, Inc., Andover, MA) was used as the internal standard on the gas chromatography/mass spectrometry (GC/MS) system. Triphenyl phosphate-D15 (98% purity, Sigma Aldrich, St. Louis, MO, USA) was used as the extraction recovery check standard (RCS). Chromatography-grade methylene chloride (Burdick and Jackson, Muskegon, MI, USA) and ethyl acetate (OmniSoly, Billerica, MA, USA) were used as solvents in extraction and cleaning without further purification. The solvents were regularly analyzed to monitor potential contamination with OPEFRs.
2.2. Materials
PIR foam materials made by ICL-IP America Inc. (Gallipolis Ferry, WV, USA) were used as the OPEFR emission source materials. As specified by the manufacturer, the TCEP, TCIPP, and TDCIPP contents in the foam material are 2%, 4%, and 14% by weight, respectively. Extraction measurements were also conducted to determine the material phase concentrations of TCEP, TCIPP, and TDCIPP prior to the chamber tests. The foam materials were wrapped in aluminum foil and stored at room temperature before use.
2.3. Microchamber emission test
Two pieces of PIR foam (4.5 cm diameter and 3.97 mm thickness) were placed at the bottom and top of a 44 mL Silicosteel®-coated stainless steel microchamber (Model μ-CTE, Markes International, Llantrisant, UK). Prior to the test, the empty microchamber was connected to dry clean air and background samples were collected to determine any evidence of contamination. Duplicate tests were conducted under each test condition using two micro chambers simultaneously with dry clean air at approximate 200 mL/min for 500 – 600 h. Polyurethane foam (PUF) (pre-cleaned and certified, Supelco, St. Louis, MO, USA) samples were collected at the exhaust of each chamber to monitor the gas-phase concentrations of emitted OPEFRs. The tests were conducted at controlled temperatures of 23 ± 1, 35 ± 1, and 55 ± 1 °C separately. For each emission test, newly cut foam materials from the original PIR foam were used. A more detailed configuration of the emission tests can be found in SI and a previous study (Liang et al., 2018a).
2.4. Sample extraction and analysis
After PUF sample collection, each PUF cartridge was capped in a glass holder, wrapped in aluminum foil, placed in a sealable plastic bag, and refrigerated at 4 °C until analysis. PUF samples and PIR foam extraction and analysis followed the methods described in Liang et al. (2018a).
2.5. Determination of Dm and Kma from microchamber emission test
A schematic representation of the foam placed in the microchamber is shown in Fig. 1. With reference to Fig. 1, assuming the microchamber air is well mixed and a boundary layer exists adjacent to the sorption surfaces, the gas-phase OPEFR accumulation in the microchamber obeys the following mass balance:
| (1) |
where V (m3) is the volume of the microchamber, Q (m3/h) is the ventilation rate, y (μg/m3) is the gas-phase OPEFR concentration in the chamber, yin (μg/m3) is the inlet gas-phase OPEFR concentration, hm and hs (m/h) are the mass transfer coefficients across the material surface and chamber surface, respectively, A0 (m2) is the total emission area of the top and bottom foam in the chamber, As (m2) is the area of the chamber wall, y0 (μg/m3) is the gas-phase concentration in the layer above the foam material, and ys (μg/m3) is the gas-phase OPEFR concentration in the boundary layer adjacent to the chamber surface. The accumulation rate of surface-phase OPEFR concentration on the chamber wall is
| (2) |
where qs (μg/m2) is the surface-phase OPEFR concentration on the chamber wall. To simplify the equation, we assume a linear equilibrium relationship between the surface-phase concentration and the gas-phase concentration in the boundary layer adjacent to the chamber surface (Xu et al., 2012), or
| (3) |
where Ksa (m) is the chamber surface/air partition coefficient.
Fig. 1.
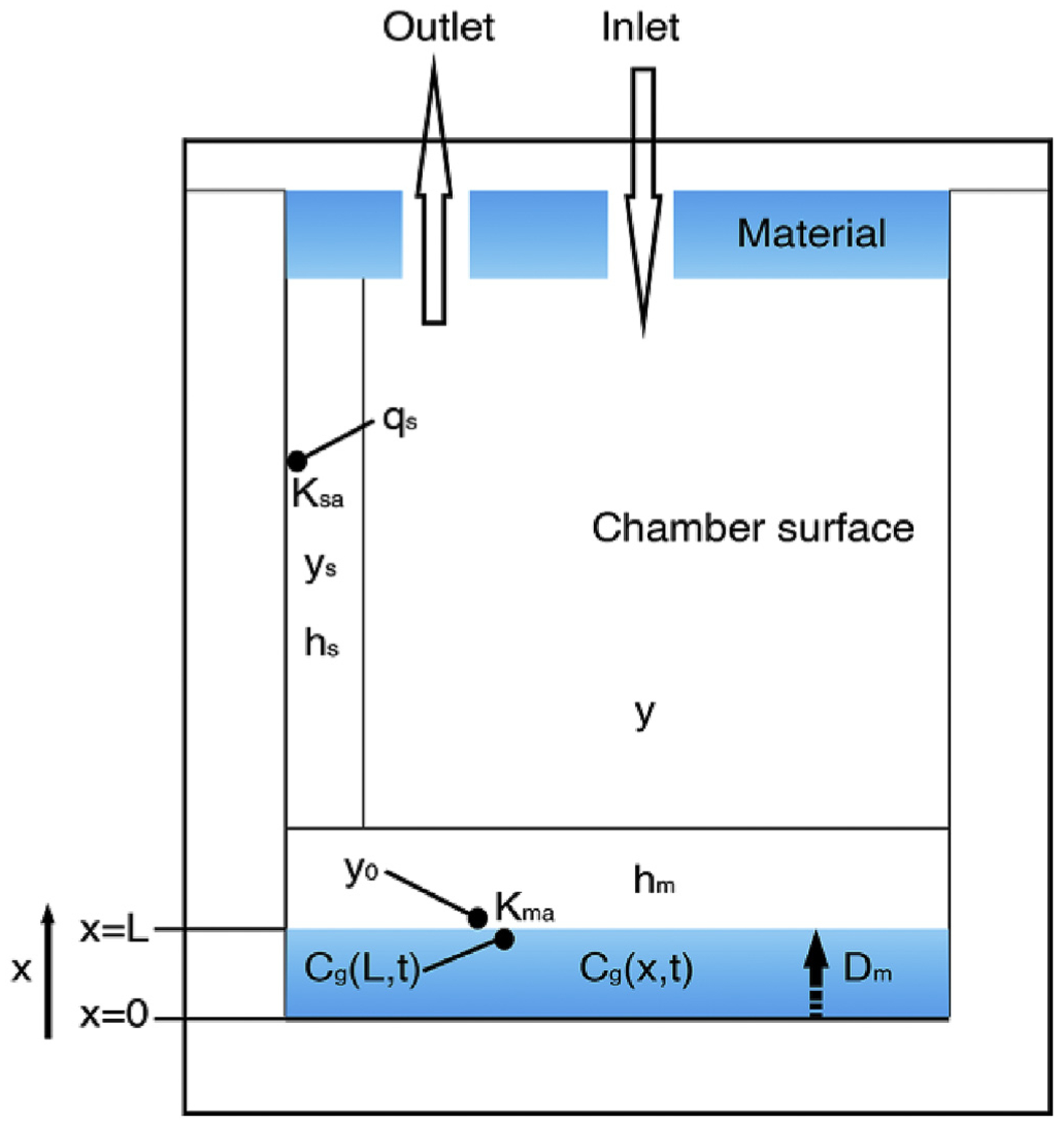
Schematic of the emissions of OPEFRs from the PIR foam in the microchamber.
The governing equation for the transient diffusion through the foam is
| (4) |
where C (x,t) (μg/m3) is the OPEFR concentration in the foam, t (h) is the time, x (m) is the distance from the bottom to the top of the material surface, and Dm is assumed to be independent of the concentration in the foam. The initial condition assumes that the OPEFRs are uniformly distributed throughout the foam, or
| (5) |
where L (m) is the thickness of the foam and C0 (μg/m3) is the initial OPEFR concentration. Given that the lower surface of the foam is attached to the top and bottom of the microchamber, the lower surface boundary condition is
| (6) |
The upper surface of the foam is exposed to the chamber air; therefore, the upper boundary condition can be described as
| (7) |
Equilibrium is assumed to exist between the OPEFR in the upper layer of the foam and the air immediately adjacent to the surface, or
| (8) |
Previous study (Xu and Little, 2006) has shown that Kma can be considered constant if C0 remains effectively constant; therefore, in this study, Kma is also assumed to be independent of the OPEFR concentration.
Equations (1) – (8)characterize the emission of OPEFRs from the foam to the chamber air and sorption on the chamber surface. In this study, the gas-phase OPEFR concentrations (y) in the microchamber over time were measured and fitted to obtain Dm and Kma simultaneously. The non-linear regression fitting with equations (1) – (8)was performed using MATLAB (Mathworks, Inc. Natick, MA).
2.6. Quality assurance (QA) and quality control (QC)
A quality assurance project plan (QAPP) that describes the project objectives, scientific approaches, measurement procedures, and QA/QC activities was prepared, reviewed, and approved by the EPA QA officer prior to taking any measurements in this study. The calibration of the GC/MS system was completed and verified using an internal audit program. Extraction method blank and field blank samples were prepared and analyzed for each test to check contamination. Acceptance criteria for PUF sample and foam material extraction and analysis were that the recovery check standards had to be within 100 ± 25% recovery, and precision of the duplicate samples within ±25%. Quality control samples were included in each batch of samples analyzed. For extraction samples, if the measured concentrations of OPEFRs were above the highest calibration level, the extract was diluted and reanalyzed.
3. Results and discussion
3.1. OPEFRs in PIR foam
The content of OPEFRs in the PIR foam materials was extracted and measured following the procedure described previously. The measured concentrations listed in Table 1 and Table S1 were used as initial material-phase concentrations (C0) of OPEFRs for the chamber tests.
Table 1.
Summary of parameters used for model fitting of the microchamber emission tests and obtained Dm and Kma values at different temperatures.
| Unit | TCEP | TCIPP | TDCIPP | |
|---|---|---|---|---|
| Chamber conditions | ||||
| Chamber volume (V)a | m3 | 4.4 × 10−5 | ||
| Average air velocity in chamberb | m/s | 0.09 | ||
| Emission surface area (A0)a | m2 | 3.2 × 10−3 | ||
| Chamber surface area (As)a | m2 | 3.5 × 10−3 | ||
| Thickness of foam (L)a | m | 3.97 × 10−3 | ||
| Density of foam (ρ)a | g/m3 | 3.04 × 104 | ||
| Parameters at 23°C (296K) | ||||
| Ventilation rate (Q)a | m3/h | 1.2 × 10−2 | ||
| Air change rate (ACH)a | h−1 | 273 | ||
| Initial material-phase concentration in foam (C0)a | μg/m3 | 1.08 × 109 | 5.69 × 108 | 3.45 × 109 |
| Mass transfer coefficient (hm, hs)c | m/h | 4.92 | 4.55 | 4.28 |
| Chamber surface/area partition coefficient (Ksa)d | m | 29.4 | 6.2 | N/A |
| Steady-state gas-phase concentration ± STD (yss, Chamber 1)a | μg/m3 | 5.02 ± 0.54 | 2.02 ± 0.27 | N/A |
| Steady-state gas-phase concentration ± STD (yss, Chamber 2)a | μg/m3 | 4.65 ± 0.9 | 2.02 ± 0.2 | N/A |
| Material-phase diffusion coefficient (Dm, chamber 1)e | m2/h | 8.08 × 10−12 | 6.30 × 10−12 | N/A |
| Material-phase diffusion coefficient (Dm, chamber 2)e | m2/h | 9.68 × 10−12 | 6.72 × 10−12 | N/A |
| Material/air partition coefficient (Kma, Chamber 1)e | 9.22 × 108 | 1.37 × 108 | N/A | |
| Material/air partition coefficient (Kma, Chamber 2)e | 9.96 × 108 | 1.37 × 108 | N/A | |
| Parameters at 35°C (308 K) | ||||
| Ventilation rate (Q)a | m3/h | 1.21 × 10−2 | ||
| Air change rate (ACH)a | h−1 | 275 | ||
| Initial material-phase concentration in foam (C0)a | μg/m3 | 1.08 × 108 | 5.69 × 108 | 3.45 × 109 |
| Mass transfer coefficient (hm, hs)c | m/h | 5.17 | 4.71 | 4.49 |
| Chamber surface/area partition coefficient (Ksa)f | m | 0 | 0 | N/A |
| Steady-state gas-phase concentration ± STD (yss, Chamber 1)a | μg/m3 | 6.48 ± 1.05 | 8.18 ± 2.91 | N/A |
| Steady-state gas-phase concentration ± STD (yss, Chamber 2)a | μg/m3 | 6.80 ± 1.24 | 8.21 ± 2.14 | N/A |
| Material-phase diffusion coefficient (Dm, Chamber 1)e | m2/h | 1.81 × 10−11 | 9.38 × 10−12 | N/A |
| Material-phase diffusion coefficient (Dm, chamber 2)e | m2/h | 1.62 × 10−11 | 8.20 × 10−12 | N/A |
| Material/air partition coefficient (Kma, Chamber 1)e | 7.27 × 107 | 1.98 × 107 | N/A | |
| Material/air partition coefficient (Kma, Chamber 2)e | 6.66 × 107 | 1.42 × 107 | N/A | |
| Parameters at 55°C (328 K) | ||||
| Ventilation rate (Q)a | m3/h | 1.17 × 10−2 | ||
| Air change rate (ACH)a | h−1 | 265 | ||
| Initial material-phase concentration in foam (C0)a | μg/m3 | 1.08 × 109 | 5.69 × 108 | 3.45 × 109 |
| Mass transfer coefficient (hm, hs)c | m/h | 5.47 | 4.99 | 4.75 |
| Chamber surface/area partition coefficient (Ksa)f | m | 0 | 0 | 0 |
| Steady-state gas-phase concentration ± STD (yss, after 200 h, Chamber 1)a | μg/m3 | 59.90 ± 3.1 | 27.35 ± 4.9 | 8.99 ± 1.04 |
| Steady-state gas-phase concentration ± STD (yss, after 200 h, Chamber 2)a | μg/m3 | 56.18 ± 4.2 | 28.69±6.7 | 8.38 ± 1.37 |
| Material-phase diffusion coefficient (Dm, Chamber 1)e | m2/h | 1.81 × 10−11 | 9.38 × 10−12 | 9.86 × 10−12 |
| Material-phase diffusion coefficient (Dm, chamber 2)e | m2/h | 1.62 × 10−11 | 8.20 × 10−12 | 1.04 × 10−11 |
| Material/air partition coefficient (Kma, Chamber 1)e | 7.27 × 107 | 1.98 × 107 | 2.20 × 109 | |
| Material/air partition coefficient (Kma, Chamber 2)e | 6.66 × 107 | 1.42 × 107 | 2.20 × 109 |
Measured in the test.
Air velocity measured following the method in Liang et al. (2018b).
Calculated with the measured air velocity in microchamber using program PARAMS (Guo, 2005). hm and hs in Equation (6) have the same value at a temperature as a result of the same measured air velocity in microchamber.
Obtained from Liang et al. (2018b).
Determined in this study.
Because the adsorption of SVOCs with low volatility (i.e. phthalates) to stainless steel surface decreased greatly as temperature increased (Clausen et al., 2012), partitioning of TCEP and TCIPP between microchamber wall and air was ignored at 35 and 55 °C.
3.2. Emissions of OPEFRs at different temperatures
Fig. 2 shows that temperature had a significant influence on OPEFR emissions. When the temperature was increased from 23 to 35 °C, the average air concentrations of TCEP and TCIPP in microchamber 1 increased by approximately 40% and 150%, respectively (TDCIPP results at 23 and 35 °C are not presented because the measured concentrations were below the lowest calibration concentration). Similar results were observed in microchamber 2 (duplicate test, Fig. S2 in SI). When the temperature was increased from 35 to 55 °C, the average air concentration of TCEP, TCIPP, and TDCIPP in the chamber increased up to an order of magnitude. In addition, the gas-phase concentrations of TCEP and TCIPP decayed gradually over time at 55 °C. As a comparison, the gas-phase concentrations of TCEP and TCIPP at lower temperatures (23 and 35 °C) reached steady-state within 50 h during the test. For TDCIPP, the trend at 55 °C are not very clear due to the concentration fluctuation.
Fig. 2.
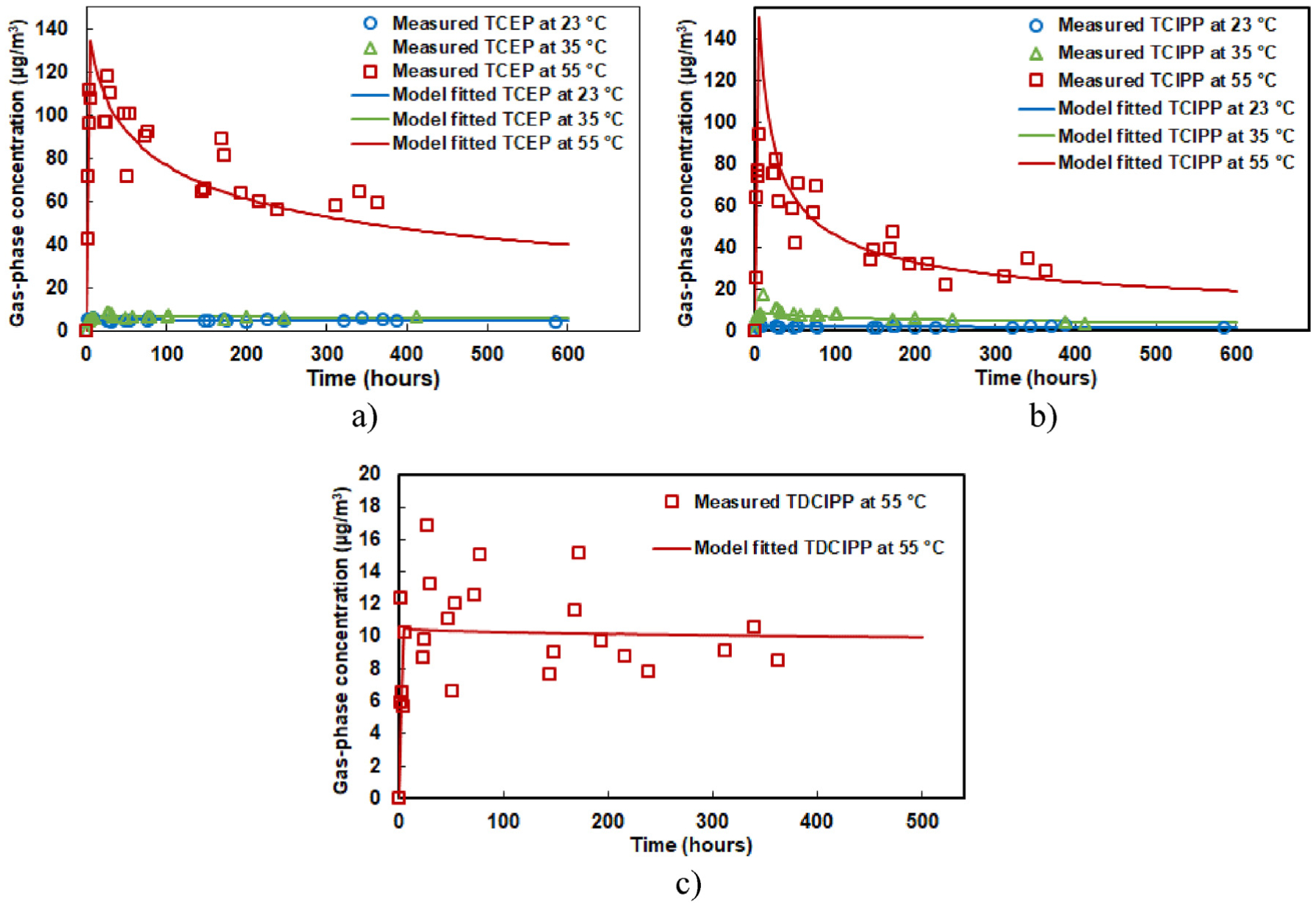
Measurement of gas-phase concentrations of OPEFRs emitted from PIR foam and model fit in microchamber 1 at different temperatures. a) TCEP, b) TCIPP, and c) TDCIPP.
Liu et al. (2016) developed an improved dual small chamber testing method for estimating Dm and Kma. The estimated values of Dm and Kma for TCEP in polyurethane foam at 23 °C in the study were approximately 2.0 × 10−9 m2/h and 4.5 × 106, respectively. The value of Dm for TCEP at 23 °C in this study was over two orders of magnitude lower and that of Kma over two orders of magnitude higher compared with Liu et al.’s estimations (2016) (Table 2). The difference is also pronounced for TCIPP. Moreover, in a very recent study, Liang et al. (2018a) determined the values of Dm and Kma for TCEP, TCIPP, and TDCIPP through the same type of dual small chamber test in PIR foam materials that were free of OPEFRs. As a comparison, the values of Dm for TCEP and TCIPP determined in this study are an order of magnitude lower and values for Kma are two orders of magnitude higher than data reported at 23 °C in Liang et al. (2018a). The differences revealed the substantial difficulty in obtaining the accurate values of Dm and Kma for SVOCs in materials. Both the emission test method in this study and the sorption test method in Liang et al. (2018a) involved determining Dm and Kma simultaneously from nonlinear regression based on measurements. The accuracy of such regression approach is often questioned by researchers. For example, Liu et al. (2016) provided a thorough discussion on the starting value issue associated with nonlinear regressions. There is an urgent need to independently determine Dm and Kma with higher accuracy in future studies. Another possible reason for the Dm and Kma differences between different studies may be that C0 in Table 1 was measured through material extraction and represents the total OPEFR mass contained in the foam material for each OPEFR. Xiong and Zhang (2010) pointed out that the initial emittable concentration of formaldehyde is less than the total concentration in the material and can increase significantly with increasing temperature. With this consideration, C0 used in this study may be higher than the initial emittable concentration, especially at low temperatures (i.e. 23 and 35 °C), which may result in the underestimation of Dm and the overestimation of Kma. In summary, the discrepancy of Dm and Kma values can attribute to experimental and computational differences in different methods.
Table 2.
Comparison of Dm and Kma values of OPEFRs in different foam materials at 23 °C.
| Material | Chemical | Dm (m2/h) | Kma (dimensionless) | Method | Comment |
|---|---|---|---|---|---|
| PIR foam | TCEP | 8.88 × 10−12 | 9.59 × 108 | Microchamber emission test | This study |
| TCIPP | 6.51 × 10−12 | 1.37 × 108 | |||
| TDCIPP | N/A | N/A | |||
| PIR foam | TCEP | 2.01 × 10−10 | 7.76 × 106 | Small chamber sorption test | Liang et al. (2018a) |
| TCIPP | 8.25 × 10−11 | 6.85 × 106 | |||
| TDCIPP | 1.39 × 10−11 | 1.90 × 108 | |||
| PUF | TCEP | 1.22 × 10−9 | 5.28 × 106 | Small chamber sorption test | Liu et al. (2016) |
| TCIPP | 5.01 × 10−10 | 4.99 × 106 | |||
| TDCIPP | 8.43 × 10−11 | 2.26 × 107 | |||
| PUF | TCEP | 2.95 × 10−9 | 3.87 × 106 | Small chamber sorption test | Liu et al. (2016) |
| TCIPP | 1.21 × 10−9 | 3.70 × 106 | |||
| TDCIPP | 2.03 × 10−10 | 1.23 × 107 |
Xu et al. (2012) proposed that for SVOCs with very low volatility and high material-phase concentration (C0), the diffusion within the source material can be ignored and y0 can be considered as a constant during the emission process. This simplification was adopted in several studies on characterizing emissions of phthalates from building materials (Liang and Xu, 2014b; Wu et al., 2016; Cao et al., 2016). In the study on temperature influence on phthalate emissions, Liang and Xu (2014a) observed that y0 remained constant for DEHP and DINP even at 55 °C (Fig. 1 in Liang and Xu (2014a)). However, it is difficult to extend the validation of the simplification to other SVOCs without the knowledge of C0, Dm and Kma (Liang et al. (2018a)). Indeed, the results in Fig. 2 clearly showed that y0 was decreasing over time for TCEP and TCIPP at 55 °C, though y0 remained relatively stable at lower temperatures (23 and 35 °C). This observation indicates that temperature increase may result in a behavioral change from SVOC-type to VOC-type for OPEFRs and other SVOCs with similar properties. This is consistent with the fact that the vapor pressures of compounds will increase when temperature is increased, and thus less volatile compounds will become more volatile at a higher temperature. In future research, it is important to know the critical temperature for SVOCs, above which y0 can no longer assumed to be constant throughout the emission process.
3.3. Relationship between OPEFR emission rate and temperature
Fig. 2 shows that temperature increase can significantly enhance the emissions of OPEFRs from the foam material. Therefore, we would like to further investigate the relationship between the emission rate and temperature for OPEFRs in this study. Xiong et al. (2013) studied the relationship between the emission rate and the temperature for VOCs and SVOCs in building materials and found that it can be generally represented by
| (9) |
where E (μg/m2/h) is the steady-state emission rate, G1 and G2 are constants for a given chemical and material. The steady-state emission rate of OPFERs from the foam material in the microchamber can be described as
| (10) |
where yss (μg/m3) is the steady-state gas-phase OPEFR concentration in the microchamber. At steady state, dy/dt = zero and y = yss = ys in Equation (1). If we substitute Equation (10) into Equation (1), the steady-state emission rate can be represented based on the OPEFR mass conservation in the microchamber:
| (11) |
The steady-state gas-phase concentrations of OPEFRs in the microchamber were calculated by averaging the measured concentrations during the whole test period in the chamber at 23 and 35 °C. At 55 °C, the gas-phase concentrations of OPEFRs kept decaying during the emission process, and the average gas-phase concentrations for t > 200 h was used to approximate the steady-state concentrations. We further determined the steady-state emission rates of TCEP and TCIPP with Equation (11) and plotted E/T0.25 against 1/T with reference to Equation (9). The results are presented in Fig. 3.
Fig. 3.
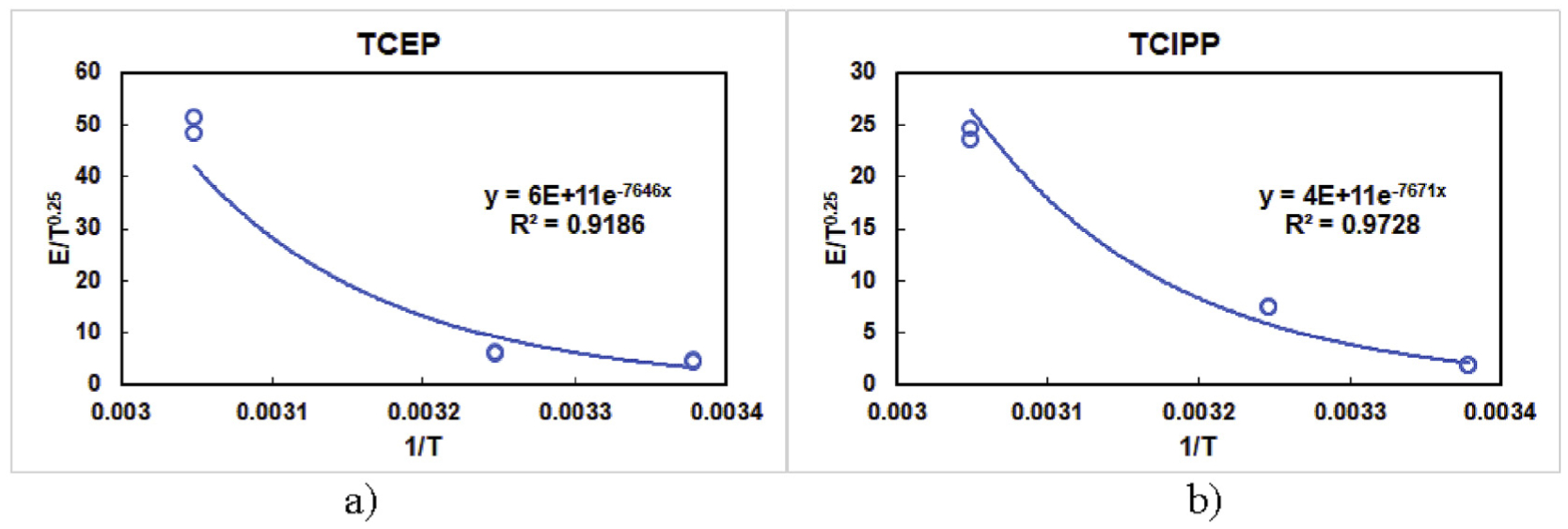
Correlation of steady-stead emission rate and temperature for OPEFRs. a) TCEP (G1 = 6 × 1011 and G2 = 7646) and b) TCIPP (G1 = 4 × 1011 and G2 = 7671).
Fig. 3 shows that the relationship between the steady-state emission rate and temperature for TCEP and TCIPP agreed well with Equation (9) (R2 > 0.9). The equations in Fig. 3 can thus be used to predict the steady-state emission rates of OPEFRs from foam materials at the temperature range of 23–55 °C for future research. The results indicate that temperature plays an important role on the emission behaviors of OPEFRs. A change in temperature can result in a corresponding deviation in the steady-state emission rate and the gas phase concentration. Therefore, it is important to accurately control the temperature to maintain a high measurement accuracy for their source emission tests. It is worth noting that the correlations between the steady-state emission rate and temperature for TCEP and TCIPP were fitted against temperature from three different data points. The applicability of the obtained correlations is thus limited considering that both OPEFRs behaved very differently when temperature reached 55 °C. It can be improved by conducting emission tests at higher temperatures (>55 °C), which is one of the objectives in our future studies.
3.4. Relationships between Dm, Kma, and temperature
Based on theoretical analysis and experimental data, Deng et al. (2009) proposed correlation relationships between Dm, Kma and temperature for VOCs in several porous building materials based on the assumption that molecular diffusion is dominant. According to their results, the relationships can be expressed as
| (12) |
| (13) |
where A1, A2 and B1, B2 are constants for a given chemical and material. Following the forms of equations (12) and (13), we can obtain these constants for TCEP and TCIPP in PIR foam by fitting Dm and Kma reported in Table 1 with temperature. The fitting results are shown in Fig. 4 and the obtained correlations are listed in Table 3.
Fig. 4.
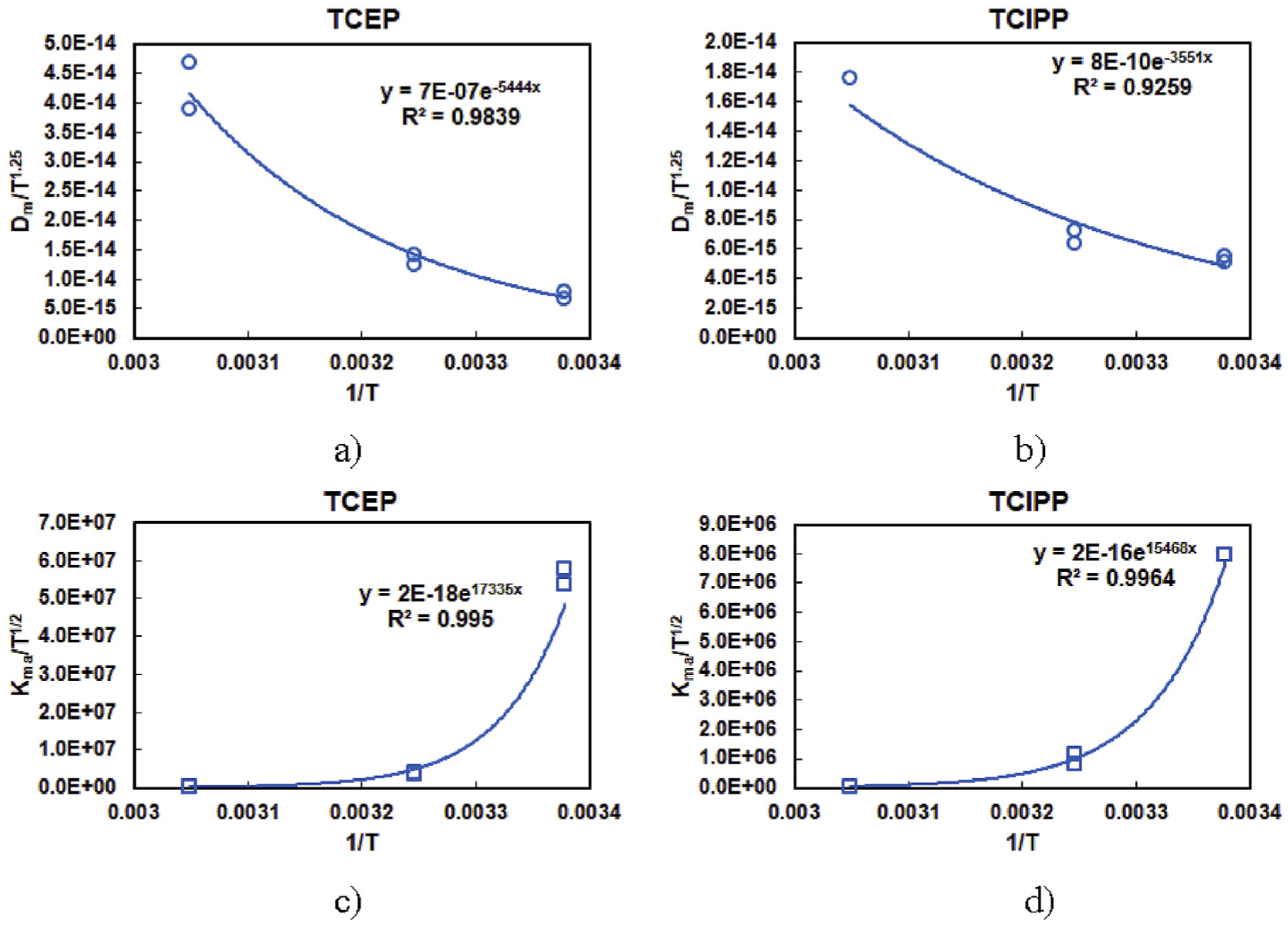
Determined Dm and Kma of OPEFR emission from PIR foam under different temperatures. a) Dm of TCEP, b) Dm of TCIPP, c) Kma of TCEP, and c) Kma of TCIPP.
Table 3.
The correlations between Dm, Kma of OPEFR emission from the PIR foam and temperature.
| Chemical | Correlations | R2 |
|---|---|---|
| TCEP | Dm = 7 × 10−7 × T1.25 × e−5444/T | 0.984 |
| Kma = 2 × 10−18 × T0.5 × e17335/T | 0.995 | |
| TCIPP | Dm = 8 × 10−10 × T1.25 × e−3551/T | 0.926 |
| Kma = 2 × 10−16 × T0.5 × e15468/T | 0.996 |
The correlations listed in Table 3 can be used to estimate Dm and Kma for TCEP and TCIPP in PIR foam materials at different temperatures. However, cautions should be taken when using these correlations. First, Equations (12) and (13) proposed by Deng et al. (2009) were used to characterize the emission of VOCs from building materials (particle board, vinyl flooring, etc.), and their application to SVOCs, such as OPEFRs, need more systematic research. We adopted the equations for TCEP and TCIPP because these two compounds behaved closer to VOCs as the temperature increased from 23 °C to 55 °C. Second, each set of Dm and Kma listed in Table 1 were simultaneously determined from model fitting, and therefore the accuracy of the obtained values is impacted by the starting value issue associated with non-linear regressions. Third, the measurements in this study were conducted at three different temperatures, whether the relationship can be applied to conditions beyond the temperature range needs further investigation.
Supplementary Material
HIGHLIGHTS.
Emissions of organophosphate ester flame retardants from foam were measured.
Values of emission parameters at different temperatures were determined.
Correlations between emission parameters and temperature were obtained.
Temperature influences on the emission rates of OPEFRs were elucidated.
Acknowledgments
We thank Gary Folk from Jacobs Technology Inc. for helping with the GC/MS instrument operation and microchamber tests and ICL Industrial Products America Inc. for providing the PIR foam.
Footnotes
Publisher's Disclaimer: Disclaimer
Publisher's Disclaimer: The views expressed in this article are those of the authors and do not necessarily represent the views or policies of the U.S. EPA. Mention of trade names or commercial products does not constitute endorsement or recommendation for use by the U.S. EPA.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.chemosphere.2019.05.289.
References
- Bergh C, Torgrip R, Emenius G, Ostman C, 2011. Organophosphate and phthalate esters in air and settled dust - a multi-location indoor study. Indoor Air 21, 67–76. [DOI] [PubMed] [Google Scholar]
- Besis A, Christia C, Covaci G, Samara C, 2017. Legacy and novel brominated flame retardants in interior car dust-Implications for human exposure. Environ. Pollut 230, 871–881. [DOI] [PubMed] [Google Scholar]
- Bradman A, Castorina R, Gaspar F, Nishioka M, Colén M, Weathers W, Egeghy PP, Maddalena R, Williams J, Jenkins PL, McKone TE, 2014. Flame retardant exposures in California early childhood education environments. Chemosphere 116, 61–66. [DOI] [PubMed] [Google Scholar]
- Brommer S, Harrad S, Van den Eede N, Covaci A, 2012. Concentrations of organophosphate esters and brominated flame retardants in German indoor dust samples. J. Environ. Monit 14, 2482–2487. [DOI] [PubMed] [Google Scholar]
- Cao J, Xiong J, Wang L, Xu Y, Zhang Y, 2016. Transient method for determining indoor chemical concentrations based on SPME: model development and calibration. Environ. Sci. Technol 50, 9452–9459. [DOI] [PubMed] [Google Scholar]
- Christia C, Poma G, Besis A, Samara C, Covaci A, 2018. Legacy and emerging organophosphorus flame retardants in car dust from Greece: implications for human exposure. Chemosphere 196, 231–239. [DOI] [PubMed] [Google Scholar]
- Clausen PA, Liu Z, Kofoed-Sørensen V, Little J, Wolkoff P, 2012. Influence of temperature on the emission of di-(2-ethylhexyl)phthalate (DEHP) from PVC flooring in the emission cell FLEC. Environ. Sci. Technol 46, 909–915. [DOI] [PubMed] [Google Scholar]
- Deng Q, Yang X, Zhang J, 2009. Study on a new correlation between diffusion coefficient and temperature in porous building materials. Atmos. Environ 43, 2080–2083. [Google Scholar]
- Eichler CMA, Wu Y, Cao J, Shi S, Little JC, 2018. Equilibrium relationship between SVOCs in PVC products and the air in contact with the product. Environ. Sci. Technol 52, 2918–2925. [DOI] [PubMed] [Google Scholar]
- Ekelund M, Azhdar B, Gedde UW, 2010. Evaporative loss kinetics of di(2-ethylhexyl)phthalate (DEHP) from pristine DEHP and plasticized PVC. Polym. Degrad. Stabil 95, 1789–1793. [Google Scholar]
- Ekelund M, Azhdar B, Hedenqvist MS, Gedde UW, 2008. Long-term performance of poly(vinyl chloride) cables, Part 2: migration of plasticizer. Polym. Degrad. Stabil 93, 1704–1710. [Google Scholar]
- Fujii M, Shinohara N, Lim A, Otake T, Kumagai K, Yanagisawa Y, 2003. A study on emission of phthalate esters from plastic materials using a passive flux sampler. Atmos. Environ 37, 5495–5504. [Google Scholar]
- Greaves AK, Letcher RJ, 2017. A Review of organophosphate esters in the environment from biological effects to distribution and fate. Bull. Environ. Contam. Toxicol 98, 2–7. [DOI] [PubMed] [Google Scholar]
- Guo Z, 2005. Program PARAMS Users Guide. U.S. Environmental Protection Agency, Washington, DC: EPA/600/R-05/066 (NTIS PB2006–102000). [Google Scholar]
- Hartmann PC, Bürgi D, Giger W, 2004. Organophosphate flame retardants and plasticizers in indoor air. Chemosphere 57, 781–787. [DOI] [PubMed] [Google Scholar]
- Hoffman K, Butt CM, Webster TF, Preston EV, Hammel SC, Makey C, Lorenzo AM, Cooper EM, Carignan C, Meeker JD, Hauser R, Soubry A, Murphy SK, Price TM, Hoyo C, Mendelsohn E, Congleton J, Daniels JL, Stapleton HM, 2017. Temporal trends in exposure to organophosphate flame retardants in the United States. Envrion. Sci. Technol. Lett 4, 112–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemmlein S, Hahn O, Jann O, 2003. Emissions of organophosphate and brominated flame retardants from selected consumer products and building materials. Atmos. Environ 37, 5485–5493. [Google Scholar]
- Liang Y, Liu X, Allen MR, 2018a. Measurements of parameters controlling the emissions of organophosphate flame retardants in indoor environments. Environ. Sci. Technol 52, 5821–5829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y, Liu X, Allen MR, 2018b. Measuring and modeling surface sorption dynamics of organophosphate flame retardants on impervious surfaces. Chemosphere 193, 754–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y, Xu Y, 2014a. Emission of phthalates and phthalate alternatives from vinyl flooring and crib mattress covers: the influence of temperature. Environ. Sci. Technol 48, 14228–14237. [DOI] [PubMed] [Google Scholar]
- Liang Y, Xu Y, 2014b. Improved method for measuring and characterizing phthalate emissions from building materials and its application to exposure assessment. Environ. Sci. Technol 48, 4475–4484. [DOI] [PubMed] [Google Scholar]
- Liu X, Allen MR, Roache NF, 2016. Characterization of organophosphorus flame retardants’ sorption on building materials and consumer products. Atmos. Environ 140, 333–341. [Google Scholar]
- Liu Z, Ye W, Little JC, 2013. Predicting emissions of volatile and semivolatile organic compounds from building materials: a review. Build. Environ 64, 7–25. [Google Scholar]
- Mao Y, Li Z, Zhang Y, He Y, Tao W, 2018. A review of mass-transfer models and mechanistic studies of semi-volatile organic compounds in indoor environments. Indoor Built Environ. 27, 1307–1321. [Google Scholar]
- McLaren C, Null J, Quinn J, 2005. Heat stress from enclosed vehicles: moderate ambient temperatures cause significant temperature rise in enclosed vehicles. Pediatrics 116, e109–e112. [DOI] [PubMed] [Google Scholar]
- Ni Y, Kumagai K, Yanagisawa Y, 2007. Measuring emissions of organophosphate flame retardants using a passive flux sampler. Atmos. Environ 41, 3235–3240. [Google Scholar]
- Staaf T, Ostman C, 2005. Organophosphate triesters in indoor environments. J. Environ. Monit 7, 883–887. [DOI] [PubMed] [Google Scholar]
- Stapleton HM, Klosterhaus S, Eagle S, Fuh J, Meeker JD, Blum A, Webster TF, 2009. Detection of organophosphate flame retardants in furniture foam and U.S. house dust. Environ. Sci. Technol 43, 7490–7495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- U.S. Department of Health, September 2012. Human Services, Agency for Toxic Substances and Disease Registry, Toxicology Profile for Phosphate Ester Flame Retardants. http://www.atsdr.cdc.gov/toxprofiles/tp.asp?id=1119&tid=239. (Accessed 26 May 2019). [PubMed]
- Van den Eede N, Dirtu AC, Neels H, Covaci A, 2011. Analytical developments and preliminary assessment of human exposure to organophosphate flame retardants from indoor dust. Environ. Int 37, 454–461. [DOI] [PubMed] [Google Scholar]
- Van der Veen I, de Boer J, 2012. Phosphorus flame retardants: properties, production, environmental occurrence, toxicity and analysis. Chemosphere 88, 1119–1153. [DOI] [PubMed] [Google Scholar]
- Wei GL, Li DQ, Zhuo MN, Liao YS, Xie ZY, Guo TL, Li JJ, Zhang SY, Liang ZQ, 2015. Organophosphorus flame retardants and plasticizers: sources, occurrence, toxicity and human exposure. Environ. Pollut 196, 29–46. [DOI] [PubMed] [Google Scholar]
- Wensing M, Uhde E, Salthammer T, 2005. Plastics additives in the indoor environment—flame retardants and plasticizers. Sci. Total Environ 339, 19–40. [DOI] [PubMed] [Google Scholar]
- Weschler CJ, Nazaroff WW, 2008. Semivolatile organic compounds in indoor environments. Atmos. Environ 42, 9018–9040. [Google Scholar]
- Wu Y, Xie M, Cox SS, Marr LC, Little JC, 2016. A simple method to measure the gas-phase SVOC concentration adjacent to a material surface. Indoor Air 26, 903–912. [DOI] [PubMed] [Google Scholar]
- Xiong J, Wei W, Huang S, Zhang Y, 2013. Association between the emission rate and temperature for chemical pollutants in building materials: general correlation and understanding. Environ. Sci. Technol 47, 8540–8547. [DOI] [PubMed] [Google Scholar]
- Xiong J, Zhang Y, 2010. Impact of temperature on the initial emittable concentration of formaldehyde in building materials: experimental observation. Indoor Air 20, 523–529. [DOI] [PubMed] [Google Scholar]
- Xu Y, Little JC, 2006. Predicting emissions of SVOCs from polymeric materials and their interaction with airborne particles. Environ. Sci. Technol 40, 456–461. [DOI] [PubMed] [Google Scholar]
- Xu Y, Liu Z, Park J, Clausen PA, Benning JL, Little JC, 2012. Measuring and predicting the emission rate of phthalate plasticizer from vinyl flooring in a specially-designed chamber. Environ. Sci. Technol 46, 12534–12541. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.