

Abstract
Juvenile papillomatosis (JP) of the breast is a rare benign mass‐forming lesion occurring in young women, which is histologically characterized by a constellation of proliferative changes and large cysts, giving it the gross appearance of Swiss cheese. A subset of patients with JP report a family history of breast carcinoma and/or coexisting or subsequent breast carcinoma. We performed whole‐exome sequencing of the hyperplastic epithelial component of three JPs, including one with coexisting ductal carcinoma in situ (DCIS) and invasive ductal carcinoma of no special type (IDC‐NST). JPs harbored clonal somatic PIK3CA hotspot mutations in two cases. In the JP with coexisting DCIS and IDC‐NST, these lesions were clonally related to the associated JP, sharing a clonal PIK3CA E542K somatic hotspot mutation. JP showed a paucity of copy number alterations, whereas the associated DCIS and IDC‐NST showed concurrent 1q gains/16q losses, hallmarks of estrogen receptor (ER)‐positive breast cancers. We observed JP to harbor a dominant aging‐related mutational signature, whereas coexisting DCIS and IDC‐NST showed greater exposure to an APOBEC signature. Taken together, our findings suggest that, at least in a subset of cases, JP might constitute the substrate from which DCIS and invasive breast carcinomas develop.
Keywords: juvenile papillomatosis, PIK3CA, breast carcinoma
Introduction
Juvenile papillomatosis of the breast (JP) is a rare benign mass‐forming lesion usually occurring in women in their 20s to 30s that often mimics fibroadenoma clinically [1, 2]. Histologically, JP is characterized by a constellation of proliferative and nonproliferative changes, the most distinct being abundant large cysts, giving the gross appearance of Swiss cheese [1]. Since the description of JP as a distinct entity by Rosen [1], multiple studies have reported a significant family history of breast carcinoma cancer in patients developing JP [2, 3, 4], suggesting a relationship between JP and breast carcinoma. Furthermore, patients with JP have coexisting breast carcinoma or subsequently develop carcinoma in approximately 10% of cases [4, 5]. Recently, Guillet et al reported recurrent PIK3CA or AKT1 hotspot mutations in benign JP lesions in the first published study of the molecular features of JP, expanding the spectrum of benign breast lesions with these mutations [3]. Nonetheless, the genetic relationship between JP and coexisting breast carcinoma has not yet been documented. Herein, we sought to characterize the repertoire of genetic alterations of JP by whole‐exome sequencing (WES) and to study the clonal relationship between JP, ductal carcinoma in situ (DCIS), and invasive carcinoma in one case where these components coexisted.
Materials and methods
Subjects and samples
Following approval from the Institutional Review Boards (IRB), formalin‐fixed paraffin‐embedded tissue blocks were retrieved from the pathology archives of the authors' institutions. Patient consents were obtained if required by the IRB protocols of the different institutions. Samples were anonymized prior to tissue processing. Our cohort included three JP lesions (JuP1, JuP2, and JuP3), including one case (JuP3) with coexisting invasive ductal carcinoma of no special type (IDC‐NST) and DCIS. Slides were reviewed by three pathologists (TMD, FP, and MV). The hyperplastic epithelial components of JP, and DCIS and IDC‐NST components of case JuP3 were separately microdissected from 10 to 15 8‐μm thick histological sections under a stereomicroscope (Olympus SZ61, Tokyo, Japan), as previously described [6]. DNA was extracted from lesion/tumor and matched benign non‐hyperplastic breast parenchyma away from the JP using the DNAeasy Blood and Tissue Kit (Qiagen, Hilden, Germany), following manufacturers' instructions. DNA samples were subjected to WES at the Integrated Genomics Operation at Memorial Sloan Kettering Cancer Center and analyzed.
Whole‐exome sequencing analysis
Reads were aligned to the reference human genome GRCh37 using the Burrows‐Wheeler Aligner (BWA v0.7.15) [7]. The Genome Analysis Toolkit (GATK. V3.1.1) [8] was employed for local realignment, duplicate removal and base quality recalibration. Somatic single nucleotide variants (SNVs) were detected with MuTect (v1.0) [9], indels with Strelka (v2.0.15) [10], Varscan2 (v2.3.7) [11], Scalpel (v0.5.3) [12], and Lancet (v1.0.0) [13] as previously described [6]. SNVs and indels outside of the target regions were filtered out, as were SNVs and indels for which the variant allele fraction (VAF) in the tumor sample was <5 times that of the paired normal VAF, and SNVs and indels found at >5% global minor allele frequency of dbSNP (build 137), as previously described [14]. Only somatic mutations with a depth ≥20 reads in the respective normal samples were considered [14]. All mutations were manually inspected using the Integrative Genomics Viewer (IGV) [15]. The cancer cell fraction (CCF) of each mutation was inferred using ABSOLUTE (v1.0.6) [16], as previously described [17, 18]. Mutations were cataloged as clonal if their probability of being clonal was >50% [19], or if the lower bound of the 95% CI of its CCF was >90% [20]. Copy number alterations and loss of heterozygosity were determined using FACETS [21]. Mutations targeting hotspot loci [22] were annotated as previously described [20]. Mutational signatures were inferred using SigMA [23] based on all synonymous and nonsynonymous somatic mutations, as previously reported [24]. A mutation‐based phylogenetic tree of the JP and associated DCIS and IDC‐NST of case JuP3 was constructed using Treeomics [25], based on all synonymous and nonsynonymous mutations identified. Mutations were considered shared when present in different histologic components, and private when present in only one of the components. Clonal relatedness between the JP and coexisting DCIS and IDC‐NST of case JuP3 were assessed using ‘clonality index’ (CI), which is the probability of the mutations shared between different lesions not co‐occurring by chance [26, 27]. We used a previously established method [26, 27], where CI is defined as follows: The probability of identifying a given mutation in two samples is defined by the binomial probability P(X) = C k n p k(1 − p)n−k, n = 2, k = 2, where p represents the frequency of a given mutation and n represents the number of shared mutations in different components or the average number of mutations identified in the samples. Hence, the probability of identifying a certain set of M identical mutations in the different samples is given by .
Results
Case JuP1 was a 43‐year‐old woman without a history of breast carcinoma who presented with a palpable breast mass. She underwent lumpectomy that showed JP. Case JuP2 was a 31‐year‐old woman with a family history of breast carcinoma who was found to have clustered calcifications on screening mammogram. Following biopsies showing fibrocystic changes, she underwent lumpectomy showing JP. JuP3 was a 34‐year‐old woman with a family history of breast carcinoma and a history of fibrocystic changes. Ultrasound revealed an irregular hypoechoic mass that was biopsied revealing IDC‐NST and DCIS. She underwent lumpectomy showing JP in association with the carcinoma.
All cases showed classic histologic features of JP including abundant large cysts, ducts with lipid‐laden histiocytes, florid usual ductal hyperplasia, and apocrine metaplasia/hyperplasia (Figure 1A,B). Stromal sclerosis, papilloma/papillary hyperplasia, adenosis, and microcalcifications were present in varying degrees among cases. JuP3 showed a 1.5 cm estrogen receptor (ER)‐positive/HER2‐negative moderately‐differentiated IDC‐NST and DCIS with intermediate to high nuclear grade and necrosis that was intimately associated with benign JP components (Figure 2A–D).
Figure 1.
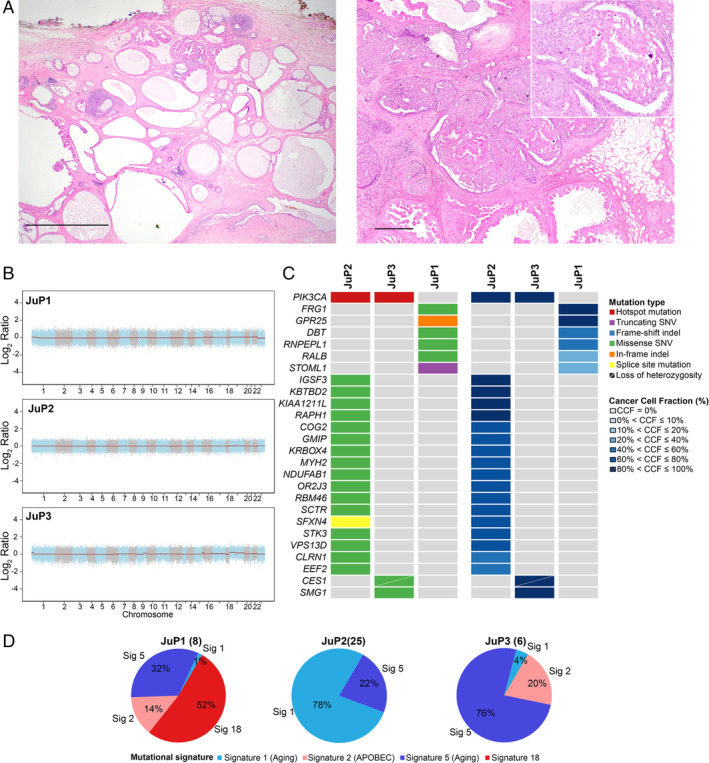
Histologic features and repertoire of genetic alterations of JP. (A) Representative micrographs of H&E stained sections of JP illustrating the characteristic cystic cut surface (left) and florid hyperplasia with papillary and apocrine features with calcifications (right). Scale bars: left, 2 mm; right, 500 μm. (B) Genome plots depicting Log2 ratios (y‐axis) plotted according to their genomic positions (x‐axis). (C) Heatmaps depicting the nonsynonymous somatic mutations identified by whole‐exome sequencing (left) and cancer cell fraction (CCF) (right). Cases are shown in columns and genes in rows. Mutations and CCF are color‐coded according to the legend. (D) Pie charts showing the mutational signatures as inferred by SigMA. The numbers of synonymous and nonsynonymous mutations of each JP lesion are shown in parentheses.
Figure 2.
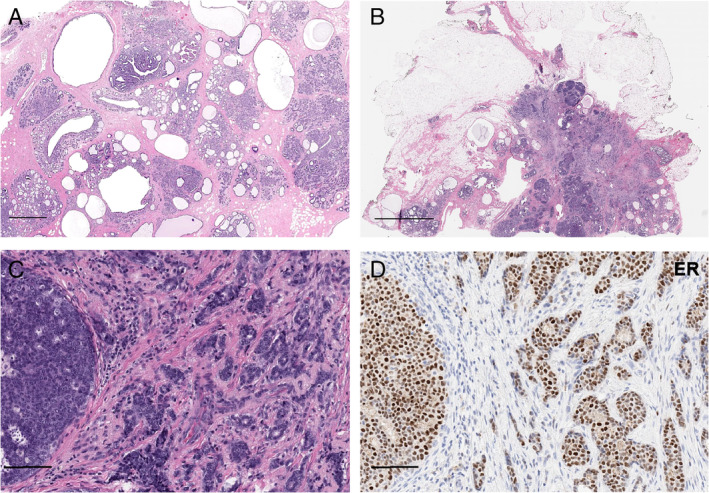
Histologic images of JP and synchronously identified invasive breast carcinoma and DCIS. (A) Representative micrographs of H&E stained sections of JP showing the cystic cut surface and (B) synchronously identified DCIS and IDC‐NST of case JuP3. (C) Higher power magnification shows DCIS adjacent to the IDC‐NST, showing a moderately differentiated IDC‐NST and intermediate‐grade DCIS. (D) Representative micrograph of the ER immunohistochemical analysis showing strong positive staining in IDC‐NST and DCIS. Scale bars: A: 1 mm; B: 3 mm; C, D: 100 μm.
To determine the repertoire of genetic alterations underpinning JP we subjected these three cases to WES. Our analysis revealed that JP shows a paucity of copy number alterations (CNAs) and somatic mutations (Figure 1B,C). Benign JP showed a median of 9 (range, 6–25) total and 6 (range, 3–18) nonsynonymous mutations. We identified somatic clonal PIK3CA hotspot mutations in cases JuP2 (H1047R) and JuP3 (E542K). Interestingly, JuP1 was found to harbor a germline PIK3CA I391M variant, whose significance is uncertain. No other genes were found to be recurrently mutated. We inferred the mutational signatures using SigMA [23] using all synonymous and nonsynonymous somatic mutations. We observed that mutational signatures ascribed to aging (signatures 1 and 5) were dominant in JuP2 and JuP3 (Figure 1D). In JuP1, signature 5 (aging) was the second most dominant following signature 18, of cryptic origin (Figure 1D).
As a hypothesis generating aim, we sought to determine whether JP could constitute a substrate for the development of breast carcinoma and conducted WES analysis of the separately microdissected JP and coexisting DCIS and IDC‐NST of JuP3. Our analyses revealed that, although the JP component of JuP3 displayed a paucity of CNAs, the synchronously identified DCIS and IDC‐NST showed concurrent 1q gains/16q losses, the hallmark genetic alteration of ER‐positive breast cancer [28] (Figure 3A). We observed that the DCIS and IDC‐NST of JuP3 were clonally related to adjacent JP and shared a clonal PIK3CA E542K hotspot mutation and CES1 (S12A) and SMG1 (R420Q) missense mutations (Figure 3B,C). DCIS and IDC‐NST shared 29 synonymous and nonsynonymous mutations not seen in JP, including a truncating mutation in the chromatin remodeling gene ARID1A, missense mutations in KMT2C and PIK3CB, and an E14K hotspot mutation affecting NUP93, a nucleoporin implicated in cell migration [29] (Figure 3B,C). A subset of mutations found to be subclonal in the DCIS became dominant in the IDC‐NST, including a PIK3C2A missense mutation, suggesting that a minor subclone of the DCIS became the dominant clone in the progression from DCIS to invasive breast cancer (Figure 3B,C). The DCIS and IDC‐NST subsequently acquired somatic mutations, with the acquisition of a truncating mutation in the tumor suppressor STAG2 in DCIS and an FGF12 missense mutation in IDC‐NST (Figure 3B,C).
Figure 3.
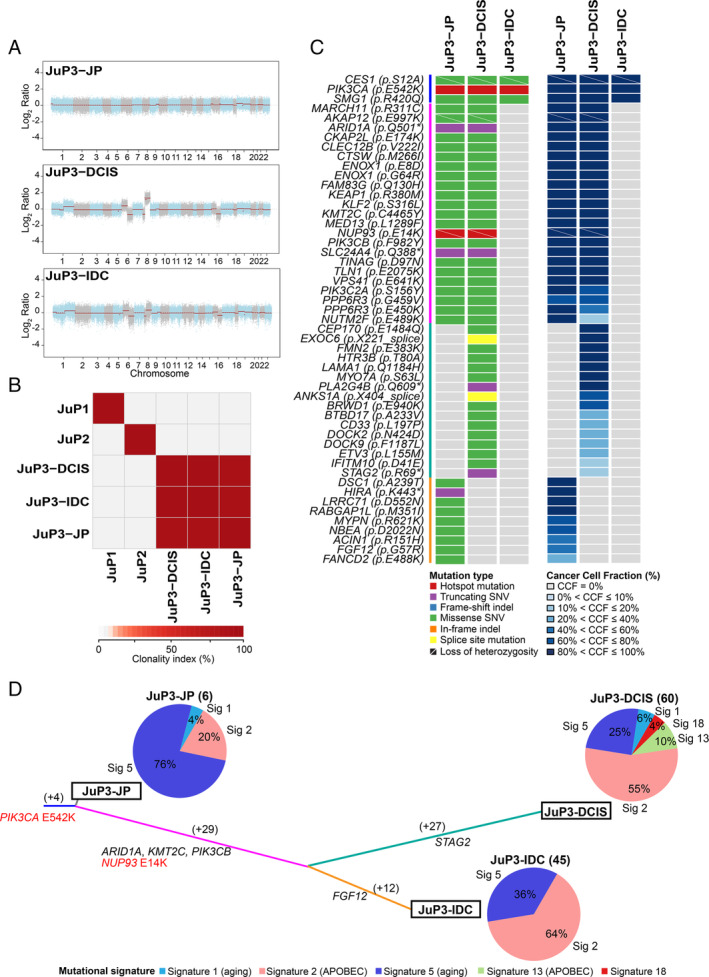
JP and coexisting breast cancer are clonally related. (A) Genome plots depicting Log2 ratios (y‐axis) plotted according to their genomic positions (x‐axis). (B) Pairwise comparison of the clonality index (CI) based on somatic mutations identified in JP lesions JuP1, JuP2 and JuP3 and ductal carcinoma in situ (JuP3‐DCIS) and invasive ductal carcinoma (JuP3‐IDC) coexisting with JuP3 identified by whole exome sequencing. (C) Heatmaps depicting the nonsynonymous somatic mutations identified in the different histologic components of case JuP3 (left) and their cancer cell fraction (CCF) (right). Cases are shown in rows and genes in columns. Mutations and CCF are color‐coded according to the legend. (D) Mutation‐based phylogenetic tree of case JuP3 depicting the clonal evolution of JP and synchronously identified DCIS and IDC‐NST. The lengths of the trunk and branches are proportional to the number of synonymous and nonsynonymous mutations shared or private to a given histologic component. Mutational signatures identified in the different histologic components of case JuP3 using SigMA are depicted in pie charts. The numbers of synonymous and nonsynonymous mutations of each histologic component are shown in parentheses.
Mutational signature analysis revealed that JP harbored a dominant aging‐related signature 5 (signature exposure of 76%) followed by signature 2 (APOBEC; signature exposure of 20%). The DCIS and IDC‐NST from JuP3, however, displayed a greater number of mutations than benign JP as well as a greater mutational signature 2 exposure (DCIS: 55%; IDC‐NST: 64%), suggesting that the development of carcinoma in this case might be associated with an increase in APOBEC mutagenesis (Figure 3D).
Discussion
Here we provide evidence that JP shows recurrent PIK3CA mutations, and that it may constitute the substrate from which DCIS and invasive breast cancers develop, given that we documented clonal relatedness between JP and associated carcinoma.
Mutations in the PI3K‐AKT pathway, which are commonly seen in invasive breast carcinoma, have also been identified in a variety of benign proliferative epithelial lesions, including papillary neoplasms, usual ductal hyperplasia, and columnar cell change, among others [30, 31]. WES analysis of the proliferative components of JP resulted in the identification of clonal PIK3CA hotspot mutations in 2 of 3 cases studied. We also observed a germline PIK3CA variant affecting the C2 domain in the third case. Although mutations affecting the C2 domain of PIK3CA have been reported to increase its kinase activity [32], the I391M germline mutation has a frequency of 6% in the normal population. Hence, its significance is uncertain. Our study confirms the findings of Guillet et al [3], who recently reported PIK3CA and AKT1 mutations in 5 of 10 and 2 of 10 cases of JP, respectively. In our study and that of Guillet et al [3], it was the epithelial hyperplastic component of JP that was sequenced. It is unclear whether these mutations would be evident in other components of JP. Nonetheless, these studies expand the spectrum of benign breast lesions harboring mutations affecting PI3K‐AKT pathway‐related genes to include JP.
Interestingly, there are rare reports of JP occurring in patients with Cowden syndrome and Proteus syndrome [5], two syndromes affecting genes in the PI3K pathway. Patients with Cowden syndrome, caused by a germline PTEN mutation, develop hamartomatous lesions and are at risk of multiple cancers [33]. Proteus syndrome, characterized by progressive asymmetric growth of multiple tissue types, is caused by somatic mosaicism for an activating AKT1 mutation [34]. The presence of PI3K pathway mutations among these disorders suggests a possible link to the development of JP. Study of such lesions, however, is challenging due to their rarity.
Our study not only confirms the presence of PIK3CA hotspot mutations in JP, but also expands our understanding of the molecular relationship between JP and breast cancer, given that here we demonstrated in one case the presence of a clonal E542K PIK3CA hotspot mutation shared by separate components of JP, DCIS, and IDC‐NST. The IDC‐NST component was ER‐positive/HER2‐negative with no distinctive histologic features and displayed CNAs typical of ER‐positive disease. Other types of breast carcinoma including invasive lobular carcinoma [5] and secretory carcinoma [1, 35] have been reported in association with JP. Classic lobular carcinomas are usually ER‐positive/HER2‐negative tumors driven by CDH1 alterations and PIK3CA mutations [36], whereas secretory carcinoma is a low‐grade triple‐negative breast cancer with a recurrent ETV6‐NTRK3 fusion gene and is not driven by PI3K pathway alterations [37]. Whether JP shows similar genetic alterations as or is clonally related to associated special types of breast carcinoma driven by other molecular mechanisms is uncertain.
Finally, we observed a greater exposure to an APOBEC signature in in situ and invasive carcinoma, compared with JP, which showed a dominant exposure to an aging signature. Our observations suggest that APOBEC mutational processes might underpin the evolution of JP to the associated DCIS and IDC‐NST components, in a way akin to the shifts from aging‐related signatures to APOBEC‐related mutagenesis in the progression from lobular carcinoma in situ to invasive lobular cancer [7] and from primary tumors to metastasis [38].
This study has several limitations, including its small sample size due to the rarity of JP and only one of three JPs analyzed was associated with in situ and invasive carcinoma. Although we provide a proof‐of‐principle that JP may constitute the substrate from which DCIS and invasive breast cancers may develop, further studies are required to ascertain the frequency of this phenomenon. Despite these limitations, here we confirm the presence of recurrent PIK3CA mutations in JP and provide evidence that JP and coexisting carcinoma are clonally related, further expanding our understanding of the relationship between JP and breast cancer.
Author contributions statement
TMD, FP, EB, BW and JSR‐F conceived the study. TMD, EC and SAH provided samples. TMD, FP, MV, EC, SAH, YHW, EB and JSR‐F performed histopathological review. MV conducted the sample processing. ADCP, AG and LF performed the bioinformatics analysis. TMD, FP, ADCP, MV, AG, EMdS, BW and JSR‐F discussed and interpreted the results. TMD, FP and MV wrote the first draft. ADCP, AG, LF, EMdS, EC, LS, SC, HZ, SAH, YHW, EB, BW and JSR‐F read, edited, and approved the final manuscript.
Acknowledgements
This study was funded by the Breast Cancer Research Foundation. Research reported in this publication was funded in part by a Cancer Center Support Grant of the National Institutes of Health/National Cancer Institute (Grant No. P30CA008748). FP is partially funded by a National Institutes of Health/National Cancer Institute K12 CA184746 grant and BW by Cycle for Survival and Stand Up To Cancer grants. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest statement: JSR‐F reports receiving personal/consultancy fees from Goldman Sachs and REPARE Therapeutics, membership of the scientific advisory boards of VolitionRx and Paige.AI, and ad hoc membership of the scientific advisory boards of Roche Tissue Diagnostics, Ventana Medical Systems, Novartis, Genentech and InVicro, outside the scope of this study. SC has received research support from Daichi Sankyo and consulting fees from Novartis, Sermonix, BMS, Context Therapeutics, Revolution Medicine, Paige AI, and Eli Lilly. HZ reports receiving consultancy fee from Genentech/Roche. The remaining authors declare no conflicts of interest.
Contributor Information
Fresia Pareja, Email: parejaf@mskcc.org.
Jorge S Reis‐Filho, Email: reisfilj@mskcc.org.
Data availability statement
Whole‐exome sequencing data that support the findings of this study are available for visualization and download in cBioPortal for Cancer Genomics https://www.cbioportal.org/study/summary?id=brca_jup_msk_2020
References
- 1. Rosen PP, Cantrell B, Mullen DL, et al Juvenile papillomatosis (Swiss cheese disease) of the breast. Am J Surg Pathol 1980; 4: 3–12. [PubMed] [Google Scholar]
- 2. Rosen PP, Lyngholm B, Kinne DW, et al Juvenile papillomatosis of the breast and family history of breast carcinoma. Cancer 1982; 49: 2591–2595. [DOI] [PubMed] [Google Scholar]
- 3. Guillet C, Rechsteiner M, Bellini E, et al Juvenile papillomatosis of the breast (Swiss cheese disease) has frequent associations with PIK3CA and/or AKT1 mutations. Hum Pathol 2020; 98: 64–73. [DOI] [PubMed] [Google Scholar]
- 4. Rosen PP, Holmes G, Lesser ML, et al Juvenile papillomatosis and breast carcinoma. Cancer 1985; 55: 1345–1352. [DOI] [PubMed] [Google Scholar]
- 5. Cheng E, D'Alfonso T, Patel A, et al Mammary juvenile papillomatosis ("Swiss cheese" disease): study of 121 cases reiterates need for long‐term follow‐up. Breast J 2018; 24: 1136–1137. [DOI] [PubMed] [Google Scholar]
- 6. Pareja F, Brown DN, Lee JY, et al Whole‐exome sequencing analysis of the progression from non‐low‐grade ductal carcinoma in situ to invasive ductal carcinoma. Clin Cancer Res 2020; 26: 3682–3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li H, Durbin R. Fast and accurate short read alignment with burrows‐wheeler transform. Bioinformatics 2009; 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. McKenna A, Hanna M, Banks E, et al The genome analysis toolkit: a MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Res 2010; 20: 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cibulskis K, Lawrence MS, Carter SL, et al Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 2013; 31: 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Saunders CT, Wong WS, Swamy S, et al Strelka: accurate somatic small‐variant calling from sequenced tumor‐normal sample pairs. Bioinformatics 2012; 28: 1811–1817. [DOI] [PubMed] [Google Scholar]
- 11. Koboldt DC, Zhang Q, Larson DE, et al VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 2012; 22: 568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Narzisi G, O'Rawe JA, Iossifov I, et al Accurate de novo and transmitted indel detection in exome‐capture data using microassembly. Nat Methods 2014; 11: 1033–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Narzisi G, Corvelo A, Arora K, et al Genome‐wide somatic variant calling using localized colored de Bruijn graphs. Commun Biol 2018; 1: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ashley CW, Da Cruz Paula A, Kumar R, et al Analysis of mutational signatures in primary and metastatic endometrial cancer reveals distinct patterns of DNA repair defects and shifts during tumor progression. Gynecol Oncol 2019; 152: 11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative genomics viewer (IGV): high‐performance genomics data visualization and exploration. Brief Bioinform 2013; 14: 178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Carter SL, Cibulskis K, Helman E, et al Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol 2012; 30: 413–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Geyer FC, Li A, Papanastasiou AD, et al Recurrent hotspot mutations in HRAS Q61 and PI3K‐AKT pathway genes as drivers of breast adenomyoepitheliomas. Nat Commun 2018; 9: 1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Weigelt B, Bi R, Kumar R, et al The landscape of somatic genetic alterations in breast cancers from ATM Germline mutation carriers. J Natl Cancer Inst 2018; 110: 1030–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Landau DA, Carter SL, Stojanov P, et al Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell 2013; 152: 714–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ng CKY, Piscuoglio S, Geyer FC, et al The landscape of somatic genetic alterations in Metaplastic breast carcinomas. Clin Cancer Res 2017; 23: 3859–3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shen R, Seshan VE. FACETS: allele‐specific copy number and clonal heterogeneity analysis tool for high‐throughput DNA sequencing. Nucleic Acids Res 2016; 44: e131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chang MT, Asthana S, Gao SP, et al Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat Biotechnol 2016; 34: 155–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gulhan DC, Lee JJ, Melloni GEM, et al Detecting the mutational signature of homologous recombination deficiency in clinical samples. Nat Genet 2019; 51: 912–919. [DOI] [PubMed] [Google Scholar]
- 24. Smith ES, Da Cruz Paula A, Cadoo KA, et al Endometrial cancers in BRCA1 or BRCA2 Germline mutation carriers: assessment of homologous recombination DNA repair defects. JCO Precision Oncology 2019: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Reiter JG, Makohon‐Moore AP, Gerold JM, et al Reconstructing metastatic seeding patterns of human cancers. Nat Commun 2017; 8: 14114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lee JY, Schizas M, Geyer FC, et al Lobular carcinomas in situ display Intralesion genetic heterogeneity and clonal evolution in the progression to invasive lobular carcinoma. Clin Cancer Res 2019; 25: 674–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schultheis AM, Ng CK, De Filippo MR, et al Massively parallel sequencing‐based Clonality analysis of synchronous Endometrioid endometrial and ovarian carcinomas. J Natl Cancer Inst 2016; 108: djv427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Natrajan R, Lambros MBK, Geyer FC, et al Loss of 16q in high grade breast cancer is associated with estrogen receptor status: evidence for progression in tumors with a luminal phenotype? Genes Chromosomes Cancer 2009; 48: 351–365. [DOI] [PubMed] [Google Scholar]
- 29. Bersini S, Lytle NK, Schulte R, et al Nup93 Regulates breast tumor growth by modulating cell proliferation and Actin cytoskeleton remodeling. Life Sci Alliance 2020; 3: e201900623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ang DC, Warrick AL, Shilling A, et al Frequent phosphatidylinositol‐3‐kinase mutations in proliferative breast lesions. Mod Pathol 2014; 27: 740–750. [DOI] [PubMed] [Google Scholar]
- 31. Troxell ML, Levine J, Beadling C, et al High prevalence of PIK3CA/AKT pathway mutations in papillary neoplasms of the breast. Mod Pathol 2010; 23: 27–37. [DOI] [PubMed] [Google Scholar]
- 32. Gymnopoulos M, Elsliger MA, Vogt PK. Rare cancer‐specific mutations in PIK3CA show gain of function. Proc Natl Acad Sci U S A 2007; 104: 5569–5574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bubien V, Bonnet F, Brouste V, et al High cumulative risks of cancer in patients with PTEN hamartoma tumour syndrome. J Med Genet 2013; 50: 255–263. [DOI] [PubMed] [Google Scholar]
- 34. Lindhurst MJ, Sapp JC, Teer JK, et al A mosaic activating mutation in AKT1 associated with the Proteus syndrome. N Engl J Med 2011; 365: 611–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tokunaga M, Wakimoto J, Muramoto Y, et al Juvenile secretory carcinoma and juvenile papillomatosis. Jpn J Clin Oncol 1985; 15: 457–465. [PubMed] [Google Scholar]
- 36. Buttitta F, Felicioni L, Barassi F, et al PIK3CA mutation and histological type in breast carcinoma: high frequency of mutations in lobular carcinoma. J Pathol 2006; 208: 350–355. [DOI] [PubMed] [Google Scholar]
- 37. Krings G, Joseph NM, Bean GR, et al Genomic profiling of breast secretory carcinomas reveals distinct genetics from other breast cancers and similarity to mammary analog secretory carcinomas. Mod Pathol 2017; 30: 1086–1099. [DOI] [PubMed] [Google Scholar]
- 38. Angus L, Smid M, Wilting SM, et al The genomic landscape of metastatic breast cancer highlights changes in mutation and signature frequencies. Nat Genet 2019; 51: 1450–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Whole‐exome sequencing data that support the findings of this study are available for visualization and download in cBioPortal for Cancer Genomics https://www.cbioportal.org/study/summary?id=brca_jup_msk_2020