

Abstract
Protein arginine methyltransferase 5 (PRMT5) has previously been reported to be upregulated in many malignant tumors. This study investigated the significance of PRMT5 in endometrial carcinoma (EC) and explored its function in tumorigenesis. Immunohistochemistry was performed to evaluate PRMT5 expression in 62 EC and 66 endometrial hyperplasia samples. The functions of PRMT5 were investigated by cell counting kit‐8, plate colony formation, wound healing, and transwell and flow cytometry assays. Quantitative reverse transcription‐polymerase chain reaction and western blotting were used to measure the expression of PRMT5, changes in estrogen receptor α (ERα), and related functional proteins. Coimmunoprecipitation was performed to examine the interaction of PRMT5 with ERα and its coactivator steroid receptor coactivator‐1 (SRC1). Compared to endometrial hyperplasia tissue, PRMT5 was overexpressed in endometrioid adenocarcinoma (EAC) but not overexpressed in mucinous EC. The main expression pattern of PRMT5 in EAC was cytoplasmic. However, the positive cases of endometrial hyperplasia showed both cytoplasmic and nuclear positivity in the endometrial glands or were mainly positive in stromal cells. Knockdown of PRMT5 significantly inhibited the growth and migration ability of EAC cells and promoted their apoptosis by regulating cyclin D1, c‐myc, p53, and Bcl2 proteins. Furthermore, PRMT5 could form a complex with ERα and SRC1 to promote the expression of ERα. In conclusion, PRMT5 plays a significant role in the progression of EAC by interacting with ERα and impacting the cell cycle signaling pathways.
Keywords: endometrial cancer, epigenetics, PRMT5, ERα, cell function
Introduction
Endometrial carcinoma (EC) is one of the most common malignancies of the female reproductive tract [1], with increasing incidence and mortality rates [2]. According to Bockham's classification in 1983, EC can be divided into estrogen‐dependent (type I) and nonhormone‐dependent (type II) types. Nearly 70–80% of ECs are designated as type I, including endometrioid adenocarcinoma (EAC) and mucinous EC (MEC), showing well‐differentiated features, obesity, and hyperlipidemia. In contrast, those women (20–30%) who present with nonobesity‐related, poorly differentiated tumors and who are estrogen receptor (ER)‐negative are usually defined as having type II EC, which mainly includes cases of serous carcinoma and clear cell carcinoma [3, 4]. Types I and II ECs have distinct molecular characteristics. Type I EC is often related to mutations in the PTEN, POLE, K‐RAS, PIK3CA, and CTNNB1 (β‐catenin) genes. Type II EC usually exhibits alterations of TP53 or HER2/neu or loss of heterozygosity on several chromosomes [5, 6]. Molecular detection is of great significance for the classification of various tumors. The Cancer Genome Atlas database, and subsequent molecular detection based on it, revealed four subgroups of EAC: group 1, with somatic inactivating mutations in POLE and very high mutation rates; group 2, with microsatellite instability; group 3, with low copy number alterations; and group 4, serous‐like or copy number‐high alterations, with low mutation rate but frequent TP53 mutation. Groups 1, 2, and 3 are associated with good prognosis, but group 4 has a worse prognosis. This molecular classification forms the modern basis of our understanding of EC.
Both genetic changes and epigenetic modifications contribute to the development of EC [7, 8]. Histone methylation, as a main epigenetic modification, plays a crucial role in regulating gene expression and is involved in the development of various cancers [9, 10, 11]. Through histone methyltransferases, methyl groups can be transferred from S‐adenosyl methionine to lysine (K) or arginine (R) residues of histone proteins [12], and this process can be reversed by histone demethylases [13]. In recent years, increasing evidence has shown that protein arginine methyltransferase 5 (PRMT5), a type II arginine methyltransferase (PRMT), plays an important role in cancer progression [14, 15]. PRMT5 mediates the symmetric dimethylation of H2A, H2AR3, H3R8, H4R3, and several nonhistone substrates such as Sm proteins [16, 17]. PRMT5 is overexpressed in a variety of cancers, such as glioblastoma, colon cancer, lung cancer, breast cancer, and prostate cancer [18, 19, 20, 21, 22]. PRMT5 usually functions in the context of forming multimeric complexes with other components (e.g. MEP50 and Sp1), which can regulate PRMT5 localization and substrate specificity [23]. PRMT5 has distinct effects on stem cell development and cancer cell proliferation depending on whether it is located in the cytoplasm or the nucleus [24, 25]. It has been shown that cytoplasmic PRMT5 is essential for the growth of prostate cancer cells, whereas nuclear PRMT5 inhibits prostate cancer cell growth [24]. Deng et al reported that, in the nucleus, PRMT5 can be recruited to the androgen receptor (AR) promoter through the interaction with Sp1 and acts as an epigenetic activator of AR in prostate cancer [22].
Thus far, very little is known about the function of PRMT5 in EC. In our previous study, we found that PRMT5 can form a complex with UHRF1 in EAC cells [26]. In the present study, we detected the expression of PRMT5 in type I EC samples, including EAC and MEC, and compared them with a series of hyperplastic endometrial tissues. Our data showed that PRMT5 is overexpressed in EAC but not in MEC. We subsequently evaluated the correlation between PRMT5 expression and the clinicopathological features of EAC. In addition, the biological functions of PRMT5 were further investigated by knockdown of PRMT5 in Ishikawa cells. Given that EAC is an estrogen‐dependent disease, we also explored the regulatory effect of PRMT5 on ERα and its coactivator steroid receptor coactivator‐1 (SRC1). Our study reported, for the first time, that PRMT5 plays an important role in EAC, but not MEC, and revealed its possible mechanism in EAC development.
Materials and methods
Patients and tissue samples
A total of 128 human endometrial samples were collected from the Department of Pathology of Shandong University Qilu Hospital between January 2006 and January 2017. They include 42 cases of EAC (aged 33–78 years, mean 57 years), 20 cases of MEC (aged 41–73 years, mean 59 years), 11 cases of atypical hyperplasia (AH) (aged 29–66 years, mean 44 years), 22 cases of complex hyperplasia, and 33 cases of simple hyperplasia. All the cases of EC were diagnosed for the first time, and the samples were collected after surgery. We excluded patients with a history of other tumors, radical surgery, radiotherapy, and chemotherapy treatment prior to the surgery. TP53 mutant type I EC cases were also excluded. All sections of selected cases were reexamined by two experienced gynecological pathologists. Tumor grade and stage were established according to the World Health Organization Classification (2014) and the International Federation of Gynecology Obstetrics (FIGO, 2018), respectively. This study was approved by the Shandong University Ethics Committee (201302036).
Immunohistochemistry
According to peroxidase–antiperoxidase protocols, the sections (3 μm) were dewaxed and dehydrated in xylene and gradient alcohols successively, followed by antigen retrieval with ethylenediaminetetraacetic acid (EDTA) (pH = 9; Zsbio, Beijing, PR China). Samples were incubated with anti‐PRMT5 (1:80; ab109451, Abcam, Cambridge, MA, USA) overnight at 4 °C. After reacting with the polymer enhancer and secondary antibody (PV9000; Zsbio) for 30 min, the sections were stained with 3,3'‐diaminobenzidine (DAB) (Boster, Wuhan, PR China) for 3 min. An automated immunohistochemistry system (Bond‐III; Leica Biosystems, Vista, CA, USA) was used for the detection of estrogen receptor (ER) (Kit‐0012; Maxim Biotechnologies, Fuzhou, PR China), progesterone receptor (PR) (Kit‐0013; Maxim Biotechnologies, Fuzhou, PR China), Ki‐67 (ZM‐0166; Zsbio), and p53 (ZA‐0501; Zsbio) antibodies without dilution. ER and PR antigen retrieval was performed with a citrate buffer (pH = 6; Maxim Biotechnologies), and Ki‐67 antigens are retrieved with EDTA (pH = 8; Zsbio). All sections were counterstained with hematoxylin and sealed with a neutral resin.
A semiquantitative method was used to evaluate immunohistochemistry results. The criteria for judging the results of PRMT5, ER, and PR were as follows: The intensity (set as X) and the proportion (set as Y) of DAB cell staining was taken into consideration. In terms of staining intensity, 0 points: no staining in neoplastic cells; 1 point: weak staining; 2 points: moderate staining; and 3 points: strong staining. In terms of the proportion of stained cells, 0 points: no positive cells were counted in neoplastic cells; 1 point: <20% positive cells were counted, 2 points: the percentage of stained cells was ≥20 and <50%, 3 points: the percentage of stained cells was ≥50 and <80%, and 4 points: ≥80% positive cells were counted. The two scores were multiplied to obtain the final score, which ranged from 0 to 12 (final score = X × Y). Regarding p53 expression, all 42 cases of EAC showed nonaberrant expression of p53, and the proportion of positive cells was >0 and <70%.
Cell culture and estrogen treatments
The endometrioid carcinoma cell line Ishikawa was obtained from Procell LifeScience & Technology Company (Wuhan, PR China) and authenticated by short tandem repeat profiling. To observe the effect of estrogen (β‐estradiol, E2) (Sigma‐Aldrich, Santa Clara, CA, USA) on PRMT5 expression, the Ishikawa cells were cultured in phenol‐free medium RPMI 1640 (Macgene, Beijing, PR China) containing 10% activated carbon‐filtered fetal bovine serum (Biological industries, Beit HaEmek, Israel) in 6‐well plates for 24 h. Six concentration gradients of E2 were prepared and added to the Ishikawa cells once a day. The final concentrations of E2 were 0, 10−7, 10−6, 10−5, 10−4, and 10−3 mm. Total proteins were collected after 4 days of E2 treatment, and western blotting was performed to detect the PRMT5 expression.
Plasmid construction and transfection
Scrambled control (SC) and PRMT5‐specific knockdown (pGPU6/GFP/Neo‐PRMT5‐homo) plasmids were provided by GenePharma (Shanghai, PR China). The functional sequence of the short hairpin RNA (shRNA) was GCCATCACTCTTCCATGTTCT (#743). shRNA‐PRMT5 was transfected into cells using Lipofectamine™ 3000 Reagent (Thermo Scientific, Rockford, IL, USA) diluted in Opti‐MEM (Gibco, Carlsbad, CA, USA) in accordance with the manufacturer's instructions. The ratio of Lipofectamine 3000 to P3000™ to plasmid was 5:5:2. The transfected cells were incubated for 48 h (for mRNA) or 72 h (for protein).
Quantitative reverse transcription‐polymerase chain reaction
To predict the downstream target gene of PRMT5, shRNA‐PRMT5 was transfected into Ishikawa cells for 48 h, and total RNA was extracted with a TRIzol reagent (Life Technologies, Carlsbad, CA, USA). cDNA was obtained by reverse transcription using a First Strand cDNA Synthesis Kit (Millipore, Billerica, MA, USA) as per the manufacturer's instructions. Quantitative reverse transcription‐polymerase chain reaction (PCR) was performed using a Mastercycler X50 (Eppendorf, Westbury, NY, USA), with Real‐Time PCR SYBR Green (Toyobo, Kobe, Japan) to measure mRNA. GAPDH was used as the internal reference. Primer sequences were as follows: GAPDH, forward, 5'‐GCACCGTCAAGGCTGAGAAC‐3'; reverse, 5'‐TGGTGAAGACGCCAGTGGA‐3'; PRMT5, forward, 5'‐CCGTGGTGACGCTAGAGAAC‐3'; reverse, 5'‐CAGCAAATGAGCCCAGAAGC‐3'; and ESR1, forward, 5'‐TCTTGGACAGGAACCAGGGA‐3'; reverse, 5'‐TGGTGAAGACGCCAGTGGA‐3'.
Western blotting
Equal amounts of protein were loaded, and different molecular‐weight proteins were isolated by means of a PAGE Gel Fast Preparation Kit (EpiZyme, Shanghai, PR China). Antibodies against PRMT5, ERα, p53, Bcl‐2, and cyclin D1 were procured from Abcam, and antibodies against GAPDH, SRC1, and c‐myc were obtained from Abways (Shanghai, PR China). The polyvinylidene fluoride (PVDF) membrane was incubated in a second antibody (goat anti‐rabbit/mouse IgG (H + L)/horseradish peroxidase (HRP) [Zsbio]) for 40 min and was then exposed in a chemiluminescence imaging system (Clinx, Shanghai, PR China) and developed with Immobilon Western Chemilum HRP Substrate (Millipore). The band intensity was analyzed by Image‐J (National Institutes of Health, Bethesda, MD, USA), and GAPDH served as an internal reference.
Immunoprecipitation of PRMT5 with SRC1 and ERα
A Pierce Co‐Immunoprecipitation (Co‐IP) Kit (26149) (Thermo Scientific) was used according to the manufacturer's directions to determine whether PRMT5 could form a complex with ERα or SRC1. In brief, AminoLink Plus Coupling Resin was first immobilized with anti‐PRMT5/anti‐ERα (Abcam) antibody in the coupling buffer. The cell sediments were lysed with IP lysis/wash buffer and incubated with control agarose resin for 30 min at 4 °C. Half of the sample was taken as input after denaturation, and the other half was incubated with the resin fixed with anti‐PRMT5 on a rotary culture instrument overnight. The compound was mixed with elution buffer in a spin column and centrifuged. The flow‐through was collected for sodium dodecyl sulfate polyacrylamide gel electrophoresis analysis.
Cell proliferation assay
A cell counting kit (CCK)‐8 kit (BestBio, Shanghai, PR China) was used to determine the effect of PRMT5 knockdown on cell proliferation function. The transfected cells were placed into 96‐well plates, with 2000 cells in 100 μl of culture medium per well. Next, 10 μl of the CCK‐8 reagent was added at 0, 24, 48, 72, and 96 h, followed by further incubation at 37 °C for 90 min. The absorbance was measured using an ultraviolet spectrophotometer (Eppendorf, Hamburg, Germany) with the wavelength set at 450 nm. Three auxiliary wells were set for each assay, and the assays were repeated thrice.
Cell scratch assay
To study the effect of the loss of PRMT5 expression on cell migration ability, a straight line was drawn at the center of the plate when the degree of cell aggregation was greater than 90%. The samples were observed and photographed under the microscope at 0, 24, and 48 h. The distance of cell migration to the central line was then analyzed. This experiment was repeated thrice.
Cell migration and invasion assay
A chamber (Corning, New York, NY, USA) experiment was conducted to observe the effect of PRMT5 on the migration and invasion ability of cancer cells. Transfected cells were suspended in serum‐free Dulbecco's modification of Eagle's medium (DMEM), and 5 × 105 cells were counted in the migration assay (8 × 105 cells were counted in the cell invasion assay). Cells were thoroughly mixed with 200 μl of serum‐free DMEM and dropped into the chamber. After 22 h, cells were fixed for 20 min with 4% paraformaldehyde (Solarbio, Beijing, PR China) and then treated with crystal violet (Guangfu, Tianjin, PR China) solution for 20 min. The cells were then counted on the polycarbonate membrane.
Colony formation assay
Transfected cells were added to a 6‐well plate (800 cells per well) with 2 ml of normal serum medium. After 18 days, the cells were fixed for 20 min in 4% paraformaldehyde and then treated with crystal violet solution for 20 min. The number of colonies was observed and counted by naked eye.
Flow cytometry to detect cell apoptosis and cell cycles
The effect of PRMT5 knockdown on apoptotic function was detected by flow cytometry (Cytomics™ FC 500; Beckman Coulter, Brea, CA, USA) using an annexin V‐PE/7‐AAD double‐staining cell apoptosis assay kit (KeyGEN, Jiangsu, PR China) per the manufacturer's instructions. Three days after transfection, approximately 1–5 × 105 cells were collected and washed with cold phosphate‐buffered saline. The cells were suspended in a mixture of 7‐AAD and binding buffer and were fully reacted at room temperature in the dark for 15 min. In addition, binding buffer and annexin V‐PE were added to an aliquot of cells, and the cells were incubated at room temperature for 15 min in the dark. The cells were then detected by flow cytometry.
The effect of PRMT5 inhibition on the cell cycle was detected by flow cytometry (Cytomics FC 500) using a 401033 kit (BestBio) per the manufacturer's instructions. The excitation wavelength was set at 488 nm to obtain the cell cycle results.
Statistical analysis
All in vitro experiments were repeated in triplicate. The quantitative data are presented as the mean ± SD. The significance of the difference between groups was evaluated with the t‐test and repeated‐measure analysis of variance. The Mann–Whitney U‐test and Kruskal–Wallis H‐test were used to analyze the relationship between clinicopathological factors and PRMT5 expression in EAC. All calculations were performed using SPSS 22.0 for Windows (SPSS, Inc., Chicago, IL, USA). A P value of <0.05 was considered to be statistically significant.
Results
PRMT5 expression in EC and its clinicopathological significance in EAC
To test whether PRMT5 can act as a diagnostic or progression marker for EC, its expression was examined in a series of endometrial tissues, including 66 endometrial hyperplasia tissues and 62 endometrial cancer tissues. PRMT5 expression was found in 39% (13/33) of simple hyperplasia, 77% (17/22) of complex hyperplasia, 90% (10/11) of AH, and 95% (40/42) of EAC cases, but it was not found in any MEC case (0/20). Interestingly, PRMT5 showed different subcellular localization patterns in EAC and hyperplasia endometrial tissues (Figure 1). In the 40 PRMT5‐positive EAC cases, PRMT5 expression showed cytoplasmic staining, with limited nuclear localization (Figure 1F). However, in 15 of 22 positive cases of complex hyperplasia, 6 of 33 positive cases of simple hyperplasia, and all AH‐positive cases, PRMT5 was expressed both in the glandular cytoplasm and in the nucleus. In two other positive cases of complex hyperplasia and seven positive cases of simple hyperplasia, PRMT5 was detected only in endometrial stromal cells and not in endometrial glands.
Figure 1.
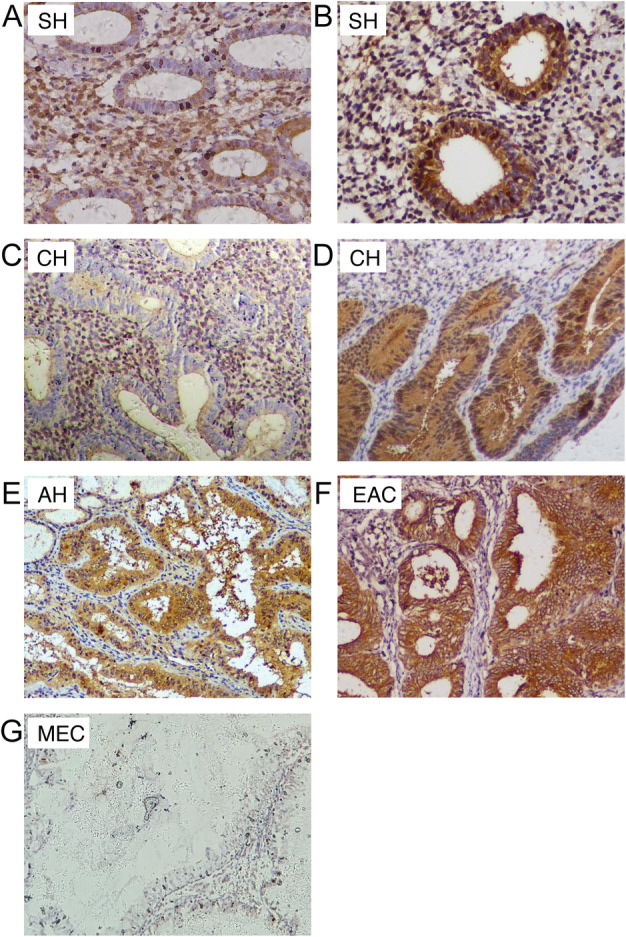
The expression pattern of PRMT5 in EC and a series of endometrial hyperplasia. (A) PRMT5 was mainly expressed in stromal cells and in the cytoplasm and nuclei of some glandular cells in some cases of SH; (B) a PRMT5‐positive case of SH (both glandular cytoplasmic and nuclear positivity, with stromal cell positivity); (C) PRMT5 was mainly expressed in stromal cells in some cases of CH; (D) PRMT5‐positive case of CH (both glandular nuclear and cytoplasmic positivity); (E) PRMT5‐positive case of AH (moderate intensity cytoplasmic positivity, with a small number of positive nuclei); (F) PRMT5 expression showed strong cytoplasmic staining and limited nuclear localization in EAC; (G) PRMT5 was barely expressed in MEC. AH, atypical hyperplasia; CH, complex hyperplasia; EAC, endometrioid adenocarcinoma; MEC, mucinous endometrial carcinoma; SH, simple hyperplasia.
We also analyzed the relationship between PRMT5 expression and the clinicopathological features of EAC (Table 1). This analysis revealed that PRMT5 was more frequently expressed in the ERα group with high scores compared to the group with low scores (p = 0.007). The expression of PRMT5 was higher in grades 1 and 2 groups than that in the grade 3 group. In addition, our data also demonstrated that PRMT5 expression was positively correlated with the Ki‐67 index in EAC tissues (p = 0.015). No statistically significant differences were found between PRMT5 expression and patient age, depth of myometrial invasion, cervical involvement, or FIGO stage.
Table 1.
The correlation between PRMT5 expression and the clinicopathological features in EAC.
| Clinicopathological feature | Number of patients | PRMT5 expression (score) | P value | ||
|---|---|---|---|---|---|
| 0 | 1–4 | 5–12 | |||
| Age (years) | 0.206 | ||||
| ≤45 | 10 | 1 | 4 | 5 | |
| >45 | 32 | 1 | 8 | 23 | |
| Myometrial invasion | 0.897 | ||||
| <1/2 | 28 | 0 | 10 | 18 | |
| ≥1/2 | 14 | 2 | 2 | 10 | |
| Cervical involvement | 1.000 | ||||
| Uninvolved | 37 | 2 | 10 | 25 | |
| Involved | 5 | 0 | 2 | 3 | |
| Lymph node metastasis | 0.093 | ||||
| − | 39 | 1 | 11 | 27 | |
| + | 3 | 1 | 1 | 1 | |
| Adnexal metastasis | 1.000 | ||||
| − | 36 | 2 | 10 | 24 | |
| + | 6 | 0 | 2 | 4 | |
| ER (score) | 0.007* | ||||
| 0–4 | 18 | 2 | 8 | 8 | |
| 5–12 | 24 | 0 | 4 | 20 | |
| PR (score) | 0.136 | ||||
| 0–4 | 11 | 0 | 6 | 5 | |
| 5–12 | 31 | 2 | 6 | 23 | |
| p53 | 0.941 | ||||
| Wild type (≥1 and <30%) | 38 | 2 | 11 | 25 | |
| Wild type (≥30 and <70%) | 4 | 0 | 1 | 3 | |
| Ki‐67 | 0.015* | ||||
| ≤30% | 19 | 2 | 8 | 9 | |
| >30% | 23 | 0 | 4 | 19 | |
| Grade | 0.016* | ||||
| G1 | 21 | 0 | 4 | 17 | 1.000 † |
| G2 | 14 | 0 | 4 | 10 | 0.017 ‡ |
| G3 | 7 | 2 | 4 | 1 | 0.054 § |
| FIGO stage | 0.762 | ||||
| I | 32 | 1 | 9 | 22 | |
| II | 3 | 0 | 1 | 2 | |
| III | 7 | 1 | 2 | 4 | |
| IV | 0 | 0 | 0 | 0 | |
The expression of PRMT5 in G1, G2, and G3 EAC was compared in pairs.
P < 0.05.
G1 versus G2.
G2 versus G3.
G1 versus G3.
Knockdown of PRMT5 inhibits EAC cell proliferation, migration, and invasion in vitro
To investigate the function of PRMT5 in EAC, we constructed a knockdown plasmid pGPU6/GFP/Neo‐PRMT5‐homo and transfected it into Ishikawa cells. A CCK‐8 assay was performed to test the proliferation of the EAC cells. The results showed that cell proliferation was inhibited after 48 h (p = 0.002) (Figure 2A). We then performed a plate colony formation assay to investigate the influence of PRMT5 on the clonality and cell cycle of EAC cells. Compared with SC, knockdown of PRMT5 decreased the colony formation ability of Ishikawa cells after 18 days (p = 0.001) (Figure 2B).
Figure 2.
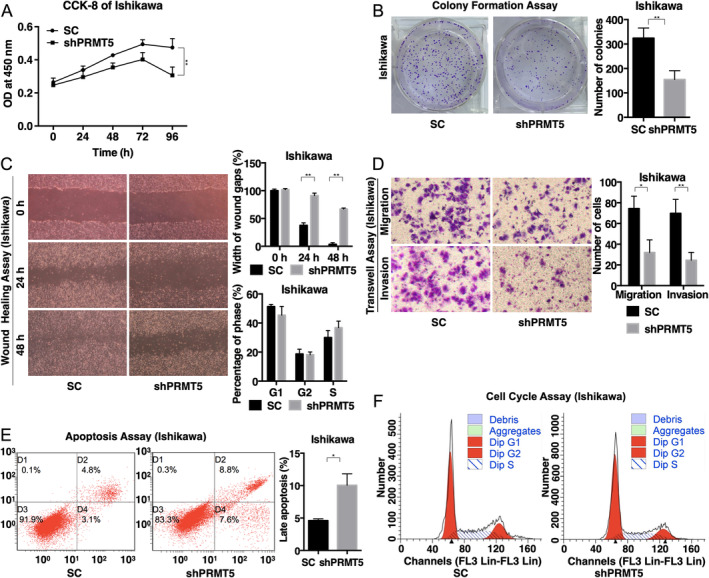
Knockdown of PRMT5 inhibited EAC cell proliferation, migration, and invasion in vitro. (A) CCK‐8 assay showed that the proliferation of Ishikawa cells was significantly inhibited after 48 h; (B) plate colony formation assay showed that silencing PRMT5 impaired the colony formation ability of Ishikawa cells after 18 days; (C) wound‐healing assays revealed that the width of wound gaps in PRMT5‐knockdown cells was markedly wider than that in SC cells at 48 h; (D) transwell assays indicated that the number of cells passing through the chamber (or matrix for the invasion test) was significantly reduced after transfection for 22 h; (E) the number of apoptotic cells increased significantly when PRMT5 was stably suppressed; (F) no significant difference was found for the effect of PRMT5 knockdown on the cell cycle by flow cytometry. SC, scrambled control; shPRMT5, short hairpin PRMT5; *p < 0.05 and **p < 0.01.
Invasion and metastasis are the main biological characteristics of malignant tumors. Therefore, we carried out wound‐healing and transwell assays to measure the migration and invasion ability of EAC cells after PRMT5 knockdown. The results showed that the width of wound gaps in PRMT5‐knockdown cells was markedly wider than that in SC after 48 h (p < 0.001) (Figure 2C). Transwell assays were also conducted to detect the migration and invasion ability of PRMT5‐knockdown EAC cells. It showed that the number of cells passing through the chamber (or matrix for the invasion test) was significantly reduced after transfection for 22 h (p = 0.013 for migration and p = 0.001 for invasion of Ishikawa cells) (Figure 2D). Taken together, these findings revealed that the knockdown of PRMT5 could significantly inhibit the migration ability of EAC cells.
Flow cytometry was applied to detect apoptosis and the cell cycle of Ishikawa cells 72 h after PRMT5 knockdown. As shown in Figure 2E, the number of apoptotic cells increased significantly when PRMT5 was stably suppressed. However, no significant difference was found for the cell cycle regarding PRMT5 knockdown (Figure 2F).
PRMT5 can form a complex with ERα and SRC1
Type I endometrial cancers usually express ERα, and Ishikawa cells are an ideal cell line that stably expresses ERα. To explore whether PRMT5 actually participates in the ERα signaling pathway, we detected changes in the ERα mRNA and protein levels in Ishikawa cells after PRMT5 knockdown. ERα transcription was suppressed at both the mRNA and protein levels after knockdown of PRMT5 (Figure 3A), indicating that ERα is probably a downstream target gene of PRMT5.
Figure 3.
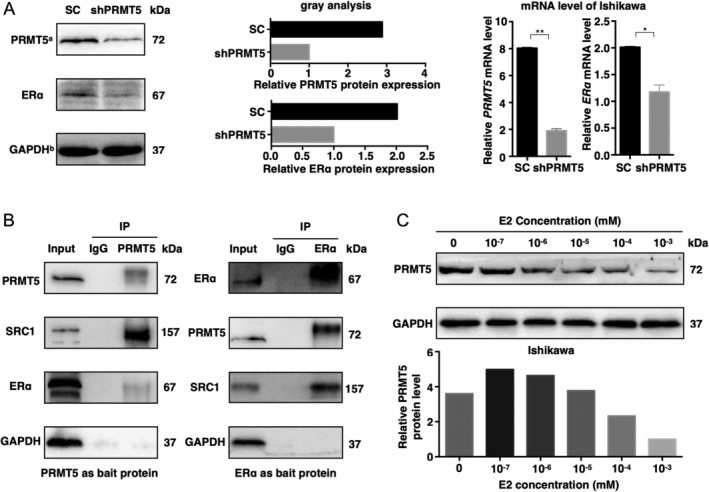
PRMT5 forms a complex with ERα and SRC1, and knockdown of PRMT5 inhibits ERα expression. (A) ERα transcription is suppressed at both the mRNA and protein levels after PRMT5 knockdown. PRMT5a and GAPDHb: the bands of PRMT5 and GAPDH (SC and shPRMT5) shown here are the same as those in Figure 4 but for different purposes; we aim to show the relationship between PRMT5 and ERα here. (B) Using PRMT5 and ERα as bait proteins, PRMT5, SRC1, and ERα could form a complex in EAC. (C) PRMT5 expression decreased with increasing E2 (β‐estradiol) dosage concentration (10−7–10−3 mm). SC, scrambled control; shPRMT5, short hairpin PRMT5.
To further investigate how PRMT5 regulates ERα expression, we examined the interaction between PRMT5 and SRC1, a transcriptional coregulatory protein that contains several nuclear receptor‐interacting domains and intrinsic histone acetyltransferase activity, which is crucial to the transcriptional activation of ERα by binding to the proximal promoter region of the downstream gene of ERα. PRMT5 was found to be coimmunoprecipitated with SRC1 and ERα in the Ishikawa cells, suggesting that PRMT5, SRC1, and ERα work together to form a complex in EAC (Figure 3B). This finding provides additional evidence that PRMT5 is required to promote EAC through ERα.
Type I EC arises in the background of unopposed estrogen exposure. To explore the effect of estrogen (E2) on PRMT5, Ishikawa cells were subjected to gradient E2 dosing. PRMT5 expression decreased in a dose‐dependent manner as the E2 dosage concentration increased (10−7–10−3 mm) (Figure 3C).
PRMT5 is involved in cyclin D1, c‐myc, p53, and Bcl2 signaling pathways
To further determine the pathways that PRMT5 may be involved in during EAC malignant transformation, we detected the proteins after PRMT5 knockdown. Western blotting analysis showed that cyclin D1 and c‐myc proteins were significantly reduced after knockdown (Figure 4). Both c‐myc and cyclin D1 are the target genes of estrogen [27] and are involved in the regulation of cell proliferation. Correspondingly, CCK‐8 and plate colony formation assays were used to validate that PRMT5‐silenced cell growth was markedly suppressed in vitro.
Figure 4.
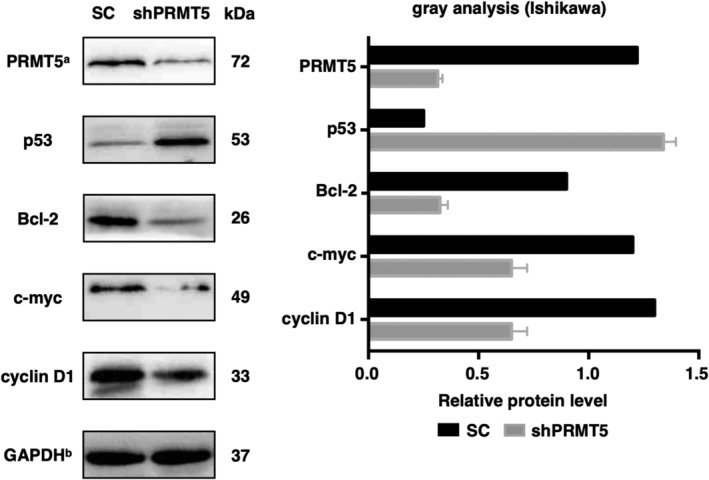
The changes of related proteins after PRMT5 knockdown. Cyclin D1, Bcl‐2, and c‐myc proteins were significantly reduced, while p53 expression was increased after knockdown of PRMT5 in Ishikawa cells. PRMT5a and GAPDHb: the bands of PRMT5 and GAPDH (SC and shPRMT5) shown here are the same as those in Figure 3A but for different purposes; we aim to show the relationship between PRMT5 and involved signaling pathways here. SC, scrambled control; shPRMT5, short hairpin PRMT5.
Our study also showed that PRMT5 inhibition in Ishikawa cells led to a marked upregulation of p53 expression, which is responsible for cellular proapoptosis. We also compared the levels of Bcl‐2 protein in PRMT5‐knockdown cells and untreated cells. Bcl‐2 expression decreased by 60% in Ishikawa cells after PRMT5 knockdown. This result is consistent with our finding that PRMT5 inhibited Ishikawa cell apoptosis.
Discussion
Previous studies have shown that PRMT5 is overexpressed in various cancers and is involved in signaling pathways affecting apoptosis and malignant transformation [28, 29, 30, 31, 32], but little is known about the clinical significance of PRMT5 in EC. In our study, PRMT5 was highly expressed in EAC, but not in MEC, compared with endometrial hyperplasia tissues, indicating that PRMT5 may act as a biomarker for distinguishing EAC from MEC.
Different subcellular localizations of PRMT5 have different biological functions. Gu et al discovered that PRMT5 was localized in the nucleus in benign prostate epithelium, whereas it was localized in the cytoplasm in prostate premalignancy and cancer [24]. In our case, the expression of PRMT5 was localized in the nucleus and cytoplasm in complex hyperplasia and simple hyperplasia tissues, but it was only localized in the cytoplasm in EC cells, consistent with the observations in the prostate. The relationship between PRMT5 expression and the clinicopathological features of EAC revealed that PRMT5 expression correlated with ERα, tumor grade, and Ki‐67 index. It should be pointed out that PRMT5 expression correlated positively with Ki‐67 index, but the expression of PRMT5 was decreased in poorly differentiated cases. One of the possible reasons may be that most of the biomarkers, including PRMT5, were lost due to the poor tumor differentiation. We further evaluated the biological significance of PRMT5 in EAC cells. After knockdown of PRMT5 in Ishikawa cells, the proliferation, metastasis, and invasion abilities of EAC cells were inhibited, confirming that PRMT5 participated in the progression of EAC cells.
EAC is an estrogen‐dependent cancer and highly expresses ERα. Increased expression of ERα is associated with EAC development and progression by activating downstream oncogenes [33, 34]. Studies have indicated that, in human prostate cancer cells, PRMT5 regulates prostate cancer cell growth in an AR‐dependent manner. Through interactions with Sp1 and Brg1 in the AR promoter region, PRMT5 activates AR transcription epigenetically [22]. In our study, PRMT5 expression was found to correlate with ERα expression; we therefore hypothesized that PRMT5 might be involved in the development of endometrial cancer cells by regulating ERα expression. Our results demonstrated that knockdown of PRMT5 could inhibit ERα expression, consistent with our previous immunohistochemistry result, where PRMT5 expression correlated positively with ERα expression in tissues, indicating that PRMT5 may activate ERα in EAC. However, whether PRMT5 regulates ERα transcription epigenetically or via the regulation of ERα transcriptional regulators requires further analysis. Co‐IP assays revealed that PRMT5 could form a complex with ERα and SRC1. SRC1 contains multiple nuclear receptor interaction domains at the AD1 or AD2 domain that can bind with ERα and then be recruited to the DNA promoter region, acting as a primary coactivator. It has been reported that histone methyltransferases such as CARM1/PRMT4 and PRMT1 can interact with SRC1 to enhance the transcription activity of nuclear receptors as a secondary coactivator of ERα [35]. In addition to SRC1, PRMT2 was also found to directly interact with AF‐1 (one subunit of ERα) to regulate ERα activation [36]. Here, we demonstrated that PRMT5 could also bind to SRC1 and promote the transcriptional function of ERα, just as the other PRMTs. In addition, we also treated Ishikawa cells with E2 and found that E2 could inhibit PRMT5 expression in a dose‐dependent manner upon increasing dosage concentrations of E2, indicating a feedback loop between PRMT5 expression and the ER signaling pathway.
In our study, knockdown of PRMT5 could markedly decrease the expression of cyclin D1, c‐myc, and Bcl2 while upregulating p53 expression in EAC cells. Chung et al found that PRMT5 inhibition with either shRNA‐mediated knockdown or a specific small‐molecule PRMT5 inhibitor not only leads to the derepression of WNT antagonists, but also results in decreased transcription of WNT/β‐catenin target genes, cyclin D1, c‐myc, and survivin and enhanced lymphoma cell death [37]. Li et al reported that inactivation of PRMT5 inhibits colony‐forming activity by multiple oncogenic drivers, including cyclin D1, c‐myc, Notch1, and MLL‐AF9 [38]. Furthermore, they demonstrated that PRMT5 overexpression specifically cooperates with cyclin D1 to drive lymphomagenesis in a mouse model. The tumor suppressor p53 functions as a transcription factor that regulates the expression of target genes linked to cell cycle arrest, apoptosis, senescence, and DNA repair [39, 40, 41, 42]. Tan et al found that loss of PRMT5 activity leads to endogenous DNA damage that triggers p53 activation and induces apoptosis in hematopoietic stem cells [43]. Consistent with these findings, our data demonstrated that PRMT5 promotes the development of EAC by regulating cyclin D1, c‐myc, p53, and Bcl2.
In conclusion, our study has demonstrated for the first time that PRMT5 is highly expressed in EAC but not in MEC compared with endometrial hyperplasia, providing a helpful biomarker for the early diagnosis of EAC and for the differential diagnosis between EAC and MEC. PRMT5 can form a complex with SRC1 and ERα to promote EAC development by regulating cyclin D1, c‐myc, p53, and Bcl2, implying that PRMT5 may serve as a potential therapeutic target for EAC.
Author contributions statement
SM performed the majority of the experiments and wrote the initial manuscript together with CZ. JW and HL were responsible for patient sample collection. KL was responsible for immunochemistry. SG was responsible for the analysis and interpreting data. XJ, XZ and CX were responsible for the cell experiments. TZ was responsible for experimental design. All authors contributed to writing and revising the manuscript.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 81372810) and the Natural Science Foundation of Shandong (No. ZR2018MH020).
No conflicts of interest were declared.
References
- 1. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin 2012; 62: 10–29. [DOI] [PubMed] [Google Scholar]
- 2. Balch C, Matei DE, Huang TH, et al Role of epigenomics in ovarian and endometrial cancers. Epigenomics 2010; 2: 419–447. [DOI] [PubMed] [Google Scholar]
- 3. Bokhman JV. Two pathogenetic types of endometrial carcinoma. Gynecol Oncol 1983; 15: 10–17. [DOI] [PubMed] [Google Scholar]
- 4. Talhouk A, McAlpine JN. New classification of endometrial cancers: the development and potential applications of genomic‐based classification in research and clinical care. Gynecol Oncol Res Pract 2016; 3: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Piulats JM, Guerra E, Gil‐Martin M, et al Molecular approaches for classifying endometrial carcinoma. Gynecol Oncol 2017; 145: 200–207. [DOI] [PubMed] [Google Scholar]
- 6. Karnezis AN, Leung S, Magrill J, et al Evaluation of endometrial carcinoma prognostic immunohistochemistry markers in the context of molecular classification. J Pathol Clin Res 2017; 3: 279–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zysman MA, Chapman WB, Bapat B. Considerations when analyzing the methylation status of PTEN tumor suppressor gene. Am J Pathol 2002; 160: 795–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Semczuk A, Boltze C, Marzec B, et al p16INK4A alterations are accompanied by aberrant protein immunostaining in endometrial carcinomas. J Cancer Res Clin Oncol 2003; 129: 589–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Xu Z, Taylor JA. Genome‐wide age‐related DNA methylation changes in blood and other tissues relate to histone modification, expression and cancer. Carcinogenesis 2014; 35: 356–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schrump DS, Nguyen DM. Novel molecular targeted therapy for esophageal cancer. J Surg Oncol 2005; 92: 257–261. [DOI] [PubMed] [Google Scholar]
- 11. Carvalho S, Freitas M, Antunes L, et al Prognostic value of histone marks H3K27me3 and H3K9me3 and modifying enzymes EZH2, SETDB1 and LSD‐1 in colorectal cancer. J Cancer Res Clin Oncol 2018; 144: 2127–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rice JC, Briggs SD, Ueberheide B, et al Histone methyltransferases direct different degrees of methylation to define distinct chromatin domains. Mol Cell 2003; 12: 1591–1598. [DOI] [PubMed] [Google Scholar]
- 13. Bannister AJ, Schneider R, Kouzarides T. Histone methylation: dynamic or static? Cell 2002; 109: 801–806. [DOI] [PubMed] [Google Scholar]
- 14. Shailesh H, Zakaria ZZ, Baiocchi R, et al Protein arginine methyltransferase 5 (PRMT5) dysregulation in cancer. Oncotarget 2018; 9: 36705–36718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Saha K, Fisher ML, Adhikary G, et al Sulforaphane suppresses PRMT5/MEP50 function in epidermal squamous cell carcinoma leading to reduced tumor formation. Carcinogenesis 2017; 38: 827–836. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 16. Huang L, Liu J, Zhang XO, et al Inhibition of protein arginine methyltransferase 5 enhances hepatic mitochondrial biogenesis. J Biol Chem 2018; 293: 10884–10894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pesiridis GS, Diamond E, Van Duyne GD. Role of pICLn in methylation of Sm proteins by PRMT5. J Biol Chem 2009; 284: 21347–21359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mongiardi MP, Savino M, Bartoli L, et al Myc and Omomyc functionally associate with the protein arginine methyltransferase 5 (PRMT5) in glioblastoma cells. Sci Rep 2015; 5: 15494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rastetter RH, Blomacher M, Drebber U, et al Coronin 2a (CRN5) expression is associated with colorectal adenoma‐adenocarcinoma sequence and oncogenic signalling. BMC Cancer 2015; 15: 638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gu Z, Gao S, Zhang F, et al Protein arginine methyltransferase 5 is essential for growth of lung cancer cells. Biochem J 2012; 446: 235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yang F, Wang J, Ren HY, et al Proliferative role of TRAF4 in breast cancer by upregulating PRMT5 nuclear expression. Tumour Biol 2015; 36: 5901–5911. [DOI] [PubMed] [Google Scholar]
- 22. Deng X, Shao G, Zhang HT, et al Protein arginine methyltransferase 5 functions as an epigenetic activator of the androgen receptor to promote prostate cancer cell growth. Oncogene 2017; 36: 1223–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Greenblatt SM, Liu F, Nimer SD. Arginine methyltransferases in normal and malignant hematopoiesis. Exp Hematol 2016; 44: 435–441. [DOI] [PubMed] [Google Scholar]
- 24. Gu Z, Li Y, Lee P, et al Protein arginine methyltransferase 5 functions in opposite ways in the cytoplasm and nucleus of prostate cancer cells. PLoS One 2012; 7: e44033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tee WW, Pardo M, Theunissen TW, et al Prmt5 is essential for early mouse development and acts in the cytoplasm to maintain es cell pluripotency. Genes Dev 2010; 24: 2772–2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sheng Y, Wang H, Liu D, et al Methylation of tumor suppressor gene CDH13 and SHP1 promoters and their epigenetic regulation by the UHRF1/PRMT5 complex in endometrial carcinoma. Gynecol Oncol 2016; 140: 145–151. [DOI] [PubMed] [Google Scholar]
- 27. Suzuki T, Abe K, Inoue A, et al Expression of c‐MYC in nuclear speckles during mouse oocyte growth and preimplantation development. J Reprod Dev 2009; 55: 491–495. [DOI] [PubMed] [Google Scholar]
- 28. Fedoriw A, Rajapurkar SR, O'Brien S, et al Anti‐tumor activity of the type I PRMT inhibitor, GSK3368715, synergizes with PRMT5 inhibition through MTAP loss. Cancer Cell 2019; 36: 100–114.e25. [DOI] [PubMed] [Google Scholar]
- 29. Zhu F, Guo H, Bates PD, et al PRMT5 is upregulated by B‐cell receptor signaling and forms a positive‐feedback loop with PI3K/AKT in lymphoma cells. Leukemia 2019; 33: 2898–2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen X, Chen RX, Wei WS, et al PRMT5 circular RNA promotes metastasis of urothelial carcinoma of the bladder through sponging miR‐30c to induce epithelial‐mesenchymal transition. Clin Cancer Res 2018; 24: 6319–6330. [DOI] [PubMed] [Google Scholar]
- 31. Banasavadi‐Siddegowda YK, Russell L, Frair E, et al PRMT5‐PTEN molecular pathway regulates senescence and self‐renewal of primary glioblastoma neurosphere cells. Oncogene 2017; 36: 263–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li Q, Jiao J, Li H, et al Histone arginine methylation by Prmt5 is required for lung branching morphogenesis through repression of BMP signaling. J Cell Sci 2018; 131: jcs217406. [DOI] [PubMed] [Google Scholar]
- 33. Fujimoto J, Sato E. Sex steroids in uterine endometrial cancers. Horm Mol Biol Clin Investig 2011; 5: 143–151. [DOI] [PubMed] [Google Scholar]
- 34. Tian W, Teng F, Gao J, et al Estrogen and insulin synergistically promote endometrial cancer progression via crosstalk between their receptor signaling pathways. Cancer Biol Med 2019; 16: 55–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Koh SS, Chen D, Lee YH, et al Synergistic enhancement of nuclear receptor function by p160 coactivators and two coactivators with protein methyltransferase activities. J Biol Chem 2001; 276: 1089–1098. [DOI] [PubMed] [Google Scholar]
- 36. Qi C, Chang J, Zhu Y, et al Identification of protein arginine methyltransferase 2 as a coactivator for estrogen receptor alpha. J Biol Chem 2002; 277: 28624–28630. [DOI] [PubMed] [Google Scholar]
- 37. Chung J, Karkhanis V, Baiocchi RA, et al Protein arginine methyltransferase 5 (PRMT5) promotes survival of lymphoma cells via activation of WNT/beta‐catenin and AKT/GSK3beta proliferative signaling. J Biol Chem 2019; 294: 7692–7710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li Y, Chitnis N, Nakagawa H, et al PRMT5 is required for lymphomagenesis triggered by multiple oncogenic drivers. Cancer Discov 2015; 5: 288–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Uxa S, Bernhart SH, Mages CFS, et al DREAM and RB cooperate to induce gene repression and cell‐cycle arrest in response to p53 activation. Nucleic Acids Res 2019; 47: 9087–9103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zajac A, Smolarz B, Stachowiak G, et al TP53 and MDM2 polymorphisms and the risk of endometrial cancer in postmenopausal women. Med Oncol 2014; 31: 286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kaseta MK, Khaldi L, Gomatos IP, et al Prognostic value of bax, bcl‐2, and p53 staining in primary osteosarcoma. J Surg Oncol 2008; 97: 259–266. [DOI] [PubMed] [Google Scholar]
- 42. Kobel M, Piskorz AM, Lee S, et al Optimized p53 immunohistochemistry is an accurate predictor of TP53 mutation in ovarian carcinoma. J Pathol Clin Res 2016; 2: 247–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tan DQ, Li Y, Yang C, et al PRMT5 modulates splicing for genome integrity and preserves proteostasis of hematopoietic stem cells. Cell Rep 2019; 26: 2316–2328.e6. [DOI] [PubMed] [Google Scholar]