

Abstract
Rationale:
Diabetic cardiomyopathy (DbCM) is a major complication in type-1 diabetes (T1D), accompanied by altered cardiac energetics, impaired mitochondrial function and oxidative stress. Previous studies indicate that T1D is associated with increased cardiac expression of Krüppel-like factor-5 (KLF5) and Peroxisome Proliferator Activated Receptor (PPAR)α that regulate cardiac lipid metabolism.
Objective:
In this study, we investigated the involvement of KLF5 in DbCM and its transcriptional regulation.
Methods and Results:
KLF5 mRNA levels were assessed in isolated cardiomyocytes from cardiovascular patients with diabetes and was higher compared with non-diabetic individuals. Analyses in human cells and diabetic mice with cardiomyocyte-specific FOXO1 deletion showed that FOXO1 bound directly on the KLF5 promoter and increased KLF5 expression. Diabetic mice with cardiomyocyte-specific FOXO1 deletion had lower cardiac KLF5 expression and were protected from DbCM. Genetic, pharmacologic gain and loss of KLF5 function approaches and AAV-mediated Klf5 delivery in mice showed that KLF5 induces DbCM. Accordingly, the protective effect of cardiomyocyte FOXO1 ablation in DbCM was abolished when KLF5 expression was rescued. Similarly, constitutive cardiomyocyte-specific KLF5 overexpression caused cardiac dysfunction. KLF5 caused oxidative stress via direct binding on NADPH oxidase (NOX)4 promoter and induction of NOX4 expression. This was accompanied by accumulation of cardiac ceramides. Pharmacologic or genetic KLF5 inhibition alleviated superoxide formation, prevented ceramide accumulation and improved cardiac function in diabetic mice.
Conclusion:
Diabetes-mediated activation of cardiomyocyte FOXO1 increases KLF5 expression, which stimulates NOX4 expression, ceramide accumulation and causes DbCM.
Keywords: Type 1 diabetes, oxidative stress, lipotoxicity, transcriptional regulation, KLF5, diabetic cardiomyopathy, diabetes mellitus, Kruppel-like factors
Subject Terms: Basic Science Research, Cell Signaling/Signal Transduction, Diabetes, Type 1, Metabolism, Myocardial Biology
Graphical Abstract
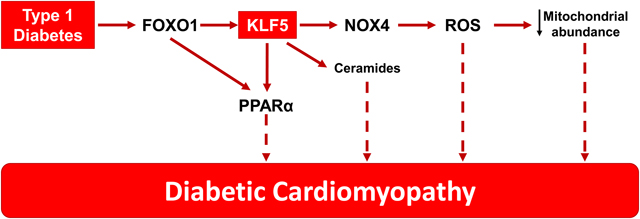
INTRODUCTION
Diabetes mellitus is anticipated to affect 10% of the global population by 20401. One of the major complications of diabetes is diabetic cardiomyopathy (DbCM)2, which represents heart failure that occurs in the absence of coronary artery disease2. Until the recent use of inhibitors for the sodium glucose co-transporter 2 (SGLT2), blood-lowering glucose treatments did not alleviate heart failure in patients with diabetes3, 4. Numerous hypotheses have been proposed to explain the underlying pathophysiology of DbCM. These include excess fatty acid oxidation, reactive oxygen species formation, and glucolipid toxicity5, all of which have been found in hearts from mice with DbCM6.
Impaired insulin signaling compromises glucose uptake by peripheral tissues and activates hepatic gluconeogenesis, leading to hyperglycemia7. Concurrently, it increases triglyceride hydrolysis and free fatty acid (FA) release by the adipose tissue5. In diabetic mice and humans, cardiac FA oxidation (FAO) is increased to compensate for the higher energetic demands of the heart that cannot use glucose8. Greater cardiac FAO is accompanied by increased expression of Peroxisome Proliferator-Activated Receptor alpha (PPARα)9. Accordingly, cardiomyocyte-specific overexpression of PPARα alone replicates DbCM10. We previously identified cardiomyocyte Krüppel-like factor 5 (KLF5) as a direct activator of the Pparα promoter in mice with streptozotocin (STZ)-induced type-1 diabetes (T1D)11.
In this study, we investigated how cardiomyocyte KLF5 is regulated in diabetes and if it is involved in DbCM either via stimulation of PPARα expression or independently. Impaired insulin signaling reduces phosphorylation of AKT and its downstream target, Forkhead box protein O1 (FOXO1), which promotes its nuclear retention. FOXO1 overexpression impairs cardiac function in diabetic mice12. Now, we show that FOXO1 drives KLF5 expression during DbCM and that inhibition of either cardiomyocyte-KLF5 or cardiomyocyte-FOXO1 can alleviate DbCM. Moreover, constitutive cardiomyocyte KLF5 expression caused cardiac dysfunction that was associated with higher NOX4 expression, cardiac ROS content and ceramide accumulation in a PPARα-independent manner. Thus, KLF5 emerges as a potential therapeutic target for cardiomyopathy in diabetes.
METHODS
An expanded Methods section is included in the Online Supplement. Please, see also the Major Resources Table in the Online Materials.
Data availability.
The authors avail all supporting data and methods within the present article. Additional information will be available upon request to the corresponding author.
Animal care.
All animal experiments were conducted in male mice and were approved by the Institutional Animal Care and Use Committee at Temple University and carried out in accordance with the NIH guidelines. Wild type C57BL/6, FOXO1fl/fl and Pparα−/− mice were obtained from Jackson Laboratory. The R26-lsl-rtTA-TRE-Klf5 mice were provided by Jeffrey Whitsett13 and Inderpreet Sur14. αMHC-rtTA-Klf5 mice were generated by crossing αMHC-Cre with R26-lsl-rtTA-TRE-Klf5 mice, respectively. The αMHC-Klf5−/− and αMHC-FoxO1−/− mice were generated by our group11, 15. Insulin deficiency was induced by injecting i.p. streptozotocin (STZ, Sigma-Aldrich) dissolved in saline (50mg/kg) for 5 consecutive days. LGM260516 was dissolved in saline and administered orally (100mg/kg b.w.; daily gavage). ML264 (10mg/kg b.w. i.p.) was dissolved in 80% saline, 10% dimethyl-sulfoxide (DMSO) and 10% Tween 80 and was administered twice per day. Type 2 diabetes was induced in C57BL/6 mice by feeding them on high-fat diet (Online Table I) for 32 weeks.
Glucose and insulin tolerance tests were conducted after 6 hours of fasting. For glucose tolerance test, mice received i.p. 20% glucose solution (2g of glucose/kg body mass). For insulin tolerance test, mice received intraperitoneal injection of insulin (0.75 U/kg). Glucose levels were determined in blood collected from the tail vein at designated time points.
Cell culture.
The AC1617 human ventricular cardiomyocyte line was maintained in complete DMEM/F-12 medium (ThermoFischer Scientific, Carlsbad, CA) at 37°C and 5%CO2.
Generation of AAV and adenoviruses and infection of adult mice or cells.
AAV9-cTnT-Klf5 was generated by cloning the human KLF5 cDNA in the pAAV9-cTnT receiving vector18 (Addgene). The AAVs were administered in C57BL/6 mice via retro-orbital injection (7×1011viral particles/100μl). Adenoviruses expressing KLF5 or shKLF519 (under H1 promoter) were generated by cloning the coding sequence of human KLF5 into the pAdTrackCMV vector or pAdTrack respectively. Ad-wtFOXO1 and Ad-caFOXO1 were provided by Dr. D. Accili20. Ad-caFOXO1 has Akt phosphorylation sites Thr24, Ser316 substituted to Ala and Ser253 to Asp20. Ad-shNOX4 was previously described21.
Echocardiography.
Cardiac function was assessed by transthoracic echocardiography using the VisualSonics Vevo 2100 system (VisualSonics, Toronto, ON). Mice were sedated via inhalation of 3% isoflurane and maintained on 0.5% isoflurane during image acquisition. Echocardiography images were processed to perform off-line LV trace analysis or B-mode speckle-tracking analysis by a single observer blinded to the respective treatments of mice with VevoLab software (3.1.1). More information is provided in the Online Supplement.
Protein and RNA purification and analysis.
Protein and RNA purification from isolated hearts or cells were performed as described previously22. Antibodies used are listed in Online Table II. Primer sequences are described in Online Table III.
Chromatin Immunoprecipitation (ChIP).
ChIP experiments were performed in AC16 cells as described previously11 and in the Online Supplement.
Transient transfection and luciferase assay.
For transfection and luciferase assay analyses human KLF5 (-1757/-263bp), PPARα (-1913/-1468bp) and NOX4 (−1547/+54bp) promoters and were cloned into pGL3 basic vector (pGL3-BV, Promega) as described in the Online Supplement.
Adult Murine and Human Cardiomyocyte Isolation
Adult murine cardiomyocytes were isolated as described previously22, 23. Human cardiomyocytes were isolated as previously described, using interventricular septum biopsies of 18 aortic stenosis patients undergoing aortic valve replacement24. Among these patients, 12 were non-diabetic and were used as control group, 5 had type-2 diabetes and 1 had type-1 diabetes (Online Table IV). The study was approved by the local ethics committee, followed the principles outlined in the Declaration of Helsinki and written consent was obtained from all patients.
Measurement of superoxide and NAD(P)H in cardiac tissue.
Heart was perfused with PBS and excised from the thoracic cavity. Similarly, gastrocnemius skeletal muscle was obtained following removal of the skin and the surrounding fascia. Small pieces (1mm × 1mm) of freshly isolated cardiac tissue and gastrocnemius skeletal muscle were incubated for 30min in 20μM solution of dihydroethidium (DHE) (ThermoFischer Scientific) at room temperature and imaged on a Zeiss Axio Observer Z1 microscope (560nm). Cardiac NAD(P)H levels were assessed by NAD(P)H autofluorescence at 350/460 nm (excitation/emission)25, 26.
Measurement of oxygen consumption rate in cells.
AC16 cells (105 cells per well) were plated in XF96 Seahorse plates that were pre-coated with 10μg/ml laminin (ThermoFischer Scientific). Oxygen consumption rate in AC16 cells was measured using oligomycin, carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP), and antimycinA/rotenone. Detailed information is provided in the Online Supplement.
Assessment of mitochondrial abundance.
Mitochondrial abundance was estimated quantified by calculating the ratio of mitochondrial gene copy number (COXII, tRNALeu) to nuclear gene copy number (β-globin) as described previously22, 27
Measurement of reactive oxygen species, mitochondrial membrane potential and Ca2+ uptake.
Reactive Oxygen Species were measured in AC16 cells that were incubated with DHE, MitoSOX and TMRM dyes (Invitrogen) as previously described28. Extramitochondrial Ca2+ ([Ca2+]out) clearance and ΔΨm were determined with a multi-wavelength excitation dual-wavelength emission fluorimeter (Delta RAM, PTI) as previously29.
Mitochondrial DNA damage.
Mitochondrial DNA damage was assessed with the DNA damage analysis kit (Detroit R&D) according to the instructions of the manufacturer.
Assessment of cardiac fibrosis.
Paraffin-embedded sections of myocardial tissue (0.6μm) were deparaffinized and stained using Picrosirius red (PolySciences, Inc) according to manufacturer’s protocol. Sections were scanned with a Nikon iEclipse microscope. Images of 20X magnification were analyzed in ImageJ software.
Lipidomic analysis.
Total cardiac lipidome was extracted via chloroform-methanol extraction, spiked with appropriate internal standards, and analyzed using a 6490 Triple Quadrupole LC-MS/MS system (Columbia University)30.
Statistical analysis and selection of representative images.
GraphPad Prism6 software was used to perform statistical analyses. All groups with N≥6 were assessed for normal distribution with Shapiro-Wilk Test (p<0.05). Upon confirmation of normal distribution, non-paired two-tailed Student’s t-test or 1- or 2-way ANOVA analysis with Tukey multiple comparisons were performed. When normal distribution was not confirmed, Mann-Whitney and Kruskal-Wallis (followed by Dunn’s multiple comparison test) non-parametric tests were performed. Mann-Whitney and Kruskal-Wallis tests were also applied for experimental groups of N<6. Graphs represent means ± SEM. Outliers were tested with the ESD method (extreme studentized deviate; Grubbs’ test, two-sided, p<0.05) and removed from the comparison analysis. A p-value<0.05 was considered statistically significant. Exact p-values are reported for all groups with statistically significant differences except for p<0.0001 in multiple comparison tests (ANOVA or Kruskal Wallis) that the GraphPad Prism6 software does not specify. ANOVA or Kruskal Wallis tests generated adjusted p-values. Detailed statistical analysis information for all main and online figures can be found on Online Table V)
We selected representative images based on their quality and accurate representation of similarity with the average value of each experimental group.
RESULTS
Patients and mice with diabetes have higher cardiac KLF5 expression.
In accordance with our previous findings showing increased cardiac KLF5 expression in T1D11, cardiomyocytes obtained from aortic stenosis patients with T1D (n=1) or Type-2 diabetes (T2D; n=5) showed higher KLF5 mRNA levels compared with patients without diabetes (Fig. 1A).
Fig. 1:
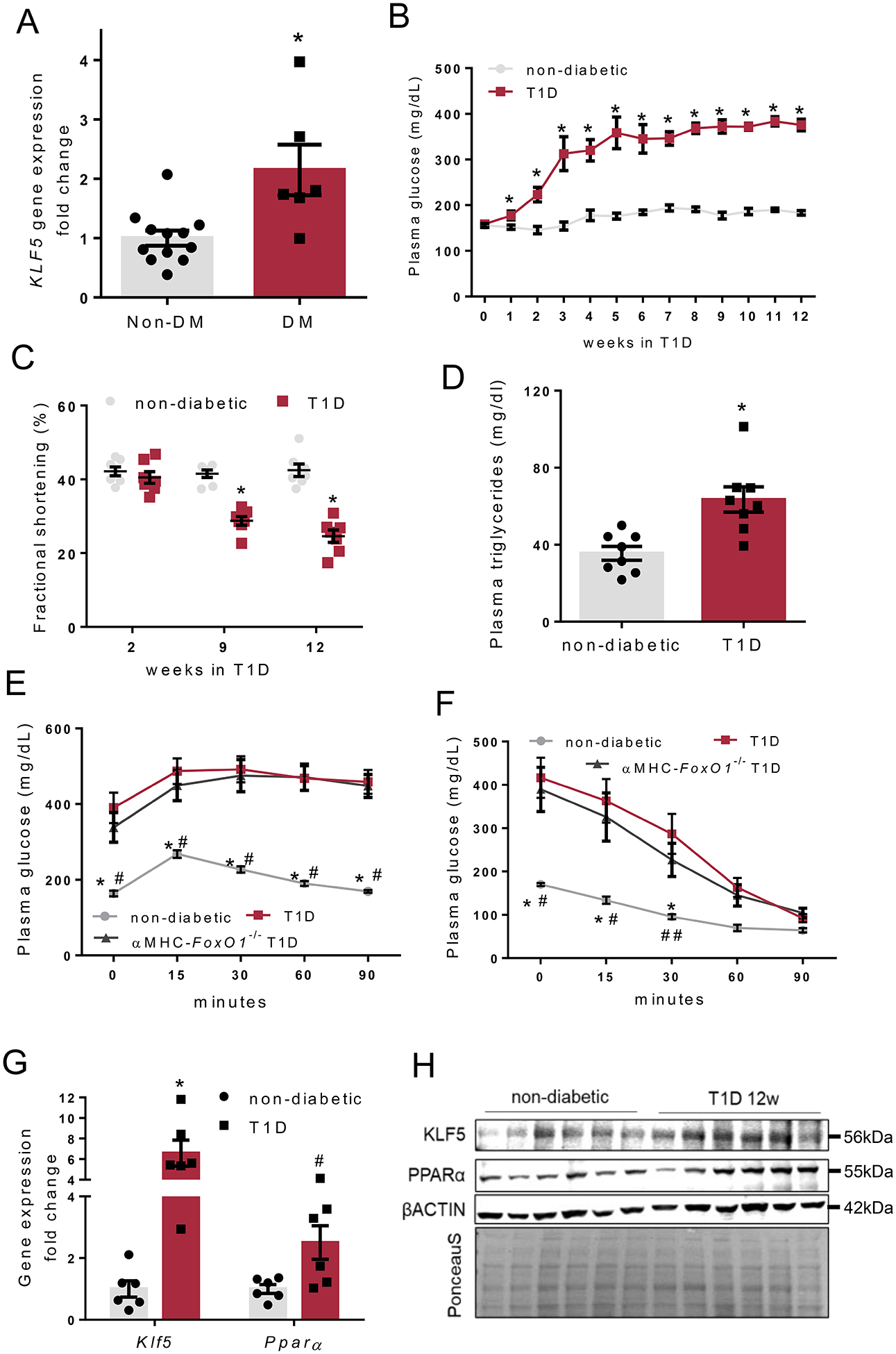
A: KLF5 gene expression in cardiomyocytes of aortic stenosis patients with diabetes or without diabetes. B-C: Plasma glucose levels (B) and fractional shortening (C) in non-diabetic and type-1 diabetic C57Bl/6 mice. D: Plasma triglyceride levels in non-diabetic and diabetic C57BL/6 mice 12 weeks post-STZ administration. E, F: Plasma glucose levels in non-diabetic and diabetic FOXO1fl/fl and diabetic αMHC-FoxO1−/− mice 12 weeks post-STZ administration that were subjected to glucose (E) or insulin (F) tolerance test. G: Cardiac KLF5 and PPARα mRNA levels in non-diabetic and diabetic C57BL/6 mice, 12 weeks post-STZ administration. H: Immunoblots of KLF5, PPARα, β-ACTIN and PonceauS staining (analysis shown in Online Figure I-F), in cardiac protein lysates from non-diabetic and diabetic C57BL/6 mice, 12 weeks post-STZ injections.
To explore if cardiac KLF5 expression is increased in animal models of diabetes, we induced T1D and T2D in C57BL/6 mice. T1D was induced by 5 daily i.p. injections of STZ. Diabetic mice had lower plasma insulin levels (Online Figure I-A) and higher blood glucose levels (Fig. 1B) as early as 2 weeks post-STZ injections and developed both systolic (Fig. 1C, Online Figure I-B) and diastolic (Online Figure I-C) cardiac dysfunction later (9 and 12 weeks; Online Table VI). Furthermore, mice with T1D had higher plasma triglyceride levels (Fig. 1D), and impaired glucose tolerance (Fig 1E, Online Figure I-D) although they maintained their insulin sensitivity following exogenous administration of insulin (Fig 1F, Online Figure I-E). Cardiac KLF5 and PPARα mRNA (Fig. 1G) and protein levels (Fig. 1H, Online Figure I-F) were increased 12 weeks post-injection.
To investigate if cardiac KLF5 is also increased in mice with T2D, C57BL/6 mice fed on high-fat diet for 32 weeks. These mice gained weight faster than control mice fed on chow (Online Figure II-A), had higher plasma insulin levels (Online Figure II-B), hyperglycemia, impaired glucose tolerance (Online Figures II-C, II-D), and cardiac dysfunction (Online Figures II-E, II-F, Online Table VII). In accordance with patient data, mice with T2D also had increased cardiac KLF5 protein levels (Online Figure II-G). Thus, cardiac KLF5 expression is increased in both T1D and T2D.
FOXO1 is a positive transcriptional regulator of KLF5 in diabetes.
In accordance with development of insulin deficiency, phosphorylation of Ser473 of AKT and Ser256 of FOXO1 (Fig. 2A, Online Figure III-A) was decreased in cardiac extracts from C57BL/6 mice with T1D for 12 weeks compared with non-diabetic C57BL/6 mice. This indicates inhibition of insulin signaling and increased nuclear translocation of FOXO1. Dephosphorylation of FOXO1 in hearts of diabetic mice was accompanied by increased expression of FOXO1 gene targets, such as pyruvate dehydrogenase kinase-4 (Pdk4), Bcl-2-like protein 11 (Bim), catalase (Cat) and heme oxygenase-1 (Ho1)12, 31, 32 (Online Figure III-B). On the other hand, expression of histone deacetylase 2 (Hdac2) and p300, which are not regulated by FOXO1, was not altered (Online Figure III-B). Cardiac expression of Myh6 was decreased and Myh7 expression was increased (Online Figure III-C). Expression of fibrosis-related gene Opn exhibited a trend for increase whereas Acta1 and Col1a1 were not changed (Online Figure III-D). Moreover, Nppa gene expression exhibited a trend for decrease but Nppb gene expression remained unaltered (Online Figure III-E).
Fig. 2.
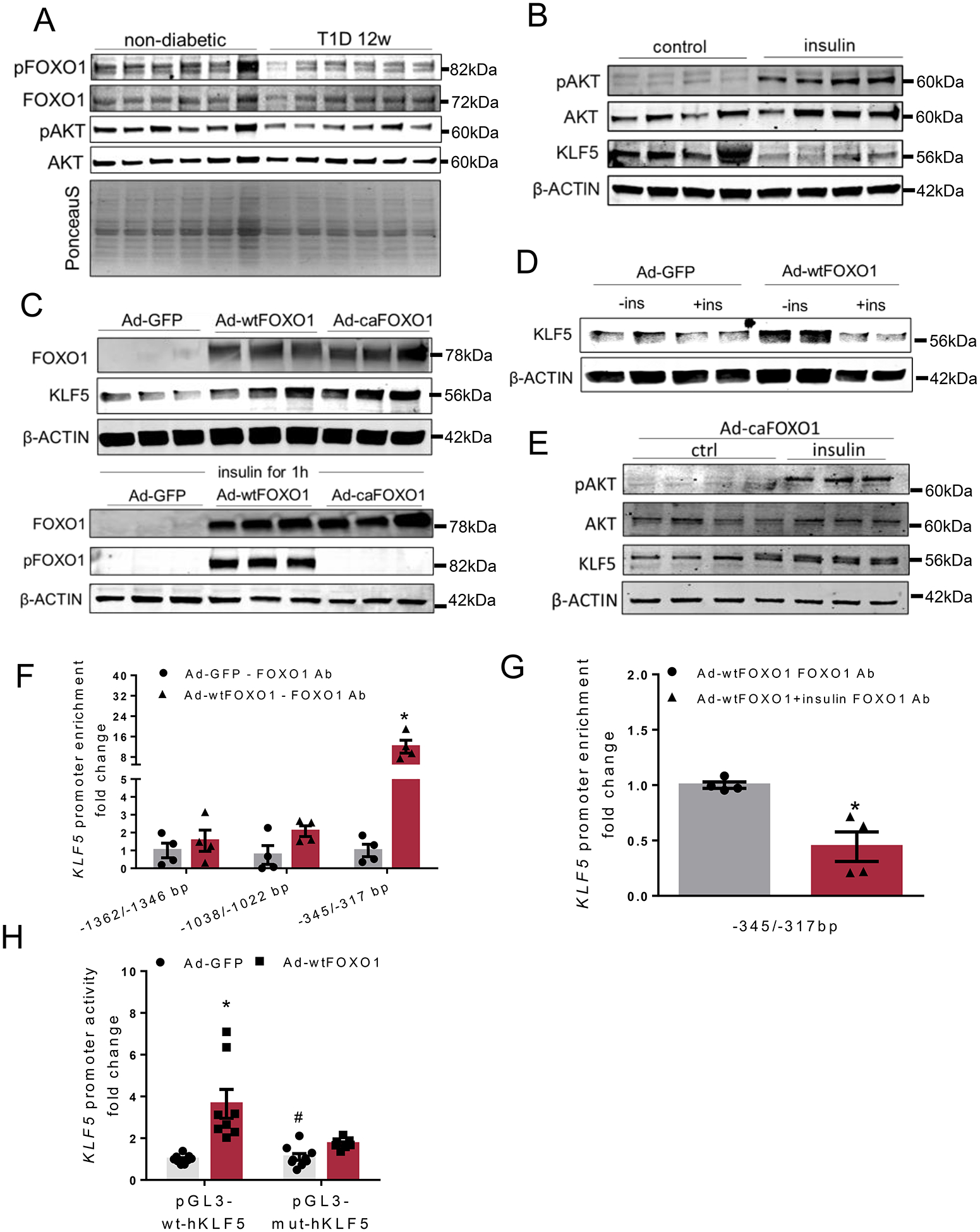
A: Immunoblots of cardiac pSer256-FOXO1, total FOXO1, pSer473-AKT and total AKT and PonceauS staining in non-diabetic and diabetic C57BL/6 mice, 12 weeks post-STZ administration (analysis shown in Online Figure III-A). B: Immunoblots of pSer473-AKT, total AKT, KLF5 and β-ACTIN proteins from control and insulin-stimulated AC16 cells (analysis shown in Online Figure IV-A). C: Immunoblots of total FOXO1, pSer256-FOXO1 and β-ACTIN proteins from AC16 cells treated with adenoviruses expressing GFP, wtFOXO1 or caFOXO1 without (analysis shown in Online Figure IV-C) or with insulin treatment. D: Immunoblots of KLF5 and β-ACTIN proteins from AC16 cells treated with adenoviruses expressing GFP and wtFOXO1 without or with insulin stimulation (analysis shown in Online Figure IV-F). E: Immunoblots of pSer473-AKT, total AKT, KLF5 and β-ACTIN proteins from AC16 cells treated with adenovirus expressing caFOXO1 (analysis shown in Online Figure IV-G) without or with insulin treatment. F: FOXO1 enrichment in KLF5 promoter sequence fragments precipitated with FOXO1 antibody from AC16 cells expressing GFP or wtFOXO1. G:FOXO1 enrichment in KLF5 promoter sequence fragments precipitated with FOXO1 antibody from AC16 cells expressing wtFOXO1 and treated with insulin. H: Luciferase activity normalized to firefly from AC16 cells transfected with plasmids containing the wild type KLF5 −1757/−263bp promoter fragment (pGL3BV-wt-hKLF5) or the mutant [-1757/-263bp](-342/-317bp)N→A (pGL3BV-mut-hKLF5) and infected with adenoviruses expressing GFP or wtFOXO1.
To investigate whether insulin signaling, which is inhibited in T1D and thus leads to FOXO1 activation, accounts for altered KLF5 expression, we treated a human cardiomyocyte-derived cell line (AC16) with insulin for 1 hour. This treatment increased phosphorylation of AKT at Ser473 and decreased KLF5 protein levels compared with control cells that were not treated with insulin (Fig. 2B, Online Figure IV-A). Treatment of AC16 cells with adenoviruses expressing either wild-type FOXO1 (Ad-wtFOXO1) or constitutively-active FOXO1 (Ad-caFOXO1) increased KLF5 mRNA (Online Figure IV-B; trend for caFOXO1) and protein levels (Fig. 2C, Online Figure IV-C). To investigate whether insulin-mediated inhibition of KLF5 expression is accounted for by FOXO1, we treated wtFOXO1- and caFOXO1-expressing AC16 cells with insulin. Insulin increased phosphorylation of AKT (Online Figures IV-D, IV-E) and FOXO1 (Fig. 2C) in AC16 cells expressing wtFOXO1. Apparently, FOXO1 phosphorylation was not induced in cells expressing the phosphorylation-resistant caFOXO1 (Fig. 2C). Accordingly, insulin treatment decreased KLF5 expression in AC16 cells infected with Ad-wtFOXO1 (Fig. 2D, Online Figure IV-F). Moreover, insulin did not suppress KLF5 protein levels in cells that express caFOXO1 (Fig. 2E, Online Figure IV-G).
To investigate if induction of KLF5 expression by FOXO1 is via direct transcriptional regulation mechanism, we performed in silico analysis of the −2000/+1 bp region of the human KLF5 promoter (Genomatix) followed by alignment (STRAP)33 of the mouse and human KLF5 promoters (obtained from the UCSC Genome Browser). This analysis of the human KLF5 promoter sequence identified 11 potential binding sites for members of the FOX protein family (Online Figures V-A, V-B). Four of these sites (−1362/−1346bp, −1038/−1022bp, −345/−329bp, and −333/−317bp) were conserved as they share 100% core sequence homology between the two species (Online Figure V-B). ChIP analysis in AC16 cells treated with Ad-wtFOXO1 (Fig. 2F, Online Figures VI-A, VI-B, VI-C) showed increased (12-fold) FOXO1 enrichment in the dual potential FOXO1 binding region (−345/−317bp consisting of −345/−329bp and −333/−317bp). On the contrary, FOXO1 binding was not detected in −1362/−1346bp and −1038/−1022bp KLF5 promoter regions. FOXO1 binding on −345/−317bp KLF5 promoter region was not observed in wtFOXO1-expressing cells that were stimulated with insulin, which precludes nuclear translocation of FOXO1 and makes it transcriptionally inactive (Fig. 2G, Online Figure VI-D).
To assess further the involvement of the −345/−317bp promoter region in the regulation of KLF5 expression by FOXO1, we performed luciferase activity assay. Specifically, AC16 were transfected with pGL3 plasmid carrying the wt −1757/−263bp KLF5 promoter region or the −1757/−263bp region with mutated −345/317bp sequence (Online Figure VI-E). Infection of the transfected cells with Ad-wtFOXO1 or control Ad-GFP showed that FOXO1 increased KLF5 promoter activity (Fig. 2H). The effect of FOXO1 in increasing KLF5 promoter activity was diminished in AC16 cells that were transfected with the vector that carried the mutant −345/−317bp region of the KLF5 promoter (Fig. 2H). Thus, the −345/−317bp region of the KLF5 promoter that contains two potential FOXO1 binding sites is critical for the regulation of KLF5 expression by FOXO1.
Cardiomyocyte–specific FOXO1 ablation prevents KLF5 upregulation and cardiomyopathy in T1D.
To test whether FOXO1-mediated induction of KLF5 is a central event that is associated with development of DbCM, we generated a mouse line with cardiomyocyte–specific FOXO1 ablation (αMHC-FoxO1−/−). These mice had lower FOXO1 and KLF5 mRNA (Online Figure VII-A) and protein levels in whole heart extracts (Online Figures VII-B, VII-C). In isolated primary cardiomyocytes we verified complete ablation for FOXO1 and decreasing trend for KLF5 (Online Figure VII-D, VII-E). Diabetic αMHC-FoxO1−/− mice, which had similar plasma insulin (Online Figure VII-F) blood glucose (Online Figure VII-G), and triglyceride (Online Figure VII-H) levels with the diabetic floxed littermates, were protected from cardiac dysfunction (Fig. 3A, Online Figures VII-I, VII-J, Online Table VIII). The improvement in cardiac function of diabetic αMHC-FoxO1−/− mice was accompanied by lower KLF5 and PPARα protein levels compared with diabetic FOXO1fl/fl mice (Fig. 3B, Online Figure VII-K). Comparison between non-diabetic and diabetic αMHC-FoxO1−/− mice showed similar KLF5 levels (Online Figure VII-L). Plasma glucose clearance was not different between diabetic αMHC-FoxO1−/− and diabetic floxed littermates (Fig. 1E, Online Figure I-D). Accordingly, no statistically significant difference was observed between the two experimental groups in insulin sensitivity following intraperitoneal administration of insulin (Fig. 1F, Online Figure I-E). Thus, improvement of cardiac function in diabetic αMHC-FoxO1−/− mice was not accompanied by altered systemic glucose management.
Fig. 3.
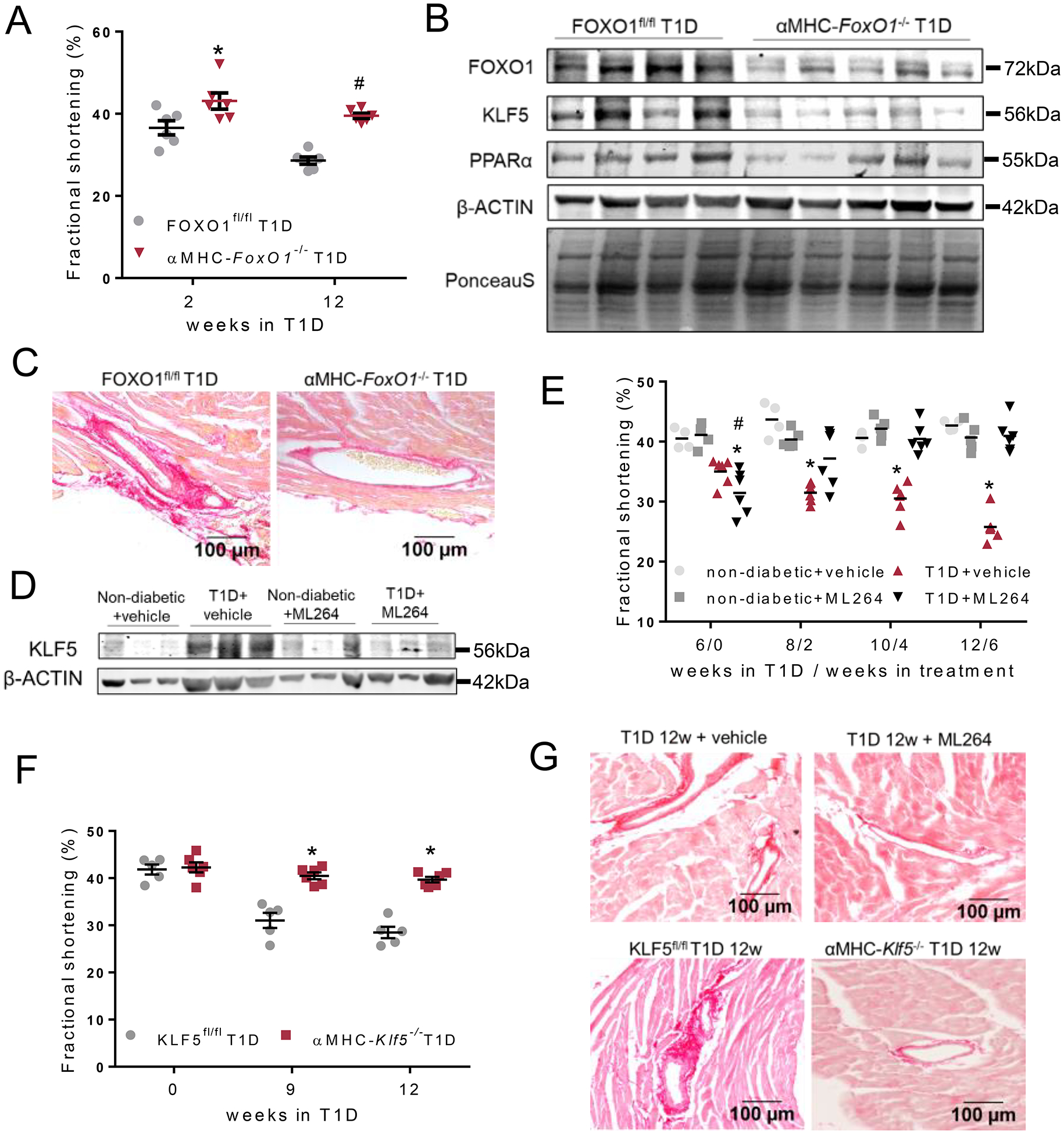
A: Left ventricular fractional shortening of diabetic FOXO1fl/fl and αMHC-FoxO1−/− mice 2 and 12 weeks post-STZ. B: Immunoblots of FOXO1, KLF5, PPARα, β-ACTIN protein and PonceauS staining in hearts obtained from diabetic FOXO1fl/fl and αMHC-FoxO1−/− mice 12 weeks post-STZ administration (analysis shown in Online Figure VII-K). C: Representative images of picrosirius red staining (20X magnification) of cardiac tissue sections of diabetic FOXO1fl/fl and αMHC-FoxO1−/− mice. Additional images and quantification in Online Figures IX-A,B. D: Immunoblot of cardiac KLF5 and β-ACTIN proteins in non-diabetic treated with 6 weeks with vehicle or ML264 or diabetic C57BL/6 mice treated with vehicle or ML264 starting 6 weeks post-STZ administration (analysis shown in Online Figure X-A). E: Left ventricular fractional shortening of diabetic or control non-diabetic mice both treated with either vehicle or ML264 for 6 weeks starting 6 weeks post-STZ administration. F: Left ventricular fractional shortening of diabetic KLF5fl/fl and αMHC-Klf5−/− mice 9 weeks and 12 weeks post-STZ administration. G: Picrosirius red staining and perivascular fibrosis analysis of cardiac tissue from diabetic C57BL/6 mice treated with ML264 or control vehicle and diabetic KLF5fl/fl or αMHC-Klf5−/− mice 12 weeks post-STZ administration. Additional images and quantification analysis in Online Figures XII-A,B or Online Figures XIII–A,B.
In agreement with previous findings showing that FOXO1 is a direct transcriptional activator of Myh7 in mice with insulin resistance34, diabetic αMHC-FoxO1−/− mice had lower Myh7 and unaltered Myh6 gene expression compared with diabetic littermates (Online Figure VII-M). At the same time, Nppa mRNA levels were increased in diabetic αMHC-FoxO1−/− mice compared with diabetic controls (Online Figure VIII-A). On the contrary, we did not observe hypertrophy in either diabetic FOXO1fl/fl or diabetic αMHC-FoxO1−/− mice compared with non-diabetic mice (Online Figures VIII-B, VIII-C, VIII-D). Improvement in cardiac function of diabetic αMHC-FoxO1−/− mice was associated with lower perivascular fibrosis (Fig. 3C, Online Figures IX-A, IX-B), although OPN mRNA levels were increased compared with diabetic littermates (Online Figure IX-C).
Cardiomyocyte KLF5 accounts for DbCM.
To explore the therapeutic potential of KLF5 inhibition in T1D we treated non-diabetic and diabetic C57BL/6 mice with ML264, which suppressed KLF5 expression35, 36 compared to mice with diabetes that were treated with control vehicle (Fig 3D, Online Figure X-A) Treatment started 6 weeks after induction of T1D, when hyperglycemia (Online Figure X-B) and cardiac dysfunction (Fig. 3E, Online Figure X-C, Online Table IX) had occurred. Despite persistent hyperglycemia and hypoinsulinemia (Online Figure X-D), 6 weeks of treatment with ML264 normalized both systolic and diastolic function as shown by fractional shortening levels and reverse global longitudinal strain rate (Fig. 3E, Online Figures X-C, X-D, X-E). The improvement in cardiac function was accompanied by lower plasma triglycerides (Online Figure X-F), cardiac KLF5 mRNA (Online Figure X-G) and protein levels (Fig. 3D, Online Figure X-A).
To exclude non-KLF5-related effects of ML264, we assessed whether cardiomyocyte KLF5 genetic ablation would protect mice from DbCM. To do this, we induced T1D in αMHC-Klf5−/− mice11 for 12 weeks. Plasma glucose, triglyceride and insulin levels did not differ between diabetic αMHC-Klf5−/− and floxed mice (Online Figures XI-A, XI-B, XI-C). Despite hyperglycemia, cardiac systolic (Fig. 3F, Online Figures XID, XI-E, XI-F, Online Table X) and diastolic (Online Figure XI-G) function of diabetic αMHC-Klf5−/− mice was improved compared with floxed mice with T1D. In a similar manner with diabetic αMHC-FoxO1−/− mice, cardiomyocyte-KLF5 ablation did not improve either glucose tolerance (Online Figures XI-H, XI-I) or responsiveness to insulin treatment (Online Figures XI-J, XI-K) compared with the diabetic floxed littermates.
Hearts from diabetic ML264-treated (Fig. 3G and Online Figures XII-A, XII-B) and αMHC-Klf5−/− (Fig. 3G and Online Figures XIII-A, XIII-B) mice had decreased fibrosis. Although treatment of diabetic C57BL/6 mice with ML264 decreased Acta1 and Col1a1 gene expression but not Opn (Online Figure XIV-A), diabetic αMHC-Klf5−/− mice had elevated Acta1 and Opn gene expression but not Col1a1 (Online Figure XIV-B) and so had diabetic αMHC-FoxO1−/− mice. This indicates distinct effects between systemic pharmacologic KLF5 inhibition and cardiomyocyte-specific genetic ablation on fibrosis-related gene expression. In both pharmacologic and genetic inhibition of KLF5 in diabetic mice, Myh6 and Myh7 gene expression did not differ compared with the respective diabetic controls (Online Figures XIV-C, XIV-D). Accordingly, heart size, Nppa and Nppb gene expression were not affected in C57BL/6 mice treated with ML264 (Online Figures XV-A, XV-B, XV-C, XV-D) and in diabetic αMHC-Klf5−/− mice (Online Figures XVI-A, XVI-B, XVI-C, XVI-D).
FOXO1 activates PPARα expression via a KLF5-independent mechanism.
We have shown previously that cardiomyocyte-KLF5 is a transcriptional regulator of PPARα11, which has been associated with DbCM37. Therefore, we tested whether the protective effect of Klf5 or FoxO1 ablation is associated with altered cardiac PPARα expression. Induction of diabetes in αMHC-Klf5−/− mice increased cardiac PPARα protein levels 12 weeks post-STZ injections (Fig. 4A, Online Figure XVII). As KLF5 is ablated in αMHC-Klf5−/− mice, we aimed to delineate whether there are KLF5-independent transcriptional regulatory mechanisms that mediate cardiac PPARα upregulation.
Fig. 4.
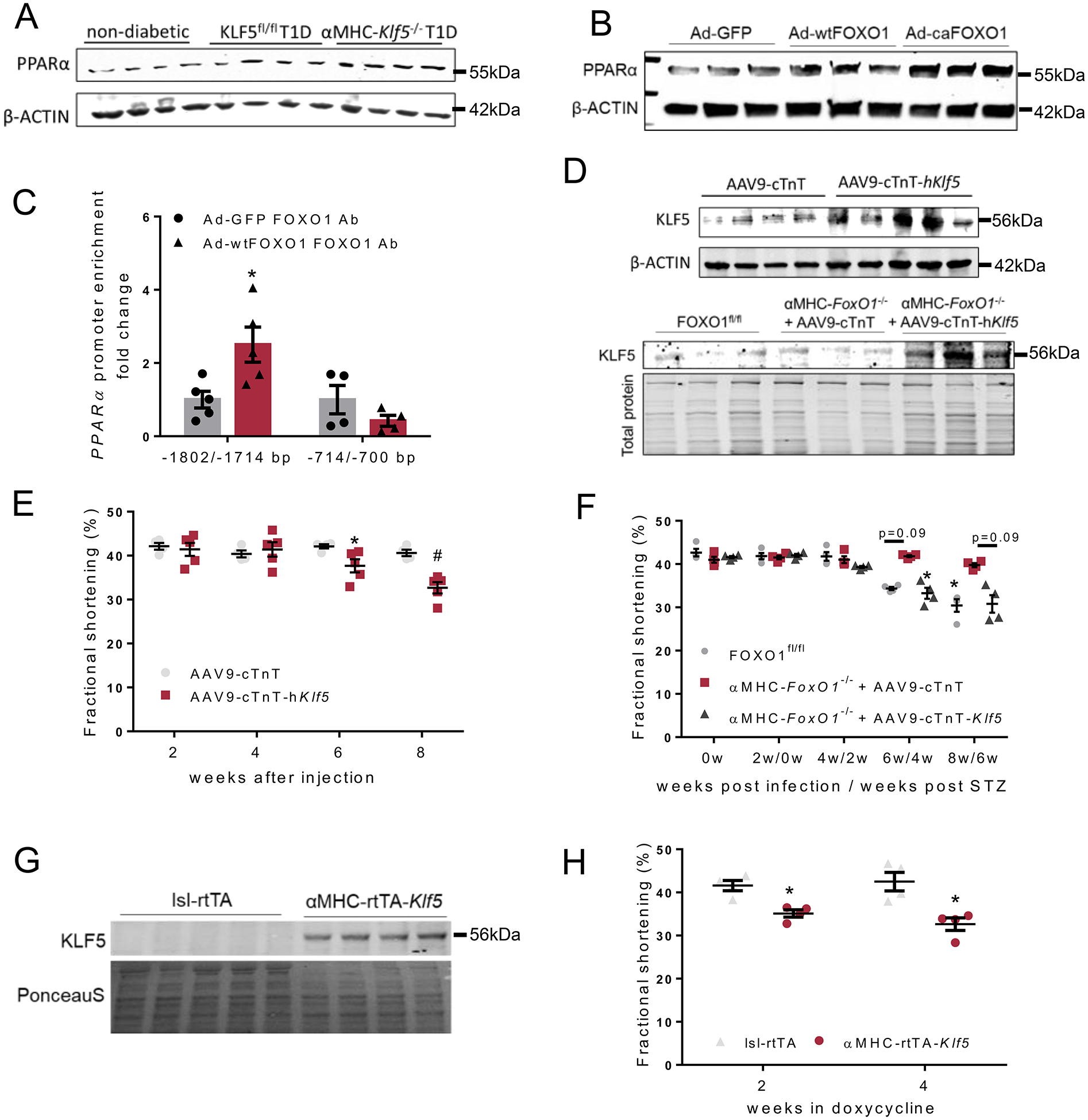
A: Immunoblots of cardiac PPARα and β-ACTIN proteins in non-diabetic, diabetic KLF5fl/fl and diabetic αMHC-Klf5−/− mice 12 weeks post-STZ administration (analysis shown in Online Figure XVII). B: Immunoblots of PPARα and β-ACTIN proteins in AC16 cells infected with adenoviruses expressing GFP, wtFOXO1 or caFOXO1 (analysis shown in Online Figure XIX–B). C: FOXO1 enrichment in PPARα promoter sequence fragments precipitated with FOXO1 antibody from AC16 cells expressing GFP or wtFOXO1. D: Immunoblot of cardiac KLF5 and β-ACTIN proteins in C57BL/6 mice infected with AAV9-cTnT or AAV9-cTnT-hKlf5 (analysis shown in Online Figure XXIV–A), and immunoblot of cardiac KLF5 and total protein stain of diabetic FOXO1fl/fl mice and diabetic αMHC-FoxO1−/− mice infected with control AAV9-cTnT or AAV9-cTnT-hKlf5 (analysis shown in Online Figure XXIV–B). E: Left ventricular fractional shortening of C57BL/6 mice infected with AAV9-cTnT or AAV9-cTnT-hKlf5 2, 4, 6 and 8 weeks post-AAV9 administration. F: Left ventricular fractional shortening of diabetic FOXO1fl/fl mice and diabetic αMHC-FoxO1−/− mice infected with AAV9-cTnT or AAV9-cTnT-hKlf5. G: Immunoblot of cardiac KLF5 and PonceauS staining of lsl-rtTA-TRE-Klf5 and αMHC-rtTA-Klf5 mice fed on doxycycline enriched diet for 2 weeks (analysis shown in Online Figure XXV–D). H: Left ventricular fractional shortening of lsl-rtTA-TRE-Klf5 and αMHC-rtTA-Klf5 mice fed on doxycycline enriched diet for 2 and 4 weeks.
Along these lines, we performed in silico analysis (Genomatix) of human PPARα promoter followed by alignment of both human and murine promoters (STRAP)33. This analysis identified 2 conserved potential FOX protein binding sites at −1802/−1788bp and −714/−700bp (Online Figures XVIII-A, XVIII-B). To assess the capacity of FOXO1 to act as a transcriptional activator of the PPARα gene, we infected AC16 cells with Ad-wtFOXO1. FOXO1 overexpression increased PPARα mRNA (Online Figure XIX-A) and protein (Fig. 4B, Online Figure XIX-B) levels. The effect of FOXO1 on PPARα expression was abrogated by treatment with insulin in cells expressing wtFOXO1 (Online Figures XIX-C, XIX-D) but not the phosphorylation-resistant caFOXO1 (Online Figures XIX-E, XIX-F). Chromatin immunoprecipitation in AC16 cells overexpressing wt-FOXO1 revealed increased FOXO1 enrichment of the −1802/−1788bp region of the PPARα promoter (Fig. 4C, Online Figure XIX-G). As we did for KLF5 promoter, we wanted to explore further whether FOXO1 binding site at −1802/−1788bp region of the PPARα is critical for FOXO1 to regulate PPARα expression. For that we performed luciferase activity assay by transfecting pGL3 plasmids carrying the wt −1913/−1468bp sequence of human PPARα promoter or the promoter with deleted the −1805/−1787bp region (Online Figure XIX-H). Subsequently, transfected AC16 cells were infected with adenoviruses expressing wtFOXO1 or GFP. AC16 overexpressing FOXO1 showed increased PPARα promoter activity (Online Figure XIX-I). Increased promoter activity was abrogated in AC16 cells transfected with the vector carrying the mutated PPARα promoter region (Online Figure XIX-I).
In agreement with these findings, cardiac PPARα protein levels were significantly lower in diabetic αMHC-FoxO1−/− mice compared with control diabetic floxed mice (Fig. 3B). Thus, FOXO1 can directly bind to −1802/−1788bp region of PPARα promoter and activate its gene expression independently of KLF5.
Improved cardiac function in diabetic αMHC-FoxO1−/− and αMHC-Klf5−/− mice is independent of PPARα and its target genes.
To assess whether lower expression of cardiac PPARα that we observed in diabetic αMHC-FoxO1−/− mice, accounts for the alleviation of DbCM, we induced T1D in Pparα−/− mice. These mice developed hyperglycemia (Online Figure XX-A) and cardiac dysfunction (Online Figures XX-B, XX-C, XX-D, XX-E, XX-F, Online Table XI) to the same extent as diabetic C57BL/6 mice although their plasma triglycerides showed a trend of reduction (Online Figure XX-G) and so did plasma insulin levels (Online Figure XX-H). Cardiac dysfunction was accompanied by increased cardiac KLF5 protein levels compared with either C57BL/6 mice (Online Figures XX-I, XX-J) or Pparα−/− mice without T1D (Online Figures XX-K, XX-L).
We then tested, whether FOXO1 or KLF5 ablation in T1D affects the expression of a PPARα target, uncoupling protein 3 (UCP3)38. Cardiac UCP3 mRNA and protein levels were increased in C57BL/6 mice with T1D (Online Figures XXI-A, XXI-B, XXI-C) that had higher cardiac PPARα expression, whereas Ucp2 expression was not altered (Online Figure XXI-A). Both precursor and cleaved (activated) sterol regulatory element binding protein 1 (SREBP-1), which is a negative regulator of UCP3 in hyperinsulinemia39, had lower levels compared with non-diabetic mice (Online Figures XXI-C, XXI-D, XXI-E, XXI-F). At the same time, FOXO1 (Online Figures XXII-A, XXII-D, XXII-E, XXII-F, XXII-G) or KLF5 ablation (Online Figures XXII-B,XXII-C, XXII-H to O) in diabetic mice did not alter UCP3 expression. Surprisingly, active cardiac SREBP-1 levels were increased in diabetic αMHC-FoxO1−/− mice (Online Figures XXII-D, XXII-G) and diabetic C57BL/6 mice treated with ML264 (Online Figure 22L,N) but not in diabetic αMHC-Klf5−/− mice (Online Figures XXII-H, XXII-J). Thus, improved cardiac function in diabetic mice with KLF5 or FOXO1 inhibition seems to be independent of cardiac UCP3 and SREBP-1 changes.
Metabolic changes in diabetic hearts have been associated with altered expression of MEF2C that regulates GLUT4 expression40. MEF2C and its transcriptional target Glut4 is decreased during diabetes40,41. We observed that C57BL/6 mice with T1D had lower MEF2C mRNA and protein levels (Online Figures XXIII-A to C). The effect of T1D in cardiac MEF2C expression does not seem to be reversed in diabetic αMHC-FoxO1−/−(Online Figures XXIII-D to F), αMHC-Klf5−/− (Online Figures XXIII-G to I) and ML264-treated C57BL/6 mice (Online Figures XXIII-J to L) that had improved cardiac function. Conclusively, KLF5 causes DbCM via a pathway that does not involve PPARα, UCP3, SREBP-1, and MEF2C.
Cardiomyocyte KLF5 causes cardiac dysfunction itself.
To investigate further the cardiotoxic effect of KLF5, we induced constitutive cardiomyocyte-KLF5 expression in non-diabetic C57BL/6 mice and diabetic αMHC-FoxO1−/− mice that do not develop DbCM (Fig. 3A, Online Figures VII-I, VII-J) and have lower cardiac KLF5 protein levels compared with diabetic floxed mice (Fig. 3B, Online Figure VII-K). We performed cardiomyocyte-specific induction of KLF5 expression by injecting mice with AAV9 that carries KLF5 under the cardiomyocyte-specific cardiac troponin T (cTnT). It was shown previously that gene delivery with AAV9-cTnT ensures selective cardiac tropism42. Injection of AAV9-cTnT-Klf5 in non-diabetic C57BL/6 mice (Fig. 4D, Online Figure XXIV-A) and diabetic αMHC-FoxO1−/− mice (Fig. 4D, Online Figures XXIV-B to F) increased cardiac KLF5 expression. C57BL/6 mice injected with AAV9-cTnT-Klf5 developed cardiac dysfunction compared with control C57BL/6 mice treated with empty AAV9-cTnT (Fig. 4E, Online Figures XXIV-G, XXIV-H, Online Table XII). In line with the above, αMHC-FoxO1−/− mice injected with AAV9-cTnT-Klf5 and STZ 2 weeks later (Online Figure XXV-A) developed cardiac systolic (Fig. 4F, Online Figure XXV-B, Online Table XIII) and diastolic dysfunction (Online Figure XXV-C) opposite to diabetic αMHC-FoxO1−/− mice that were protected from DbCM (Fig. 3A, Online Figures VII-I, VII-J).
To assess the cardiotoxic effect of cardiomyocyte-KLF5 further, we generated a mouse line with doxycycline-inducible cardiomyocyte-specific constitutive expression of KLF5 (αMHC-rtTA-Klf5) (Fig. 4G, Online Figure XXV-D). After 2 weeks on doxycycline-enriched diet, αMHC-rtTA-Klf5 mice developed cardiac dysfunction, which was further aggravated by week 4 (Fig. 4H, Online Figures XXV-E, XXV-F, Online Table XIV).
KLF5 promotes oxidative stress.
Previous studies have shown that oxidative stress is one of the components of the pathophysiology of DbCM43. Cardiac tissue from diabetic C57BL/6 mice exhibited higher DHE staining for superoxide (O−2) 12 weeks post-STZ administration compared with non-diabetic control mice (Fig. 5A, Online Figure XXVI-A). Similarly, diabetic mice had increased DHE staining in the skeletal muscle (Online Figures XXVI-B, XXVI-C).
Fig. 5.
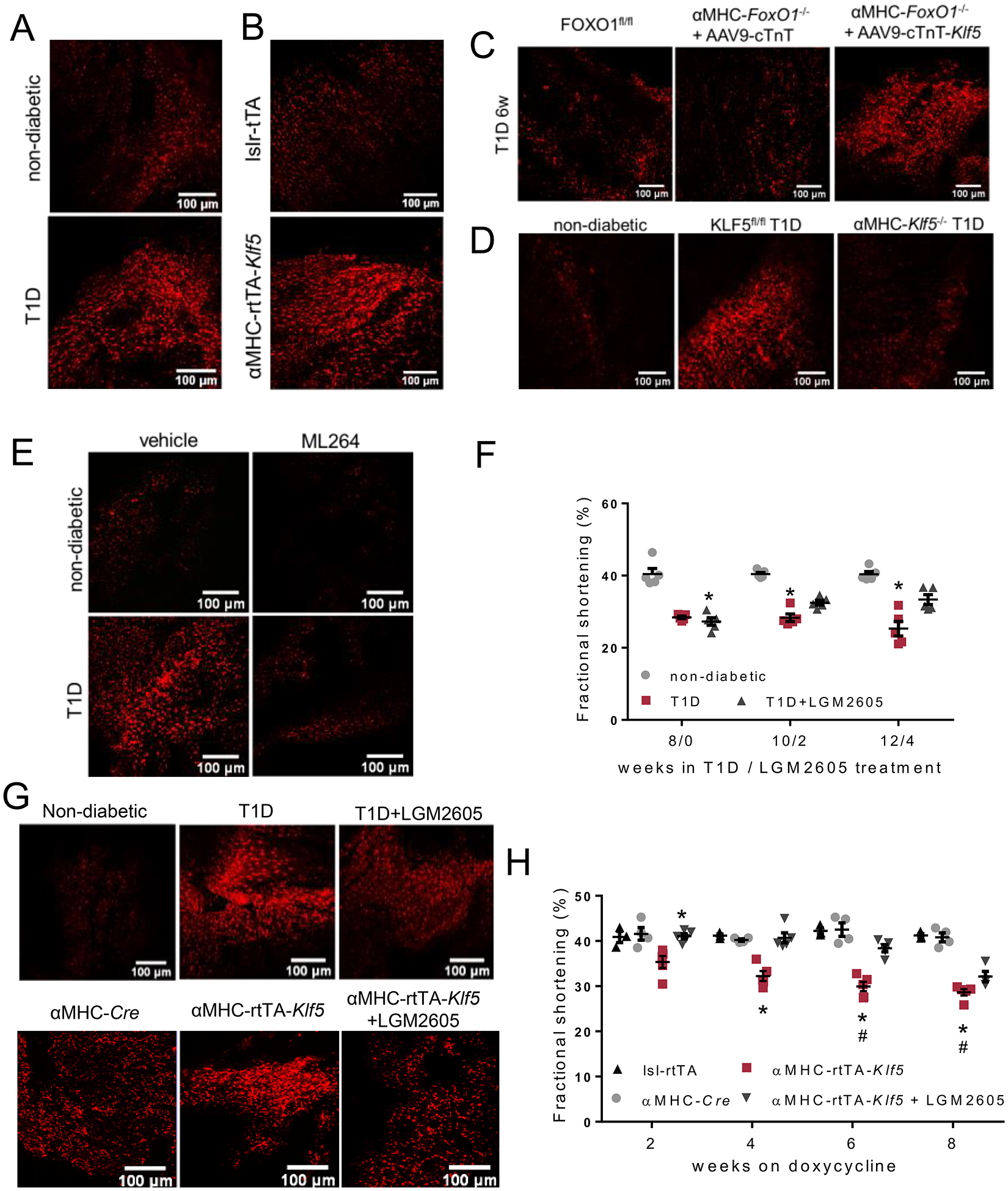
A-E: Representative DHE fluorescence images in fresh cardiac tissue of non-diabetic C57BL/6 and diabetic C57BL/6 (A) mice 12 weeks post-STZ, lsl-rtTA-TRE-Klf5 and αMHC-rtTA-Klf5 mice fed on doxycycline-enriched diet for 10 days (B), diabetic FOXO1fl/fl mice and diabetic αMHC-FoxO1−/− mice infected with AAV9-cTnT or AAV9-cTnT-hKlf5 (C), non-diabetic C57BL/6, diabetic KLF5fl/fl and diabetic αMHC-Klf5−/− mice (D), non-diabetic and diabetic mice treated with ML264 or vehicle (E). F: Left ventricular fractional shortening of non-diabetic C57BL/6, diabetic C57BL/6 and diabetic C57BL/6 mice treated with LGM2605. G: Representative DHE fluorescence images in fresh cardiac tissue of non-diabetic C57BL/6, diabetic C57BL/6 and diabetic C57BL/6 mice treated with LGM2605 and doxycycline-fed αMHC-Cre, αMHC-rtTA-Klf5 mice treated with LGM2605 or control saline. H: Left ventricular fractional shortening of doxycycline-fed lsl-rtTA mice, αMHC-Cre, αMHC-rtTA-Klf5 mice treated with LGM2605 or control saline.
To explore whether KLF5 accounts for O−2 generation independent of diabetes, we treated AC16 cells with adenovirus expressing KLF5 (Ad-KLF5; Online Figures XXVI-D, XXVI-E). Overexpression of KLF5 increased O−2 formation compared with control Ad-GFP-infected cells in the presence of either regular medium (Online Figures XXVI-F, XXVI-G) or medium enriched with either palmitate or high glucose (Online Figures XXVI-H, XXVI-I).
After 10 days on doxycycline diet, αMHC-rtTA-Klf5 mice had increased cardiac DHE fluorescence intensity (Fig. 5B, Fig. 5BOnline Figure XXVI-J) compared with doxycycline-fed control mice but not in the skeletal muscle (Online Figures XXVI-K, XXVI-L) that does not have constitutive KLF5 expression. Cardiac O−2 formation was also increased in diabetic αMHC-FoxO1−/− mice injected with AAV9-cTnT-Klf5 (Fig. 5C, Online Figure XXVII-A). In accordance with KLF5-driven oxidative stress, diabetic αMHC-Klf5−/− mice exhibited lower cardiac O−2 formation compared with diabetic C57BL/6 mice and similar when compared to non-diabetic mice (Fig. 5D, Online Figure XXVII-B). On the other hand, O−2 formation in skeletal muscle showed an increasing trend in diabetic C57BL/6 mice and a clear increase in the skeletal muscle of diabetic mice with cardiomyocyte KLF5 ablation (Online Figures XXVII-C, XXVII-D). Likewise, systemic administration of the pharmacological KLF5 inhibitor ML264 decreased O−2 production in both heart (Fig. 5E, Online Figure XXVII-E) and skeletal muscle (Online Figures XXVII-F, XXVII-G), compared with diabetic mice treated with vehicle. In accordance, KLF5 knockdown in AC16 cells treated with Ad-shKLF5 (Online Figure XXVII-H) decreased O−2 formation mediated by high glucose treatment (Online Figures XXVII-I to K). Diabetic αMHC-FoxO1−/− mice with decreased KLF5 levels had also lower cardiac O−2 formation (Online Figures XXVII-L, XXVII-M). In diabetic Pparα−/− mice, which have comparable cardiac KLF5 expression with diabetic C57BL/6 mice (Fig. 4A, Online Figure XVII–) and were not protected from DbCM, cardiac O−2 levels were increased (Online Figures XXVIII-A, XXVIII-B) while skeletal muscle O−2 levels showed an increasing trend (Online Figures XXVIII-C, XXVIII-D) compared with non-diabetic mice.
Antioxidant treatment alleviates KLF5-mediated cardiac dysfunction partially.
To evaluate the contribution of cardiac oxidative stress on KLF5-mediated cardiac dysfunction, we treated diabetic C57BL/6 mice and αMHC-rtTA-Klf5 mice with LGM2605, an antioxidant compound we developed16, 43 and tested previously22. Treatment of diabetic C57BL/6 mice with LGM2605 started 8 weeks post-STZ injection, when mice had developed cardiac dysfunction (Fig. 5F, Online Figure XXIX-A). Daily oral gavage treatment with LGM2605 for 4 weeks, alleviated cardiac dysfunction partially (Fig. 5F, Online Figures XXIX-A, XXIX-B, Online Table XV). Upon completion of the treatment with LGM2605, O−2 formation was only partially reduced in cardiac tissue (Fig. 5G, Online Figure XXIX-C) and was completely restored to normal levels in the skeletal muscle (Online Figures XXIX-D, XXIX-E).
Although treatment of non-diabetic αMHC-rtTA-Klf5 mice with LGM2605 for 8 weeks resulted in almost complete restoration of cardiac O−2 levels (Fig. 5G, Online Figure XXX-A), systolic and diastolic cardiac function of LGM2605-treated αMHC-rtTA-Klf5 mice was improved compared with untreated αMHC-rtTA-Klf5 mice but it was still worse than control lsl-rtTA and αMHC-Cre mice (Fig. 5H, Online Figures XXX-B, XXX-C, Online Table XVI). Interestingly, cardiac NAD(P)H levels of αMHC-rtTA-Klf5 mice treated with LGM2605 were decreased compared with control αMHC-Cre mice (Online Figure XXXA XXX-D). Thus, antioxidant treatment with LGM2605 does not suffice to ablate KLF5-mediated cardiac dysfunction.
Cardiomyocyte KLF5 increases NOX4 expression.
Proton leak at complex I and III of mitochondrial electron transport chain or increased NOX2 and NOX4 are major sources for cardiomyocyte O−244. To assess whether KLF5 increases O−2 formation via higher proton leak, we measured OCR in AC16 cells treated with Ad-KLF5 (Online Figure XXXI-A). KLF5 overexpression decreased slightly basal (Online Figure XXXI-B) and maximal (Online Figure XXXI-C) respiration, and lowered proton leak-related OCR (Online Figure XXXI-D) compared with control cells treated with Ad-GFP. ATP-linked OCR remained unaltered (Online Figure XXXI-E).
Then, we assessed the expression of various genes, which encode for proteins that either promote or alleviate oxidative stress and contribute to NADPH generation (Online Figure XXXII), in αMHC-rtTA-Klf5 mice fed on doxycycline-enriched diet for 10 days (Online Figures XXXIII-A, XXXIII-B). Our analysis revealed higher cardiac expression for NADPH oxidases Nox2 and Nox4 and lower expression for superoxide dismutases Sod1 and Sod2, as well as for cytosolic malate dehydrogenase (Mdh1), cardiac hexokinase 2 (Hk2), α-ketoglutare dehydrogenase (Odgc) and glutathione synthetase (Gss) (Online Figure XXXIII-B). Similar to the αMHC-rtTA-Klf5 mice, diabetic C57BL/6 mice had increased cardiac Nox4 and decreased Sod1 and Sod2 gene expression (Online Figure XXXIII-C). Conversely, diabetic αMHC-Klf5−/− mice had lower Nox4 and higher Sod1 expression, whereas Nox2 expression was increased compared with diabetic floxed littermates (Online Figure XXXIII-D). Diabetic C57BL/6 mice treated with the pharmacological KLF5 inhibitor ML264 had lower cardiac Sod1 expression but no statistically significant differences were observed in Sod2, Nox2, Nox4, Mdh1 and Hk2 expression compared with diabetic mice that were given control vehicle (Online Figure XXXIII-E). Thus, cardiac Nox4 was the only among 29 oxidative stress-related genes that we tested, the expression of which changed reciprocally with KLF5 in most of the mouse models with altered KLF5 expression that we tested.
We then assessed cardiac NOX4 and NOX2 protein levels in non-diabetic αMHC-rtTA-Klf5 mice, as well as in diabetic αMHC-FoxO1−/− mice that were treated with AAV9-cTnT-Klf5, in diabetic αMHC-Klf5−/− mice and in diabetic C57BL/6 mice treated with ML264. NOX4 protein levels were induced in αMHC-rtTA-Klf5 mice, whereas cardiac NOX2 expression was not (Fig. 6A, Online Figures XXXIII-F, XXXIII-G). Furthermore, NOX4 was increased in diabetic αMHC-FoxO1−/− mice injected with AAV9-cTnT-Klf5 (Fig. 6B, Online Figure XXXIII-H). On the other hand, NOX4 was decreased in diabetic αMHC-Klf5−/− (Fig. 6C, Online Figure XXXIII-I) and diabetic C57BL/6 mice treated with ML264 (Fig. 6C, Online Figure XXXIII-J) in comparison to their diabetic controls. AC16 cells overexpressing KLF5 also had higher NOX4 but not NOX2 protein levels (Online Figures XXXIV-A to C). Thus, KLF5 activation increased cardiac NOX4 protein levels, which was abrogated upon genetic or pharmacologic inhibition of KLF5 expression.
Fig. 6.
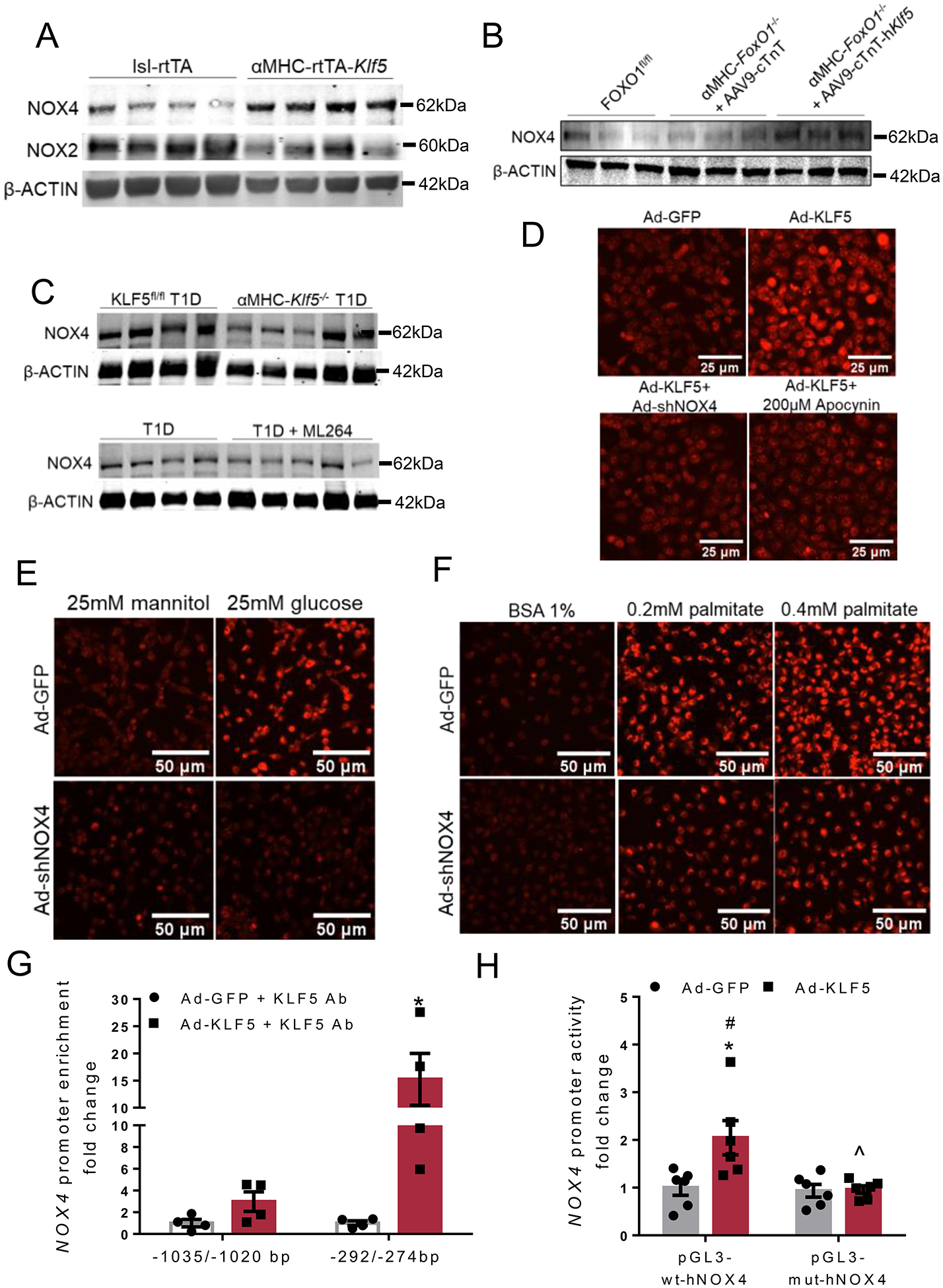
A-C: Immunoblots of cardiac NOX4 (A, B, C), NOX2 (A) and β-ACTIN (A, B, C) proteins of lsl-rtTA-TRE-Klf5 and αMHC-rtTA-Klf5 mice fed on doxycycline-enriched diet for 10 days (A; analysis shown in Online Figures XXXIII–F,G), diabetic FOXO1fl/fl mice and diabetic αMHC-FoxO1−/− mice infected with AAV9-cTnT or AAV9-cTnT-hKlf5 (B; analysis shown in Online Figure XXXIII–H), diabetic KLF5fl/fl or αMHC-Klf5−/− mice (C; analysis shown in Online Figure XXXIII–I), and diabetic mice treated with ML264 or control vehicle (C; analysis shown in Online Figure XXXIII–J). D-F: Representative DHE fluorescence images (D, E, F) of AC16 cells infected with adenovirus expressing GFP, KLF5, KLF5+shNOX4 or KLF5 followed by treatment with apocynin (D), AC16 cells infected with adenovirus expressing GFP and shNOX4 treated with 25mM mannitol or 25mM glucose (E), and AC16 cells infected with adenovirus expressing GFP and shNOX4 and treated with 0.2mM or 0.4mM palmitate (PA) dissolved in 1% BSA fraction V (F). G:KLF5 enrichment in NOX4 promoter sequence fragments precipitated with KLF5 antibody from AC16 cells expressing GFP or KLF5. H: Luciferase activity normalized to firefly in AC16 cells transfected with reporter plasmids containing the wild type NOX4 −1546/+54bp promoter fragment (pGL3-wt-hNOX4) or the mutant [−1547/+54bp](-282/-276bp)N→A (pGL3-mut-hNOX4) and infected with adenoviruses expressing GFP or KLF5.
To evaluate further the involvement of NOX4 in mediating the effect of KLF5 in O−2 formation, we performed DHE staining in AC16 cells treated with Ad-KLF5 and either Ad-shNOX4 or apocynin, an inhibitor of NADPH oxidases. Increased DHE fluorescence intensity mediated by KLF5 overexpression was completely abrogated when AC16 cells were co-infected with Ad-shNOX4 (Fig. 6D, Online Figure XXXIV-D), which specifically decreased NOX4 (Online Figures XXXIV-E to G). Also, KLF5-mediated increase of O−2 formation did not occur when cells were treated with 200μM apocynin (Fig. 6D, Online Figure XXXIV-D). NOX4 knockdown in AC16 cells also rescued O−2 accumulation mediated by FOXO1 overexpression (Online Figures XXXIV-H, XXXIV-I), which promotes KLF5 expression (Fig. 2C,D,F,H). Accordingly, high O−2 levels detected in AC16 cells treated with high glucose (Fig. 6E, Online Figure XXXIV-J) or palmitate (Fig. 6F, Online Figure XXXIV-K) were diminished when cells were treated with Ad-shNOX4 or 200μM apocynin (Online Figures XXXIV-I to L).
To examine, whether NOX4 expression is induced directly by KLF5, we performed in silico analysis (Genomatix) of the murine Nox4 promoter (UCSC Genome Browser), which revealed 11 potential KLF binding sites (Online Figure XXXV-A). Alignment (STRAP) of the mouse and human NOX4 promoters predicted one conserved potential binding site for KLF5 (-242/-224bp) which corresponds with the −292/−274bp region of the human NOX4 promoter (Online Figure XXXV-B). Chromatin immunoprecipitation in AC16 cells overexpressing KLF5 verified that KLF5 binds directly on the −292/−274bp region of NOX4 promoter (Fig. 6G, Online Figure XXXV-C).
Luciferase reporter assays in AC16 cells transfected with pGL3 plasmid carrying the wt −1547/+54bp human NOX4 promoter region and infected with Ad-KLF5 showed increased NOX4 promoter activity compared with cells infected with Ad-GFP (Fig. 6H). Substitution of the −282/−276bp region with adenosines (Online Figure XXXVI) abrogated KLF5-mediated induction of NOX4 promoter activity (Fig. 6H). Thus, KLF5 activates cardiac oxidative stress in diabetes via direct regulation of NOX4 expression.
Cardiomyocyte KLF5-mediated oxidative stress decreases mitochondrial abundance.
We then assessed whether KLF5-mediated NOX4 upregulation is associated with mitochondrial superoxide accumulation and mitochondrial abundance. AC16 cells over-expressing KLF5 had higher mitochondrial O−2 as shown by MitoSOX red staining (Online Figures XXXVII-A, XXXVII-B). Higher O−2 content in AC16 cells overexpressing KLF5 was accompanied by lower mitochondrial membrane potential (ΔΨm; Online Figure XXXVII-C), which was also confirmed with TMRM staining (Online Figures XXXVII-D to G). Furthermore, permeabilized AC16 cells that were infected with Ad-KLF5 and treated with thapsigargin, which inhibits Ca2+ uptake by endoplasmic reticulum, did not show significant differences in mitochondrial Ca+2 uptake compared with control cells treated with Ad-GFP (Online Figure XXXVII-C). The effect of KLF5 in lowering ΔΨm was also observed in AC16 cells that were treated with combination of Ad-KLF5 and palmitate (Online Figures XXXVII-D, XXXVII-E), whereas combination of Ad-KLF5 and mannitol or glucose resulted in a trend for decrease (Online Figures XXXVII-F, XXXVII-G).
Based on these findings, we explored whether KLF5 affects mitochondrial abundance. The αMHC-rtTA-Klf5 mice exhibited lower mitochondrial DNA:nuclear DNA ratio (mtDNA/nuDNA; Fig. 7A), as well as reduced cardiac protein levels of the mitochondrial proteins VDAC1 and ATP5a (Fig. 7B, Online Figures XXXVII-H, XXXXVII-I). Accordingly, AAV9-cTnT-hKlf5 infection resulted in a strong trend of mtDNA/nuDNA suppression in non-diabetic C57BL/6 mice (Fig. 7C) and a significant decrease in diabetic αMHC-FoxO1−/− (Fig. 7D). Lower mtDNA/nuDNA ratio was also detected in AC16 cells infected with Ad-KLF5 (Fig. 7E), which was reversed upon combined infection of AC16 cells with Ad-KLF5 and Ad-shNOX4 (Fig. 7E) that alleviates KLF5-mediarted oxidative stress (Fig. 6D, Online Figure XXXIV-D). Conversely, combined treatment of αMHC-rtTA-Klf5 mice with diet enriched in both doxycycline and antioxidant for 8 weeks failed to rescue mitochondrial abundance (Online Figure XXXVII-J). Similar analysis in diabetic mice, revealed that T1D incurred a decreasing trend for mtDNA:nuDNA, while no effect was observed when KLF5 was inhibited pharmacologically (Online Figure XXXVII-K) or genetically (Online Figure XXXVII-L). Subsequently, assessment of mtDNA integrity identified cardiac mtDNA damage in αMHC-rtTA-Klf5 mice (Fig. 7F), which was not observed in non-diabetic C57BL/6 and diabetic αMHC-FoxO1−/−, both of which were infected with AAV9-cTnT-hKlf5 (Fig. 7G,H). Moreover, diabetic KLF5fl/fl mice had a strong trend for reduced mtDNA integrity compared with non-diabetic mice (Online Figure XXXVII-M). On the contrary, cardiac mtDNA integrity of diabetic αMHC-Klf5−/− mice was similar to non-diabetic mice (Online Figure XXXVII-M).
Fig. 7.
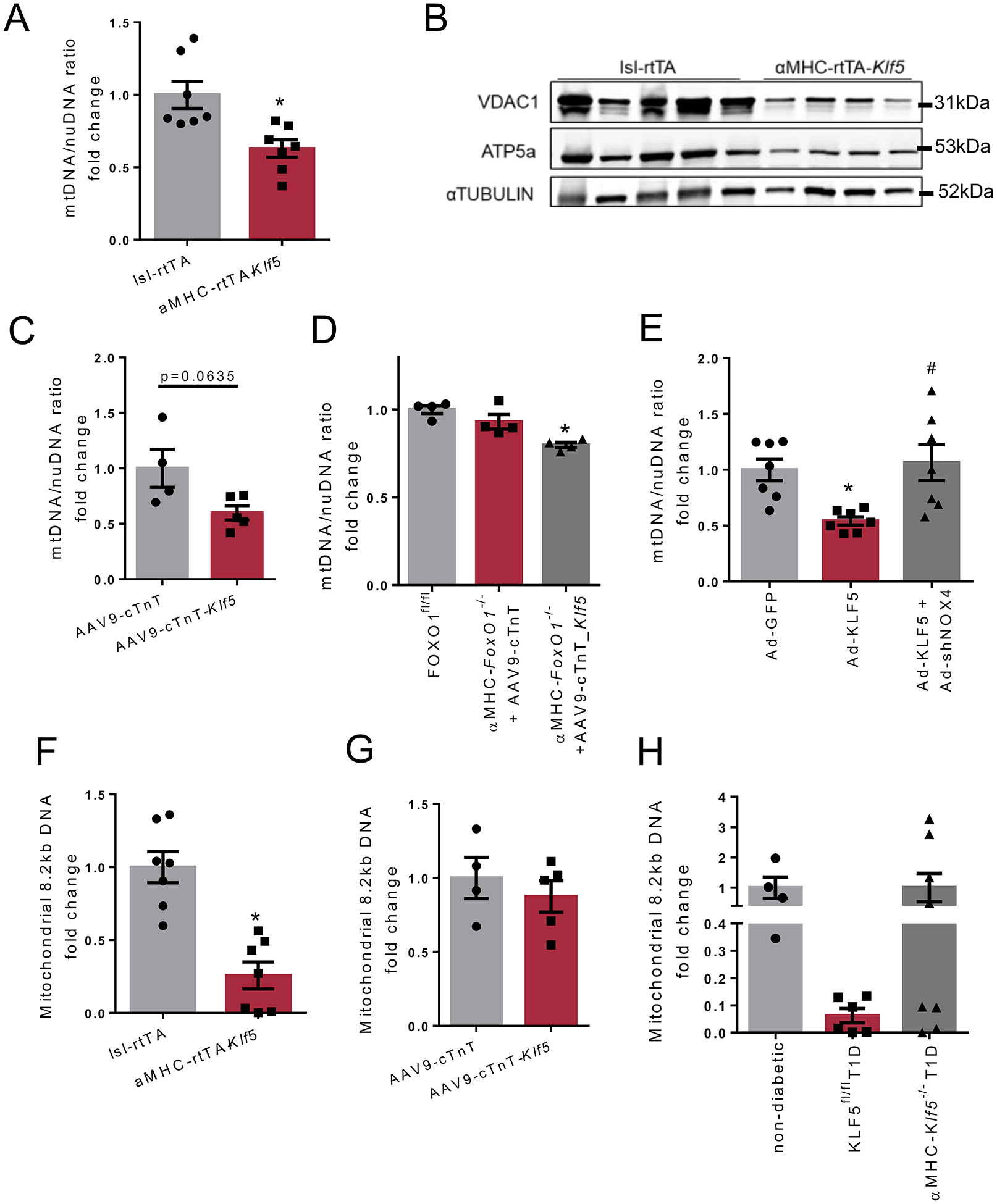
A: Mitochondrial DNA/nuclear DNA ratio in cardiac tissue of lsl-rtTA-TRE-Klf5 and αMHC-rtTA-Klf5 mice fed on doxycycline-enriched diet for 10 days. B: Immunoblots of cardiac VDAC1, ATP5a and α-TUBULIN in lsl-rtTA-TRE-Klf5 and αMHC-rtTA-Klf5 mice fed on doxycycline-enriched diet for 10 days (analysis shown in Online Figures XXXVII–H,I). C-E: Mitochondrial DNA/nuclear DNA ratio in cardiac tissue of C57BL/6 mice infected with AAV9-cTnT or AAV9-cTnT-hKlf5 (C), diabetic FOXO1fl/fl mice and diabetic αMHC-FoxO1−/− mice infected with AAV9-cTnT or AAV9-cTnT-hKlf5 (D), AC16 cells infected with adenovirus expressing GFP, KLF5 or KLF5+shNOX4 (E). F-H: Amplification of mitochondrial 8.2kb DNA sequence normalized to mitochondrial abundance of lsl-rtTA-TRE-Klf5 and αMHC-rtTA-Klf5 mice fed on doxycycline-enriched diet for 10 days (F), C57BL/6 mice infected with AAV9-cTnT or AAV9-cTnT-hKlf5 (G), and non-diabetic C57BL/6, diabetic KLF5fl/fl and diabetic αMHC-Klf5−/− mice (H).
Assessment of the expression of the transcription factor COUP-TFII, which suppresses expression of genes related to mitochondrial oxidative stress detoxification and contributes to diabetic cardiac failure45, showed unaltered mRNA and higher protein levels in diabetic C57BL/6 mice (Online Figures XXXVIII-A to C). FOXO1 or KLF5 inhibition in diabetes did not restore COUP-TFII mRNA and protein to the level observed in non-diabetic mice (Online Figures XXXVIII-D to L).
KLF5 promotes myocardial ceramides accumulation.
As antioxidant treatment incurred only partial improvement in cardiac function of αMHC-rtTA-Klf5 mice, and KLF5 has been linked to lipid metabolism46, we assessed whether altered KLF5 expression is accompanied by changes in cardiac lipid content. Lipidomic analysis in left ventricles of control non-diabetic C57BL/6 mice, C57BL/6 mice with T1D for 12 weeks that were treated either with ML264 or vehicle, diabetic αMHC-Klf5−/− mice, diabetic Pparα−/− and non-diabetic αMHC-rtTA-Klf5 mice following treatment with doxycycline for 10 days revealed distinct lipidomic profiles (euclidean clustering) (Fig. 8A, Online Table XVII). Of note, the cardiac lipidome profile of non-diabetic αMHC-rtTA-Klf5 mice resembled the profile of the diabetic C57BL/6 and diabetic Pparα−/− mice (Fig. 8A). On the other hand, cardiac lipidome of diabetic αMHC-Klf5−/− mice or diabetic C57BL/6 mice treated with ML264 was similar to non-diabetic mice (Fig. 8A).
Fig. 8.
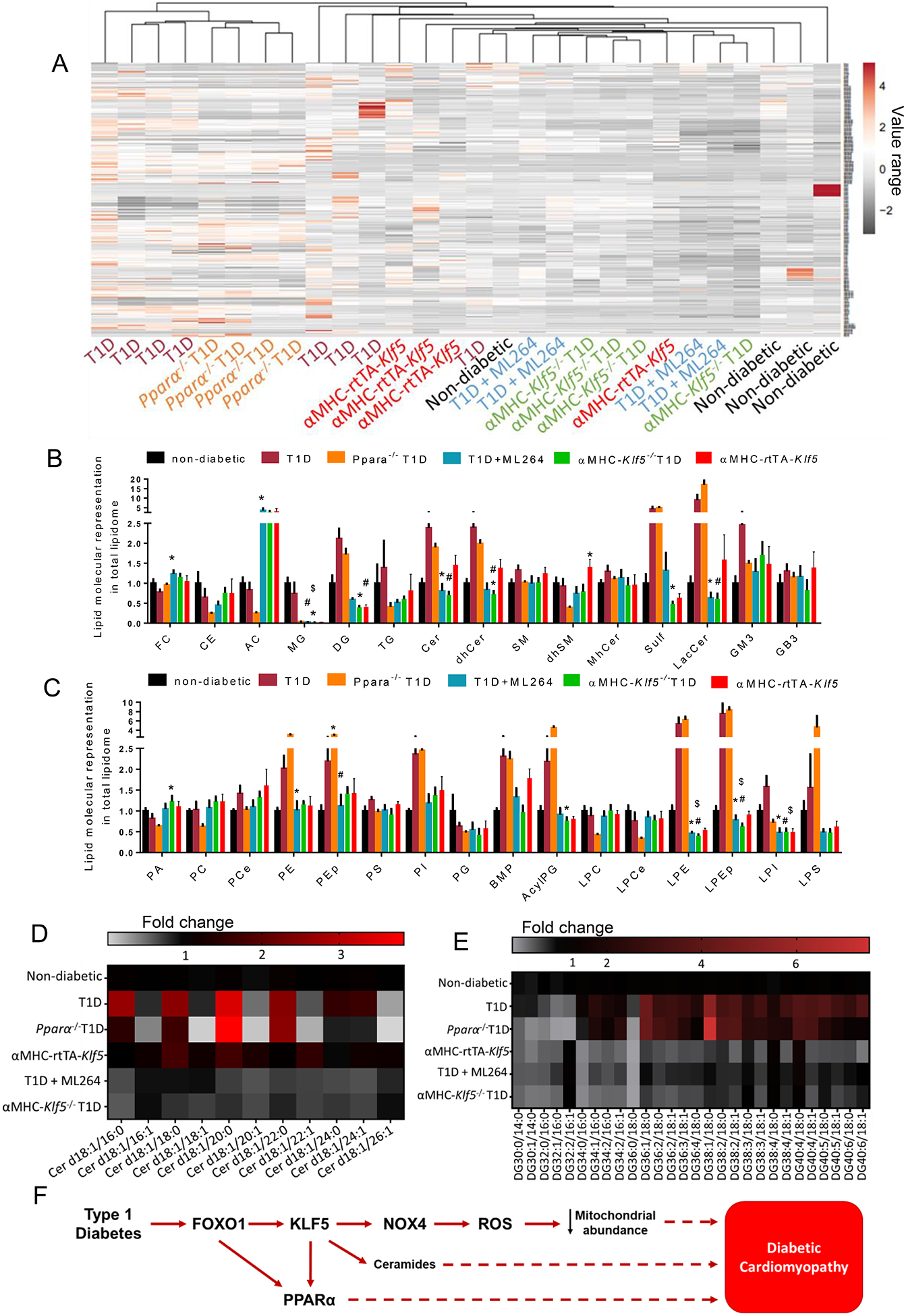
A-C: Heatmap (A) with Euclidean hierarchical clustering of lipid species (lipid species that are listed on the right side of the heat map are described in Online Table XVII). Cardiac lipid species representation to total lipidome (presented as fold-change compared to non-diabetic wt mice) in non-diabetic C57BL/6 mice, diabetic C57BL/6 mice, diabetic Pparα−/− mice, diabetic C57BL/6 mice treated with ML264, diabetic aMHC-Klf5−/− mice and aMHC-rtTA-Klf5 mice fed on doxycycline-enriched diet for 10 days; FC; free cholesterol, CE; cholesterol ester, AC; acylcarnitines, MG; monoglycerides, DG; diacylglycerols, TG; triglycerides, Cer; ceramides, dhCer; dihydroceramides, SM; sphingomyelins, dhSM; dihydrosphingomyelins, MhCer; monohexosylceramides, Sulf; sulfatides, LacCer; lactosylceramides, GM3; gangliosides, GB3; globotrioseacylceramides) (B) and PA; phosphatidic acids, PC; phosphatidylcholines, PCe; phosphatidylcholine ethers, PE; phosphatidylethanolamines, PEp; plasmalogen phosphatidylethanolamines, PS; phosphatidylserines, PI; phosphatidylinositols, PG; phosphatidylglycerols, BMP; bis(monoacylglycero)phosphates, AcylPG; acylphosphatidylglycerols, LPC; lysophosphosphatidylcholines, LPCe; lysophosphosphatidylcholine ethers, LPE; lysophosphatidylethanolamines, LPEp; plasmalogen lysophosphatidylethanolamines, LPI; lysophosphatidylinositols and LPS; lysophosphatidylserines (C). D-E; Heatmaps of ceramides family (D) and diacylglycerol family (E) in non-diabetic C57BL/6 mice, diabetic C57BL/6 mice, diabetic Pparα−/− mice, diabetic C57BL/6 mice treated with ML264, diabetic αMHC-Klf5−/− mice, and αMHC-rtTA-Klf5 mice fed on doxycycline-enriched diet for 10 days. F: Schematic representation of the proposed model about pathways that are involved in diabetic cardiomyopathy.
Among various lipid families that we measured, diabetic C57BL/6 and Pparα−/− mice exhibited similar changes compared to control non-diabetic mice (Fig. 8B–E). More specifically, they showed strong trend or significant increase for diacylglycerols (DGs), triglycerides (TGs), ceramides (Cer), dihydroceramides (dhCer), lactosylceramides (LacCer), sulfatides (Sulf), phosphatidylethanolamine (PE), plasmalogen phosphatidylethanolamines (Pep), phosphatidylinositols (PI), bis(monoacylglycero)phosphates (BMP), acyl phosphatidylglycerol (AcylPG), lysophosphosphatidylcholine ethers (LPE), and lysophosphatidylethanolamines, (LPEp). Comparing changes in cardiac lipid species of mice with elevated KLF5 expression (T1D, Pparα−/− T1D, αMHC-rtTA-Klf5) and mice that KLF5 was inhibited (T1D+ML264, αMHC-Klf5−/− T1D), showed that only Cer, dhCer, LacCer, and BMP showed reciprocal changes with KLF5 expression (Fig. 8B,C).
More focused analysis on certain ceramide species showed that d18:1/18:0 (N-stearoyl-D-erythro-sphingosine) and d18:1/20:0 (N-arachidoyl-D-erythro-sphingosine) had a trend for increase in diabetic C57BL/6, Pparα−/− mice and αMHC-rtTA-Klf5 mice (Fig. 8D, Online Figure XXXIX-A) but not in diabetic αMHC-Klf5−/− mice or ML264-treated diabetic C57BL/6 mice. Dihydroceramides of similar chain length and lactosyl-ceramides that also have been implicated in promoting mitochondrial dysfunction in diabetes47, showed a trend for increase in diabetic C57BL/6 mice and were decreased in diabetic αMHC-Klf5−/− mice or ML264-treated diabetic C57BL/6 mice (Online Figures XXXIX-B, XXXIX-C). Diacylglycerols that lead to DbCM through PKC activation8, 48, exhibited a trend for increase in diabetic C57BL/6 and Pparα−/− mice (Fig. 8B,E, Online Figure XXXIX-D) and decreased in diabetic αMHC-Klf5−/− mice and diabetic C57BL/6 mice treated with ML264 compared to diabetic Pparα−/− mice (Fig. 8B,E, Online Figure XXXIX-D). On the contrary, αMHC-rtTA-Klf5 mice did not exhibit elevated cardiac diacylglycerol levels (Fig. 8B,E, Online Figure XXXIX-D). Thus, higher KLF5 expression is associated primarily with higher cardiac ceramide content, which is prevented upon KLF5 inhibition in mice with T1D.
DISCUSSION
DbCM has been associated with activation of various pathologic mechanisms at the systemic and cellular level. Oxidative stress, cardiac lipotoxicity, metabolic rigidity and glucotoxicity are some of the complications that have been proposed to account for cardiac dysfunction in diabetes. Our previous study11 associated KLF5 with transcriptional regulation of cardiac PPARα. Studies in hearts of humans and rats with diabetes have shown lower expression of cardiac PPARα49, 50. While this is in agreement with lower cardiac PPARα expression in failing hearts41, 51, 52, constitutive cardiomyocyte PPARα expression also caused cardiac dysfunction37, 50 due to lipotoxicity. Conversely, other studies have associated DbCM with increased expression of cardiac PPARα and its target genes37, 53. As failed hearts have lower PPARα expression, the discrepancy in cardiac PPARα expression between different studies may reflect a different stage of DbCM that has been reached in each study. Future studies that will investigate systematically how cardiac PPARα expression changes over the course of DbCM progression are warranted.
As alleviation of insulin resistance in a lipotoxic diabetic heart has been suspected for causing glucolipotoxicity and cytotoxic damage54, elucidation of the mechanisms that contribute to lipotoxicity is crucial for resolving DbCM. To this end, our study focused on the interplay among inhibition of insulin signaling, FOXO1 activation, and activation of KLF5 that regulates PPARα11, which has been associated with cardiac lipotoxicity and DbCM37, 50. Our findings identified cardiac KLF5 as a unifying transcriptional factor that is regulated by FOXO1 and contributes to DbCM in a PPARα-independent manner. Our data show that the cardiotoxic effect of KLF5 is accounted for, at least in part, by stimulation of cardiac NOX4 expression and oxidative stress, and ventricular ceramides accumulation.
Inhibition of insulin signaling that occurs in both T1D and T2D, activates FOXO1, which has been associated with several pathological pathways including apoptosis, inflammation, autophagy, glucose metabolism and hypertrophy in DbCM12. Separately, high glucose levels can also activate FOXO1, which can eventually drive apoptosis in cardiomyocytes55. The cardioprotective role of FOXO1 ablation in mice with T1D shown here, which correlates with lower KLF5 expression, is in agreement with a similar effect of FOXO1 ablation in mice fed with HFD56, which is a model of T2D.
Based on our new observations about the regulation of cardiac KLF5 by FOXO1 and the higher cardiomyocyte KLF5 expression in patients with diabetes, along with our previous findings showing altered expression of cardiomyocyte KLF5 in diabetes11, we assessed potential involvement of KLF5 in DbCM. We showed that altered cardiac Klf5 expression parallels Pparα expression changes in diabetes11. Other studies have shown that KLF5 influences hepatic FA uptake and triglyceride synthesis via regulation of the Pparg2 gene57. Thus, KLF5 is important for the regulation of lipid metabolism in various tissues. Our present study shows that cardiomyocyte-KLF5 inhibition alleviates DbCM. Moreover, although inhibition of insulin signaling is linked to DbCM in both T1D and T2D58, alleviation of DbCM by KLF5 inhibition was not associated with improvements in either responsiveness to insulin or glucose uptake in our mice with T1D. In fact, it was associated with modulation of oxidative stress but not by altered expression of cardiac PPARα, which also leads to DbCM37. The lack of downregulation of cardiac PPARα in diabetic mice that underwent KLF5 inhibition may be due to compensation by FOXO1-mediated stimulation of PPARα expression that we also show or other pathways. Similarly, expression of cardiac UCP3, which is a PPARα target38, was increased in mice with T1D, and KLF5 inhibition did not reverse it. Upregulation of cardiac UCP3 expression in mice with T1D was also consistent with inhibition of SREBP-1 activation that we also observed. Nevertheless, SREBP-1 suppresses UCP3 expression in mice with hyperinsulinemia39. However, inhibition of KLF5 or FOXO1 did not restore UCP3 expression to normal levels although it did increase protein levels of active SREBP-1 in the case of cardiomyocyte FoxO1 ablation or administration of ML264 compared with control diabetic mice. Along these lines, cardiac MEF2C expression was decreased in mice with T1D, the same way it did in myocardial samples obtained from diabetic patients with non-ischemic heart failure59. However, inhibition of either FOXO1 or KLF5 did not alter MEF2C expression. Thus, the protective effect of KLF5 inhibition seems to be underlain by a separate pathway that does not involve PPARα, UCP3 or MEF2C, which have been associated with DbCM. Notably, KLF5 inhibition alleviates DbCM even with persistently high levels of expression of cardiac PPARα, which identifies its therapeutic potential.
The involvement of cardiomyocyte KLF5 in DbCM was further demonstrated by cardiac dysfunction of αMHC-rtTA-Klf5 without diabetes, as well as of diabetic αMHC-Fox01−/− mice that are treated with AAV-cTnT-Klf5. Increased Cer content and accumulation of O−2 preceded cardiac dysfunction, as shown by lipidomic analysis and DHE staining in hearts of αMHC-rtTA-Klf5 mice fed on doxycycline for 10 days. Both O−2 and Cer have been ascribed causality for DbCM60, 61. Accordingly, genetic and pharmacological inhibition of KLF5 restored normal cardiac function in diabetic mice, which was accompanied by lower myocardial O−2 and Cer content. Similarity of the ceramide content in hearts of wild type and Pparα−/− mice with T1D, both of which have higher KLF5 expression, suggests a potential regulatory role of KLF5 in accumulation of cardiac ceramides that is independent of PPARα-induced lipotoxicity.
Cardiac accumulation of O−2 in αMHC-rtTA-Klf5 mice was associated with higher expression of the pro-oxidant NOX4 and lower gene expression of the antioxidants Sod1 and Sod2. NOX4 has been linked with ROS-related cardiac dysfunction62, which can be further aggravated due to lower expression of superoxide dismutases63. Combined increase in NOX4 and lower Hk2 and Mdh1 gene expression is consistent with the lower NAD(P)H content that we observed.
NOX4 is a major source of cytosolic ROS, which can also stimulate mitochondrial oxidative stress in diabetes64. NOX4 has also been detected in mitochondria65–67. In our study, we documented that KLF5 increased O−2 levels by stimulating NOX4 expression, which decreased cardiac mitochondrial abundance. Lower mitochondrial number has also been documented in cardiac and hepatic tissues of alloxan-induced-T1D rats68. Conversely, other studies in mice with T1D69 or T2D70 revealed increased mitochondrial number. This discrepancy can be because of differences in the type of diabetes, duration of T1D, and mouse sex.
Our loss of function studies showed that the effect of KLF5 in oxidative stress is independent of cardiac COUP-TFII expression changes, which is increased in non-ischemic cardiomyopathy patients and mice with pressure overload45, 71 and contributes to mitochondrial oxidative stress. The beneficial effects of KLF5 inhibition in DbCM can only merely be attributed to the antioxidant consequences of the treatment. Nevertheless, treatment with the antioxidant vitamin E did not show any improvement in the cardiovascular adverse events of diabetic patients of the Heart Outcomes Prevention Evaluation (HOPE) trial72, although it improved some aspects of diabetic pathophysiology in the smaller Secondary Prevention with Antioxidants of Cardiovascular disease in End-stage renal disease (SPACE) trial73. This is in agreement with only partial alleviation of cardiac dysfunction following treatment of aMHC-rtTA-Klf5 and diabetic C57Bl/6 mice with LGM2605. This suggests that KLF5 is involved in additional pathways that contribute to DbCM. Lipotoxicity may be one of these mechanisms as suggested by our lipidomic analysis. Also, a previous study in a mouse model of diabetic nephropathy showed that anti-oxidant interventions have low efficacy unless they are applied in the early stage of T1D74. Thus, the timing of administration of anti-oxidants may also be critical. Future studies that will investigate the potential involvement of KLF5 in other pathophysiological mechanisms of DbCM, such as Cer metabolism are warranted.
Limitations.
This is a pre-clinical study, which identified the involvement of KLF5 in DbCM in mice with T1D. While we also found increased expression of KLF5 in cardiomyocytes from cardiac patients with diabetes, these patients do not have overt diabetic cardiomyopathy. As a therapeutic approach, we should note that prolonged complete loss of function of KLF5 leads to heart dysfunction11. So, any therapeutic approach would require partial inhibition of this transcription factor. The use of global Pparα−/− mouse model is not ideal for thorough understanding of the interplay between KLF5 and cardiomyocyte PPARα in DbCM. Nevertheless, these mice have lower plasma triglycerides in diabetes compared with wild type mice. Future studies with mice bearing cardiomyocyte-specific Pparα deletion are warranted. The variety of mouse models suggests that the development of heart failure in diabetes is likely to be via many routes.
Conclusion.
Our study identified the involvement of KLF5 in DbCM, as well as its regulation by FOXO1 in T1D. Therefore, KLF5 inhibition emerges as a potential therapeutic target for the alleviation of cardiac dysfunction, lipotoxicity and oxidative stress in T1D.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
Kruppel-like factor 5 (KLF5) increases peroxisome proliferator-activated receptor-alpha (PPARα) expression in cardiomyocytes.
Cardiomyocyte PPARα overexpression mimics diabetic cardiomyopathy/
Impaired insulin signaling activates FOXO1 and increases PPARα expression in cardiomyocytes.
What New Information Does This Article Contribute?
Inhibition of insulin signaling stimulates cardiomyocyte KLF5 expression via FOXO1 activation in both mice and humans.
Cardiomyocyte KLF5 promotes NOX4-mediated oxidative stress and ceramide accumulation.
Inhibition of KLF5 alleviates diabetic cardiomyopathy in a PPARα-independent manner.
Diabetic cardiomyopathy is diagnosed by the presence of ventricular dysfunction in patients with diabetes in the absence of coronary arterial disease. Previous studies have associated increased cardiac PPARα expression and fatty acid oxidation with diabetic cardiomyopathy. We have shown the importanceof KLF5 in regulating PPARa, a central regulator of fatty acid metabolism in cardiomyocytes. In this study, we found that insulin deficiency stimulates FOXO1, which eventually induces KLF5 expression promoting cardiomyopathy. Inhibition of cardiomyocyte KLF5 mitigated diabetic cardiomyopathy andwas associated with decreased cardiac oxidative stress and lower ceramide content. The association of KLF5 with oxidative stress was NOX4-dependent. Surprisingly, despite the association of KLF5 with stimulation of PPARα expression, the cardiotoxic effect of KLF5 was independent of PPARα changes. Thus, these findings identify a novel pathway for diabetic cardiomyopathy and demonstrates KLF5 as a potential therapeutic target.
SOURCES OF FUNDING
This study was supported by the National Heart Lung and Blood Institute HL130218, HL151924 (K.D.), HL45094, HL73029, HL135987 (IJG), and the National Institute of General Medical Sciences GM135399 (K.D.) of the NIH (Bethesda, MD), the W.W. Smith Charitable Trust (Conshohocken, PA) (K.D.), an American Heart Association (Dallas, TX) and the Kahn Family Post-Doctoral Fellowship in Cardiovascular Research (I.D.K.; 18POST34060150), an American Heart Association pre-doctoral fellowship (M.H.; 18PRE34060115) and a Ruth L. Kirschstein National Research Service Award (NRSA; Bethesda, MD) F30 predoctoral fellowship (MH; F30HL146007).
Nonstandard Abbreviations and Acronyms:
- DbCM
diabetic cardiomyopathy
- FA
fatty acid
- FAO
fatty acid oxidation
- PPARα
peroxisome proliferator-activated receptor alpha
- KLF5
Krüppel-like factor 5
- STZ
streptozotocin
- type-1
diabetes (T1D)
- FOXO1
Forkhead box protein O1
- ChIP
Chromatin Immunoprecipitation
- DHE
dihydroethidium
- ΔΨm
mitochondrial potential
- Cer
ceramides
- dhCer
dihydroceramides
- LacCer
lactosyl-ceramides
- SM
sphingomyelin
- HFD
high fat diet
Footnotes
DISCLOSURES
MCS has patents No. US 10,045,951 B2, No. US 10,030,040 B2, and No. US 9,987,321 B2 issued, patents No. PCT/US2016/049780, No. PCT/US17/35960, and No. PCT/US2008/006694 pending, and has a founder’s equity position in LignaMed, LLC. All other authors have no competing interests or conflicts to report.
Publisher's Disclaimer: This article is published in its accepted form. It has not been copyedited and has not appeared in an issue of the journal. Preparation for inclusion in an issue of Circulation Research involves copyediting, typesetting, proofreading, and author review, which may lead to differences between this accepted version of the manuscript and the final, published version.
REFERENCES
- 1.Ogurtsova K, da Rocha Fernandes JD, Huang Y, Linnenkamp U, Guariguata L, Cho NH, Cavan D, Shaw JE, Makaroff LE. Idf diabetes atlas: Global estimates for the prevalence of diabetes for 2015 and 2040. Diabetes Res Clin Pract. 2017;128:40–50 [DOI] [PubMed] [Google Scholar]
- 2.Lee MMY, McMurray JJV, Lorenzo-Almoros A, Kristensen SL, Sattar N, Jhund PS, Petrie MC. Diabetic cardiomyopathy. Heart. 2018 [DOI] [PubMed] [Google Scholar]
- 3.Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, Mattheus M, Devins T, Johansen OE, Woerle HJ, Broedl UC, Inzucchi SE, Investigators E-RO. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. 2015;373:2117–2128 [DOI] [PubMed] [Google Scholar]
- 4.Nolan CJ, Ruderman NB, Kahn SE, Pedersen O, Prentki M. Insulin resistance as a physiological defense against metabolic stress: Implications for the management of subsets of type 2 diabetes. Diabetes. 2015;64:673–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bayeva M, Sawicki KT, Ardehali H. Taking diabetes to heart--deregulation of myocardial lipid metabolism in diabetic cardiomyopathy. J Am Heart Assoc. 2013;2:e000433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tan Y, Zhang Z, Zheng C, Wintergerst KA, Keller BB, Cai L. Mechanisms of diabetic cardiomyopathy and potential therapeutic strategies: Preclinical and clinical evidence. Nat Rev Cardiol. 2020;17:585–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tokarz VL, MacDonald PE, Klip A. The cell biology of systemic insulin function. J Cell Biol. 2018;217:2273–2289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goldberg IJ, Trent CM, Schulze PC. Lipid metabolism and toxicity in the heart. Cell Metab. 2012;15:805–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pol CJ, Lieu M, Drosatos K. Ppars: Protectors or opponents of myocardial function? PPAR research. 2015;2015:835985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Finck BN, Kelly DP. Peroxisome proliferator-activated receptor alpha (pparalpha) signaling in the gene regulatory control of energy metabolism in the normal and diseased heart. J Mol Cell Cardiol. 2002;34:1249–1257 [DOI] [PubMed] [Google Scholar]
- 11.Drosatos K, Pollak NM, Pol CJ, Ntziachristos P, Willecke F, Valenti MC, Trent CM, Hu Y, Guo S, Aifantis I, Goldberg IJ. Cardiac myocyte klf5 regulates ppara expression and cardiac function. Circ Res. 2016;118:241–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kandula V, Kosuru R, Li H, Yan D, Zhu Q, Lian Q, Ge RS, Xia Z, Irwin MG. Forkhead box transcription factor 1: Role in the pathogenesis of diabetic cardiomyopathy. Cardiovasc Diabetol. 2016;15:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin SC, Wani MA, Whitsett JA, Wells JM. Klf5 regulates lineage formation in the pre-implantation mouse embryo. Development. 2010;137:3953–3963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sur I, Rozell B, Jaks V, Bergstrom A, Toftgard R. Epidermal and craniofacial defects in mice overexpressing klf5 in the basal layer of the epidermis. J Cell Sci. 2006;119:3593–3601 [DOI] [PubMed] [Google Scholar]
- 15.Pfleger J, Coleman RC, Ibetti J, Roy R, Kyriazis ID, Gao E, Drosatos K, Koch WJ. Genomic binding patterns of forkhead box protein o1 reveal its unique role in cardiac hypertrophy. Circulation. 2020;142:882–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mishra OP, Simmons N, Tyagi S, Pietrofesa R, Shuvaev VV, Valiulin RA, Heretsch P, Nicolaou KC, Christofidou-Solomidou M. Synthesis and antioxidant evaluation of (s,s)- and (r,r)-secoisolariciresinol diglucosides (sdgs). Bioorg Med Chem Lett. 2013;23:5325–5328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davidson MM, Nesti C, Palenzuela L, Walker WF, Hernandez E, Protas L, Hirano M, Isaac ND. Novel cell lines derived from adult human ventricular cardiomyocytes. J Mol Cell Cardiol. 2005;39:133–147 [DOI] [PubMed] [Google Scholar]
- 18.Lin Z, Zhou P, von Gise A, Gu F, Ma Q, Chen J, Guo H, van Gorp PR, Wang DZ, Pu WT. Pi3kcb links hippo-yap and pi3k-akt signaling pathways to promote cardiomyocyte proliferation and survival. Circ Res. 2015;116:35–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang Y, Goldstein BG, Nakagawa H, Katz JP. Kruppel-like factor 5 activates mek/erk signaling via egfr in primary squamous epithelial cells. FASEB J. 2007;21:543–550 [DOI] [PubMed] [Google Scholar]
- 20.Qiang L, Banks AS, Accili D. Uncoupling of acetylation from phosphorylation regulates foxo1 function independent of its subcellular localization. J Biol Chem. 2010;285:27396–27401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matsushima S, Kuroda J, Ago T, Zhai P, Ikeda Y, Oka S, Fong GH, Tian R, Sadoshima J. Broad suppression of nadph oxidase activity exacerbates ischemia/reperfusion injury through inadvertent downregulation of hypoxia-inducible factor-1alpha and upregulation of peroxisome proliferator-activated receptor-alpha. Circ Res. 2013;112:1135–1149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kokkinaki D, Hoffman M, Kalliora C, Kyriazis ID, Maning J, Lucchese AM, Shanmughapriya S, Tomar D, Park JY, Wang H, Yang XF, Madesh M, Lymperopoulos A, Koch WJ, Christofidou-Solomidou M, Drosatos K. Chemically synthesized secoisolariciresinol diglucoside (lgm2605) improves mitochondrial function in cardiac myocytes and alleviates septic cardiomyopathy. Journal of molecular and cellular cardiology. 2019;127:232–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kalliora C, Kyriazis ID, Oka SI, Lieu MJ, Yue Y, Area-Gomez E, Pol CJ, Tian Y, Mizushima W, Chin A, Scerbo D, Schulze PC, Civelek M, Sadoshima J, Madesh M, Goldberg IJ, Drosatos K. Dual peroxisome-proliferator-activated-receptor-α/γ activation inhibits sirt1-pgc1α axis and causes cardiac dysfunction. JCI Insight. 2019;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gaignebet L, Kańduła MM, Lehmann D, Knosalla C, Kreil DP, Kararigas G. Sex-specific human cardiomyocyte gene regulation in left ventricular pressure overload. Mayo Clin Proc. 2020;95:688–697 [DOI] [PubMed] [Google Scholar]
- 25.Rehman AU, Anwer AG, Gosnell ME, Mahbub SB, Liu G, Goldys EM. Fluorescence quenching of free and bound nadh in hela cells determined by hyperspectral imaging and unmixing of cell autofluorescence. Biomed Opt Express. 2017;8:1488–1498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dong Z, Shanmughapriya S, Tomar D, Siddiqui N, Lynch S, Nemani N, Breves SL, Zhang X, Tripathi A, Palaniappan P, Riitano MF, Worth AM, Seelam A, Carvalho E, Subbiah R, Jaña F, Soboloff J, Peng Y, Cheung JY, Joseph SK, Caplan J, Rajan S, Stathopulos PB, Madesh M. Mitochondrial ca. Mol Cell. 2017;65:1014–1028.e1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fazzini F, Schopf B, Blatzer M, Coassin S, Hicks AA, Kronenberg F, Fendt L. Plasmid-normalized quantification of relative mitochondrial dna copy number. Sci Rep. 2018;8:15347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Joseph LC, Kokkinaki D, Valenti MC, Kim GJ, Barca E, Tomar D, Hoffman NE, Subramanyam P, Colecraft HM, Hirano M, Ratner AJ, Madesh M, Drosatos K, Morrow JP. Inhibition of nadph oxidase 2 (nox2) prevents sepsis-induced cardiomyopathy by improving calcium handling and mitochondrial function. JCI Insight. 2017;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tomar D, Dong Z, Shanmughapriya S, Koch DA, Thomas T, Hoffman NE, Timbalia SA, Goldman SJ, Breves SL, Corbally DP, Nemani N, Fairweather JP, Cutri AR, Zhang X, Song J, Jana F, Huang J, Barrero C, Rabinowitz JE, Luongo TS, Schumacher SM, Rockman ME, Dietrich A, Merali S, Caplan J, Stathopulos P, Ahima RS, Cheung JY, Houser SR, Koch WJ, Patel V, Gohil VM, Elrod JW, Rajan S, Madesh M. Mcur1 is a scaffold factor for the mcu complex function and promotes mitochondrial bioenergetics. Cell reports. 2016;15:1673–1685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ji R, Akashi H, Drosatos K, Liao X, Jiang H, Kennel PJ, Brunjes DL, Castillero E, Zhang X, Deng LY, Homma S, George IJ, Takayama H, Naka Y, Goldberg IJ, Schulze PC. Increased de novo ceramide synthesis and accumulation in failing myocardium. JCI insight. 2017;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xiong S, Salazar G, Patrushev N, Alexander RW. Foxo1 mediates an autofeedback loop regulating sirt1 expression. J Biol Chem. 2011;286:5289–5299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xiong S, Salazar G, San Martin A, Ahmad M, Patrushev N, Hilenski L, Nazarewicz RR, Ma M, Ushio-Fukai M, Alexander RW. Pgc-1 alpha serine 570 phosphorylation and gcn5-mediated acetylation by angiotensin ii drive catalase down-regulation and vascular hypertrophy. J Biol Chem. 2010;285:2474–2487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gille C, Fahling M, Weyand B, Wieland T, Gille A. Alignment-annotator web server: Rendering and annotating sequence alignments. Nucleic Acids Res. 2014;42:W3–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qi Y, Zhu Q, Zhang K, Thomas C, Wu Y, Kumar R, Baker KM, Xu Z, Chen S, Guo S. Activation of foxo1 by insulin resistance promotes cardiac dysfunction and β-myosin heavy chain gene expression. Circ Heart Fail. 2015;8:198–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bialkowska A, Crisp M, Madoux F, Spicer T, Knapinska A, Mercer B, Bannister TD, He Y, Chowdhury S, Cameron M, Yang VW, Hodder P. Ml264: An antitumor agent that potently and selectively inhibits kruppel-like factor five (klf5) expression: A probe for studying colon cancer development and progression. Probe reports from the nih molecular libraries program. Bethesda (MD); 2010. [PubMed] [Google Scholar]
- 36.Ruiz de Sabando A, Wang C, He Y, Garcia-Barros M, Kim J, Shroyer KR, Bannister TD, Yang VW, Bialkowska AB. Ml264, a novel small-molecule compound that potently inhibits growth of colorectal cancer. Mol Cancer Ther. 2016;15:72–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Finck BN, Lehman JJ, Leone TC, Welch MJ, Bennett MJ, Kovacs A, Han X, Gross RW, Kozak R, Lopaschuk GD, Kelly DP. The cardiac phenotype induced by pparalpha overexpression mimics that caused by diabetes mellitus. J Clin Invest. 2002;109:121–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Young ME, Patil S, Ying J, Depre C, Ahuja HS, Shipley GL, Stepkowski SM, Davies PJ, Taegtmeyer H. Uncoupling protein 3 transcription is regulated by peroxisome proliferator-activated receptor (alpha) in the adult rodent heart. FASEB J. 2001;15:833–845 [DOI] [PubMed] [Google Scholar]
- 39.Harmancey R, Haight DL, Watts KA, Taegtmeyer H. Chronic hyperinsulinemia causes selective insulin resistance and down-regulates uncoupling protein 3 (ucp3) through the activation of sterol regulatory element-binding protein (srebp)-1 transcription factor in the mouse heart. J Biol Chem. 2015;290:30947–30961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thai MV, Guruswamy S, Cao KT, Pessin JE, Olson AL. Myocyte enhancer factor 2 (mef2)-binding site is required for glut4 gene expression in transgenic mice. Regulation of mef2 dna binding activity in insulin-deficient diabetes. J Biol Chem. 1998;273:14285–14292 [DOI] [PubMed] [Google Scholar]
- 41.Razeghi P, Young ME, Alcorn JL, Moravec CS, Frazier OH, Taegtmeyer H. Metabolic gene expression in fetal and failing human heart. Circulation. 2001;104:2923–2931 [DOI] [PubMed] [Google Scholar]
- 42.Prasad KM, Xu Y, Yang Z, Acton ST, French BA. Robust cardiomyocyte-specific gene expression following systemic injection of aav: In vivo gene delivery follows a poisson distribution. Gene Ther. 2011;18:43–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ritchie RH, Abel ED. Basic mechanisms of diabetic heart disease. Circ Res. 2020;126:1501–1525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Faria A, Persaud SJ. Cardiac oxidative stress in diabetes: Mechanisms and therapeutic potential. Pharmacol Ther. 2017;172:50–62 [DOI] [PubMed] [Google Scholar]
- 45.Wu SP, Kao CY, Wang L, Creighton CJ, Yang J, Donti TR, Harmancey R, Vasquez HG, Graham BH, Bellen HJ, Taegtmeyer H, Chang CP, Tsai MJ, Tsai SY. Increased coup-tfii expression in adult hearts induces mitochondrial dysfunction resulting in heart failure. Nat Commun. 2015;6:8245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pollak NM, Hoffman M, Goldberg IJ, Drosatos K. Kruppel-like factors: Crippling and un-crippling metabolic pathways. JACC Basic Transl Sci. 2018;3:132–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Novgorodov SA, Riley CL, Yu J, Keffler JA, Clarke CJ, Van Laer AO, Baicu CF, Zile MR, Gudz TI. Lactosylceramide contributes to mitochondrial dysfunction in diabetes. J Lipid Res. 2016;57:546–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Inoguchi T, Battan R, Handler E, Sportsman JR, Heath W, King GL. Preferential elevation of protein kinase c isoform beta ii and diacylglycerol levels in the aorta and heart of diabetic rats: Differential reversibility to glycemic control by islet cell transplantation. Proc Natl Acad Sci U S A. 1992;89:11059–11063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bugger H, Chen D, Riehle C, Soto J, Theobald HA, Hu XX, Ganesan B, Weimer BC, Abel ED. Tissue-specific remodeling of the mitochondrial proteome in type 1 diabetic akita mice. Diabetes. 2009;58:1986–1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Depre C, Young ME, Ying J, Ahuja HS, Han Q, Garza N, Davies PJ, Taegtmeyer H. Streptozotocin-induced changes in cardiac gene expression in the absence of severe contractile dysfunction. J Mol Cell Cardiol. 2000;32:985–996 [DOI] [PubMed] [Google Scholar]
- 51.Sack MN, Rader TA, Park S, Bastin J, McCune SA, Kelly DP. Fatty acid oxidation enzyme gene expression is downregulated in the failing heart. Circulation. 1996;94:2837–2842 [DOI] [PubMed] [Google Scholar]
- 52.Karbowska J, Kochan Z, Smoleński RT. Peroxisome proliferator-activated receptor alpha is downregulated in the failing human heart. Cell Mol Biol Lett. 2003;8:49–53 [PubMed] [Google Scholar]
- 53.Finck BN, Han X, Courtois M, Aimond F, Nerbonne JM, Kovacs A, Gross RW, Kelly DP. A critical role for pparalpha-mediated lipotoxicity in the pathogenesis of diabetic cardiomyopathy: Modulation by dietary fat content. Proc Natl Acad Sci U S A. 2003;100:1226–1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Taegtmeyer H, Beauloye C, Harmancey R, Hue L. Insulin resistance protects the heart from fuel overload in dysregulated metabolic states. Am J Physiol Heart Circ Physiol. 2013;305:H1693–1697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang M, Lin Y, Wang Y, Wang Y. High-glucose induces cardiac myocytes apoptosis through foxo1 /grk2 signaling pathway. Biochem Biophys Res Commun. 2019;513:154–158 [DOI] [PubMed] [Google Scholar]
- 56.Battiprolu PK, Hojayev B, Jiang N, Wang ZV, Luo X, Iglewski M, Shelton JM, Gerard RD, Rothermel BA, Gillette TG, Lavandero S, Hill JA. Metabolic stress-induced activation of foxo1 triggers diabetic cardiomyopathy in mice. J Clin Invest. 2012;122:1109–1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kumadaki S, Karasawa T, Matsuzaka T, Ema M, Nakagawa Y, Nakakuki M, Saito R, Yahagi N, Iwasaki H, Sone H, Takekoshi K, Yatoh S, Kobayashi K, Takahashi A, Suzuki H, Takahashi S, Yamada N, Shimano H. Inhibition of ubiquitin ligase f-box and wd repeat domain-containing 7alpha (fbw7alpha) causes hepatosteatosis through kruppel-like factor 5 (klf5)/peroxisome proliferator-activated receptor gamma2 (ppargamma2) pathway but not srebp-1c protein in mice. J Biol Chem. 2011;286:40835–40846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Taegtmeyer H, Young ME, Lopaschuk GD, Abel ED, Brunengraber H, Darley-Usmar V, Des Rosiers C, Gerszten R, Glatz JF, Griffin JL, Gropler RJ, Holzhuetter HG, Kizer JR, Lewandowski ED, Malloy CR, Neubauer S, Peterson LR, Portman MA, Recchia FA, Van Eyk JE, Wang TJ, Sciences AHACoBC. Assessing cardiac metabolism: A scientific statement from the american heart association. Circ Res. 2016;118:1659–1701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Razeghi P, Young ME, Cockrill TC, Frazier OH, Taegtmeyer H. Downregulation of myocardial myocyte enhancer factor 2c and myocyte enhancer factor 2c-regulated gene expression in diabetic patients with nonischemic heart failure. Circulation. 2002;106:407–411 [DOI] [PubMed] [Google Scholar]
- 60.Wilson AJ, Gill EK, Abudalo RA, Edgar KS, Watson CJ, Grieve DJ. Reactive oxygen species signalling in the diabetic heart: Emerging prospect for therapeutic targeting. Heart. 2018;104:293–299 [DOI] [PubMed] [Google Scholar]
- 61.Basu R, Oudit GY, Wang X, Zhang L, Ussher JR, Lopaschuk GD, Kassiri Z. Type 1 diabetic cardiomyopathy in the akita (ins2wt/c96y) mouse model is characterized by lipotoxicity and diastolic dysfunction with preserved systolic function. Am J Physiol Heart Circ Physiol. 2009;297:H2096–2108 [DOI] [PubMed] [Google Scholar]
- 62.Maalouf RM, Eid AA, Gorin YC, Block K, Escobar GP, Bailey S, Abboud HE. Nox4-derived reactive oxygen species mediate cardiomyocyte injury in early type 1 diabetes. Am J Physiol Cell Physiol. 2012;302:C597–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ikegami T, Suzuki Y, Shimizu T, Isono K, Koseki H, Shirasawa T. Model mice for tissue-specific deletion of the manganese superoxide dismutase (mnsod) gene. Biochem Biophys Res Commun. 2002;296:729–736 [DOI] [PubMed] [Google Scholar]
- 64.Coughlan MT, Thorburn DR, Penfold SA, Laskowski A, Harcourt BE, Sourris KC, Tan AL, Fukami K, Thallas-Bonke V, Nawroth PP, Brownlee M, Bierhaus A, Cooper ME, Forbes JM. Rage-induced cytosolic ros promote mitochondrial superoxide generation in diabetes. J Am Soc Nephrol. 2009;20:742–752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Block K, Gorin Y, Abboud HE. Subcellular localization of nox4 and regulation in diabetes. Proc Natl Acad Sci U S A. 2009;106:14385–14390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vendrov AE, Vendrov KC, Smith A, Yuan J, Sumida A, Robidoux J, Runge MS, Madamanchi NR. Nox4 nadph oxidase-dependent mitochondrial oxidative stress in aging-associated cardiovascular disease. Antioxid Redox Signal. 2015;23:1389–1409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shanmugasundaram K, Nayak BK, Friedrichs WE, Kaushik D, Rodriguez R, Block K. Nox4 functions as a mitochondrial energetic sensor coupling cancer metabolic reprogramming to drug resistance. Nat Commun. 2017;8:997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Marinari UM, Monacelli R, Cottalasso D, Novelli A. Effects of alloxan diabetes and insulin on morphology and certain functional activities of mitochondria of the rat liver and heart. Acta Diabetol Lat. 1974;11:296–314 [DOI] [PubMed] [Google Scholar]
- 69.Marciniak C, Marechal X, Montaigne D, Neviere R, Lancel S. Cardiac contractile function and mitochondrial respiration in diabetes-related mouse models. Cardiovasc Diabetol. 2014;13:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Boudina S, Sena S, Theobald H, Sheng X, Wright JJ, Hu XX, Aziz S, Johnson JI, Bugger H, Zaha VG, Abel ED. Mitochondrial energetics in the heart in obesity-related diabetes: Direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes. 2007;56:2457–2466 [DOI] [PubMed] [Google Scholar]
- 71.Kittleson MM, Minhas KM, Irizarry RA, Ye SQ, Edness G, Breton E, Conte JV, Tomaselli G, Garcia JG, Hare JM. Gene expression analysis of ischemic and nonischemic cardiomyopathy: Shared and distinct genes in the development of heart failure. Physiol Genomics. 2005;21:299–307 [DOI] [PubMed] [Google Scholar]
- 72.Yusuf S, Dagenais G, Pogue J, Bosch J, Sleight P. Vitamin e supplementation and cardiovascular events in high-risk patients. N Engl J Med. 2000;342:154–160 [DOI] [PubMed] [Google Scholar]
- 73.Boaz M, Smetana S, Weinstein T, Matas Z, Gafter U, Iaina A, Knecht A, Weissgarten Y, Brunner D, Fainaru M, Green MS. Secondary prevention with antioxidants of cardiovascular disease in endstage renal disease (space): Randomised placebo-controlled trial. Lancet. 2000;356:1213–1218 [DOI] [PubMed] [Google Scholar]
- 74.Tan SM, Sharma A, Stefanovic N, de Haan JB. Late-intervention study with ebselen in an experimental model of type 1 diabetic nephropathy. Free Radic Res. 2015;49:219–227 [DOI] [PubMed] [Google Scholar]
- 75.Kolb H Mouse models of insulin dependent diabetes: Low-dose streptozocin-induced diabetes and nonobese diabetic (nod) mice. Diabetes Metab Rev. 1987;3:751–778 [DOI] [PubMed] [Google Scholar]
- 76.Wu KK, Huan Y. Streptozotocin-induced diabetic models in mice and rats. Curr Protoc Pharmacol. 2008;Chapter 5:Unit 5.47 [DOI] [PubMed] [Google Scholar]
- 77.Cheng Z, Shen X, Jiang X, Shan H, Cimini M, Fang P, Ji Y, Park JY, Drosatos K, Yang X, Kevil CG, Kishore R, Wang H. Hyperhomocysteinemia potentiates diabetes-impaired edhf-induced vascular relaxation: Role of insufficient hydrogen sulfide. Redox Biol. 2018;16:215–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Vedantham S, Noh H, Ananthakrishnan R, Son N, Hallam K, Hu Y, Yu S, Shen X, Rosario R, Lu Y, Ravindranath T, Drosatos K, Huggins LA, Schmidt AM, Goldberg IJ, Ramasamy R. Human aldose reductase expression accelerates atherosclerosis in diabetic apolipoprotein e−/− mice. Arterioscler Thromb Vasc Biol. 2011;31:1805–1813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Drosatos K, Pollak NM, Pol CJ, Ntziachristos P, Willecke F, Valenti MC, Trent CM, Hu Y, Guo S, Aifantis I, Goldberg IJ. Cardiac myocyte klf5 regulates ppara expression and cardiac function. Circulation research. 2016;118:241–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lin SC, Wani MA, Whitsett JA, Wells JM. Klf5 regulates lineage formation in the pre-implantation mouse embryo. Development. 2010;137:3953–3963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sur I, Rozell B, Jaks V, Bergstrom A, Toftgard R. Epidermal and craniofacial defects in mice overexpressing klf5 in the basal layer of the epidermis. Journal of cell science. 2006;119:3593–3601 [DOI] [PubMed] [Google Scholar]
- 82.Pfleger J, Coleman RC, Ibetti J, Roy R, Kyriazis ID, Gao E, Drosatos K, Koch WJ. Genomic binding patterns of forkhead box protein o1 reveal its unique role in cardiac hypertrophy. Circulation. 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kalliora C, Kyriazis ID, Oka SI, Lieu MJ, Yue Y, Area-Gomez E, Pol CJ, Tian Y, Mizushima W, Chin A, Scerbo D, Schulze PC, Civelek M, Sadoshima J, Madesh M, Goldberg IJ, Drosatos K. Dual peroxisome-proliferator-activated-receptor-α/γ activation inhibits sirt1-pgc1α axis and causes cardiac dysfunction. JCI Insight. 2019;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mishra OP, Simmons N, Tyagi S, Pietrofesa R, Shuvaev VV, Valiulin RA, Heretsch P, Nicolaou KC, Christofidou-Solomidou M. Synthesis and antioxidant evaluation of (s,s)- and (r,r)-secoisolariciresinol diglucosides (sdgs). Bioorganic & medicinal chemistry letters. 2013;23:5325–5328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lin Z, Zhou P, von Gise A, Gu F, Ma Q, Chen J, Guo H, van Gorp PR, Wang DZ, Pu WT. Pi3kcb links hippo-yap and pi3k-akt signaling pathways to promote cardiomyocyte proliferation and survival. Circulation research. 2015;116:35–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gray SJ, Choi VW, Asokan A, Haberman RA, McCown TJ, Samulski RJ. Production of recombinant adeno-associated viral vectors and use in in vitro and in vivo administration. Current protocols in neuroscience. 2011;Chapter 4:Unit 4 17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.de Lucia C, Wallner M, Eaton DM, Zhao H, Houser SR, Koch WJ. Echocardiographic strain analysis for the early detection of left ventricular systolic/diastolic dysfunction and dyssynchrony in a mouse model of physiological aging. The journals of gerontology. Series A, Biological sciences and medical sciences 2019;74:455–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Schnelle M, Catibog N, Zhang M, Nabeebaccus AA, Anderson G, Richards DA, Sawyer G, Zhang X, Toischer K, Hasenfuss G, Monaghan MJ, Shah AM. Echocardiographic evaluation of diastolic function in mouse models of heart disease. Journal of molecular and cellular cardiology. 2018;114:20–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hoffman M, Kyriazis ID, Lucchese AM, de Lucia C, Piedepalumbo M, Bauer M, Schulze PC, Bonios MJ, Koch WJ, Drosatos K. Myocardial strain and cardiac output are preferable measurements for cardiac dysfunction and can predict mortality in septic mice. Journal of the American Heart Association. 2019;8:e012260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Davidson MM, Nesti C, Palenzuela L, Walker WF, Hernandez E, Protas L, Hirano M, Isaac ND. Novel cell lines derived from adult human ventricular cardiomyocytes. Journal of molecular and cellular cardiology. 2005;39:133–147 [DOI] [PubMed] [Google Scholar]
- 91.Yang Y, Goldstein BG, Nakagawa H, Katz JP. Kruppel-like factor 5 activates mek/erk signaling via egfr in primary squamous epithelial cells. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2007;21:543–550 [DOI] [PubMed] [Google Scholar]
- 92.Qiang L, Banks AS, Accili D. Uncoupling of acetylation from phosphorylation regulates foxo1 function independent of its subcellular localization. The Journal of biological chemistry. 2010;285:27396–27401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Matsushima S, Kuroda J, Ago T, Zhai P, Ikeda Y, Oka S, Fong GH, Tian R, Sadoshima J. Broad suppression of nadph oxidase activity exacerbates ischemia/reperfusion injury through inadvertent downregulation of hypoxia-inducible factor-1alpha and upregulation of peroxisome proliferator-activated receptor-alpha. Circulation research. 2013;112:1135–1149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative pcr and the 2(-delta delta c(t)) method. Methods. 2001;25:402–408 [DOI] [PubMed] [Google Scholar]
- 95.Kokkinaki D, Hoffman M, Kalliora C, Kyriazis ID, Maning J, Lucchese AM, Shanmughapriya S, Tomar D, Park JY, Wang H, Yang XF, Madesh M, Lymperopoulos A, Koch WJ, Christofidou-Solomidou M, Drosatos K. Chemically synthesized secoisolariciresinol diglucoside (lgm2605) improves mitochondrial function in cardiac myocytes and alleviates septic cardiomyopathy. Journal of molecular and cellular cardiology. 2019;127:232–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gaignebet L, Kańduła MM, Lehmann D, Knosalla C, Kreil DP, Kararigas G. Sex-specific human cardiomyocyte gene regulation in left ventricular pressure overload. Mayo Clin Proc. 2020;95:688–697 [DOI] [PubMed] [Google Scholar]
- 97.Rehman AU, Anwer AG, Gosnell ME, Mahbub SB, Liu G, Goldys EM. Fluorescence quenching of free and bound nadh in hela cells determined by hyperspectral imaging and unmixing of cell autofluorescence. Biomed Opt Express. 2017;8:1488–1498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Dong Z, Shanmughapriya S, Tomar D, Siddiqui N, Lynch S, Nemani N, Breves SL, Zhang X, Tripathi A, Palaniappan P, Riitano MF, Worth AM, Seelam A, Carvalho E, Subbiah R, Jaña F, Soboloff J, Peng Y, Cheung JY, Joseph SK, Caplan J, Rajan S, Stathopulos PB, Madesh M. Mitochondrial ca. Mol Cell. 2017;65:1014–1028.e1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Joseph LC, Kokkinaki D, Valenti MC, Kim GJ, Barca E, Tomar D, Hoffman NE, Subramanyam P, Colecraft HM, Hirano M, Ratner AJ, Madesh M, Drosatos K, Morrow JP. Inhibition of nadph oxidase 2 (nox2) prevents sepsis-induced cardiomyopathy by improving calcium handling and mitochondrial function. JCI insight. 2017;2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tomar D, Dong Z, Shanmughapriya S, Koch DA, Thomas T, Hoffman NE, Timbalia SA, Goldman SJ, Breves SL, Corbally DP, Nemani N, Fairweather JP, Cutri AR, Zhang X, Song J, Jana F, Huang J, Barrero C, Rabinowitz JE, Luongo TS, Schumacher SM, Rockman ME, Dietrich A, Merali S, Caplan J, Stathopulos P, Ahima RS, Cheung JY, Houser SR, Koch WJ, Patel V, Gohil VM, Elrod JW, Rajan S, Madesh M. Mcur1 is a scaffold factor for the mcu complex function and promotes mitochondrial bioenergetics. Cell reports. 2016;15:1673–1685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ji R, Akashi H, Drosatos K, Liao X, Jiang H, Kennel PJ, Brunjes DL, Castillero E, Zhang X, Deng LY, Homma S, George IJ, Takayama H, Naka Y, Goldberg IJ, Schulze PC. Increased de novo ceramide synthesis and accumulation in failing myocardium. JCI insight. 2017;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors avail all supporting data and methods within the present article. Additional information will be available upon request to the corresponding author.
Animal care.
All animal experiments were conducted in male mice and were approved by the Institutional Animal Care and Use Committee at Temple University and carried out in accordance with the NIH guidelines. Wild type C57BL/6, FOXO1fl/fl and Pparα−/− mice were obtained from Jackson Laboratory. The R26-lsl-rtTA-TRE-Klf5 mice were provided by Jeffrey Whitsett13 and Inderpreet Sur14. αMHC-rtTA-Klf5 mice were generated by crossing αMHC-Cre with R26-lsl-rtTA-TRE-Klf5 mice, respectively. The αMHC-Klf5−/− and αMHC-FoxO1−/− mice were generated by our group11, 15. Insulin deficiency was induced by injecting i.p. streptozotocin (STZ, Sigma-Aldrich) dissolved in saline (50mg/kg) for 5 consecutive days. LGM260516 was dissolved in saline and administered orally (100mg/kg b.w.; daily gavage). ML264 (10mg/kg b.w. i.p.) was dissolved in 80% saline, 10% dimethyl-sulfoxide (DMSO) and 10% Tween 80 and was administered twice per day. Type 2 diabetes was induced in C57BL/6 mice by feeding them on high-fat diet (Online Table I) for 32 weeks.
Glucose and insulin tolerance tests were conducted after 6 hours of fasting. For glucose tolerance test, mice received i.p. 20% glucose solution (2g of glucose/kg body mass). For insulin tolerance test, mice received intraperitoneal injection of insulin (0.75 U/kg). Glucose levels were determined in blood collected from the tail vein at designated time points.
Cell culture.
The AC1617 human ventricular cardiomyocyte line was maintained in complete DMEM/F-12 medium (ThermoFischer Scientific, Carlsbad, CA) at 37°C and 5%CO2.
Generation of AAV and adenoviruses and infection of adult mice or cells.
AAV9-cTnT-Klf5 was generated by cloning the human KLF5 cDNA in the pAAV9-cTnT receiving vector18 (Addgene). The AAVs were administered in C57BL/6 mice via retro-orbital injection (7×1011viral particles/100μl). Adenoviruses expressing KLF5 or shKLF519 (under H1 promoter) were generated by cloning the coding sequence of human KLF5 into the pAdTrackCMV vector or pAdTrack respectively. Ad-wtFOXO1 and Ad-caFOXO1 were provided by Dr. D. Accili20. Ad-caFOXO1 has Akt phosphorylation sites Thr24, Ser316 substituted to Ala and Ser253 to Asp20. Ad-shNOX4 was previously described21.
Echocardiography.
Cardiac function was assessed by transthoracic echocardiography using the VisualSonics Vevo 2100 system (VisualSonics, Toronto, ON). Mice were sedated via inhalation of 3% isoflurane and maintained on 0.5% isoflurane during image acquisition. Echocardiography images were processed to perform off-line LV trace analysis or B-mode speckle-tracking analysis by a single observer blinded to the respective treatments of mice with VevoLab software (3.1.1). More information is provided in the Online Supplement.
Protein and RNA purification and analysis.
Protein and RNA purification from isolated hearts or cells were performed as described previously22. Antibodies used are listed in Online Table II. Primer sequences are described in Online Table III.
Chromatin Immunoprecipitation (ChIP).
ChIP experiments were performed in AC16 cells as described previously11 and in the Online Supplement.