

Abstract
The comprehensive characterization of human leukocyte antigen (HLA) genomic sequences remains a challenging problem. Despite the significant advantages of next-generation sequencing (NGS) in the field of Immunogenetics, there has yet to be a single solution for unambiguous, accurate, simple, cost-effective, and timely genotyping necessary for all clinical applications. This report demonstrates the benefits of nanopore sequencing introduced by Oxford Nanopore Technologies (ONT) for HLA genotyping. Samples (n = 120) previously characterized at high-resolution three-field (HR-3F) for 11 loci were assessed using ONT sequencing paired to a single-plex PCR protocol (Holotype) and to two multiplex protocols OmniType (Omixon) and NGSgo®-MX6-1 (GenDx). The results demonstrate the potential of nanopore sequencing for delivering accurate HR-3F typing with a simple, rapid, and cost-effective protocol. The protocol is applicable to time-sensitive applications, such as deceased donor typings, enabling better assessments of compatibility and epitope analysis. The technology also allows significantly shorter turnaround time for multiple samples at a lower cost. Overall, the nanopore technology appears to offer a significant advancement over current next-generation sequencing platforms as a single solution for all HLA genotyping needs.
Keywords: Human Leukocyte Antigens, NGS, Oxford Nanopore sequencing, Transplantation, HLA genotyping
1. Introduction
The need for accurate, thorough, and timely characterization of Human Leukocyte Antigen (HLA) genes for several clinical applications is well-documented [1-6]. High-resolution typing is necessary in pharmacogenomics, disease association testing, and is the gold standard for bone marrow transplantation [5-7]. While HLA typing for solid organ transplantation has historically been limited to lower resolution methods, newer approaches for donor-recipient compatibility assessment call for a shift to high-resolution methods [8,9]. Virtual crossmatching, commonly performed using recipient antibody data of known specificity with single antigen beads, can be more accurately performed with donor HLA typing at the two-field level [10]. Furthermore, an increasing number of retrospective studies highlight the role of epitope mismatch load on clinical outcomes [11-16]. Epitope analysis depends on complete and unambiguous protein sequences, which require high-resolution two-field typing (HR-2F) [8,17]. Such tools have been applied to living donor solid organ pre-transplant assessments [10,18,19]. While they may also benefit over 100,000 United Network of organ Sharing (UNOS) wait list candidates, deceased donor HLA typing remains limited to lower resolution methods, such as sequence-specific oligonucleotide probes (SSOP) or real-time polymerase chain reaction due to the time constraints of these transplants [9,20]. Virtual crossmatches are therefore performed using lower resolution or inferred high-resolution donor typings, introducing uncertainty to compatibility assessments for patients with allele-specific antibodies or high sensitization [10,21]. HR-2F of deceased donors would clarify potential donor-specific antibodies (DSA) and permit epitope analysis for improved donor selection [8,9]. However, a cost-effective method with a short turnaround time (less than six hours) and without ambiguities has proven an elusive target.
For over fifty years, HLA genotyping has remained a challenge. The hallmark of these genes is their extreme and non-static polymorphism leading to continually revised catalogs, as new alleles are discovered [22]. This issue is exacerbated by allele-specific characteristics and pairings, requiring phasing along the length of the genes, and naturally-occurring heterozygosity, generating infinite scenarios of heterozygous polymorphisms. The above has, thus far, defied efforts to develop a single methodology that is simple, comprehensive, robust, and cost-effective to address research and clinical needs adequately.
The advent of Next Generation Sequencing (NGS) in the last decade allowed for clonal sequencing and high throughput. Given that sequencing of single DNA molecules permits setting phase between distant polymorphic positions, and high-throughput enables more extensive and thorough characterization of genes, NGS became relevant for the characterization of HLA genomic sequences. As with legacy methods, however, initial use of NGS was limited to sequencing critical exons, thereby limiting its potential [23,24]. Longer characterized DNA pieces in a continuum was essential, and amplifying the entire HLA gene as a single amplicon proved to be a more effective and meaningful way of using NGS to characterize HLA polymorphisms, further revealing its potential for HLA typing [25].
The community has since adopted NGS of targeted long PCR amplicons, including most of the genomic sequences of each HLA locus, as the method of choice for HLA typing; almost all commercial protocols are based on this approach. However, while available NGS platforms (Illumina and Ion Torrent) can provide phasing of fragments ranging from 400 to 900 bp, they are unable to sequence very long fragments (3–15 Kb) as a continuum or as a single read. This limitation hinders the potential of this technology as the number of HLA alleles continues to increase, and the inability to phase distal polymorphic positions results in ambiguities. Additionally, a longer turnaround time (2–3 days) and a need to queue multiple samples per run, for cost effectiveness and capacity utilization, preclude the use of this method for time-sensitive applications such as deceased donor HLA genotyping [9] or for low throughput needs.
PacBio platform offered a better solution than short-read platforms, enabling the sequencing of longer fragments like intact HLA amplicons. Even though the platform was characterized by a high error rate in the past, most recently it has been reported that by generating circular consensus sequences you attain high accuracy upwards of Phred 30 [26]. However, primarily because of its cost, size, maintenance requirements, and the lack of a commercial HLA typing kit with a comprehensive software solution, the platform has not been adopted widely by clinical laboratories. Its utilization, nevertheless, in a hematopoietic cell transplantation study has demonstrated the benefits of the complete genomic characterization of HLAs [27]. The search continues for alternative platforms offering accurate and simple sequencing of a single 3–15 kb molecule as a single read, while remaining cost-effective.
One such advancement within the last several years is DNA sequencing through nanopores, as introduced by Oxford Nanopore Technologies (ONT) on the MinION platform. This portable and relatively inexpensive device generates long sequence reads in real-time as molecules of single-stranded DNA or RNA pass through protein nanopores, resulting in characteristic disruptions in ion current. While earlier versions of this technology had high error rates that made it clinically unsuitable, recent improvements have elevated the platform to the point that a reevaluation is warranted.
Earlier reported efforts to utilize the nanopore technology for HLA genotyping fell short of providing a comprehensive solution of characterizing all loci within a time frame necessary for all of our clinical needs [28]. Most recently, however, De Santis et al. using a combination of a multiplex PCR and ONT reported successful characterization of all 11 HLA loci [29]. Our report introduces nanopore technology as a viable alternative to existing methods for reliable clinical HLA typing of all eleven loci within a significantly shorter time. Single samples can be genotyped in less than six hours using the Flongle adapter for the MinION device, and multiple indexed samples, sets of 24, can be analyzed using a single MinION flow cell in less than 24 h. This significant advancement presents new opportunities and a step towards a complete solution to the challenge of HLA typing.
2. Methods and materials
2.1. Sample selection
Assessing ONT (Oxford, UK) for HLA typing required the selection of diverse samples. Samples were selected to include alleles for HLA-A, −B, −C, −DQB1, −DRB1 and −DRB3/4/5 such that the frequencies cumulatively would comprise greater than 95% of the Caucasian and African American populations in the United States, shown in Supplemental Table 1a-g, and includes alleles and their relevant frequencies from five populations [30]. Several samples were included that present genotyping challenges for different loci. A total of 120 samples were identified. All samples had been previously HLA genotyped at 11 loci by NGS using Omixon Holotype V2 kits (Omixon, Budapest, Hungary) on the Illumina MiSeq (Illumina, San Diego, CA, USA), and reported at 3-field resolution. All samples were reanalyzed with NGSengine (GenDx, Utrecht, Netherlands) version 2.16.2 using the IMGT/HLA database v. 3.38 to minimize any discrepancies that could occur due to differences in the IMGT/HLA database version. This MiSeq-typed dataset served as the reference HLA genotyping.
2.2. DNA preparation
DNA was extracted from blood using the Qiagen EZ1®DNA extraction platform with Qiagen EZ1®Blood 350 Kits (Qiagen, Hilden, Germany) for the majority of the samples included in these experiments, n = 93. These samples are a collection of clinical samples, included after institutional approval, and an internal reference panel. DNA samples from the Coriell Institute (n = 12; Coriell Cell Repositories, Camden, NJ, USA), and from the International Histocompatibility Working Group (n = 6; Fred Hutchinson, Seattle, WA, USA) were also used. The remainder of the samples (n = 9) were from four African populations (Ethiopia, Tanzania, Cameroon and Botswana) with challenging HLA typings. These particular samples had loci with a high degree of variation that is quite distinct from what is present in the current version of the IMGT/HLA database and includes unpublished novel alleles. The written informed consent was obtained from all participants and research/ethics approval and permits were obtained from the following institutions prior to sample collection: COSTECH, NIMR and Muhimbili University of Health and Allied Sciences in Dar es Salaam, Tanzania; the University of Botswana and the Ministry of Health in Gaborone, Botswana; the University of Addis Ababa and the Federal Democratic Republic of Ethiopia Ministry of Science and Technology National Health Research Ethics Review Committee; the Cameroonian National Ethics Committee and the Cameroonian Ministry of Public Health.
DNA concentration was quantified with Qubit BR assay (ThermoFisher, Waltham, MA, USA). Sample concentration was adjusted to that suggested by the manufacturers’ PCR protocols, when possible.
2.3. PCR conditions utilized for the project
Three different protocols of PCR were used. The set of 120 samples, sequenced on the ONT MinION platform with R9.4 flow cells in five sets of 24, were amplified using the Holotype V2 protocol by Omixon, whereby 11 loci were amplified individually and pooled before library preparation and sequencing. Omixon primers amplify the entire gene for HLA-A, B, C, DQA1, DQB1, and DPA1 loci, with priming sites in the UTR regions. DRB1/3/4/5 amplicons include exons 2, 3, and 4 and DPB1 is amplified from exon 2 through the entire 3′UTR. The manufacturer’s procedure for amplification, quantitation, and pooling was performed on a Hamilton STARlet (Hamilton Robotics, Reno, NV, USA). Thirty-five μl of the diluted amplicon pools were treated with 4 μl of ExoSAP Express (ThermoFisher) incubating for 4 min at 37 °C followed by 1 min at 80 °C.
The other two protocols were rapid multiplexed PCR protocols from Omixon and GenDx. Nine samples were amplified with Omixon OmniType kits according to manufacturer’s PCR procedure and a different set of 9 samples were amplified with the NGSgo®-MX6-1 PCR (GenDx). All 18 samples being a subset of the 120 samples used for this study. The OmniType protocol amplified all eleven major HLA loci in 2 h and 10 min in a single tube using the same primers as the Holotype kit. The PCR product was then treated with 3 μl of ExoSAP Express as described above. GenDx NGSgo®-MX6-1 PCR amplified A, B, C, DRB1, DQB1, and DPB1 in 3 h and 15 min in a single tube. The NGSgo®-MX6-1 primers amplify the entire gene for HLA-A, B and C, whereas the Class II amplifications all start upstream of exon 2 and extend through exon 3 (DRB1 and DQB1) or exon 5 (DPB1). The amplifications, performed according to manufacturer’s procedure, have a total reaction volume of 10 μl. These were performed in duplicate to ensure 800 ng of starting material for sequencing. Each 10 μl reaction was treated with 1 μl of ExoSAP Express and incubated as described above. The eighteen samples amplified with these multiplexed protocols were sequenced individually on ONT Flongles (R9.4), adaptors for the MinION that enable DNA sequencing on smaller, single-use flow cells.
2.4. Library preparation
Library preparation for MinION and Flongle libraries used gentle mixing of reagents at each step to avoid shearing the amplicons.
2.4.1. MinION
Each set of 24 indexed libraries were prepared for MinION sequencing with ONT 1D Native barcoding DNA procedure v109_revH with EXP-NBD104, EXPNBD114, and SQK-LSK109 kits with minor revisions to the procedure. Briefly, 2 μg of sample was treated with NEBNext End Repair/dA-tailing Module reagents (NEB, Ipswich, MA, USA), substituting water for NEBNext FFPE Repair reagents. Native barcodes (ONT NBD1-24) were ligated to 700 ng of dA-tailed amplicons with NEB Blunt/TA Ligase Master Mix. Samples were then pooled approximately equimolar. ONT sequencing adapters from the Barcoding Expansion kit were ligated to 900 ng of the pooled library using NEBNext Quick Ligation Reaction Buffer and T4 Ligase (NEB). Libraries were cleaned after each enzymatic reaction with AMPure XP beads (Beckman Coulter, Indianapolis, IN) using the ONT Long Fragment Buffer for wash steps after sequencing adapters were ligated. Libraries were quantitated after each cleanup step with Qubit BR assay.
2.4.2. Flongle
Libraries for eighteen individual multiplexed samples were prepared for sequencing on Flongles using ONT 1D Genomic DNA by Ligation protocol v109_revL starting at the End-Prep step with SQK-LSK109 kits with minor revisions to the procedure. Briefly, 800 ng of sample was dA-tailed with NEBNext End Repair/dA-tailing Module reagents (NEB), substituting water for NEBNext FFPE Repair reagents, and cleaned with AMPure XP beads. ONT sequencing adapters were then ligated to the amplicons using ONT Ligation Buffer with NEB Quick T4 ligase and cleaned again with AMPure XP beads using the ONT Long Fragment Buffer for the wash steps. Libraries were quantitated after each cleanup step with Qubit BR assay. The Flongle library preparation process takes approximately 1 h and 45 min.
2.5. Sequencing
The number of active pores on each sensory array was assessed before loading libraries on flow cells for each sequencing run to confirm flow cells met manufacturer’s quality control metrics.
2.5.1. MinION
Indexed libraries were loaded onto the MinION SpotON flow cells following ONT procedure with reagents from SQK-LSK109 and EXP-FLP002 kits. To summarize, flow cells were primed with ONT Flush Buffer and Flush Tether reagents before loading library mixed with ONT Loading Beads and Sequencing Buffer in a dropwise fashion onto the SpotON sample port. After each run was terminated, MinION flow cells were washed according to the manufacturer’s protocol v1_revB with ONT Flow Cell Wash kits (EXP-WSH003). In these experiments, flow cells were re-used a maximum of four times.
2.5.2. Flongle
Individual sample libraries were loaded onto the Flongle flow cells following the ONT procedure with reagents from SQK-LSK109 and EXP-FLP002 kits. To summarize, flow cells were primed with ONT Flush Buffer and Flush Tether reagents applied directly to the SpotON port before loading approximately 20 fmol library mixed with ONT Loading Beads and Sequencing Buffer pipetted directly onto the SpotON sample port.
2.6. Data analysis
ONT raw signal data was basecalled and demultiplexed using Guppy (ONT software v3.4.3). Fastq files were then analyzed with a custom pipeline available at http://nanopore-hla.chop.edu. The fasta output of the web application was then submitted to NGSEngine® to determine the HLA genotyping (GenDx, V2.16.2). In NGSEngine, the following parameters were selected: sequencing platform type was set to PacBio-Consensus; for Holotype and OmniType, the amplicon region was set to ‘auto’ for all loci except for DRB1, which was set to ‘DRB1 All Exon’. Regions outside of the amplicon, including primers, were added to the ‘Ignored Regions’ list. For the NGSgo®-MX6-1 amplicons, the default parameters corresponding to this PCR protocol were chosen for analysis.
The error rate of sequencing has been calculated based upon the reads that were selected for genotyping and includes mismatches, insertions and deletions.
3. Results
3.1. Sequencing strategy
The objective of the study was two-fold: 1) Evaluate ONT sequencing technology for HLA typing taking advantage of the long reads, thereby eliminating ambiguities, and assess other aspects such as accuracy of sequencing and cost. 2) Evaluate pairing this nanopore sequencing technology with a multiplexed PCR protocol amplifying all 11 HLA genes that would provide high-resolution two-field (HR-2F) HLA typing in less than six hours. This shorter turn-around time would enable HLA HR-2F typing of deceased donors and potentially optimize compatibility assessments.
3.2. Objective 1
Our current clinical HLA typing protocol uses the Holotype V2 kit for PCR where each locus is amplified separately; this approach was used for the first objective. The intent was to assess the post-PCR process, including library preparation, sequencing, and analysis without interference from the targeting and amplification of the HLA genes. Our goal was to minimize unknown variables and complexities introduced by alternative PCR protocols that would obscure an independent and objective assessment of the analysis pipeline and genotyping. All MinION experiments utilized the maximum number of barcodes available and were performed as sets of 24 samples.
3.2.1. Library preparation and sequencing; metrics of relevance
A summary of the timeline for post-PCR library preparation and sequencing steps on the MinION platform is found in Table 1. Library preparation steps take approximately 6.5 h after PCR amplification. Depending on the amount of data desired, sequencing and analysis takes between 14 and 28 h, which is largely dictated by the amount of time spent sequencing. Generally, no more than 4–6 h are necessary to collect enough data to genotype 24 samples at 11 HLA loci. Given the 6.5 h for library prep, sequencing was often run overnight and terminated the following morning for convenience, generating a surplus of data. The total time for the entire post-PCR process ranged from 20 to 34 h.
Table 1.
Timing information for post-PCR to sequencing. NA: Not applicable
| Step | MinION | Flongle | |
|---|---|---|---|
| Library Preparation | Post-Amplification DNA prep | 65 min | 15 min |
| DNA End prep | 2.0 h | 37 min | |
| Native barcode ligation | 2.5 h | NA | |
| Adapter Ligation and Clean-up | 1.0 h | 52 min | |
| Total Library Prep Time | 6.5 h | 1.75 h | |
| Sequencing & Analysis | Flow cell check | 15 min | 10 min |
| Priming and loading flow cell | 10 min | 5 min | |
| Sequencing | 4.6–18.4 h | 1.0 h | |
| Base calling | 2.5 h | 4.5 min | |
| Demultiplexing | 17 min | NA | |
| Consensus | 5.4 h (13.5 min/sample) | 11.5 min | |
| Genotyping | 1.0 h (2.5 min/sample) | 2.1 min | |
| Total Sequencing & Analysis Time | 14.2–28.0 h | 1.5 h | |
| Total Time | 20.6–34.4 h | 3.3 h |
Using R9.4 flowcells, the current stable version, five experiments were performed, sequencing 120 diverse samples (Supplemental Table 1). The amplicons ranged from 2.7 kb (HLA-B) to 6.6 kb (HLA-DPB1), with amplicons from each sample pooled and barcoded. The number of active pores varied by flow cell, ranging from 1,225 to 1,714, impacting the total data generated for each experiment (Table 2). When washed as recommended with the most recent ONT wash kit (EXP-WSH003), a flow cell can be used for multiple sequencing experiments with minimal carryover of intact full-length amplicons from the previous experiment(s). Approximately 10% of active pores are lost with each successive experiment, allowing for 3–4 experiments per flow cell. The accuracy after four experiments was decreased minimally, by 0.48%, when compared to the first experiment (experiment 1 = 93.07%; experiment 4 = 92.59%). Sequencing ranged from 4.6 to 18.4 h (Table 2). Although run time was variable, it was only necessary to analyze 800,000 reads per run, which were collected in the first 3.5–4.5 h, to obtain reliable genotypes. After demultiplexing, an average of approximately 30,000 reads per sample were identified. While there was variation in the overall representation of each barcoded sample, in general, this variation was not extreme (Fig. 1). The third sequencing experiment (Set 3) had two technical failures that were repeated independently and successfully genotyped. Approximately 15,000 high-quality reads were used for genotyping. The majority of reads excluded from analysis were due to 1) low alignment score, often short reads not spanning the entire amplicon, the result of incomplete amplification or DNA breakage during the experiment, or 2) because the read did not map to an HLA gene of interest.
Table 2.
MinION sequencing experiment metrics.
| Sequencing Run | Total run time (h) |
Number of active pores |
Total # of reads per run (millions) |
Number of reads per hour (thousands) |
Total reads analyzed |
Time to reach 800 k reads (h) |
Total # of reads assigned to barcodes |
Number of samples |
Average # of reads per sample (demultiplexed) |
Average # of usable reads per sample |
Average # of reads per locus |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 16.6 | 1,714 | 2.78 | 167.4 | 800,000 | 4.42 | 722,199 | 24 | 30,092 | 17,125 | 1,818 |
| 2 | 4.6 | 1,267 | 0.83 | 180.4 | 800,000 | 4.45 | 685,527 | 24 | 28,564 | 13,827 | 1,474 |
| 3* | 18.4 | 1,225 | 2.51 | 136.4 | 800,000 | 4.57 | 679,949 | 24 | 29,170 | 14,510 | 1,611 |
| 4 | 6.1 | 1,460 | 1.01 | 165.6 | 800,000 | 4.70 | 701,794 | 24 | 29,241 | 16,131 | 1,661 |
| 5 | 15.6 | 1,688 | 3.28 | 210.3 | 800,000 | 3.28 | 723,768 | 24 | 30,157 | 11,795 | 1,281 |
| Average | 12.3 | 1,471 | 2.08 | 169.8 | 800,000 | 4.28 | 702,647 | 24 | 29,445 | 14,678 | 1,569 |
Two samples failed to amplify on this run. Upon repeat the samples were successfully sequenced.
Fig. 1.
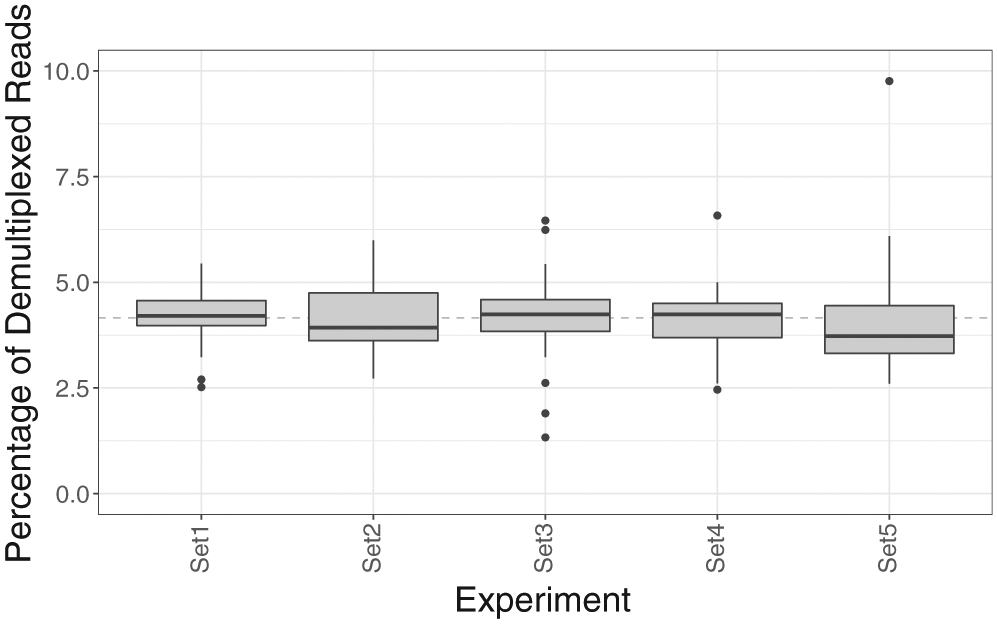
Distribution of reads per sample for the five MinION experiments. The dashed line represents the expected representation of a sample if all 24 samples in an experiment are equally represented.
Library preparation is initiated with quantitation of the amplicons, followed by course adjustment of amplicon concentration and amplicon pooling so that the samples could be barcoded for identification. Variation in the locus balance is expected since amplicons are not pooled to exact concentrations (Fig 2A and B). After accounting for only high-quality reads, on average approximately 1,570 reads per locus were used for genotyping (Table 2).
Fig. 2.
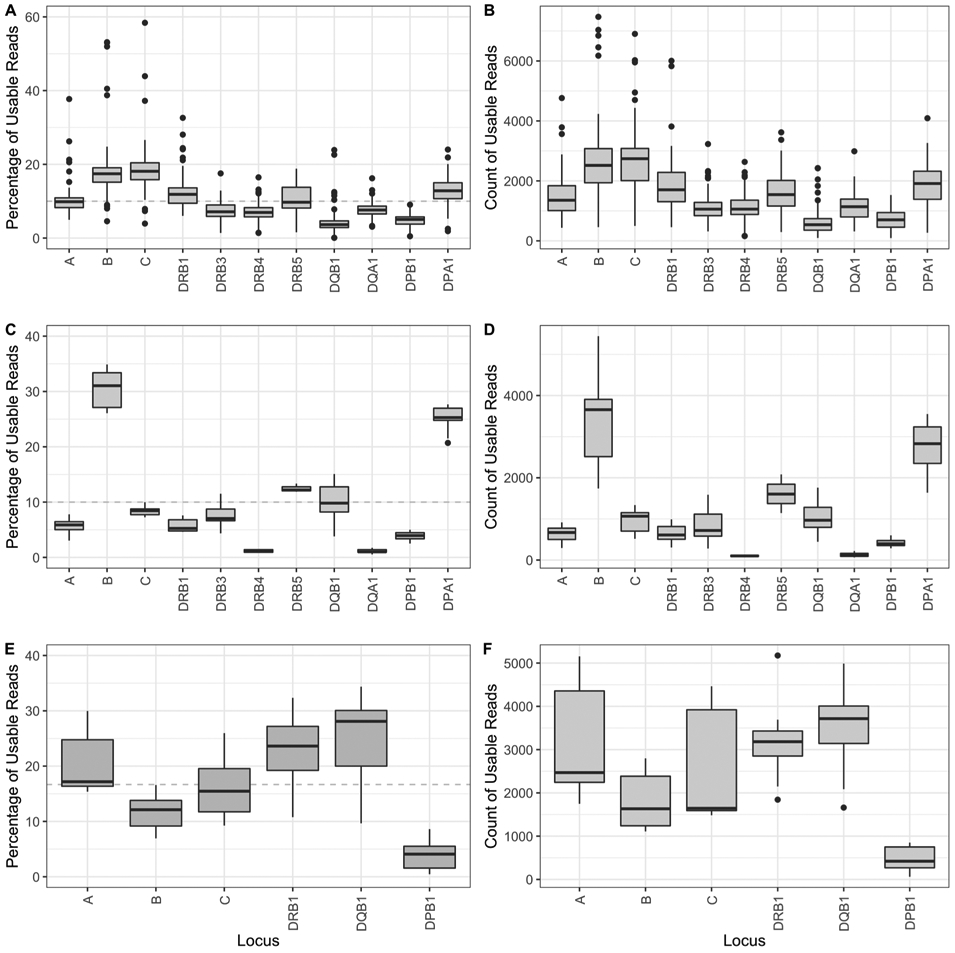
Distribution of reads among the 11 HLA loci for the MinION and Flongle experiments. A) Percent Usable Reads – MinION; B) Count of Usable Reads – MinION; C) Percent usable reads – OmniType; D) Count of usable Reads – OmniType; E) Percent usable reads – NGSgo®-MX6-1; F) Count of usable reads – NGSgo®-MX6-1. The y-axis represents the percentage of the reads assigned that were used for analysis for each locus out of the total reads that were used for the sample (A, C and E) or the count of reads used for analysis per locus (B, D, and F).
3.2.2. Genotypic analysis
A consensus sequence was determined for each allele sequenced on the ONT MinION platform. In brief, on a per locus basis, a fully characterized allele(s), as defined in the IMGT/HLA database, with minimal differences to the ONT reads is chosen as reference(s) for alignment, the ONT reads are aligned to the reference allele(s) where variants were then called to produce a consensus sequence for each allele. Genotyping is performed using GenDx NGSengine® as described in the methods. The ONT-based HLA genotypes were compared to the reference genotypes generated by the Holotype V2 protocol using the Illumina MiSeq platform (HR-3F) (Supplemental Table 2). The performance metric summary for the ONT method is found in Table 3 and the specific calculations can be found in Supplemental Tables 3a-3i. Overall the method is highly accurate at 99.98%, with a sensitivity of 99.63% and specificity of 99.99%. Genotyping at HLA-A, B, C, DPA1, DPB1, DQA1, DRB3 and DRB5 had 100% accuracy. For the alleles that did not match the known reference genotype, the majority of the problems were related to the amplification step. In DQB1, there were four differences from known typings. In one sample, the DQB1 locus failed to amplify, causing 2 allele discrepancies. In two samples, there was allele dropout of DQB1*03:01:01 when in combination with DQB1*06:01:01 and DQB1*06:03:01, where the DQB1*03:01:01 was detected at 3.29% and 0.64% depth of coverage respectively. Additionally, three samples failed to amplify the DRB4*01:01:01 allele. In two cases, only 1 allele was expected at DRB4 and no genotype was called for these samples. A third sample genotyped homozygous DRB4*01:03:01 instead of heterozygous DRB4*01:01:01 with DRB4*01:03:01, where the DRB4*01:01:01 allele was found at 3.07% depth of coverage. The aforementioned discrepancies were all due to the low representation of the particular alleles and below our internal threshold for detecting minor allele species. For the majority of heterozygous loci, alleles were generally well balanced (Fig. 3A). In the case of DRB4, which has known preferential amplification in the Holotype kit, the minor allele in heterozygous samples typically varied between 10% and 30% of the total reads. In all the cases presented above, whenever there is a missing allele, the genotyping anomaly would have been detected in our system through the use of haplotype analysis, and rectified upon further evaluation and repeat testing.
Table 3.
Performance specifications for genotyping on the ONT MinION. PPV: Positive Predictive Value; NPV: Negative Predictive Value.
| Metric | HLA-A | HLA-B | HLA-C | HLA-DPA1 | HLA-DPB1 | HLA-DQA1 | HLA-DQB1 | HLA-DRB1 | HLA-DRB3/4/5 | Total |
|---|---|---|---|---|---|---|---|---|---|---|
| Sensitivity | 100.00% | 100.00% | 100.00% | 100.00% | 100.00% | 100.00% | 98.33% | 99.58% | 98.75% | 99.63% |
| Specificity | 100.00% | 100.00% | 100.00% | 100.00% | 100.00% | 100.00% | 99.96% | 99.99% | 99.93% | 99.99% |
| PPV (Precision) | 100.00% | 100.00% | 100.00% | 100.00% | 100.00% | 100.00% | 99.16% | 99.58% | 98.75% | 99.72% |
| NPV | 100.00% | 100.00% | 100.00% | 100.00% | 100.00% | 100.00% | 99.92% | 99.99% | 99.93% | 99.99% |
| Accuracy | 100.00% | 100.00% | 100.00% | 100.00% | 100.00% | 100.00% | 99.89% | 99.98% | 99.86% | 99.98% |
Fig. 3.
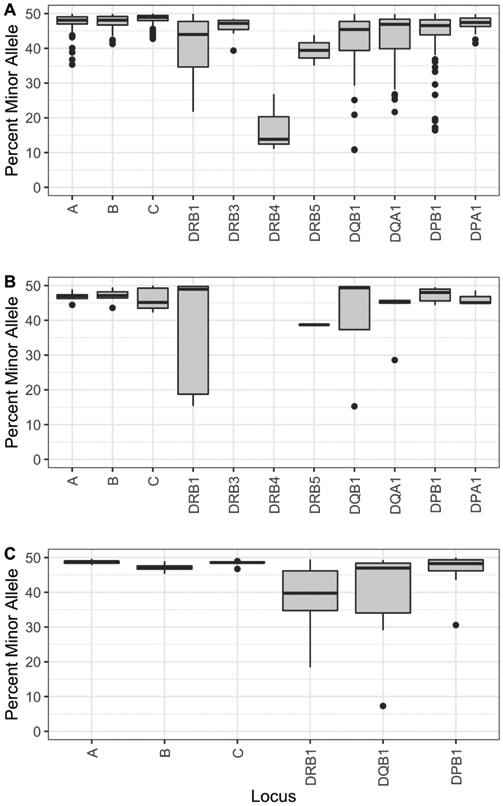
Percentage of the minor allele for all heterozygous loci sequenced for the MinION and Flongle experiments. A) Holotype PCR on MinION; B) OmniType multiplex PCR on Flongle. All samples with DRB3 and DRB4 were hemizygous. C) NGSgo®-MX6-1 multiplex PCR on Flongle.
Among the 2,126 expected allele calls, there was only one (1) discrepancy attributed directly to the ONT sequencing and subsequent analysis, occurring within the DRB1 locus. The reference genotype of this sample was DRB1*13:08 and DRB1*15:02:02, where the DRB1*13:08 allele was mistakenly called a novel DRB1*11 allele upon ONT genotyping. The particular problem persisted after reanalysis through the pipeline with different parameters. Considering that the DRB1*13:08 allele is incomplete in the IMGT/HLA database, and that there were sufficient reads for the locus we are convinced it is a bioinformatics issue. Additional development efforts regarding the analysis of this data are being made to optimally address this problem, and includes the addition of a step using exons only to better address incomplete alleles, however these improvements are not yet validated. For the 29 other allele determinations made for incomplete alleles in the IMGT/HLA database, encompassing 17 unique alleles, all were genotyped successfully.
3.2.3. Assessing ambiguity
Two forms of ambiguity are common with HLA NGS methods: 1) the inability to phase the length of the amplicon using short reads, and 2) exclusion of distal exons from the amplicon (e.g. DPB1 and DRB1 loci). Of the 2,126 allele calls, the reference genotype was ambiguous for 119 alleles, and were limited to the DRB1 and DPB1 loci. Sequencing with the long reads of the ONT platforms resolved all of the ambiguities caused by a lack of phase, which is 41.7% (49/119) of the total ambiguities. The remaining 70 ambiguous allele calls were all due to incomplete characterization of all exonic regions during amplification.
3.2.4. Novel alleles
Samples with known exonic novelty were included to test the ONT-based sequencing as these alleles present certain challenges and yet are a somewhat common occurrence. Of the 2,126 alleles included, 15 alleles had a known exonic novelty and distributed across all loci except DRB4 and DRB5. After genotyping, 16 alleles were called with exonic novelty, where all 15 cases of known novelty were properly detected. However, the pipeline falsely identified an extra novel allele as described in section 3.2.2 above. While this method is sensitive to novelty and allows for proper detection in known cases, further refinement of the algorithm will be necessary to minimize the opportunity for false novel alleles.
3.3. Objective 2
The first objective of this study was to assess the sequencing and analysis of nanopore data for credible HLA typing using ONT MinION data. After having shown the ONT-based method is credible in Objective 1, we now aim for a protocol that is less than 6 h to obtain HLA typing for deceased donors. For that purpose, two commercially available multiplex PCR protocols were used, GenDx and Omixon coupled with the use of the ONT Flongle for sequencing.
To assess the turn-around time of a single sample on the Flongle, nine experiments were run with each of the rapid PCR protocols. The Flongle utilized the same R9.4 pores as the MinION flow cells. The time required for each multiplexed PCR protocol is reduced from the Holotype amplification protocol in Objective 1: OmniType takes about 2 h and NGSgo®-MX6-1 takes 3.2 h when following the manufacturer’s protocol. The post-PCR library preparation process for a single sample on the Flongle is reduced to 1.75 h as compared to 6.5 h for 24 samples on the MinION (Table 1 and Fig. 4). The preparation of a single sample and the elimination of barcoding of multiple samples account for the reduced time. Sequencing and analysis take an additional 1.5 h, whereby sequencing is limited to 1 h. The total post-PCR process takes a total of 3.3 h (Table 1). The total time for all steps from DNA extraction through to the final genotype result is 5.6 h (ranging from 5.47 to 5.63 h) when using OmniType (Fig. 4).
Fig. 4.

Timing Diagram for Flongle Experiments. * Amplification time varies based on method. Shown here is 2 h for the OmniType protocol, whereas the NGSgo®-MX6-1 multiplex takes 3 h and 15 min.
The details of the 18 Flongle experiments, 9 for each multiplexed PCR method, are shown in Table 4. The number of active pores varied between the experiments ranging from 58 to 92 active pores, resulting in 14,000 to 36,000 reads, of which 56.0% are usable (Table 3). The NGSgo®-MX6-1 multiplex targeting 6 loci averaged 2,460 reads per locus, whereas the OmniType multiplex targeting 11 loci averaged was 1,200 reads per locus.
Table 4.
Flongle Sequencing Metrics.
| Multiplexed PCR Method | Sequencing Run | Total Run Time (min) | Number of Active Pores | Total Reads per run | Number of Usable reads | Average reads per locus |
|---|---|---|---|---|---|---|
| NGSgo®-MX6-1 | 1 | 60 | 76 | 25,000 | 10,289 | 1,715 |
| 2 | 60 | 85 | 28,000 | 13,473 | 2,246 | |
| 3 | 60 | 71 | 30,000 | 17,195 | 2,866 | |
| 4 | 60 | 74 | 24,000 | 13,504 | 2,251 | |
| 5 | 60 | 83 | 33,000 | 17,116 | 2,853 | |
| 6 | 60 | 90 | 36,000 | 18,597 | 3,100 | |
| 7 | 60 | 77 | 24,000 | 16,001 | 2,667 | |
| 8 | 60 | 58 | 19,000 | 13,745 | 2,291 | |
| 9 | 60 | 73 | 23,000 | 13,130 | 2,188 | |
| Average | 76 | 26,889 | 14,783 | 2,464 | ||
| OmniType | 1 | 60 | 63 | 15,000 | 6,418 | 642 |
| 2 | 60 | 84 | 21,000 | 13,035 | 1,448 | |
| 3 | 60 | 78 | 21,000 | 13,155 | 1,462 | |
| 4 | 60 | 68 | 17,000 | 9,623 | 1,069 | |
| 5 | 60 | 86 | 23,000 | 13,789 | 1,532 | |
| 6 | 60 | 92 | 29,000 | 15,605 | 1,734 | |
| 7 | 60 | 67 | 14,000 | 8,102 | 810 | |
| 8 | 60 | 66 | 16,000 | 9,480 | 948 | |
| 9 | 60 | 79 | 20,000 | 11,699 | 1,170 | |
| Average | 76 | 19,556 | 11,212 | 1,202 |
Balancing the representation of all loci in a multiplexed reaction is challenging, and may compromise genotyping given the limited time for sequencing to keep the whole protocol under 6 h. As such, locus balance was evaluated. For both OmniType and NGSgo®-MX6-1, the locus balance is reproducible within each assay and we find certain loci are less represented in particular DPB1 in NGSgo®-MX6-1 (Fig. 2E and F) and DQA1 and DRB4 in OmniType (Fig. 2C and D). Based on our experience of sequencing for one hour on the Flongle, even with the lowest number of reads obtained for the DRB4 locus (74–109 reads), we generated accurate genotyping. Considering that the DRB4 locus is hemizygous, we can derive that approximately 150 reads would be necessary for credible genotyping a heterozygous locus.
Genotyping results on the Flongle experiments were highly similar to the MinION experiments. Using the OmniType, only 1 allele out of the 161 allele determinations was discrepant compared to the reference. The locus was expected to genotype homozygous for DQA1*01:02:01 and instead was genotyped as DQA1*01:02:01 + DQA1*01:NEW, with an artificial novelty in exon 3 in one of the two alleles. The initial analysis of this sample had 43% of reads with the incorrect base, leading to the novel allele call. In this situation, only 100 reads were available for analysis of the locus, when a typical analysis of a homozygous sample uses a minimum of 400 reads. Upon reanalysis of this sample with more reads, even with as few as 150 reads, the locus genotypes correctly. It is to be noted that the frequency of the incorrect base did not change significantly (41%), however introduction of additional reads in the new analysis revealed a strand bias of the incorrect base, which was accurately detected as noise. When the NGSgo®-MX6-1 amplification protocol was utilized, all 108 allele determinations matched the expected reference genotypes. None of the loci amplified with either multiplexed PCR protocol had allelic imbalance below the threshold for detection, allowing for proper genotyping (Fig. 3B and C).
Regarding the error rate on the Flongles, we observed that there is an increased error rate when compared to the MinION (Fig. 5). Overall, independent of the type of PCR method utilized, multiplexed or not, the Class I loci exhibit a higher error rate than the Class II loci.
Fig. 5.

Percent error of sequencing on the two different ONT platforms: MinION and Flongle. The error rate is broken down and colored by locus and includes substitutions, insertions and deletions. For the MinION experiments, the error rate is combined for the five sequencing experiments (n = 120 samples). For the Flongle, the error rate is grouped by the PCR method (n = 9 samples each).
4. Discussion
Given the inadequacies of current NGS methodologies for HLA typing, we have utilized ONT to resolve individual challenging HLA genotyping scenarios and for complete characterization of the MICA gene [31,32]. In the current study, we demonstrate that ONT sequencing technology has improved sufficiently to be utilized for the credible characterization of all 11 HLA loci, providing distinct advantages. Amplification protocols for targeting HLA genes were assessed independently from library preparation, sequencing, and genotyping analysis. The intent is to introduce a flexible protocol whereby users can select the targeting of the genes, through different strategies of amplification (individual locus vs. multiplex), approaches for selection and targeting of HLA genomic sequences, or scales of typing (single vs. multiple samples; selected vs. all 11 HLA loci). It was critical for each pre-PCR protocol to be assessed independently from the rest of the process because anomalies in PCR, particularly with multiplexing, may eventually affect the genotyping. Issues such as preferential amplification, drop-outs, and challenging homopolymer genomic segments within the gene, may influence genotyping accuracy.
The careful selection of samples covering frequent HLA allele specificities in five populations in the US, along with samples/novel alleles with idiosyncrasies that challenge our analysis pipeline, provides credibility to our post-PCR protocol for HLA genotyping utilizing the ONT platform MinION (R9.4 version). For the first phase of this work, we used a well-tested protocol [33], whereby individual HLA genes are amplified separately, to sequence 120 samples. This credible PCR approach enables an objective assessment of the post-PCR components, including library preparation, sequencing, and analysis of nanopore reads. The data obtained from each of the five sequencing runs were reproducible and comparable (Fig. 1). The minor allele frequency was also comparable among the 11 loci, except DRB4 (Fig. 3). The different metrics assessed (Table 3) demonstrate excellent performance. Overall, our data demonstrate an impressive accuracy and total lack of phase ambiguities (due to distant polymorphisms). Other ambiguities due to the absence of some genomic segments (i.e., exon 1 in some amplicons, such as DRB1 or DPB1) persist and they are unrelated to the performance of the nanopore technology. Amplification protocols or other approaches targeting the totality of HLA genomic sequences may eventually eliminate all ambiguities.
The few DQB1 and DRB4 discrepancies observed with the typing of 120 samples (2,126 allele calls) were all related to PCR issues, whereby some alleles were minimally amplified and did not exceed our threshold set by the analysis program, resulting in no call. The threshold level is an internal value set in our analysis pipeline, and differentiates heterozygous versus potentially homozygous samples. Conceptually, this threshold is not identical to the minor allele percentage used for variant calling currently employed by software programs designed for Illumina data. Of note, given our practice of assessing HLA haplotypes before reporting, these discrepant cases would be detected. Haplotype anomalies trigger an investigation to reveal minimally amplified alleles and repeat testing by NGS and another DNA-based method (SSOP) to detect the presence of another allele. The single DRB1 discrepancy observed in this study arose from a software problem in which certain alleles have incomplete sequences in IMGT or very close resemblance to others, and is an active area of development. The detection of a “new” allele, however, would have triggered a further investigation resulting in the detection of the problem. The remaining 15 “new” alleles included in the reference set of samples were detected accurately by this new sequencing approach. High-resolution typing at the fourth-field level (HR-4F) using this technology also appears realistic. A remaining challenge involves the confident characterization of alleles with homopolymers, as it is unclear whether PCR, sequencing or a combination of both is the culprit.
Considering the total elimination of ambiguities due to phasing, and the very few detected discrepancies, this method is impressively accurate, simple, with short turnaround time and low cost. Using the flow sequencing cells multiple times (3–4) reduces the cost of sequencing significantly. Indexing of 24 samples may reduce the cost further. Of note is the very low cost of the MinION device at $1,000, which is far less expensive as compared to other NGS platforms.
The technology continually develops as the ONT company introduces improvements to address existing limitations. Although we do not present the data generated with the R10.0 and R10.3 flow cells (most recent versions of flow cells) in this report, the improvements for the sequencing of homopolymers further enhance the potential of this technology for HLA typing. As ONT develops technical improvements to library preparation, the time of sample preparation and, therefore, turnaround time, will decrease. Analysis software improvements for synchronous sequencing, analysis, and genotyping may expedite reporting. The sequencing platform need not continue sequencing if enough reads are generated, and genotyping is secured. Our protocol did not incorporate this dynamic approach, but sequencing was discontinued when we estimated that an adequate number of reads had been obtained. With the incorporation of the described improvements, genotyping of 24 samples, which presently takes approximately 14 h, will be further reduced.
The second phase of our work assessed the pairing of two multiplexing PCR protocols with our post-PCR nanopore sequencing and analysis protocol. The objective was to take advantage of the Flongle adapter for the MinION, with a reduced number of pores and, therefore, of sequencing capacity, designed by ONT to be a cost-effective solution for single-use experiments. The Flongle happens to be able to accommodate all 11 HLA amplicons from a single sample and provide enough reads for credible genotyping in a relatively short period (approximately one hour). Potentially, however the platform could sequence more than one sample, if the sequencing time could be extended. We used two multiplex PCR protocols to assess whether different protocols have different performance after nanopore sequencing. The GenDx protocol did not amplify all 11 HLA loci, but we understand that the GenDx company will soon have an 11-locus multiplexed PCR (NGSgo®-MX11-3). To examine as many samples as possible, the samples selected for the assessment of the two protocols were not the same. There was only one discrepancy (1 out of 161 allele calls) with the OmniType, whereby a reference homozygous DQA1 locus was typed as heterozygous, with the second allele being a “new” allele. The initial analysis of this sample was restricted to the data generated in the first hour of sequencing. However, upon reanalysis, using more reads from the same sequencing run that were available, the novel allele was found to be a false-positive, and the sample genotyped correctly. In the absence of reference typing, this “new” allele would have been further evaluated after initial typing to confirm the call, and the error would have been identified. Regarding the number of reads needed for credible genotyping on the Flongle flow cells, we found that a heterozygous sample would need more than 150 reads per locus and this number of reads can be collected in the period of one hour.
There is room for improvement in the relative balance of the different loci by these two multiplex PCRs. With regards to NGSgo®-MX6-1 multiplexed PCR, it appears that DPB1 is underrepresented among the six amplified loci. The OmniType protocol appears to have a low representation of DQA1 and DRB4 loci, while the B locus is overrepresented. Despite the low representation of certain loci by these two protocols, the genotyping performance was uncompromised. The locus imbalance can have an increased effect when sequenced on a Flongle flow cell, which is characterized by fewer active pores than a MinION and when coupled with a required short sequencing time, potentially may not generate enough reads for accurate genotyping. The Omixon company is in the process of further optimizing its multiplex PCR protocol; we look forward to its most recent improvements. We are also interested in exploring emerging techniques to apply during sequencing to mitigate the effects of locus imbalance with adaptive subsampling during sequencing, which may also further reduce the sequencing time [34].
Another interesting observation is that the error rate in substitutions, deletions, insertions, between the MinION and the Flongle is different and significant. It is unclear whether this observation is due to the PCR products or the sequencing platforms by ONT, and further investigation is warranted. These errors may be readily resolved if the same PCR products are on different platforms. Additionally, the class I loci have higher error rates than class II loci, an observation that is reproducible and independent of the sequencing platforms. These differences in error rate had no impact on genotyping, as class I and II loci were all accurately genotyped. As the technical sensitivities of the assay are likely to become relevant, an awareness of the limitations of this new system of HLA typing is warranted.
The combination of features of this nanopore technology, paired to multiplex protocols, form the basis for an accurate methodology with a turnaround time of approximately six hours, likely to shorten in the future, for a single sample.
This development is extremely relevant to the transplant community, given the generated typing can be HR-2F. HR-2F typing will facilitate better characterization of recipient antibody profiles for DSA. With an increasing number of studies linking the epitope mismatch loads to clinical outcomes, HR-2F typing for deceased donors has the potential to permit such analyses to optimize donor selection [11-16]. The technology may also reduce the burden on transplant center labs that routinely type deceased donors at the high-resolution level. This information would become available upon the initial offer, which may translate into savings for the overall health system.
The turnaround times of HLA genotyping for a single sample on the Flongle or multiple samples on a MinION may be further reduced as individual steps in the process are optimized. Shortened multiplex PCR protocols paired to post-PCR protocols would improve the time for processing many samples in parallel. The number of commercially available indexes is the primary barrier, which we anticipate can readily be increased. Additionally, automated library preparation currently underway by ONT can expedite this step. Finally, modifications to permit simultaneous data processing and sequencing on the flow cell can further expedite HLA genotyping. The benefits of this technology for HLA characterization are bound to extend its reach beyond existing clinical and research applications.
Supplementary Material
Acknowledgments
We thank the Omixon company for making the OmniType PCR protocol available for our study through their Early Access program. We also thank the CHOP Immunogenetics Clinical lab for their work on generating the reference HLA typing for the 120 samples. CHOP institutional funds supported this work (DSM) and partially supported by NIH grants 1R01DK104339-01 (SAT) and R35 GM134957-01 (SAT) and American Diabetes Association Pathway to Stop Diabetes grant #1-19-VSN-02 (SAT).
Abbreviations:
- HLA
Human leukocyte antigen
- HR-2F
high-resolution two-field
- NGS
next generation sequencing
- ONT
Oxford Nanopore Technologies
- PCR
polymerase chain reaction
Footnotes
Disclosures
D.S. Monos is a consultant to, and owns options in Omixon. D.S. Monos, D. Ferriola and J.L. Duke receive royalties from Omixon. The other authors of this manuscript have no conflicts of interest to disclose.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.humimm.2020.06.004.
References
- [1].Lee SJ, Klein J, Haagenson M, Baxter-Lowe LA, Confer DL, Eapen M, Fernandez-Vina M, Flomenberg N, Horowitz M, Hurley CK, Noreen H, Oudshoorn M, Petersdorf E, Setterholm M, Spellman S, Weisdorf D, Williams TM, Anasetti C, High-resolution donor-recipient HLA matching contributes to the success of unrelated donor marrow transplantation, Blood 110 (2007) 4576–4583, 10.1182/blood-2007-06-097386. [DOI] [PubMed] [Google Scholar]
- [2].Pidala J, Lee SJ, Ahn KW, Spellman S, Wang H-L, Aljurf M, Askar M, Dehn J, Fernandez Vina M, Gratwohl A, Gupta V, Hanna R, Horowitz MM, Hurley CK, Inamoto Y, Kassim AA, Nishihori T, Mueller C, Oudshoorn M, Petersdorf EW, Prasad V, Robinson J, Saber W, Schultz KR, Shaw B, Storek J, Wood WA, Woolfrey AE, Anasetti C, Nonpermissive HLA-DPB1 mismatch increases mortality after myeloablative unrelated allogeneic hematopoietic cell transplantation., Blood. 124 (2014) 2596–2606. 10.1182/blood-2014-05-576041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Williams RC, Opelz G, Weil EJ, McGarvey CJ, Chakkera HA, The risk of transplant failure With HLA mismatch in first adult kidney allografts 2: living donors, summary, guide, Transplant Direct e152 (2017), 10.1097/TXD.0000000000000664 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Williams RC, Opelz G, McGarvey CJ, Weil EJ, Chakkera HA, The risk of transplant failure with HLA mismatch in first adult kidney allografts from deceased donors, Transplantation 100 (2016) 1094–1102, 10.1097/TP.0000000000001115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Illing PT, Purcell AW, McCluskey J, The role of HLA genes in pharmacogenomics: unravelling HLA associated adverse drug reactions, Immunogenetics 69 (2017) 617–630, 10.1007/s00251-017-1007-5. [DOI] [PubMed] [Google Scholar]
- [6].Matzaraki V, Kumar V, Wijmenga C, Zhernakova A, The MHC locus and genetic susceptibility to autoimmune and infectious diseases, Genome Biol. 18 (2017) 76, 10.1186/s13059-017-1207-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Dehn J, Spellman S, Hurley CK, Shaw BE, Barker JN, Burns LJ, Confer DL, Eapen M, Fernandez-Vina MA, Hartzman R, Maiers M, Marino SR, Mueller C, Perales M-A, Rajalingam R, Pidala J, Selection of Unrelated Donors and Cord Blood Units for Hematopoietic Cell Transplantation: Guidelines from NMDP/CIBMTR, Blood. (2019) blood.2019001212. 10.1182/blood.2019001212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Duquesnoy RJ, Kamoun M, Baxter-Lowe LA, Woodle ES, Bray RA, Claas FHJ, Eckels DD, Friedewald JJ, Fuggle SV, Gebel HM, Gerlach JA, Fung JJ, Middleton D, Nickerson P, Shapiro R, Tambur AR, Taylor CJ, Tinckam K, Zeevi A, Should HLA mismatch acceptability for sensitized transplant candidates be determined at the high-resolution rather than the antigen level? Am. J. Transplant 15 (2015) 923–930, 10.1111/ajt.13167. [DOI] [PubMed] [Google Scholar]
- [9].Vogiatzi P, Some considerations on the current debate about typing resolution in solid organ transplantation, Transplant. Res 5 (2016) 1–6, 10.1186/s13737-016-0032-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Huang Y, Dinh A, Heron S, Gasiewski A, Kneib C, Mehler H, Mignogno MT, Morlen R, Slavich L, Kentzel E, Frackelton EC, Duke JL, Ferriola D, Mosbruger T, Timofeeva OA, Geier SS, Monos D, Assessing the utilization of high-resolution 2-field HLA typing in solid organ transplantation, Am. J. Transplant 19 (2019) 1955–1963, 10.1111/ajt.15258. [DOI] [PubMed] [Google Scholar]
- [11].Wiebe C, Pochinco D, Blydt-Hansen TD, Ho J, Birk PE, Karpinski M, Goldberg A, Storsley LJ, Gibson IW, Rush DN, Nickerson PW, Class II HLA epitope matching - a strategy to minimize de novo donor-specific antibody develpment and improve outcomes, Am. J. Transplant 13 (2013) 3114–3122, 10.1111/ajt.12478. [DOI] [PubMed] [Google Scholar]
- [12].Wiebe C, Nevins TE, Robiner WN, Thomas W, Matas AJ, Nickerson PW, The synergistic effect of class II HLA epitope-mismatch and nonadherence on acute rejection and graft survival, Am. J. Transplant 15 (2015) 2197–2202, 10.1111/ajt.13341. [DOI] [PubMed] [Google Scholar]
- [13].Wiebe C, Rush DN, Nevins TE, Birk PE, Blydt-Hansen T, Gibson IW, Goldberg A, Ho J, Karpinski M, Pochinco D, Sharma A, Storsley L, Matas AJ, Nickerson PW, Class II eplet mismatch modulates tacrolimus trough levels required to prevent donor-specific antibody development, J. Am. Soc. Nephrol 28 (2017) 3353–3362, 10.1681/ASN.2017030287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Sapir-Pichhadze R, Tinckam K, Quach K, Logan AG, Laupaeis A, John R, Beyene J, Kim SJ, HLA-DR and -DQ eplet mismatches and transplant glomerulopathy: a nested case-control study, Am. J. Transplant 15 (2015) 137–148, 10.1111/ajt.12968. [DOI] [PubMed] [Google Scholar]
- [15].Walton DC, Hiho SJ, Cantwell LS, Diviney MB, Wright ST, Snell GI, Paraskeva MA, Westall GP, HLA matching at the eplet level protects against chronic lung allograft dysfunction, Am. J. Transplant 16 (2016) 2695–2703, 10.1111/ajt.13798. [DOI] [PubMed] [Google Scholar]
- [16].Sapir-Pichhadze R, Zhang X, Ferradji A, Madbouly A, Tinckam KJ, Gebel HM, Blum D, Marrari M, Kim SJ, Fingerson S, Bashyal P, Cardinal H, Foster BJ, Epitopes as characterized by antibody-verified eplet mismatches determine risk of kidney transplant loss, Kidney Int. 97 (2020) 778–785, 10.1016/j.kint.2019.10.028. [DOI] [PubMed] [Google Scholar]
- [17].Duquesnoy RJ, HLA epitope based matching for transplantation, Transpl. Immunol 31 (2014) 1–6, 10.1016/j.trim.2014.04.004. [DOI] [PubMed] [Google Scholar]
- [18].Sullivan HC, Dean CL, Liwski RS, Biswas S, Goodman AL, Krummey S, Gebel HM, Bray RA, (F)Utility of the physical crossmatch for living donor evaluations in the age of the virtual crossmatch, Hum. Immunol 79 (2018) 711–715, 10.1016/j.humimm.2018.08.001. [DOI] [PubMed] [Google Scholar]
- [19].Pandey S, Harville TO, Epitope analysis aids in transplant decision making by determining the clinical relevance of apparent pre-transplant donor specific antibodies (DSA), Ann. Clin. Lab. Sci 49 (2019) 50–56. [PubMed] [Google Scholar]
- [20].Transplant Trends, United Netw. organ Shar. (2020). https://unos.org/data/transplant-trends/ (accessed April 19, 2020).
- [21].Kaur N, Pinelli D, Kransdorf EP, Pando MJ, Smith G, Murphey CL, Kamoun M, Bray RA, Tambur A, Gragert L, A precision virtual crossmatch decision support system for interpretation of ambiguous molecular HLA typing data, BioRxiv. (2019) 756809. 10.1101/756809. [DOI] [Google Scholar]
- [22].Hurley CK, Kempenich J, Wadsworth K, Sauter J, Hofmann JA, Schefzyk D, Schmidt AH, Galarza P, Cardozo MBR, Dudkiewicz M, Houdova L, Jindra P, Sorensen BS, Jagannathan L, Mathur A, Linjama T, Torosian T, Freudenberger R, Manolis A, Mavrommatis J, Cereb N, Manor S, Shriki N, Sacchi N, Ameen R, Fisher R, Dunckley H, Andersen I, Alaskar A, Alzahrani M, Hajeer A, Jawdat D, Nicoloso G, Kupatawintu P, Cho L, Kaur A, Bengtsson M, Dehn J, Common, intermediate and well-documented HLA alleles in world populations: CIWD version 3.0.0, HLA. (2020) tan.13811. 10.1111/tan.13811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Gabriel C, Danzer M, Hackl C, Kopal G, Hufnagl P, Hofer K, Polin H, Stabentheiner S, Pröll J, Rapid high-throughput human leukocyte antigen typing by massively parallel pyrosequencing for high-resolution allele identification, Hum. Immunol 70 (2009) 960–964, 10.1016/j.humimm.2009.08.009. [DOI] [PubMed] [Google Scholar]
- [24].Bentley G, Higuchi R, Hoglund B, Goodridge D, Sayer D, Trachtenberg EA, Erlich HA, High-resolution, high-throughput HLA genotyping by next-generation sequencing, Tissue Antigens 74 (2009) 393–403, 10.1111/j.1399-0039.2009.01345.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Lind C, Ferriola D, Mackiewicz K, Heron S, Rogers M, Slavich L, Walker R, Hsiao T, McLaughlin L, D’Arcy M, Gai X, Goodridge D, Sayer D, Monos D, Next-generation sequencing: the solution for high-resolution, unambiguous human leukocyte antigen typing, Hum. Immunol 71 (2010) 1033–1042, 10.1016/j.humimm.2010.06.016. [DOI] [PubMed] [Google Scholar]
- [26].Wenger AM, Peluso P, Rowell WJ, Chang P-C, Hall RJ, Concepcion GT, Ebler J, Fungtammasan A, Kolesnikov A, Olson ND, Töpfer A, Alonge M, Mahmoud M, Qian Y, Chin C-S, Phillippy AM, Schatz MC, Myers G, DePristo MA, Ruan J, Marschall T, Sedlazeck FJ, Zook JM, Li H, Koren S, Carroll A, Rank DR, Hunkapiller MW, Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome, Nat. Biotechnol 37 (2019) 1155–1162, 10.1038/s41587-019-0217-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Mayor NP, Hayhurst JD, Turner TR, Szydlo RM, Shaw BE, Bultitude WP, Sayno J-R, Tavarozzi F, Latham K, Anthias C, Robinson J, Braund H, Danby R, Perry J, Wilson MC, Bloor AJ, McQuaker IG, MacKinnon S, Marks DI, Pagliuca A, Potter MN, Potter VT, Russell NH, Thomson KJ, Madrigal JA, Marsh SGE, Recipients receiving better HLA-matched hematopoietic cell transplantation grafts, uncovered by a novel HLA typing method, have superior survival: a retrospective study, Biol. Blood Marrow Transplant 25 (2019) 443–450, 10.1016/j.bbmt.2018.12.768. [DOI] [PubMed] [Google Scholar]
- [28].Liu C, Xiao F, Hoisington-Lopez J, Lang K, Quenzel P, Duffy B, Mitra RD, Accurate typing of human leukocyte antigen class I genes by oxford nanopore sequencing, J. Mol. Diagnostics 20 (2018) 428–435, 10.1016/j.jmoldx.2018.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].De Santis D, Truong L, Martinez P, D’Orsogna L, Rapid high resolution HLA genotyping by MinION oxford nanopore sequencing for deceased donor organ allocation, HLA (2020), 10.1111/tan.13901. [DOI] [PubMed] [Google Scholar]
- [30].Gragert L, Madbouly A, Freeman J, Maiers M, Six-locus high resolution HLA haplotype frequencies derived from mixed-resolution DNA typing for the entire US donor registry, Hum. Immunol. 74 (2013) 1313–1320, 10.1016/j.humimm.2013.06.025. [DOI] [PubMed] [Google Scholar]
- [31].Duke JL, Mosbruger TL, Ferriola D, Chitnis N, Hu T, Tairis N, Margolis DJ, Monos DS, Resolving MiSeq-generated ambiguities in HLA-DPB1 typing by using the oxford nanopore technology, J. Mol. Diagn 21 (2019) 852–861, 10.1016/j.jmoldx.2019.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Duke JL, Zou Y, Ferriola D, Luo Q, Wasserman J, Mosbruger TL, Luo W, Cai L, Zou K, Tairis N, Damianos G, Pagkrati I, Kukuruga D, Huang Y, Monos D, (in press), Genomic characterization of MICA gene using multiple next generation sequencing platforms: A validation study, HLA. (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Duke JL, Lind C, Mackiewicz K, Ferriola D, Papazoglou A, Gasiewski A, Heron S, Huynh A, McLaughlin L, Rogers M, Slavich L, Walker R, Monos DS, Determining performance characteristics of an NGS-based HLA typing method for clinical applications, HLA 87 (2016) 141–152, 10.1111/tan.12736. [DOI] [PubMed] [Google Scholar]
- [34].Kovaka S, Fan Y, Ni B, Timp W, Schatz MC, Targeted nanopore sequencing by real-time mapping of raw electrical signal with UNCALLED, BioRxiv. (2020) 2020. 02.03.931923. 10.1101/2020.02.03.931923. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.