

Abstract
Compared with root-associated habitats, little is known about the role of microbiota inside other rice organs, especially the rhizome of perennial wild rice, and this information may be of importance for agriculture. Oryza longistaminata is perennial wild rice with various agronomically valuable traits, including large biomass on poor soils, high nitrogen use efficiency, and resistance to insect pests and disease. Here, we compared the endophytic bacterial and archaeal communities and network structures of the rhizome to other compartments of O. longistaminata using 16S rRNA gene sequencing. Diverse microbiota and significant variation in community structure were identified among different compartments of O. longistaminata. The rhizome microbial community showed low taxonomic and phylogenetic diversity as well as the lowest network complexity among four compartments. Rhizomes exhibited less phylogenetic clustering than roots and leaves, but similar phylogenetic clustering with stems. Streptococcus, Bacillus, and Methylobacteriaceae were the major genera in the rhizome. ASVs belonging to the Enhydrobacter, YS2, and Roseburia are specifically present in the rhizome. The relative abundance of Methylobacteriaceae in the rhizome and stem was significantly higher than that in leaf and root. Noteworthy type II methanotrophs were observed across all compartments, including the dominant Methylobacteriaceae, which potentially benefits the host by facilitating CH4-dependent N2 fixation under nitrogen nutrient-poor conditions. Our data offers a robust knowledge of host and microbiome interactions across various compartments and lends guidelines to the investigation of adaptation mechanisms of O. longistaminata in nutrient-poor environments for biofertilizer development in agriculture.
Introduction
In natural environments, plants share their habitats with diverse microbiota, such as bacteria, archaea, fungi, oomycetes and viruses [1–3]. Many plant microbiota have substantial beneficial effects on their plant host, including improved nutrient uptake, accelerated growth, resilience against pathogens, and tolerance to biotic and abiotic stresses [1, 4, 5]. Plant microbiota can be found on the exterior of plants (the rhizosphere or the phyllosphere) and the interior of plants (the endosphere) [6]. Plant endophytes are microorganisms that live within plant tissues without causing disease [7, 8]. Plant endophytes are particularly fascinating given their plant growth-promoting (PGP) abilities, such as nitrogen fixation, the production phytohormone, ACC deaminase, phosphate solubilization, siderophore secretion and pathogen suppression [9–12]. The precise identification of plant endophytes is necessary to determine their distribution and biofunctions in the host and their beneficial effects.
Rice (Oryza sativa) is the most economically important crop in the world. The genus Oryza consists of cultivated species, O. sativa and O. glaberrima, and more than 22 wild species [13, 14]. The wild rice genomes provide an important pool of useful genes for rice improvement [15, 16]. Red rice, Oryza longistaminata, is a perennial wild rice with strong rhizomes. Rhizomes are horizontal, underground plant stems and essential organs that enable the plant to rapidly growth at the beginning of the next growing season [8, 17]. O. longistaminata has been used as the model species for genetic and molecular dissection of rhizome development and in breeding efforts to transfer rhizome-related traits into annual rice species [18, 19]. Moreover, O. longistaminata contains various valuable traits, including long anther, large biomass on poor soils, high nitrogen use efficiency, and resistance to insect pests and disease [15]. It has been hypothesized that survival of O. longistaminata in the nutrient-poor environments is associated with its microbiota. Flooded rice paddy fields represent a large contributor to agricultural CH4 emissions via biogeochemical processes that are mediated by soil and plant microbial communities [20–22]. Paddy-rice microbiome paly important role in the biogeochemical processes of C, N, P, and Fe, and the rice rhizosphere is one of the most dynamic habitats for element cycling due to carbon (C) input from roots [23]. Nitrogen is one of the most important nutrients for rice growth. Assessment of the diversity and community compositions of microbiota of O. longistaminata, especially the rhizome microbiota, is a fascinating area of research.
Whether the rhizome endophytes are mores similar to stems or roots remains unknown. Despite the significance of rhizomes in red rice [17, 24], little is known about the endophytic community structure and function of the rhizome. Endophytic communities of perennial O. longistaminata in rhizome may be different than those in the other compartments, given the role of rhizomes in adaption to nutrient-poor environments and functional differentiation [24]. In this study, we sequenced the root, rhizome, stem, and leaf endophytic microbiota in O. longistaminata and bulk soil. We compared the microbial communities and network structures in different compartments to investigate rhizome specificity and identify microbiomes that appeared to play important roles in rhizome function. Our work provides a basis for potential agricultural improvement of rice by endophytic microbiota modification.
Materials and methods
Sample collection and processing
The site for rice experiment is located at Nanchang city, China (28°40′04′′N, 115°49′31′′E). In May, 60 germinated seeds of Oryza longistaminata were transplanted into well-mixed red soil with 5 seeds in each tub. Rice was watered properly with tap water to ensure plant health and keep soil under flooding conditions, and harvested in October.
The roots, rhizomes, stems, and leaves of 12 Oryza longistaminata individuals, and 12 bulk soil were chosen for sampling, giving a total of 60 samples. The bulk soil samples were collected from unplanted pots approximately 2 inches below the soil surface. The soil was manually shaken from the roots. 5 cm long stem and rhizome were cut from 5 cm away from the node. Fragments of the roots, rhizomes, stems and leaves were washed with sterile water and separated. Then, the rice samples were treated with ultrasound. Ethanol-sodium hypochlorite was used for plant surface sterilization [25–27]. All the samples were washed successively in 70% ethanol for 1 min and 0.3% sodium hypochlorite with 0.01% Tween 20 for 15 min to further clean living microorganisms from the surfaces and subsequently washed the rice in sterile water. Finally, the sterile filter paper was used to absorb extra moisture. Water used for the final wash was spread on the Luria Broth (LB) and potato dextrose agar (PDA) plates to examine the surface sterilization effect.
Characterization of the root and rhizome phenotype
The plants, roots and rhizomes were photographed using a Nikon E995 digital camera. The roots and rhizomes of O. longistaminata were dissected and vacuum infiltered with 4% (v/v) paraformaldehyde in phosphate-buffered saline (pH 7.0) for 30 min, and then renewed with fresh paraformaldehyde solution and incubated overnight at 4°C. Then, the fixed samples were dehydrated in a graded ethanol series, and embedded in Paraplast Plus (Sigma-Aldrich, St. Louis, MO, USA). Subsequently, materials were sectioned into 10-lm-thick sections, and stained with 0.1% aniline blue. 6 sections of each root and rhizome were viewed and photographed with an Olympus BX51 microscope [28, 29].
Soil chemical properties analysis
Soil organic carbon (SOC) was measured with a TOC analyzer (Analytikjena HT1300, Germany) after removing soil carbonates using 1M HCl [22]. Inorganic nitrogen (NH4+, NO3-) was extracted and measured using 2 M KCl and a discrete auto analyzer (Smartchem 200, Westco, France). Soil pH was measured on soil slurry at a 2.5:1 water: soil ratio using a glass electrode [30].
DNA extraction
Plant tissues were fully ground into powder in a mortar with liquid nitrogen. Then DNA was extracted from rice samples and bulk soil using the PowerSoil DNA isolation kit (Mo Bio Laboratories) as per the manufacturer’s instructions [31, 32]. DNA was quantified with a Qubit Fluorometer by using the Qubit dsDNA BR Assay kit (Life technologies, USA) and the quality was assessed by running an aliquot on a 1% agarose gel.
16S rRNA library construction
We performed 16S rRNA gene amplification for archaea and bacteria. Barcoded primers targeting the variable V4 regions of 16S rRNA genes were used for amplification with universal primer pairs, 515F (GGACTACNVGGGTWTCTAAT) and 806R (GGACTACHVGGGTWTCTAAT) [21]. Both forward and reverse primers were tagged with an Illumina adapter, pad and linker sequences. PCR enrichment was performed in a 50-μL reaction containing 30ng template, fusion PCR primer and PCR master mix. PCR cycling conditions were as follows: 95°C for 3 minutes, 30 cycles of 95°C for 45 seconds, 56°C for 45 seconds, 72°C for 45 seconds and final extension for 10 minutes at 72°C for 10 minutes. The PCR products were purified using Agencourt AMPure XP beads and eluted in Elution buffer. Libraries were qualified using the Agilent Technologies 2100 bioanalyzer. The validated libraries were used for sequencing on the Illumina HiSeq 2500 platform (BGI, Shenzhen, China) following the standard pipelines of Illumina, and generating 2 × 250 bp paired-end reads.
Bioinformatics processing and statistical analysis
Bioinformatic processing steps and statistical analyses were conducted using QIIME 1.9.1, QIIME 2, usearch 11 and R versions 3.5.1 [33–35]. All amplicon sequence variants (ASVs) identified as belonging to chloroplast and mitochondria were removed from the data set. Rarefaction curves were calculated using QIIME 2. Sample metadata were predicted with random forest classification and regression models in QIIME 2 [36]. Principal coordinate analyses were calculated using the pcoa() function from the R package Ape. Permutational MANOVA was carried out to use Vegan’s function Adonis() to measure effect size and significances on β-diversity [37]. CAP analysis was performed using the function capscale() from the R package Vegan [38]. Variance partitioning and significances for experimental factors were performed by running Vegan’s permutest() function over the CAP model using a maximum of 500 permutations. Co-occurrence networks were inferred based on the Spearman correlation matrix constructed with R using the igraph package [39]. The Sankey diagram was plot in the R gallery (imageGP). Potential ecological functions of microbiota based on 16s rRNA gene sequences were predicted with FAPROTAX [40].
Ethical approval
This article does not contain any studies with human participants performed by any of the authors.
Results
Compartment-specific microbiome taxonomic community structure of the wild rice microbiota
The morphological characteristics of O. longistaminata showed strong rhizomes (Fig 1A and 1B), the anatomy structure of the rhizome was completely different from that of the root (Fig 1C–1F). We analyzed the bacterial and archaeal microbiomes from four separate compartments: the roots, rhizomes, stems, and leaves. For our study, the endophytic microbiomes, which are composed of the microbes inhabiting the interior of four compartments, were isolated from the four compartments after ultrasound-treatment and surface sterilization.
Fig 1. Wild rice (Oryza longistaminata) plant and rhizome.
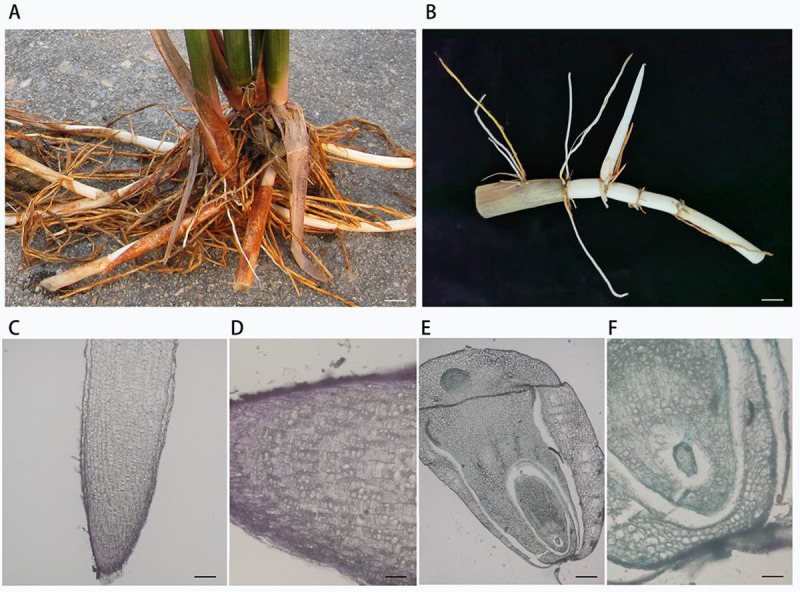
(A) Plants with strong rhizomes. Scale bar: 2 cm. (B) Rhizome and rhizome apical tips with sheath. Scale bar: 1 cm. (C-D) The vertical-section of the root apical tip. Scale bar: 160 and 40 μm. (E-F) The vertical-section of the rhizome apical tip. Scale bar: 160 and 40 μm.
A total of 4,648,671 raw reads were identified among 48 samples of O. longistaminata (roots, rhizomes, stems, and leaves) and 12 bulk soil with a median read count per sample of 80,345 (range: 57,057–88,0877). Rarefaction curves were constructed for each sample showing that sufficient observations have been made (S1 Fig). Archaea were only detected in the bulk soil and root samples, and the relative abundance of archaea was significantly higher in soil than roots (S2A Fig). Notable differences in the proportions of various phyla are observed across the compartments (S3 Fig). At the phylum level, Proteobacteria (55.2%), Firmicutes (26.9%) and Bacteroidetes (7.0%) dominated microbial assemblages among the four compartments. A higher proportion of alpha-Proteobacteria (Kruskal-Wallis, p = 0.043) and Bacteroidetes (Kruskal-Wallis, p = 0.028) were observed in the aboveground parts (stems and leaves) than that in the belowground parts (roots and rhizomes), whereas the proportions of Firmicutes (wilcox, p = 5.24e-3) and Actinobacteria (wilcox, p = 1.12e-4) are higher in the belowground parts than the aerial parts (S3A Fig). Rhizobium (30.1%), Streptococcus (11.1%) and Methylobacteriaceae (7.4%) were the dominant genera in the leaves. Methylobacteriaceae (19.1%), Neisseria (11.4%) and Staphylococcus (10.0%) were the most prevalent genera in the stems. Streptococcus (27.7%), Bacillus (20.5%) and Methylobacteriaceae (15.3%) were the major genera in the rhizome. Escherichia (8.2%), Treponema (6.0%), Pleomorphomonas (5.3%), Sulfurospirillum (4.8%) and Methylobacteriaceae (2.0%) were the dominant genera in the roots (S3B Fig). This study reveals that the dominant Methylobacteriaceae was common in all four compartments of the wild rice, and the relative abundance of Methylobacteriaceae was 7.4%, 19.2%, 15.3% and 2.0% in leaves, stems, rhizomes and roots, respectively (S3B Fig). Other bacteria genera, including Streptococcus (7.8~27.7%), Acinetobacter (1.1~3.9%) and Prevotella (0.03~1.2%), were detected in all four niches.
The microbial alpha diversity within each sample was analyzed based on the species richness estimates (Chao1 and Richness) and diversity indices (Shannon and Simpson) (Fig 2A). Measures of alpha diversity revealed root communities had the highest α-diversity, and the α-diversity of roots was significantly higher than the other three compartments (rhizomes, stems and leaves) (Kruskal-Wallis test, P<0.01). However, the differences in α-diversity among the other three compartments (stems, rhizomes and leaves) were not be considered as statistically significant (Kruskal-Wallis test, p>0.05).
Fig 2. Microbiome alpha- and beta-diversity estimates.

(A) Species richness estimates (Chao and richness) and diversity indices (Shannon and Simpson) for the 48 samples from roots, rhizomes, stems, and leaves of O. longistaminata. (B-E) Unconstrained principal coordinate analyses (PCoAs) of microbial community composition among four different compartments of Oryza longistaminata at the ASV level based on the unweighted UniFrac (UUF), weighted UniFrac (WUF), Bray-Curtis and Jaccard distance matrix. (F-G) PCoAs and CAP of microbial community composition among three different compartments of Oryza longistaminata at the ASV level based on the Bray-Curtis distance matrix.
Unconstrained principal coordinate analyses (PCoAs) based on the unweighted UniFrac (UUF), weighted UniFrac (WUF), Bray-Curtis and Jaccard distance matrix were performed to investigate patterns of separation between microbial communities within different compartments. In the UUF and Jaccard PCoAs, the root compartments separate across the first and second principal coordinate respectively, indicating that the largest source of variation in microbial communities is proximity to the roots (PERMANOVA, P<0.001) (Fig 2B–2E). To more clearly observe patterns of separation between microbial communities within rhizomes, stems and leaves compartments, we performed PCoAs and partial canonical analysis of principal coordinates (CAP) based on Bray-Curtis distance matrix. PCoAs analysis indicated that microbial communities vary significantly between three compartments (PERMANOVA, P<0.01) (Fig 2F). We found that consistent with the PERMANOVA results, CAP analysis indicated that microbial communities were well separated among different compartments (P<0.05) (Fig 2G).
Microbiome co-occurrence networks of ASVs in compartment-specific patterns
We used significant correlations to construct co-occurrence networks, consisting only of positive correlations. Root co-occurrence networks were the largest capturing 16172 associations among 537 microbial ASVs with a higher average degree (60.23) (Fig 3). Rhizome co-occurrence networks were the smallest capturing 8 associations among 8 microbial ASVs with a lower average degree (2.00) (Fig 3).
Fig 3. ASV co-occurrence network interactions of leaves, roots, stems and rhizomes.

Co-occurrence relationships with strong Spearman’s correlation values (P-value < 0.5 and abs(r) > 0.6) are depicted for each compartment. The nodes represented unique ASV in the data sets.
Classification and biomarker ASVs within different compartments
To identify ASVs that correlated with the microbiome community in different compartments, we conducted differential ASV abundance analysis. Specific ASVs were identified in each compartment, and overlaps in differentially abundant ASVs was noted between the compartments (Fig 4A).
Fig 4. Distinctive taxa of the different compartments.

(A) Numbers of differentially ASVs among each compartment in Sankey diagram. (B) Feature table heatmap of distinctive taxa in different compartments using random forest analysis. Methanotrophs are labeled by red stars.
To gain insights into the biomarker ASVs of each compartment, we performed supervised learning with random forest methods to predict niche-specific ASVs. When the number of trees to grow for estimation was set to 500, the number of seeds used by the random number generator was set to 500, and the accuracy prediction was 75% (S4 Fig). ASVs belonging to the Rhizobium, AB2 and Leptonema were specifically presented in leaves. ASVs belonging to the Neisseria, Methylobacterium and Nevskia were specifically presented in stems. ASVs belonging to the Enhydrobacter, YS2 and Roseburia were specifically presented in rhizomes (Fig 4B). Some members within the Methylobacteriaceae and Streptococcus were common in all four compartments (Fig 4B). Noteworthy methanotrophs were observed across all compartments, including Methylobacteriaceae, Pleomorphomonas, Methylobacterium, and Methylococcaceae (Fig 4B).
Phylogenetic community structure
We computed phylogenetic diversity (PD) for each compartment. PD was highest for roots (Kruskal-Wallis, p = 1.007e-05), and the average PD of rhizomes was lower than that of stems, but there was no significant difference (Wilcoxon, p = 0.09) (Fig 5). The vast majority of ses.pd, ses.mpd, and ses.mntd were negative pointing to an increased tendency towards phylogenetic clustering. Leaf and root ASVs exhibited the more phylogenetic clustering than rhizome and stem, leaf and root had lower ses.pd (wilcox, p = 0.002), ses.mpd (wilcox, p = 0.007), and ses.mntd (wilcox, p = 0.014) than rhizome and stem, and the results presented high consistency among the three metrics.
Fig 5. Phylogenetic community structure of Oryza longistaminata.

PD, phylogenetic diversity; pd.obs.z, the standard effective size of phylogenetic diversity; mpd.obs.z, the standard effect size of mean pairwise distance among distinct taxa; mntd.obs.z, the standard effective size of mean nearest taxon distance among distinct taxa.
Microbes associated methane–nitrogen (CH4–N) cycle interactions
Root endophytic microbiota root communities exhibited the highest α-diversity and the most complex co-occurrence network compared with the other three compartments of Oryza longistaminata (Figs 2 and 3). It’s well known that soil microbiota plays a crucial role in biogeochemical processes and has a profound effect on soil functions. Using ASV counts from bulk soil as a control, we conducted a comparative analysis of root microbiota of O. longistaminata and paddy soil. The chemical properties of bulk soil was analyzed (S1 Table). The results showed that root communities had a lower α-diversity than soil (p<0.001) (Fig 6A), and the microbial community structure was significantly differed between roots and soil (PERMANOVA, P<0.001) (Fig 6B).
Fig 6. Microbiota diversity, community structure and functional analysis between the roots and bulk soil.

(A) Alpha diversity estimates. (B) Beta diversity estimates. (C) ASVs involved in potential ecologically functions in roots and bulk soil. (D) ASVs involved in the potential CH4–N cycle between roots and bulk soil. Statistical significance was identified by the wilcoxon test with a false discovery rate (FDR)-corrected pairwise P values.
In order to compare the potential ecological functions between root and soil microbiota, we identified the potential biogeochemical functions of the endophytic microbiota of O. longistaminata using FAPROTAX. Our data showed that soil microbiota performed a complete set of biogeochemical processes, including methanogenesis, methanotrophy, nitrogen-cycling, sulfur-cycling, carbohydrates metabolism, metal metabolism and photoautotrophy (Fig 6C). However, root microbiota was involved in nitrogen-cycling, methanotrophy and sulfur-cycling (Fig 6C). Here we focused on the CH4–N cycle interactions. A large difference in methanogenic archaea was noted between the root and soil microbiota, the microbiome involved in methanogenic archaea in root was significantly lower than that of soil (Wilcoxon.test, p = 1.8e-5) (Fig 6D). Both root and soil microbiome had similar effect on the methanotrophy and nitrogen-fixation process (Wilcoxon.test, p>0.05) (Fig 6D).
Discussion
Annually, irrigated rice production consumes approximately 10% of global fertilizer N and contributes to up to 19% of the global CH4 emissions [41]. How to reduce greenhouse gas emissions and N fertilizer use and improve rice yield has become an urgent problem that needs to be solved. Microbiota may help rice overcome these challenges. Although root-associated microbiota have been extensively studied, the role of microbiota living inside rice remains largely unexplored [42].
Plant genotype effects were significant on the plant microbiome [43–45]. Wild rice microbiota may contribute to the host’s adaptation to adverse environmental conditions. Thus, the study of the microbiota in wild rice is important for rice production and breeding. It has been reported that the rhizomicrobiota community structures differed between wild and cultivate rice [21, 46–49]. Here, we provide insights into the microbiota within the rhizomes, roots, stems and leaves of wild rice (Oryza longistaminata).
Each compartment represents a distinctive ecological niche for endophytic microbiota
Annual and perennial habits are two different strategies by which rice adapt to seasonal environmental change [17, 50]. Rhizomes are a critical component of perenniality and allow wild rice to survive in a harsh environment [24]. Rhizomes are the original stem of plants, and plants rapidly regrow from rhizomes at the beginning of the next growing season [15]. Rhizomes are essentially underground modified stems. We are interested in the difference between the microbes of underground and aboveground stems. In addition, rhizomes and roots are underground tissues and have similar soil/rhizosphere environments. We would like to know if the microbial diversity and structure in the rhizomes are more similar to stems or roots. To answer this question, a comparison to the roots, rhizomes, stems, and leaves microbial assemblages was conducted.
Our data revealed enrichment of members of the phyla Proteobacteria, Firmicutes and Bacteroidetes and the genus Methylobacteriaceae in all four compartments of the wild rice (S2 Fig). In each of the four compartments, we detected marked “compartment-specific” structural and phylogenetic diversification. The rhizome microbiota is unique and possessed niche-specific microbial taxa (Enhydrobacter, YS2, and Roseburia) (Fig 4). Although rhizomes are underground tissues like roots, the microbial diversity and community network structures of the rhizomes were surprisingly significantly reduced compared to the roots (Figs 2, 3 and 5). Meanwhile, the microbial community of the rhizomes exhibited similar taxonomic and phylogenetic diversity compared with that of stems, and even lower network complexity compared with that of stems (Figs 2, 3 and 5). The data showed that the rhizome endophytic microbiota of O. longistaminata was significantly different from other tissues, especially the root, supporting the notion that endophytic microbiota of the rhizome are more similar to that of stems compared with that of roots. Ruifeng et al compared the transcriptome, proteome, and metabolome of the rhizome to other tissues of O. longistaminata, and found that metabolite profiling of the rhizomes was significantly different from the roots but similar with that of stems [15]. Taken together, we hypothesized that the rhizome endophytic microbiota was indeed more similar to that of stems compared with that of roots partly due to the metabolite characteristics of the rhizomes.
Compared with wild rice, Proteobacteria dominated the root endophyte community in cultivated rice (O. sativa) [41, 46]. In our data, the phyla Proteobacteria dominated all four compartments of O. longistaminata, which in agreement with cultivated rice (O. sativa). However, the microbial community structure of other wild rice reported in the literature was different from that of O. longistaminata. To investigate the relationship between rice genotype and the root microbiome, Joseph et al., compared Asian cultivated rice (O. sativa japonica and indica varieties) and African cultivated rice (O. glaberrima), the results demonstrated that microbial communities were influenced by rice genotype [21]. To compare of the root-associated microbiomes in wild (O. rufipogon) and cultivated rice varieties (O. sativa L. ssp. japonica), Lei et al., found that the relative abundance of Streptomyces, Enterobacteriaceae, Leptolyngbya, Planctomycetes, Acidovorax, Ohtaekwangia, Planctomycetaceae, and Comamonadaceae were higher in wild rice, the relative abundance of Acidovorax, Azospirillum, Ohtaekwangia, Actinoplanes, Comamonadaceae, Oxalobacteraceae, Myxococcales, and Acidobacteria were higher in cultivated rice [47]. Recently, Veronica et al., profiled the leaf microbial community composition across 3024 rice (O. sativa), the result showed that Proteobacteria, Firmicutes, Actinobacteria, Cyanobacteria, Tenericutes, and Euryarchaeota were the most abundant phyla [51].
Symbiotic type II methanotrophs may represent key microbes for Oryza longistaminata survival under the nutrient-poor condition
The relationships between biogeochemical processes and microbial functions in paddies have received considerable. Metagenomic analysis showed that root endophytes of cultivated rice (O. sativa) might be involved in the entire nitrogen cycle, as N2-fixation, denitrification, and nitrification [41, 52]. Our data suggested that root microbiota are involved in the nitrogen cycling, methanotrophy and sulfur cycling (Fig 6). Soil microbiota performed a complete set of biogeochemical processes, including methanogenesis, sulfur cycling, nitrogen cycling, carbohydrate metabolism, metal metabolism and photoautotrophy (Fig 6). Methane is mostly synthesized by methanogenic archaea in paddies. We found a large difference in the abundance of methanogenic archaea between the rice paddy soil and roots. Methanogenic archaea exhibit increased higher ASVs relative abundance in the soil compared with the roots (Wilcoxon, p = 2.234e-5) (S2 Fig). The function prediction confirms the ASV relative abundance results, suggesting that soil microbiota but not root microbiota are involved in methanogenesis (Fig 6C). Under flooded conditions, paddy soil exhibits anoxic conditions [53]. Oxygen is subsequently transported from the atmosphere into roots; thus, rice roots grow under partially oxic conditions [54, 55]. Methanogenic archaea require anoxic conditions; thus, we observed that methanogens inhabit flooded rice soil, but not in rice roots (S2 Fig).
CH4 produced from the anoxic zone of paddy soils by methanogenic archaea is transported into the rice via the aerenchyma tissues in the rhizomes, roots and stems [56, 57]. The rice tissues provide a microenvironment full of inorganic and organic substances, including C1 compounds such as methane, which allows the growth of methanotrophy bacteria that utilize CH4 and methanol as carbon sources [20, 58]. Thus, a considerable number of methanotrophs are present in the rice tissues due to adequate CH4 supply from paddies (Figs 4 and 6). Previous studies reported that type I methanotrophs were enriched in cultivated rice (Oryza sativa) roots compared to type II methanotrophs [54]. Moreover, type II methanotrophs in paddies dominate in environments with high concentrations of CH4 but limited nitrogen compared with the type I methanotrophs possibly due to the ability of type II methanotrophs to mediate CH4 oxidation and fix nitrogen (N2) [20, 59]. Nitrogen is one of the most important nutrients for rice growth. It was found that type II methanotrophs are the predominant root-associated microbiota in the rice roots with no N input [52, 58, 60]. Oryza longistaminata broadly distributed throughout tropical Africa exhibits high biomass production on poor soil [17]. In the study, noteworthy methanotrophs were identified across all compartments, including Methylobacteriaceae, Pleomorphomonas, Methylobacterium, and Methylococcaceae (Fig 4B). Most of these methanotrophs belong to type II methanotrophs [54, 61, 62]. Taken together, we may hypothesize that the wild rice (Oryza longistaminata) exhibits the potential for CH4-dependent N2 fixation by type II methanotrophs as a mechanism to adapt the CH4-enriched environment under nitrogen nutrient-poor conditions. The ecological function of this mechanism requires further research.
Conclusions
The phytobiome plays a crucial role in plant nutrition and fitness. Wild rice is an important model to study plant-microbe interactions given their adaption to arid and infertile environments. Here, diverse diversity microbiota and significant variation of community structure were observed among different compartments of O. longistaminata. Rhizome, a critical component of perenniality, harbored specific microbiomes. Rhizome endophytic microbiota of O. longistaminata are significantly different from other tissues, especially the root, supporting the notion that endophytic microbiota of the rhizome are more similar to that of stems compared with that of roots. We found that the relative abundance of Methylobacteriaceae in rhizome and stem is significantly high. The dominant microbiome of O. longistaminata, type II methanotrophs, may benefit the host by facilitating CH4-dependent N2 fixation under nitrogen nutrient-poor conditions. Overall, this study provides an opportunity to understand plant-microbiome interactions in different compartments of red rice.
Supporting information
The distributions of microbial phylum (A) and genus (B) at different compartments of Oryza longistaminata.
(TIF)
(A) Observed ASVs. (B) Shannon index.
(TIF)
(A) Model accuracy. (B) Receiver operating characteristic curves.
(TIF)
The distributions of the microbial domain (A) and phylum (B) in roots and bulk soil.
(TIF)
(XLSX)
Data Availability
All sequence data and sample metadata are publicly available under the NCBI SRA database with the accession number as No. PRJNA606404.
Funding Statement
We wish to acknowledge funding from National Natural Science Foundation of China (31560041, 81960364, 81501162, 31960124), Jiangxi Province Outstanding Young Talents Funding Scheme (20171BCB23020, 20171BCB23021), Interdisciplinary Innovation Fund of Nanchang University (9166-27060003-YB11).
References
- 1.Rodriguez PA, Rothballer M, Chowdhury SP, Nussbaumer T, Gutjahr C, Falter-Braun P. Systems Biology of Plant-Microbiome Interactions. Mol Plant. 2019;12(6):804–21. Epub 2019/05/28. 10.1016/j.molp.2019.05.006 . [DOI] [PubMed] [Google Scholar]
- 2.Berendsen RL, Pieterse CM, Bakker PA. The rhizosphere microbiome and plant health. Trends Plant Sci. 2012;17(8):478–86. Epub 2012/05/09. 10.1016/j.tplants.2012.04.001 . [DOI] [PubMed] [Google Scholar]
- 3.Levy A, Conway JM, Dangl JL, Woyke T. Elucidating Bacterial Gene Functions in the Plant Microbiome. Cell Host Microbe. 2018;24(4):475–85. Epub 2018/10/12. 10.1016/j.chom.2018.09.005 . [DOI] [PubMed] [Google Scholar]
- 4.Duran P, Thiergart T, Garrido-Oter R, Agler M, Kemen E, Schulze-Lefert P, et al. Microbial Interkingdom Interactions in Roots Promote Arabidopsis Survival. Cell. 2018;175(4):973–83 e14. Epub 2018/11/06. 10.1016/j.cell.2018.10.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Muller DB, Vogel C, Bai Y, Vorholt JA. The Plant Microbiota: Systems-Level Insights and Perspectives. Annu Rev Genet. 2016;50:211–34. Epub 2016/09/21. 10.1146/annurev-genet-120215-034952 . [DOI] [PubMed] [Google Scholar]
- 6.Brader G, Compant S, Vescio K, Mitter B, Trognitz F, Ma LJ, et al. Ecology and Genomic Insights into Plant-Pathogenic and Plant-Nonpathogenic Endophytes. Annu Rev Phytopathol. 2017;55:61–83. Epub 2017/05/11. 10.1146/annurev-phyto-080516-035641 . [DOI] [PubMed] [Google Scholar]
- 7.Uroz S, Courty PE, Oger P. Plant Symbionts Are Engineers of the Plant-Associated Microbiome. Trends Plant Sci. 2019;24(10):905–16. Epub 2019/07/11. 10.1016/j.tplants.2019.06.008 . [DOI] [PubMed] [Google Scholar]
- 8.Reinhold-Hurek B, Hurek T. Living inside plants: bacterial endophytes. Curr Opin Plant Biol. 2011;14(4):435–43. Epub 2011/05/04. 10.1016/j.pbi.2011.04.004 . [DOI] [PubMed] [Google Scholar]
- 9.Pacifico D, Squartini A, Crucitti D, Barizza E, Lo Schiavo F, Muresu R, et al. The Role of the Endophytic Microbiome in the Grapevine Response to Environmental Triggers. Front Plant Sci. 2019;10:1256 Epub 2019/10/28. 10.3389/fpls.2019.01256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Olanrewaju OS, Glick BR, Babalola OO. Mechanisms of action of plant growth promoting bacteria. World J Microbiol Biotechnol. 2017;33(11):197 Epub 2017/10/08. 10.1007/s11274-017-2364-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stringlis IA, Yu K, Feussner K, de Jonge R, Van Bentum S, Van Verk MC, et al. MYB72-dependent coumarin exudation shapes root microbiome assembly to promote plant health. Proc Natl Acad Sci U S A. 2018;115(22):E5213–E22. Epub 2018/04/25. 10.1073/pnas.1722335115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gibert A, Tozer W, Westoby M. Plant performance response to eight different types of symbiosis. New Phytol. 2019;222(1):526–42. Epub 2019/01/31. 10.1111/nph.15392 . [DOI] [PubMed] [Google Scholar]
- 13.Vaughan DA, Morishima H, Kadowaki K. Diversity in the Oryza genus. Curr Opin Plant Biol. 2003;6(2):139–46. Epub 2003/04/02. 10.1016/s1369-5266(03)00009-8 . [DOI] [PubMed] [Google Scholar]
- 14.Wang M, Yu Y, Haberer G, Marri PR, Fan C, Goicoechea JL, et al. The genome sequence of African rice (Oryza glaberrima) and evidence for independent domestication. Nat Genet. 2014;46(9):982–8. Epub 2014/07/30. 10.1038/ng.3044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.He R, Salvato F, Park JJ, Kim MJ, Nelson W, Balbuena TS, et al. A systems-wide comparison of red rice (Oryza longistaminata) tissues identifies rhizome specific genes and proteins that are targets for cultivated rice improvement. BMC Plant Biol. 2014;14:46 Epub 2014/02/14. 10.1186/1471-2229-14-46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stein JC, Yu Y, Copetti D, Zwickl DJ, Zhang L, Zhang C, et al. Genomes of 13 domesticated and wild rice relatives highlight genetic conservation, turnover and innovation across the genus Oryza. Nat Genet. 2018;50(2):285–96. Epub 2018/01/24. 10.1038/s41588-018-0040-0 . [DOI] [PubMed] [Google Scholar]
- 17.Yang H, Hu L, Hurek T, Reinhold-Hurek B. Global characterization of the root transcriptome of a wild species of rice, Oryza longistaminata, by deep sequencing. BMC Genomics. 2010;11:705 Epub 2010/12/17. 10.1186/1471-2164-11-705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jin J, Long W, Wang L, Liu X, Pan G, Xiang W, et al. QTL Mapping of Seed Vigor of Backcross Inbred Lines Derived From Oryza longistaminata Under Artificial Aging. Front Plant Sci. 2018;9:1909 Epub 2019/01/10. 10.3389/fpls.2018.01909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fan F, Long W, Liu M, Yuan H, Pan G, Li N, et al. Quantitative Trait Locus Mapping of the Combining Ability for Yield-Related Traits in Wild Rice Oryza longistaminata. J Agric Food Chem. 2019;67(32):8766–72. Epub 2019/07/18. 10.1021/acs.jafc.9b02224 . [DOI] [PubMed] [Google Scholar]
- 20.Minamisawa K, Imaizumi-Anraku H, Bao Z, Shinoda R, Okubo T, Ikeda S. Are Symbiotic Methanotrophs Key Microbes for N Acquisition in Paddy Rice Root? Microbes Environ. 2016;31(1):4–10. Epub 2016/03/11. 10.1264/jsme2.ME15180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Edwards J, Johnson C, Santos-Medellin C, Lurie E, Podishetty NK, Bhatnagar S, et al. Structure, variation, and assembly of the root-associated microbiomes of rice. Proc Natl Acad Sci U S A. 2015;112(8):E911–20. Epub 2015/01/22. 10.1073/pnas.1414592112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ding X, Peng XJ, Jin BS, Xiao M, Chen JK, Li B, et al. Spatial distribution of bacterial communities driven by multiple environmental factors in a beach wetland of the largest freshwater lake in China. Front Microbiol. 2015;6:129 Epub 2015/03/15. 10.3389/fmicb.2015.00129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xiaomeng W, Zhenke Z, Liang W, Jinshui W, Tida G. Biogeochemical cycles of key elements in the paddy-rice rhizosphere: Microbial mechanisms and coupling processes. 2019;10:100144–5. [Google Scholar]
- 24.Hu F, Wang D, Zhao X, Zhang T, Sun H, Zhu L, et al. Identification of rhizome-specific genes by genome-wide differential expression analysis in Oryza longistaminata. BMC Plant Biol. 2011;11:18 Epub 2011/01/26. 10.1186/1471-2229-11-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun L, Qiu F, Zhang X, Dai X, Dong X, Song W. Endophytic bacterial diversity in rice (Oryza sativa L.) roots estimated by 16S rDNA sequence analysis. Microb Ecol. 2008;55(3):415–24. Epub 2007/08/11. 10.1007/s00248-007-9287-1 . [DOI] [PubMed] [Google Scholar]
- 26.Zhang XX, Gao JS, Cao YH, Ma XT, He JZ. Long-term rice and green manure rotation alters the endophytic bacterial communities of the rice root. Microb Ecol. 2013;66(4):917–26. Epub 2013/09/21. 10.1007/s00248-013-0293-1 . [DOI] [PubMed] [Google Scholar]
- 27.Tamai K, Ma JF. Characterization of silicon uptake by rice roots. new phytologist. 2003;158:431–6. [DOI] [PubMed] [Google Scholar]
- 28.Wang L, Chu H, Li Z, Wang J, Li J, Qiao Y, et al. Origin and development of the root cap in rice. Plant Physiol. 2014;166(2):603–13. Epub 2014/06/25. 10.1104/pp.114.240929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tedone F, Del Dottore E, Palladino M, Mazzolai B, Marcati P. Optimal control of plant root tip dynamics in soil. Bioinspir Biomim. 2020;15(5):056006 Epub 2020/06/06. 10.1088/1748-3190/ab9a15 . [DOI] [PubMed] [Google Scholar]
- 30.Meng H, Li K, Nie M, Wan JR, Quan ZX, Fang CM, et al. Responses of bacterial and fungal communities to an elevation gradient in a subtropical montane forest of China. Appl Microbiol Biotechnol. 2013;97(5):2219–30. Epub 2012/04/28. 10.1007/s00253-012-4063-7 . [DOI] [PubMed] [Google Scholar]
- 31.Staley C, Ferrieri AP, Tfaily MM, Cui Y, Chu RK, Wang P, et al. Diurnal cycling of rhizosphere bacterial communities is associated with shifts in carbon metabolism. Microbiome. 2017;5(1):65 Epub 2017/06/26. 10.1186/s40168-017-0287-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xiong W, Song Y, Yang K, Gu Y, Wei Z, Kowalchuk GA, et al. Rhizosphere protists are key determinants of plant health. Microbiome. 2020;8(1):27 Epub 2020/03/05. 10.1186/s40168-020-00799-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7(5):335–6. Epub 2010/04/13. 10.1038/nmeth.f.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. 2019;37(8):852–7. Epub 2019/07/26. 10.1038/s41587-019-0209-9 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Team RC. R: A language and environment for statistical computing. R Foundation for Statistical Computing; 2014. [Google Scholar]
- 36.Bokulich NA, Dillon MR, Bolyen E, Kaehler BD, Huttley GA, Caporaso JG. q2-sample-classifier: machine-learning tools for microbiome classification and regression. J Open Res Softw. 2018;3(30). Epub 2018/01/01. 10.21105/joss.00934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paradis E, Claude J, Strimmer K. APE: Analyses of Phylogenetics and Evolution in R language. Bioinformatics. 2004;20(2):289–90. Epub 2004/01/22. 10.1093/bioinformatics/btg412 . [DOI] [PubMed] [Google Scholar]
- 38.Anderson MJ, Willis TJ. Canonical analysis of principal coordinates: a useful method of constrained ordination for ecology. Ecology. 2003;84:511–25. [Google Scholar]
- 39.Csárdi G, Nepusz T. The igraph software package for complex network research. InterJournal, Complex Systems. 2006;11(5):1–9. [Google Scholar]
- 40.Louca S, Parfrey LW, Doebeli M. Decoupling function and taxonomy in the global ocean microbiome. Science. 2016;353(6305):1272–7. Epub 2016/09/17. 10.1126/science.aaf4507 . [DOI] [PubMed] [Google Scholar]
- 41.Sessitsch A, Hardoim P, Doring J, Weilharter A, Krause A, Woyke T, et al. Functional characteristics of an endophyte community colonizing rice roots as revealed by metagenomic analysis. Mol Plant Microbe Interact. 2012;25(1):28–36. Epub 2011/10/06. 10.1094/MPMI-08-11-0204 . [DOI] [PubMed] [Google Scholar]
- 42.Wang W, Zhai Y, Cao L, Tan H, Zhang R. Endophytic bacterial and fungal microbiota in sprouts, roots and stems of rice (Oryza sativa L.). Microbiol Res. 2016;188–189:1–8. Epub 2016/06/15. 10.1016/j.micres.2016.04.009 . [DOI] [PubMed] [Google Scholar]
- 43.Wagner MR, Lundberg DS, Del Rio TG, Tringe SG, Dangl JL, Mitchell-Olds T. Host genotype and age shape the leaf and root microbiomes of a wild perennial plant. Nat Commun. 2016;7:12151 Epub 2016/07/13. 10.1038/ncomms12151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Walters WA, Jin Z, Youngblut N, Wallace JG, Sutter J, Zhang W, et al. Large-scale replicated field study of maize rhizosphere identifies heritable microbes. Proc Natl Acad Sci U S A. 2018;115(28):7368–73. Epub 2018/06/27. 10.1073/pnas.1800918115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bulgarelli D, Garrido-Oter R, Munch PC, Weiman A, Droge J, Pan Y, et al. Structure and function of the bacterial root microbiota in wild and domesticated barley. Cell Host Microbe. 2015;17(3):392–403. Epub 2015/03/04. 10.1016/j.chom.2015.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Edwards JA, Santos-Medellin CM, Liechty ZS, Nguyen B, Lurie E, Eason S, et al. Compositional shifts in root-associated bacterial and archaeal microbiota track the plant life cycle in field-grown rice. PLoS Biol. 2018;16(2):e2003862 Epub 2018/02/24. 10.1371/journal.pbio.2003862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tian L, Shi S, Ma L, Nasir F, Li X, Tran LP, et al. Co-evolutionary associations between root-associated microbiomes and root transcriptomes in wild and cultivated rice varieties. Plant Physiol Biochem. 2018;128:134–41. Epub 2018/05/20. 10.1016/j.plaphy.2018.04.009 . [DOI] [PubMed] [Google Scholar]
- 48.Shenton M, Iwamoto C, Kurata N, Ikeo K. Effect of Wild and Cultivated Rice Genotypes on Rhizosphere Bacterial Community Composition. Rice (N Y). 2016;9(1):42 Epub 2016/08/26. 10.1186/s12284-016-0111-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang J, Liu YX, Zhang N, Hu B, Jin T, Xu H, et al. NRT1.1B is associated with root microbiota composition and nitrogen use in field-grown rice. Nat Biotechnol. 2019;37(6):676–84. Epub 2019/05/01. 10.1038/s41587-019-0104-4 . [DOI] [PubMed] [Google Scholar]
- 50.Hu FY, Tao DY, Sacks E, Fu BY, Xu P, Li J, et al. Convergent evolution of perenniality in rice and sorghum. Proc Natl Acad Sci U S A. 2003;100(7):4050–4. Epub 2003/03/19. 10.1073/pnas.0630531100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Roman-Reyna V, Pinili D, Borja FN, Quibod IL, Groen SC, Mulyaningsih ES, et al. The rice leaf microbiome has a conserved community structure controlled by complex host-microbe interactions. bioRxiv preprint. 2019. 10.1101/615278. [DOI] [Google Scholar]
- 52.Ikeda S, Sasaki K, Okubo T, Yamashita A, Terasawa K, Bao Z, et al. Low nitrogen fertilization adapts rice root microbiome to low nutrient environment by changing biogeochemical functions. Microbes Environ. 2014;29(1):50–9. Epub 2014/01/28. 10.1264/jsme2.me13110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Christensen PB, Revsbech NP, Sand-Jensen K. Microsensor Analysis of Oxygen in the Rhizosphere of the Aquatic Macrophyte Littorella uniflora (L.) Ascherson. Plant Physiol. 1994;105(3):847–52. Epub 1994/07/01. 10.1104/pp.105.3.847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu L, Ma K, Lu Y. Rice roots select for type I methanotrophs in rice field soil. Syst Appl Microbiol. 2009;32(6):421–8. Epub 2009/06/02. 10.1016/j.syapm.2009.05.001 . [DOI] [PubMed] [Google Scholar]
- 55.Bao Z, Watanabe A, Sasaki K, Okubo T, Tokida T, Liu D, et al. A rice gene for microbial symbiosis, Oryza sativa CCaMK, reduces CH4 flux in a paddy field with low nitrogen input. Appl Environ Microbiol. 2014;80(6):1995–2003. Epub 2014/01/21. 10.1128/AEM.03646-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu C, Mao L, Zheng X, Yuan J, Hu B, Cai Y, et al. Comparative proteomic analysis of Methanothermobacter thermautotrophicus reveals methane formation from H2 and CO2 under different temperature conditions. Microbiologyopen. 2019;8(5):e00715 Epub 2018/09/28. 10.1002/mbo3.715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang X, Chen J, Liu C, Luo J, Yan X, Aihua A, et al. Over-expression of a protein disulfide isomerase gene from Methanothermobacter thermautotrophicus, enhances heat stress tolerance in rice. Gene. 2019;684:124–30. Epub 2018/10/28. 10.1016/j.gene.2018.10.064 . [DOI] [PubMed] [Google Scholar]
- 58.Bao Z, Okubo T, Kubota K, Kasahara Y, Tsurumaru H, Anda M, et al. Metaproteomic identification of diazotrophic methanotrophs and their localization in root tissues of field-grown rice plants. Appl Environ Microbiol. 2014;80(16):5043–52. Epub 2014/06/15. 10.1128/AEM.00969-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shrestha M, Shrestha PM, Frenzel P, Conrad R. Effect of nitrogen fertilization on methane oxidation, abundance, community structure, and gene expression of methanotrophs in the rice rhizosphere. ISME J. 2010;4(12):1545–56. Epub 2010/07/03. 10.1038/ismej.2010.89 . [DOI] [PubMed] [Google Scholar]
- 60.Qiu Q, Conrad R, Lu Y. Cross-feeding of methane carbon among bacteria on rice roots revealed by DNA-stable isotope probing. Environ Microbiol Rep. 2009;1(5):355–61. Epub 2009/10/01. 10.1111/j.1758-2229.2009.00045.x . [DOI] [PubMed] [Google Scholar]
- 61.Krause S, Luke C, Frenzel P. Succession of methanotrophs in oxygen-methane counter-gradients of flooded rice paddies. ISME J. 2010;4(12):1603–7. Epub 2010/06/25. 10.1038/ismej.2010.82 . [DOI] [PubMed] [Google Scholar]
- 62.Luke C, Krause S, Cavigiolo S, Greppi D, Lupotto E, Frenzel P. Biogeography of wetland rice methanotrophs. Environ Microbiol. 2010;12(4):862–72. Epub 2010/01/07. 10.1111/j.1462-2920.2009.02131.x . [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The distributions of microbial phylum (A) and genus (B) at different compartments of Oryza longistaminata.
(TIF)
(A) Observed ASVs. (B) Shannon index.
(TIF)
(A) Model accuracy. (B) Receiver operating characteristic curves.
(TIF)
The distributions of the microbial domain (A) and phylum (B) in roots and bulk soil.
(TIF)
(XLSX)
Data Availability Statement
All sequence data and sample metadata are publicly available under the NCBI SRA database with the accession number as No. PRJNA606404.