

Abstract
The understanding of the evolution of variable sex determination mechanisms across taxa requires comparative studies among closely related species. Following the fate of a known master sex-determining gene, we traced the evolution of sex determination in an entire teleost order (Esociformes). We discovered that the northern pike (Esox lucius) master sex-determining gene originated from a 65 to 90 million-year-old gene duplication event and that it remained sex linked on undifferentiated sex chromosomes for at least 56 million years in multiple species. We identified several independent species- or population-specific sex determination transitions, including a recent loss of a Y chromosome. These findings highlight the diversity of evolutionary fates of master sex-determining genes and the importance of population demographic history in sex determination studies. We hypothesize that occasional sex reversals and genetic bottlenecks provide a non-adaptive explanation for sex determination transitions.
Research organism: Other
Introduction
Genetic sex determination (GSD) evolved independently and repeatedly in diverse taxa, including animals, plants, and fungi (Tree of Sex Consortium et al., 2014; Beukeboom and Perrin, 2014), but the stability of such systems varies drastically among groups. In mammals and birds, conserved male or female heterogametic sex determination (SD) systems have been maintained over a long evolutionary time with conserved master sex-determining (MSD) genes (Marshall Graves, 2008). In contrast, teleost fishes display both genetic and environmental sex determination (ESD), and a remarkable evolutionary lability driven by rapid turnovers of sex chromosomes and MSD genes (Kikuchi and Hamaguchi, 2013; Pan et al., 2017). These characteristics make teleosts an attractive group in which to study the evolution of SD systems.
In the past two decades, the identity of a variety of MSD genes has been revealed in teleosts thanks to advances in sequencing technologies. These findings generated new hypotheses on how the birth of MSD genes through allelic diversification or duplication with or without translocation may drive sex chromosome turnover in vertebrates (Kikuchi and Hamaguchi, 2013; Pan et al., 2017). These teleost MSD genes also provided empirical support for the ‘limited option’ idea that states that certain genes known to be implicated in sex differentiation pathways are more likely to be recruited as new MSD genes (Marshall Graves and Peichel, 2010). The majority of these recently discovered MSD genes, however, were phylogenetically scattered, making it challenging to infer evolutionary patterns and conserved themes of sex chromosomes and/or MSD gene turnovers. Although comparative studies have been accomplished in medakas, poeciliids, tilapiine cichlids, salmonids, and sticklebacks (Kikuchi and Hamaguchi, 2013), transitions of SD systems in relation to the fate of known MSD genes within closely related species have only been explored in medakas (Myosho et al., 2015) and salmonids (Guiguen et al., 2018).
Esociformes is a small order of teleost fishes (Figure 1) that diverged from their sister group Salmoniformes about 110 million years ago (Mya) and diversified from a common ancestor around 90 Mya (Campbell et al., 2013; Campbell and Lopéz, 2014). With two families (Esocidae, including Esox, Novumbra, and Dallia, and Umbridae, including Umbra) and 13 well-recognized species (Warren et al., 2020), Esociformes is an ecologically important group of freshwater species from the northern hemisphere (Campbell et al., 2013; Campbell and Lopéz, 2014). We demonstrated that a male-specific duplication of the anti-Müllerian hormone gene (amhby) is the MSD gene in Esociformes and that this gene is located in a small male-specific insertion on the Y chromosome of northern pike (Esox lucius) (Pan et al., 2019).
Figure 1. Species phylogeny with estimated divergence time (Campbell et al., 2013; Kumar et al., 2017) and technical approaches for investigating SD systems in Esociformes.

The two families within Esociformes, the Esocidae and the Umbridae, are highlighted with green and yellow background, respectively. Whole-genome sequencing (WGS), homology cloning (Cloning), restriction-site-associated DNA sequencing (RAD-Seq), and pooled-sequencing (Pool-Seq) approaches used in each species are shown by a black square on the right side of the figure. Fish silhouettes were obtained from phylopic.org.
Here, we took advantage of the small number of Esociformes species and their relatively long evolutionary history to explore the evolution of SD in relation to the fate of this amhby MSD gene. Generating novel draft genome assemblies and population genomic data in multiple species of Esociformes (Figure 1), we traced the evolutionary trajectory of amhby from its origin in a gene duplication event 65–90 Mya to its demise during species- or population-specific SD transitions. Our results highlight the diverse evolutionary fates experienced by an MSD gene. Among SD transitions, we uncovered a recent loss of the entire Y chromosome in some populations of northern pike. We hypothesize that drift, exacerbated by the bottleneck effect, may have facilitated the loss of the sex chromosome along with its MSD gene in these populations, a new mechanism for entire sex chromosome loss in vertebrates.
Results
The Esox lucius amhby gene originated from an ancient duplication event
Previously, we demonstrated that a male-specific duplication of the anti-Müllerian hormone gene (amhby) is the MSD gene in Esociformes and that this gene is located in a small, ~140 kb male-specific insertion on the Y chromosome of northern pike (Esox lucius) (Pan et al., 2019). To explore the evolution of the autosomal copy of amh (amha) and amhby in Esociformes, we collected phenotypically sexed samples of most species of this order (Figure 1 and Table 1). We searched for homologs of amh using homology cloning and whole-genome sequencing. We found two amh genes in all surveyed Esox and Novumbra species. In the more basally diverging species, Dallia pectoralis and Umbra pygmaea, we found only one amh gene in both species in tissue-specific transcriptome databases (Pasquier et al., 2016), while two amh transcripts were readily identified in E. lucius (Pan et al., 2019).
Table 1. Summary of the identification of amhby and its association with sex phenotypes in 11 Esociformes species, including six populations of E. lucius and three populations of Esox masquinongy.
| Species | Sampling location | Geographic region | Males with amhby | Females with amhby | p-value of amhby association with sex |
|---|---|---|---|---|---|
| Esox lucius | France | Western Europe | 161/161 (100%) | 0/60 (0%) | <2.2E-16 |
| Esox lucius | Sweden | Northern Europe | 20/20 (100%) | 0/20 (0%) | <2.2E-16 |
| Esox lucius | Poland | Central Europe | 20/20 (100%) | 0/20 (0%) | <2.2E-16 |
| Esox lucius | Xinjiang, China | Eastern Asia | 5/5 (100%) | 0/5 (0%) | 0.0114 |
| Esox lucius | Continental USA and Canada | North America | 0/88 (0%) | 0/74 (0%) | amhby absent |
| Esox lucius | Alaska, USA | North America | 17/19 (89%) | 0/19 (0%) | 1.79E-07 |
| Esox lucius | Siberia, Russia | Northern Asia | 9/9 (100%) | 0/8 (0%) | 0.0003 |
| Esox aquitanicus | France | Western Europe | 1/1 (100%) | 0/2 (0%) | 0.665 |
| Esox cisalpinus | Italy | Western Europe | 2/2 (100%) | 0/2 (0%) | 0.317 |
| Esox reichertii | China | Eastern Asia | 11/11 (100%) | 0/10 (0%) | 3.40E-05 |
| Esox masquinongy | Iowa, USA | North America | 18/27 (66.7%) | 4/18 (22%) | 2.01E-08 |
| Esox masquinongy | Quebec, Canada | North America | 9/20 (45%) | 4/20 (20%) | 0.18 |
| Esox masquinongy | Wisconsin, USA | North America | 57/61 (93%) | 22/61 (36%) | 1.17E-10 |
| Esox americanus (americanus) | Mississippi, USA | North America | 6/6 (100%) | 0/4 (0%) | 0.0123 |
| Esox niger | Quebec, Canada | North America | 5/5 (100%) | 0/8 (0%) | 0.0025 |
| Novumbra hubbsi | Washington, USA | North America | 21/23 (91%) | 0/22 (0%) | 5.28E-09 |
| Dallia pectoralis | Alaska, USA | North America | 0/30 (0%) | 0/30 (0%) | amhby absent |
| Umbra pygmaea | Belgium | Western Europe | 0/31 (0%) | 0/31 (0%) | amhby absent |
To clarify relationships among these amh homologs, we inferred phylogenetic trees from these sequences. These phylogenies provided a clear and consistent topology with two well-supported gene clusters among the amh sequences from Esocidae (Figure 2A, Appendix 1—figure 1). The first gene cluster contains the autosomal amha of E. lucius and an amh homolog from all other species of Esox, Dallia, and Novumbra. Therefore, we assigned these amh homologs as amha orthologs. The second gene cluster contains the Y-specific amhby gene of E. lucius, along with the other amh homolog from all species for which we identified two amh sequences. We assigned these amh homologs as amhby orthologs. The general topology of each cluster was in agreement with the Esociformes species taxonomy.
Figure 2. Phylogenetic analysis of amh homologs from the Esociformes revealed an ancient origin of amhby.

(A) Phylogenetic tree of amh coding sequences built with the maximum-likelihood method. Bootstrap values are provided on each node of the tree. The amha ortholog cluster is highlighted with blue background and the amhby ortholog cluster is highlighted with red background. (B) Time-calibrated species phylogeny of the Esociformes (Campbell et al., 2013). The three putative SD transitions are shown by black stars. The presence of pre-duplication amh, as well as amha and amhby along with the amhby sex linkage, is represented by colored dots at the end of each branch. In E. cisalpinus and E. aquitanicus, sex linkage is not significant (NS) due to low sample size. The earliest duplication timing of amh is denoted by a black arrow at the root of the divergence of Esocidae.
In U. pygmaea, the closest sister species to the Esocidae, we found a single amh homolog, which roots at the base of the amha/amhby clusters. This result indicates that the amh duplication happened after the divergence of Esocidae (including Esox, Dallia, and Novumbra) and Umbridae (including Umbra) lineages. In contrast to other Esocidae that have two amh orthologs, we found only one amh gene in D. pectoralis, which fell in the phylogeny within the amha ortholog cluster, suggesting that this species experienced a secondary loss of its amhby gene after the amh duplication at the root of the Esocidae lineage (Figure 2B and Appendix 1—figure 1). We confirmed the absence of an additional divergent amh gene in D. pectoralis by searching sex-specific Pool-Seq reads from 30 males and 30 females. In addition, only one amh homolog was found in an ongoing genome assembly project with long reads for a male D. pectoralis (Y. Guiguen, personal communication). Using information from an Esociformes time-calibrated phylogeny (Campbell et al., 2013), we estimated that the amhby duplicate arose between 65 and 90 Mya (Figure 2B).
Sex linkage of amhby in the Esocidae lineage
Given that amhby originated from an ancient duplication event and is the MSD gene in E. lucius, we investigated when amhby became the MSD gene and whether transitions of SD systems exist in this group. Because male sex linkage of a sequence is a strong indication that this sequence is located on a Y-chromosome-specific region, we first investigated the association of amhby with male phenotype. We found an association between male phenotype and the presence of amhby in most species of Esox and Novumbra (Table 1). This sex linkage was significant in European, Asian, and Alaskan populations of E. lucius, in two North American populations of E. masquinongy (Iowa and Wisconsin), and in all populations surveyed of E. reichertii, E. americanus, E. niger, and N. hubbsi. For two recently described species, E. cisalpinus and E. aquitanicus (Denys et al., 2014), we had insufficient samples with clear species and sex identification for a decisive result. We confirmed the presence of amhby in all males and its absence from all females in these two species, but the association was not significant due to low sample size (Table 1). Because amhby is associated with male phenotype in most species of Esox and Novumbra, it likely gained an MSD role shortly after its origin either at the root of Esocidae or before the split of Esox and Novumbra lineages (~56 Mya) (Campbell et al., 2013). Despite this global conservation of the linkage between amhby and male phenotype, however, we found some variations in amhby sex association across populations of E. lucius and E. masquinongy (Table 1). These population variations are further investigated below.
Whole-genome analyses of the evolution of sex determination systems in Esociformes
Because amhby was not completely associated with male phenotype in E. masquinongy and N. hubbsi (Table 1) and the gene was not found in D. pectoralis and U. pygmaea, we also used population genomic approaches to search for whole-genome sex-specific signatures in these species. Because of the close phylogenetic distance (45 Mya) between E. lucius and E. masquinongy, we used the E. lucius genome assembly to remap reads from E. masquinongy. We performed Pool-Seq analysis of E. masquinongy (Iowa, USA) to compare the size and location of its sex locus with that of E. lucius (Pan et al., 2019). Comparison of the sex-specific heterozygosity across the entire genome revealed that a single region of less than 50 kb, containing the highest density of male-specific single-nucleotide polymorphisms (SNPs) in E. masquinongy, is located in a region homologous to the proximal end of LG24, where the sex locus of E. lucius is located. Besides LG24, no other linkage group showed enrichment for sex-biased chromosome differentiation (Appendix 1—figure 2), suggesting that the location and the small size of the sex locus are likely conserved between E. lucius and E. masquinongy.
No high-quality genome assembly was available from a closely related species to N. hubbsi. Therefore, we performed de novo RAD-Seq analysis on 21 males and 19 females for N. hubbsi. We found two markers showing a significant association with male sex and no female-biased marker (Figure 3A). This result supports the male heterogametic SD system (XX/XY) that was suggested by the amhby sex linkage and also indicates that the N. hubbsi sex locus is likely small (41.6 restriction enzyme cutting sites/Mb across the genome, on average, Supplementary file 2).
Figure 3. Characterization of sex determination systems in different Esociformes species through RAD-Seq analyses.

Each tile plot shows the distribution of non-polymorphic RAD-Seq markers shared between phenotypic males (horizontal axis) and phenotypic females (vertical axis). The intensity of color for a tile reflects the number of markers present in the respective number of males and females. Tiles that are significantly associated with phenotypic sex (chi-square test, p<0.05 after Bonferroni correction) are highlighted with a red border. (A) In N. hubbsi, two markers were present in 19 of 21 males and in 1 of 19 females, indicating a significant male association and thus an XX/XY SD system with a small sex locus. (B) In D. pectoralis, one marker showed a significant association with females, while no marker showed association with males, indicating a ZZ/ZW SD system. (C) In U. pygmaea, 140 markers showed a significant association with males, while no marker showed association with females, indicating a XX/XY SD system with a large non-recombining sex locus. (D) In the population of E. masquinongy from Quebec (Canada), no marker was associated with either sex. (E) In a population of E. masquinongy from Iowa (USA), five markers were significantly associated with male phenotype, indicating a XX/XY SD system.
In D. pectoralis and U. pygmaea, two species in which amhby is absent, we carried out RAD-Seq analyses comparing phenotypic males and females. In D. pectoralis, we found only three female-biased RAD markers, suggesting that this species also has a small sex locus region (30.8 restriction enzyme cutting sites/Mb on average, Supplementary file 2) under a female heterogametic SD system (ZZ/ZW) (Figure 3B). This ZZ/ZW SD system was further supported by an independent Pool-Seq analysis revealing 45 times more female-specific k-mers (N = 1,081,792) than male-specific k-mers (N = 23,816). This excess of female-specific k-mers indicates that females carry genomic regions that are absent from males and thus that females are the heterogametic sex. In contrast, 140 male-biased and no female-biased RAD markers were identified in U. pygmaea, supporting that this species has a large sex locus (26.2 restriction enzyme cutting sites/Mb on average, Supplementary file 2) under a male heterogametic SD system (XX/XY) (Figure 3C).
Some populations of Esox lucius lost their Y chromosome and ancestral master sex-determining gene
Although amhby is the MSD gene in European populations of northern pike (Pan et al., 2019), this gene was absent from the genome assembly (GCA_000721915.3) of a male specimen from a Canadian population (GCA_000721915.3). To explore this discrepancy, we surveyed the sex linkage of amhby in geographically isolated populations of E. lucius. We found significant male linkage in all investigated European and Asian populations and in one Alaskan population (Table 1). In contrast, amhby was absent from both males and females in all other North American populations.
To investigate whether the loss of amhby in most North American populations coincides with large genomic changes, we compared genome-wide sex divergence patterns between a European population carrying the amhby gene (Ille-et-Vilaine, France) and a North American population that lacks amhby (Quebec, Canada) using a Pool-Seq approach. We aligned Pool-Seq reads to an improved European E. lucius genome assembly (NCBI accession number SDAW00000000; assembly metrics presented in Supplementary file 3) in which all previously identified Y-specific contigs (Pan et al., 2019) were scaffolded into a single contiguous locus on the Y chromosome (LG24). In the European population, Pool-Seq analysis confirmed, with better resolution, previous results (Pan et al., 2019), showing the presence of a small Y chromosome region (~140 kb) characterized by many male-specific sequences at the proximal end of LG24 (Figure 4 A.1 and 4B.1). In comparison, virtually no reads from either male or female pools from the Quebec population mapped to this 140 kb Y-specific region (Figure 4 A.2 and 4B.2). Furthermore, we observed no differentiation between males and females along the remainder of the genome with either Pool-Seq or reference-free RAD-Seq (Appendix 1—figure 3). Together, these results suggest that the Quebec population and likely other mainland US populations, where the MSD gene amhby was not found, lack not only amhby, but also the surrounding Y-specific region identified in European populations. Moreover, the new sex locus of these North American populations, if it exists, is too small to be detected by the RAD-Seq and Pool-Seq approaches.
Figure 4. Loss of the ancestral sex locus and master sex determination gene in North American northern pike.

(A) The numbers of male-specific SNPs in 50 kb non-overlapping windows deduced from Pool-Seq data of a European population (A.1) and North American population (A.2) of E. lucius were mapped to the Y chromosome (LG24) of the sequence of E. lucius from a European male genome (SDAW00000000). The greatest number of male-specific SNPs is found in a single window located at the proximal end of LG24 (0.95 Mb–1.0 Mb) that contains 182 male-specific SNPs (highlighted by the gray box) in the European E. lucius population. The same region, however, shows no differentiation between males and females in the North American population. (B) Relative coverage of male and female Pool-Seq reads is indicated by blue and red lines, respectively, in contigs containing amhby in the European population (B.1) and in the North American population (B.2). Only zoomed view on the European population sex-specific region is presented to facilitate the visualization of the differences in coverage between two populations. We searched for Y-specific regions in 1 kb windows. A window is considered Y specific if it is covered by few mapped female reads (<3 reads per kb) and by male reads at a depth close to half of the genome average (relative depth between 0.4 and 0.6). Based on these depth filters, in the European population we identified 70 potential Y-specific 1 kb windows located between 0.76 Mb and 0.86 Mb and 70 Y-specific 1 kb windows located between 1.0 Mb and 1.24 Mb on LG24. This region is not covered by male or female reads in the North American population, indicating the loss of the entire Y-specific region, highlighted by the gray boxes. The region between 0.86 Mb and 1.0 Mb is not sex specific in both population and is likely an assembly artifact.
Evolving sex determination systems: the case of the muskellunge, Esox masquinongy
As in E. lucius, we found variation in amhby sex linkage among different populations of E. masquinongy (Table 1). In addition, we found that two males and one female of E. masquinongy were heterozygous for amhby in the population from Quebec (Appendix 1—figure 4), a finding that conflicts with the expected hemizygous status of a Y-specific MSD gene. We thus used RAD-Seq to compare genome-wide patterns of sex differentiation in a population from Iowa (USA) where amhby was significantly associated with male phenotype and in a population from Quebec (Canada) where it was not. In the Iowa population, five RAD markers were significantly associated with male phenotype, while no marker was associated with female phenotype (Figure 3D). This result indicates a male heterogametic SD system (XX/XY) with a low differentiation between the X and Y chromosomes, as observed in northern pike (Pan et al., 2019) and N. hubbsi (this study). In contrast, we did not find a sex-specific marker in the Quebec population (Figure 3E). This result was further supported by the analysis of a publicly available dataset from another Quebec population (PRJNA512459, Appendix 1—figure 5), suggesting that the sex locus of E. masquinongy from these Quebec populations is too small to be detected with RAD-Seq (38.4 restriction enzyme cutting sites/Mb on average, Supplementary file 2) or that this population displays a multifactorial GSD or ESD system.
Evolution of the structure of the amhby gene in the Esocidae
The typical amh gene in teleost fishes comprises seven exons encoding a protein that contains 500–571 amino acids. The conserved C-terminal TGF-β domain is crucial for canonical Amh function (di Clemente et al., 2010). The predicted structures of most of the amha and amhby genes in Esociformes are consistent with this canonical structure, and most amha and amhby genes do not show signature of relaxation from purifying selection (Supplementary file 4 and Supplementary file 5). However, in both E. niger and N. hubbsi, the predicted Amhby protein is truncated in its N- or C-terminal part. In E. niger, this truncated amhby gene is flanked by repeated elements and encompasses only three of the seven conserved Amh exons, that is exons 5, 6, and 7 with a truncated TGF-β domain (Appendix 1—figure 6). In N. hubbsi, the truncated amhby gene contains the seven conserved Amh exons but with an N-terminal truncation of exon 1 encoding only eight amino acids with no homology to the conserved 50 amino acid sequence of the first Amh exon of other Esocidae (Appendix 1—figure 6). Together, these results show that in some species where amhby is sex linked, the protein sequence is strongly modified in the N- and C-terminal-conserved domains that are needed for proper protein secretion and correct conformation (di Clemente et al., 2010), leaving questions as to whether these proteins are functional.
Discussion
The northern pike MSD gene amhby substantially diverges from its autosomal copy amha, suggesting an ancient origin (Pan et al., 2019), unlike other cases of amh duplication in fishes, that appears to be comparatively young (Hattori et al., 2013; Li et al., 2015). Here, we show that the amhby duplicate emerged before the split of Esocidae and Umbridae between 65 and 90 Mya (Campbell et al., 2013) and was subsequently secondarily lost in the Dallia lineage. We also demonstrate that amhby presence is significantly associated with male phenotype in most species of Esox and in Novumbra. Although we cannot rule out the possibility that amhby was recruited independently as the MSD gene in Esox and Novumbra, our results suggest the more parsimonious hypothesis that amhby likely acquired an MSD function before the diversification of Esocidae at least 56 Mya.
Studies on SD systems of cichlid fishes and true frogs suggested that frequent turnovers can keep sex chromosomes undifferentiated (Gammerdinger et al., 2018; Jeffries et al., 2018). It was proposed that in these organisms, the turnover of sex chromosomes was facilitated by autosomes that host genes involved in sex differentiation pathways that could be readily recruited as new MSD genes, facilitating turnovers of sex chromosomes (Vicoso, 2019). Alternative evolutionary pathways could also involve conserved MSD genes that translocate onto different autosomes, as demonstrated for the salmonid sdY gene (Guiguen et al., 2018). Interestingly, this scenario does not seem to be the case with Esocidae. No highly differentiated sex chromosomes were observed in the surveyed population of E. masquinongy and N. hubbsi (this study), or in E. lucius, despite Esocidae having retained the same ancestral XX/XY system (likely controlled by amhby) for at least 56 million years. In addition, whole-genome analyses in E. masquinongy suggest that its male-specific locus on the Y chromosome is conserved in terms of size and location with that of E. lucius despite 45 million years of divergence. Such conservation is unusual in teleosts, where frequent turnover of SD systems is considered the norm (Kikuchi and Hamaguchi, 2013; Pan et al., 2017). In line with our present results, substantially undifferentiated sex chromosomes have also been maintained over relatively long evolutionary periods in some Takifugu fish species (Kamiya et al., 2012), supporting the idea that theoretical models of sex chromosome evolution cannot be generalized across taxa and that additional, as-yet unknown evolutionary forces can prevent sex chromosome decay.
Even though this MSD gene has been maintained for a long evolutionary period, our results revealed that it has been subjected to several independent SD transitions, potentially driven by different mechanisms in the Esocidae lineage. In D. pectoralis, we observed a transition from the ancestral XX/XY male heterogametic system shared by Esocidae and U. pygmaea to a ZZ/ZW female heterogametic system. This transition also coincided with the loss of amhby. This shift from male to female heterogamety could have been driven by the rise of a new dominant female determinant, leading to the obsolescence and the subsequent loss of the ancestral male MSD gene, similar to the transition of SD documented among the Oryzias and also in the housefly (Kozielska et al., 2008, Myosho et al., 2012).
The other SD transitions that we characterized, those in E. lucius and E. masquinongy, are probably more recent because these transitions are found within populations of the same species. In E. masquinongy, we observed population-specific variation in SD systems, supporting the conservation of an XX/XY system with amhby as the MSD gene in two US populations belonging to the northern lineage, but not in a Quebec population belonging to the Great Lakes lineage (Turnquist et al., 2017). This result suggests that although amhby is still present in the genome of the Great Lakes lineage, it is no longer the MSD gene and that a new population-specific SD mechanism emerged after the split of the lineages. Previously, female heterogamety was inferred for the Great Lakes lineage based on the presence of males in the offspring of gynogenetic females (Dabrowski et al., 2000). With RAD-Seq, we did not find evidence for female-specific genomic regions in the Great Lakes lineage of E. masquinongy. Higher resolution approaches are needed to identify differentiation between male and female genomes to reveal the new SD locus and its interaction with amhby in the Great Lakes lineage.
In E. lucius, we found that amhby is present and completely sex linked in European, Asian, and Alaskan regions, but is not present elsewhere in North America (Figure 5). This finding suggests that amhby functions as an MSD gene in Eurasian and Alaskan populations, but that populations from elsewhere in North America lost amhby together with the entire sex locus. The current geographic distribution of E. lucius results from a post-glacial expansion from three glacial refugia ~0.18 to 0.26 Mya. The North American populations lacking amhby belong to a monophyletic lineage with a circumpolar distribution originating from the same refugia as the other population we surveyed that carry amhby (Skog et al., 2014). Fossil records from Alaska support the idea of an ‘out of Alaska’ North American expansion of E. lucius within the last 100,000 years (Wooller et al., 2015), suggesting that the Y-specific sequences containing amhby were lost during this dispersal period (Figure 5, Appendix 1—figure 7).
Figure 5. Schematic representation of a hypothesized route of post-glacial E. lucius expansion from a Eurasian refugium ~0.18 to 0.26 Mya.
The presence (blue silhouettes)/absence (red silhouettes) of amhby in different populations, showing that this MSD gene was lost in some North American populations during an out-of-Alaska dispersal around 0.1 Mya. The hypothesized refugium in the Ponto Caspian region is highlighted in beige. An alternative route of post-glacial dispersal is shown in Figure 5—figure supplement 1.

Figure 5—figure supplement 1. Schematic representation of an alternative hypothesized route of post-glacial E. lucius expansion from a Eurasian refugium ~0.18 to 0.26 Mya.

The loss of the entire sex locus in such a short evolutionary time is unlikely to have resulted from the pseudogenization of the amhby gene, as is the case for the ancestral MSD gene of the Luzon medaka (Myosho et al., 2012). Rather, it is probably the result of colonization by a small pool of females and sex-reversed XX males. Such a founder-effect hypothesis is supported by the fact that North American populations of E. lucius display a much lower genetic diversity compared to other populations from the same lineage (Skog et al., 2014). This hypothesis is also supported by the fact that environmental influence on GSD systems is a well-documented phenomenon in fishes (Baroiller et al., 2009; Chen et al., 2014; Sato et al., 2005) and that XX sex reversal induced by environmental factors has been observed occasionally in captive E. lucius (Pan et al., 2019). Our whole-genome analyses failed to identify any sex-associated signals in these North American populations, meaning that if such a new sex locus exists, it would lack detectable signatures of sequence differentiation, similar to the one-SNP sex locus of some Takifugu species (Kamiya et al., 2012). Whether ESD, which is common among poikilothermic animals including teleosts (Navara, 2018), could be the SD system in these North American populations or whether these populations already have a new GSD system in place is still unresolved. Notably, this Y chromosome loss in natural populations of E. lucius mirrors the loss of the W chromosome in lab strains of zebrafish after just a few decades of selective breeding (Wilson et al., 2014).
A few evolutionary models have been formulated to capture the dynamics of sex chromosome turnover (Vicoso, 2019). The replacement of the ancestral SD locus by a new one could be driven by drift alone, by positive selection (Bull and Charnov, 1977), by sexual antagonistic selection (van Doorn and Kirkpatrick, 2007), or by the accumulation of deleterious mutations at the sex locus (Blaser et al., 2013). In all of these models, the ancestral sex chromosomes would revert to autosomes only when the new SD locus is fixed in the population. However, in North American populations of E. lucius, the ancestral sex chromosome was likely lost due to drift in bottlenecked populations. Interestingly, this process would not require the simultaneous emergence of a new GSD system, given the flexibility of SD mechanisms in teleosts, where environmental cues generate phenotypic sexes in the absence of a GSD system. ESD, which is easily invaded by a new genetic system (Muralidhar and Veller, 2018; van Doorn, 2014), may serve as a transitional state between sex chromosome turnovers.
We found two cases of altered amhby copies. In N. hubbsi and E. niger, amhby is strongly sex linked, which suggests that it is located in a Y-specific region of the genome. In both species, however, the amhby gene lacks parts of the conserved C- and/or N-terminal Amh regions, which are needed for proper protein secretion and interaction between Amh and its receptor (di Clemente et al., 2010). Although we cannot exclude that these are non-functional amhby copies, this hypothesis seems unlikely because the tight sex linkage of amhby in these species would require the independent emergence of a new MSD gene in close proximity to amhby. It is more likely that these copies still function as MSD genes, but with altered protein sequences. A similar example of a truncated amh duplicate acting as an MSD gene is found in the teleost cobalt silverside Hypoatherina tsurugae (Bej et al., 2017). Additional cases of truncated duplicated genes functioning as MSD genes have been described in Salmonids, yellow perch (Perca flavescens), and Atlantic herring (Clupea harengus), suggesting that the preservation of ancestral domains is not necessary for a duplicated protein to assume a novel role (Feron et al., 2020b; Rafati et al., 2020; Yano et al., 2012). These cases are all consistent with domain gains and losses contributing to new protein functions (Moore and Bornberg-Bauer, 2012).
Collectively, our results depict the evolutionary trajectories of a conserved MSD gene in an entire order of vertebrates, highlighting both the potential stability of MSD genes as well as non-differentiated sex chromosomes in some lineages, and the dynamics of species- or population-specific SD evolution in teleost fishes. Our results highlight the importance of careful consideration of the population demographic history of SD systems, and of the potential buffering role of ESD during transitions between genetic SD systems in sex chromosome evolution.
Materials and methods
Sample collection
Information on the Esociformes species, collectors, sexing method, and experiments is provided in Supplementary file 6.
Genomic DNA extraction
Fin clips were preserved in 75–100% ethanol at 4°C until genomic DNA (gDNA) extraction. For genotyping, samples were lysed with 5% Chelex and 25 mg of Proteinase K at 55°C for 2 hr, followed by incubation at 99°C for 10 min. For Illumina sequencing, gDNA was obtained using NucleoSpin Kits for Tissue (Macherey-Nagel, Duren, Germany) following the producer’s protocol. DNA concentration was quantified using Qubit dsDNA HS Assay Kit (Invitrogen, Carlsbad, CA) and a Qubit3 fluorometer (Invitrogen, Carlsbad, CA). For Pool-Seq, DNA from different samples was normalized to the same quantity before pooling for male and female libraries separately. High-molecular-weight gDNA for long-read sequencing was extracted as described by Pan et al., 2019.
Genome and population genomics sequencing
Draft genomes of northern pike, muskellunge (E. masquinongy), chain pickerel (E. niger), Olympic mudminnow (N. hubbsi), and Alaska blackfish (D. pectoralis) were sequenced using a whole-genome shotgun strategy with 2 × 250 bp Illumina reads. Libraries were built using the Truseq nano kit (Illumina, FC-121–4001) following instructions from the manufacturer. gDNA (200 ng) was briefly sonicated using a Bioruptor (Diagenode). The gDNA was end-repaired and size-selected on beads to retain fragments of ~550 bp, and these fragments were a-tailed and ligated to Illumina’s adapter. The ligated gDNA was then subjected to eight PCR cycles. Libraries were checked on a fragment analyzer (AATI) and quantified by qPCR using a library quantification kit from KAPA. Libraries were sequenced on a HIseq2500 using the paired end 2 × 250 bp v2 rapid mode according to the manufacturer’s instructions. Image analysis was performed by HiSeq Control Software, and base calling was achieved using Illumina RTA software. The output of each run is presented in Supplementary file 7. To improve the European male genome of E. lucius, we generated an extra coverage of Oxford Nanopore long reads using a higher fragment size (50 kb) library made from gDNA extracted from a different male from the same European population as the sample used for the previous genome assembly. Library construction and genome sequencing were carried out as in Pan et al., 2019 and 12.7 Gbp of new data were generated from one PromethION flowcell.
Pool-Seq was performed on E. masquinongy, D. pectoralis, and the North American populations of E. lucius. Pooled libraries were constructed using a Truseq nano kit (Illumina, FC-121–4001) following the manufacturer’s instructions. Male and female DNA Pool-Seq libraries were prepared for each species using the Illumina TruSeq Nano DNA HT Library Prep Kit (Illumina, San Diego, CA) with the same protocol as for the draft genome sequencing. The libraries were then sequenced on a NovaSeq S4 lane (Illumina, San Diego, CA) using the paired-end 2 × 150 bp mode with Illumina NovaSeq Reagent Kits following the manufacturer’s instructions. The output of each run for each sex is presented in Supplementary file 7.
RAD-Seq was performed on E. masquinongy, N. hubbsi, D. pectoralis, U. pygmaea, and the North American populations of E. lucius. RAD libraries were constructed from gDNA extracted from fin clips for each species using a single Sbf1 restriction enzyme as in Amores et al., 2011. Each library was sequenced on one lane of an Illumina HiSeq 2500. The summary of the output of each dataset is presented in Supplementary file 8.
Analysis of population genomic data for sex-specific signals
Raw RAD-seq reads were demultiplexed with the process_radtags.pl script from stacks version 1.44 (Catchen et al., 2013) with all parameters set to default. Demultiplexed reads for each species were analyzed with the RADSex computational workflow (Feron et al., 2020a) using the radsex software version 1.1.2 (10.5281/zenodo.3775206). After generating a table of markers depth with process, the distribution of markers between sexes was computed with distrib and markers significantly associated with phenotypic sex were extracted with signif using a minimum depth of 10 (−d 10) for both commands and with all other settings at the default. RAD-Seq figures were generated with sgtr (10.5281/zenodo.3773063).
For each Pool-Seq dataset, reads were aligned to the reference genome using BWA mem (version 0.7.17) (Li and Durbin, 2009). Alignment results were sorted by genomic coordinates using samtools sort (version 1.10) (Li et al., 2009), and PCR duplicates were removed using samtools rmdup. A file containing nucleotide counts for each genomic position was generated with the pileup command from PSASS (version 3.0.1b, 10.5281/zenodo.3702337). This file was used as input to compute FST between males and females, number of male- and female-specific SNPs, and male and female depth in a sliding window along the entire genome using the analyze command from PSASS with parameters: window-size 50,000, output-resolution 1000, freq-het 0.5, range-het 0.15, freq-hom 1, range-hom 0.05, min-depth 1, and group-snps. The analysis was performed with a snakemake workflow available at https://github.com/SexGenomicsToolkit/PSASS-workflow. Pool-Seq figures were generated with sgtr version 1.1.1 (10.5281/zenodo.3773063).
For k-mer analysis, 31-mers were identified and counted in the reads of the male and female pools using the ‘count’ command from Jellyfish (version 2.2.10) (Marçais and Kingsford, 2011) with the option ‘-C’ activated to count only canonical k-mers and retaining only k-mers with an occurrence >5 and <50,000,000. Tables of k-mer counts produced by Jellyfish were merged with the ‘merge’ command from Kpool (https://github.com/INRA-LPGP/kpool), and the resulting merged table was filtered using the ‘filter’ command to only retain k-mers present >25 times in one sex and <5 times in the other sex; such k-mers were considered sex biased.
Sequencing of amha and amhby genes
To survey the presence of amha (the canonical copy of amh) and amhby in Esociformes, we collected samples of 11 species (Table 1): all species in the genus Esox (E. americanus americanus, E. americanus vermiculatus, E. aquitanicus, E. cisalpinus, E. masquinongy, E. reichertii, and E. niger), N. hubbsi (the only species in its genus), D. pectoralis (the only well-recognized species in its genus), and one species from the genus Umbra (U. pygmaea). To search for amh homologs in the genomes of these 11 species, we either sequenced PCR amplicons with custom primers (Supplementary file 9) and/or sequenced and assembled the genome (Supplementary file 10) and searched in the assembly and in the raw reads for the presence of one or two amh genes.
For species closely related to E. lucius (E. aquitanicus, E. cisalpinus, and E. reichertii), amh homologs were amplified with primers designed on amha or amhby of E. lucius. Complete sequences of amh homologs were obtained from overlapping amplicons covering the entire genomic regions of both amha and amhby with primers anchored from upstream of the start codon (SeqAMH2Fw1 and SeqAMH1Fw1) and downstream of the stop codon (SeqAMH2Rev3 and SeqAMH1Rev4). All PCR amplicons were then Sanger sequenced from both directions and assembled to make consensus gene sequences.
For three other Esocidae species (E. niger, E. masquinongy, N. hubbsi), only partial amplifications of amh sequences were obtained with primers designed on amha or amhby of E. lucius. To acquire the complete amh sequences, we generated draft genome assemblies from amhby-carrying individuals.
For the two species from the more divergent genera (D. pectoralis and U. pygmaea), we were unable to amplify amhby with primers designed on regions that appear conserved on Esox species. Therefore, we also generated a genome assembly from a phenotypic male of each species.
For E. niger, E. masquinongy, N. hubbsi, and D. pectoralis, we used amha and amhby genomic sequences from E. lucius as queries and searched for amh homologs with blastn (https://blast.ncbi.nlm.nih.gov/Blast.cgi, version 2.10.0+) (Altschul et al., 1997) using the parameters ‘Max target sequences’=50 and ‘Max matches in a query range’=1.
For U. pygmaea, the most divergent species from E. lucius in this study, blasting with E. lucius amha and amhby sequences did not yield any result. We used the blastn strategy with the coding sequence of amh as well as tblastn of the protein sequence from Salmon salar, which returned only one contig.
No genome was available for the other two more distantly related species of E. lucius, E. americanus americanus, and E. americanus vermiculatus; therefore, we designed primers on regions conserved in multi-sequence alignments of amha and amhby in the other Esox species (Supplementary file 9).
To investigate whether truncations of Amhby in E. niger and N. hubbsi could be assembly artifacts, we searched for potential missing homologous sequences in raw genome reads of both species using tblastn (https://blast.ncbi.nlm.nih.gov/Blast.cgi, version 2.10.0+) (Altschul et al., 1997) using the exon 1 protein sequence from E. lucius as a query. Homologous sequences were determined with a maximum e-value threshold of 1E-10.
Validating amh gene number in D. pectoralis and U. pygmaea
To check whether the two copies of amh were not collapsed into a single sequence during assembly, we computed the total number of apparent heterozygous sites in the amh region in population genomic data with the expectation that the presence of two divergent gene copies should result in high apparent heterozygosity when remapped on the single copy assembled in the genome. We compared these heterozygosity data in species where we only identified one amh gene, that is D. pectoralis and U. pygmaea, to results observed when mapping sex-specific pooled libraries of E. lucius (n = 30 males and 30 females, respectively) to a female reference genome containing only amha (GCA_004634155.1). Because there are two amh genes in male genomes of E. lucius and one amh gene in female genomes, this result from mapping E. lucius sex-specific Pool-Seq reads serves as a reference for expected number of variant sites. Reads from the male and female pools of E. lucius and from male pool of D. pectoralis were aligned separately to the reference genome (GCA_004634155.1) and our draft genome, respectively, using BWA mem version 0.7.17 (Li and Durbin, 2009) with default parameters. Each resulting BAM file was sorted and PCR duplicates were removed using Picard tools version 2.18.2 (http://broadinstitute.github.io/picard) with default parameters. Variants were then called for each BAM file using bcftools mpileup version 1.9 (Li et al., 2009) with default parameters on genomic regions containing amh, which locates between 12,906,561 and 12,909,640 bp on LG08 of E. lucius (GCA_004634155.1: CM015581.1), between 23,447 and 25,910 bp on the flattened_line_2941 contig of the draft genome of D. pectoralis, and between 760,763 and 757,962 bp on contig 633485 of our draft genome of U. pygmaea.
Gene phylogenetic analysis
Phylogenetic reconstructions were performed on all amh homolog sequences obtained from Esociformes with the S. salar amh used as an outgroup. Full-length CDS were predicted with the FGENESH+ (Solovyev et al., 2006) suite based on the genomic sequence and Amh protein sequence from E. lucius. Sequence alignments were performed with MAFFT (version 7.450) (Katoh and Standley, 2013). Both maximum-likelihood and Bayesian methods were used for tree construction with IQ-TREE (version 1.6.7) (Minh et al., 2020) and Phylobayes (version 4.1) (Lartillot et al., 2007), respectively.
Additional analyses were carried with or without the truncated amh/Amh sequences of E. niger and N. hubbsi using the amh/Amh homolog of S. salar as an outgroup. CDS and proteins were predicted with the FGENESH+ suite (Solovyev et al., 2006), based on genomic sequences. These putative CDS and protein sequences were then aligned using MAFFT (version 7.450) (Katoh and Standley, 2013). Residue-wise confidence scores were computed with GUIDANCE 2 (Sela et al., 2015), and only well-aligned residues with confidence scores above 0.99 were retained. The resulting alignment file was used for model selection and tree inference with IQ-TREE (version 1.6.7) (Minh et al., 2020) with 1000 bootstraps and the 1000 SH-like approximate likelihood ratio test for robustness. To verify that the topology of the single amh homolog of D. pectoralis was not an artifact of long-branch attraction, we also constructed phylogenetic trees with only the first and second codons of the coding sequences (Lemey et al., 2009) as well as with complete protein sequences. For additional confirmation of the tree topology for the amh homologs, the same alignments were run in a Bayesian framework implemented in Phylobayes (version 4.1), (Lartillot et al., 2007; Lartillot and Philippe, 2006, Lartillot and Philippe, 2004) using the CAT-GTR model with default parameters, and two chains were run in parallel for approximately 1000 cycles until the average standard deviation of split frequencies remained ≤0.01.
Detection of signatures of selection on amha and amhby genes
To compare selection pressure between amha and amhby genes, dN/dS ratios were computed between each ortholog sequence and the amh sequence of U. pygmaea, which diverged from the other Esociformes prior to the amh duplication. Sequences from E. niger and N. hubbsi were excluded because their amhby orthologs were substantially shorter and could introduce bias in the analysis. The ratio of nonsynonymous to synonymous substitution (dN/dS = ) among amha and amhby sequences was estimated using PALM (version 4.7) (Yang, 2007) based on aligned full-length CDS, and the phylogenetic tree obtained with the CDS was used in the analysis. First, a relative rate test on amino acid substitution (Hughes, 1994) was performed between amha and amhby pairs with the amh of U. pygmaea used as an outgroup sequence. For each species with amh duplication, was calculated between the amha ortholog and amh of U. pygmaea and was compared to the calculated between the amhby ortholog and amh of U. pygmaea. A Wilcoxon test was used to compare the resulting values for the amh paralogs of these species. Then, several branch and site models (M0, M1a, M2a, M7, M8, free-ratio, and branch-site) implemented in the CODEML package were fitted to the data. The goodness of fit of these models was compared using the likelihood ratio test implemented in PALM.
Genome assembly
Raw Illumina sequencing reads for D. pectoralis, E. masquinongy, E. niger, and N. hubbsi were assembled using the DISCOVAR de novo software with the following assembly parameters: MAX_MEM_GB = 256, MEMORY_CHECK = False, and NUM_THREADS = 16. Assembly metrics were calculated with the assemblathon_stats.pl script (Earl et al., 2011), and assembly completeness was assessed with BUSCO (version 3.0.2) (Simão et al., 2015) using the Actinopterygii gene set (Supplementary file 10).
To facilitate the comparison of sex loci in different populations of E. lucius, we improved the continuity of the previously published assembly of an E. lucius European male (GenBank assembly accession: GCA_007844535.1). This updated assembly was performed with Flye (version 2.6) (Kolmogorov et al., 2019) using standard parameters and genome-size set to 1.1 g to match theoretical expectations (Hardie and Hebert, 2004). Two rounds of polishing were performed with Racon (version 1.4.10) (Vaser et al., 2017) using default settings and the Nanopore reads aligned to the assembly with minimap2 (version 2.17) (Li, 2018) with the ‘map-ont’ preset. Then, three additional rounds of polishing were performed with Pilon (version 1.23) (Walker et al., 2014) using the ‘fix all’ setting and the Illumina reads aligned to the assembly with BWA mem (version 0.7.17) (Li and Durbin, 2009). Metrics for the resulting assembly were calculated with the assemblathon_stats.pl script (Earl et al., 2011). The completeness of the assembly was assessed with BUSCO (version 3.0.2) (Simão et al., 2015) using the Actinopterygii gene set (4584 genes) and the default gene model for Augustus. The resulting assembly was scaffolded with RaGOO (version 1.1) (Alonge et al., 2019) using the published female genome (GenBank accession: GCA_011004845.1) anchored to chromosomes as a reference.
Acknowledgements
We are grateful to the fish facility of INRAE LPGP for support in experimental installation and fish maintenance and to the GenoToul bioinformatics platform Toulouse Midi-Pyrenees (Bioinfo GenoToul) for providing help, computing, and storage resources. This work was supported by the Agence Nationale de la Recherche, the Deutsche Forschungsgemeinschaft (ANR/DFG, PhyloSex project, 2014–2016, SCHA 408/10–1, to YG and MS), and the U.S. National Institutes of Health (R01GM085318 to JHP). The MGX and Get-Plage core sequencing facilities were supported by France Genomique National infrastructure, funded as part of the ‘Investissement d’avenir’ program managed by the Agence Nationale pour la Recherche (contract ANR-10-INBS-09). We thank the MNHN curators for providing access to the collections of E. aquitanicus and E. cisalpinus and for the ONEMA. We thank GB Delmastro for help with specimen sampling. We also thank Mackenzie Garvey and Penny Swanson for assistance with dissection of N. hubbsi.
Appendix 1
Appendix 1—figure 1. Additional phylogenetic reconstruction of amha and amhby orthologs from the Esociformes with amh sequence from Salmo salar as an outgroup.

(A) Phylogenetic tree built by Maximum likelihood method implemented in IQ-TREE putative protein sequences of all identified amh homologs. (B) Phylogenetic tree built by Maximum likelihood method with only the 1st and 2nd codon of putative coding sequence of all identified amh homologs. (C) Phylogenetic tree built by Maximum likelihood with putative protein sequences of amh homologs without one highly truncated sequence from E. niger. (D) Phylogenetic trees built by Maximum likelihood with putative coding sequences of amh homologs without one highly truncated sequence from E. niger. (E) Phylogenetic tree built by Bayesian method implemented in PhyloBayes with putative protein sequences of all identified amh homologs. (F) Phylogenetic tree built by Bayesian method with putative coding sequences of all identified amh homologs. Bootstrap values are given on each nod of the tree and all trees are rooted with amh sequence from S. salar.
Appendix 1—figure 2. Analysis of sex differentiation in the genome of a male E. masquinongy with Pool-Seq data.

(A) The relative to genome average coverage depth of male and female Pool-Seq reads on the region containing amhby. The male data are represented in blue and female in red. The 3 kb region containing amhby (unplaced_scaffold_RaGOO_chr0: 24,967,172–24,968,938) is covered by virtually no female reads and by male reads at a depth about half of the genome average depth. (B) Number of male-specific SNPs in 50 kb non-overlapping windows in each linkage group in the male genome of E. masquinongy. The window containing the highest number of male-specific SNPs on LG24 is highlighted. (C) Number of male and female-specific SNPs from Pool-Seq in 50 kb non-overlapping windows is plotted along LG. we found the highest number of male-specific SNPs (MSS) in a single window located around 100–150 kb on LG24 that contained 212 MSS and zero female-specific SNPs, while genome average was 1.57 MSS per 50 kb window (3.17 MSS per window when excluding 50 kb windows without MSS).This result indicates that the sex locus in E. masquinongy is homologous to the proximal end of LG24 in E. lucius. Besides LG24, no other linkage group contained a window enriched with MSS. Overall, this Pool-Seq analysis confirms the XX/XY SD system with a low differentiation between the X and Y chromosomes identified with the sex linkage and RAD-Seq analyses, as observed in E. lucius (Pan et al., 2019) and N. hubbsi (see above), and supports the hypothesis that the sex locus of E. masquinongy is similar to that of E. lucius.
Appendix 1—figure 3. No differentiation between male and female genomes revealed by RAD-Seq and Pool-Seq in some North American populations of E. lucius.

(A-B) Distribution of RADSex markers in males and females of two populations of E. lucius from Canada. The distribution of markers in males and females was computed with RADSex with a minimum depth of 5 to consider a marker as present in an individual for both datasets. In each tile plot, the number of males and number of females are represented on the horizontal and vertical axes respectively, and intensity of color of a tile indicates the number of markers present in the corresponding number of males and females. In both populations, we did not find any sex-linked markers among a total of 8,440,899 and 4,526,552 markers identified in each population, indicating that if there is a new sex locus in these populations its detection escapes the resolution of RAD-Seq (31.8 RAD markers per Mb on average) due to a very low differentiation between the new sex chromosome pair. (C–E) Analysis of male/female differentiation across Canadian E. lucius genome with Pool-Seq reads from 30 males and 30 females mapped to the reference genome (Accession number: GCA_004634155.1). Between sex FST (C), female-specific SNPs (D) and male-specific SNPs (E) are computed for 50 kb non-overlapping windows across the 25 linkage groups (LGs) and unplaced scaffolds. We searched for 50 kb non-overlapping windows enriched with either male or female-specific SNP. Overall, the level of differentiation between males and females was low, and the highest Fst observed across the entire genome is 0.07 located between 0.397 Mb and 0.398 Mb on linkage group 11. Very low number of sex-specific SNPs were found for both male and female pools in this population, especially when comparing to the same analysis performed with data from an European population. In the European population, we found a genome average of 3.5 male-specific and 2.7 female-specific SNPs per 50 kb window, with peak windows containing 625 male-specific SNPs near the LG24 sex locus, and 217 female-specific SNPs at the proximal end of LG5. In comparison, the Canadian pools had roughly similar values of average genome sex-specific SNPs (3.3 male-specific and 2.9 female-specific SNPs per window), but with peak windows of sex-specific SNPs of 49 and 45 for males and females, respectively. Given this low amount of differentiation between the sexes, and that no particular chromosome was enriched with windows containing a high number of sex-specific SNPs, no region stood out to be the candidate region for the sex locus. Besides that, no 1 kb window with sex-specific read depth was found. Overall, no clear signal of a sex locus was observed across the entire genome supporting the hypothesis that the mainland USA and Canadian populations lack a well-differentiated sex determining region.
Appendix 1—figure 4. Sanger sequencing results for E. masquinongy one female and one male carrying two different alleles of amhby and one male carrying only one amhby allele.
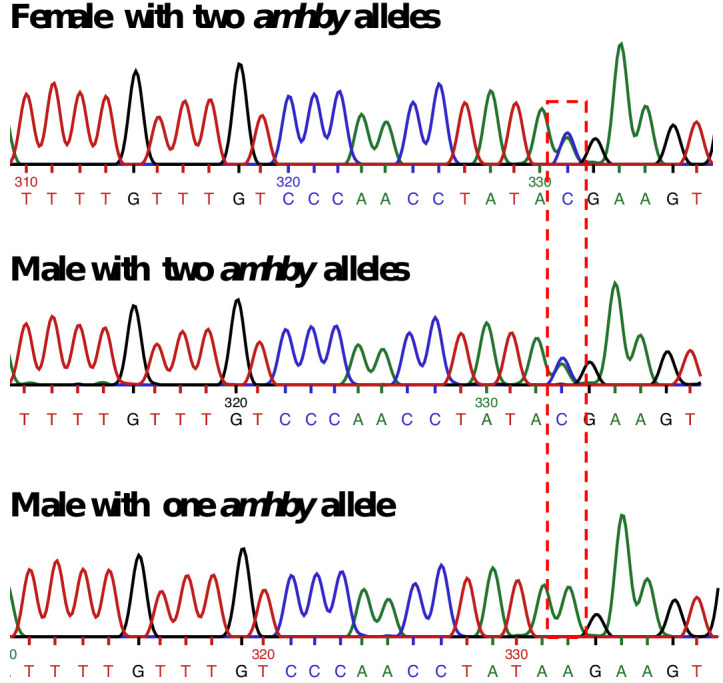
The base with a bi-allelic SNP is highlighted with the red dashed-line box.
Appendix 1—figure 5. RAD-Seq analysis in a second Esox masquinongy population from Quebec (Canada) supports the lack of sex-specific marker in this population.

Each tile plot shows the distribution of non-polymorphic RAD-Seq markers between phenotypic males (horizontal axis) and phenotypic females (vertical axis). The intensity of color for a tile corresponds to the number of markers present in the respective number of males and females. No tiles for which the association with phenotypic sex is significant (chi-square test, p<0.05 after Bonferroni correction) were detected in this analysis. RAD-Seq reads data and samples information are found under NCBI bioproject PRJNA512459.
Appendix 1—figure 6. ClustalW alignment of Esocidae Amhby putative protein sequences.

The signal peptide was predicted with SignalP (Nielsen, 2017). No signal peptide was detected in the Amhby sequences of both E. niger and N. hubbsi. N-terminal region (indicated by a black bar) and the Transforming growth factor beta like domain (indicated by a red bar) were predicted with the Motif Scan tool at ExPASy (Gasteiger et al., 2003) with the Pfam motif database (Finn et al., 2014) optimized for local alignments (pfam-fs, Pfam 32.0, September 2018). The seven exons of amhby are shown by the alternating blue and yellow colors on the sequence ruler. The seven conserved cysteines of the TGF-beta domain are indicated by red arrowheads. The region containing the putative Amh cleavage site (Cleav) is boxed in red. E. luc (Esox lucius), E. aqu (E. aquitanicus), E. cis (E. cisalpinus), E. rei (E. reichertii), E. mas (E. masquinongy), E. aam (E. americanus americanus), E. ave (E. americanus vermiculatus), E. nig (E. niger) and N. hub (Novumbra hubbsi).
Appendix 1—figure 7. Hypothesized scenarios of sex determination system transition in North American populations of E. lucius.

In the ancestral population, Linkage group 24 (LG24) was the sex chromosome (Y). Males were the heterogametic sex carrying one Y chromosome with a small (~300 kb) sex-locus region (SLR) containing amhby and one X chromosome without the SLR and amhby, while females carried two X chromosomes. During post-glaciation re-colonization of North America, the Y chromosome with amhby gene was not carried by individuals that made up the small populations that dispersed out of Alaska. These populations could still produce phenotypic males in the absence of a master sex determining gene via environment-induced sex reversal, a well-documented phenomenon among teleosts. In this transitional state, both males and females carry two X sex chromosomes (LG24). The current sex determination mechanism in these North American populations is still unknown: it could rely entirely on environmental sex determination (ESD), random sex determination, or new male (XY) or female (ZW) heterogametic genetic sex determination system with highly undifferentiated sex chromosomes. SLR: sex-locus region. ESD: environmental sex determination. NA: North America. LGN: un-specified Linkage group, that is ancestral autosomes (A) that are not LG24.
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Contributor Information
Yann Guiguen, Email: yann.guiguen@inrae.fr.
Molly Przeworski, Columbia University, United States.
Detlef Weigel, Max Planck Institute for Developmental Biology, Germany.
Funding Information
This paper was supported by the following grants:
Agence Nationale de la Recherche ANR-13-ISV7-0005 to Yann Guiguen.
Deutsche Forschungsgemeinschaft to Manfred Schartl.
Agence Nationale de la Recherche ANR-10-INBS-09 to Celine Roques, Laurent Journot.
Korea National Institute of Health R01GM085318 to John Postlethwait.
ANR SCHA 408/10–1 to Yann Guiguen, Manfred Schartl.
Additional information
Competing interests
No competing interests declared.
Author contributions
Conceptualization, Data curation, Formal analysis, Investigation, Visualization, Methodology, Writing - original draft.
Conceptualization, Data curation, Software, Formal analysis, Investigation, Visualization, Methodology, Writing - original draft.
Software, Formal analysis, Investigation, Visualization.
Investigation, Visualization, Writing - review and editing.
Supervision, Investigation, Visualization.
Resources, Supervision, Investigation.
Resources, Investigation.
Resources, Investigation.
Resources, Investigation.
Resources, Investigation.
Resources, Investigation.
Resources, Investigation.
Resources, Investigation.
Resources, Investigation.
Resources, Investigation.
Resources, Investigation.
Resources, Investigation.
Resources, Investigation.
Resources, Investigation.
Resources, Formal analysis, Investigation.
Resources, Formal analysis, Investigation.
Investigation.
Resources, Investigation.
Resources, Investigation.
Resources, Investigation.
Resources, Investigation.
Resources, Investigation.
Resources, Investigation.
Resources, Investigation.
Resources, Software, Investigation.
Resources, Software, Investigation.
Resources, Investigation.
Conceptualization, Resources, Funding acquisition, Investigation, Project administration, Writing - review and editing.
Conceptualization, Funding acquisition, Investigation, Project administration, Writing - review and editing.
Conceptualization, Supervision, Funding acquisition, Investigation, Visualization, Project administration, Writing - review and editing.
Additional files
To verify that the two copies of amh were not collapsed into a single sequence during assembly, we computed the total number of apparent heterozygous sites on the amh region in population genomic data with the expectation that the presence of two divergent gene copies should result in high apparent heterozygosity when remapped on the single copy assembled in the genome. In total, 106 variants were observed in the pool of E. lucius males on the amha region located between 12,906,561 bp and 12,909,640 bp on LG08 of E. lucius (GCA_004634155.1: CM015581.1), resulting from the mapping of reads originating from both amha and amhby, while only 12 variants (true allelic variations) were observed in the same region when mapping reads from the female pool originating only from amha. With male Pool-Seq reads from D. pectoralis, we observed only four variant sites on the ~3 kb amh region located between 23,447 bp and 25,910 bp on the flattened_line_2941 contig and zero variant sites from the female Pool-Seq reads. Compared to the ‘control’ of E. lucius Pool-Seq reads where one amh gene from female pool result in 12 variant sites and two amh gene from the male pool result in 106 variant sites, the low number of variant sites in both male and female pool of D. pectoralis support that only one amh gene is present in the genome of D. pectoralis regardless of the phenotypic sex of the individuals. Although sex-specific Pool-Seq reads were not available for U. pygmaea, we performed the same analysis with reads from the single male individual used to assemble the genome. No variant was observed in the ~3 kb region containing amh located between 760,763 bp and 757,962 bp on contig 633485 of our draft genome of U. pygmaea, supporting that only one amh gene is also present in this species.
To help estimate the size of the sex locus from sex-specific RAD markers, we determined the number of potential SbfI cleavage sites based on our draft genome assemblies for each species. For E. lucius (Canadian population), E. masquinongy (Iowa population), N. hubbsi, and D. pectoralis, we predicted the number of RAD-Seq cleavage sites present in each genome by counting the number of unambiguous matches for sequence of SbfI (CCTGCAGG), the restriction enzyme used in RAD-Seq library preparation (Herrera et al., 2015). On average, we found 31.8 RAD markers per Mb in E. lucius, 38.4 in E. masquinongy, 41.6 in N. hubbsi, 30.8 in D. pectoralis, and 26.2 in U. pygmaea. Because we do not see large species differences (26–40 RAD markers/Mb) this suggests that, apart from potential local variations of the RAD markers density in sex loci, our RAD-Seq comparative analysis could to some extent be used to compare sex locus size within species on the basis of this number of sex-specific RAD markers. We are aware of the limitation that using the number of sex-specific marker usually lead to an overestimation of the size of the sex locus. For all of our species with the exception of U. pygmaea, we identified very few sex-specific markers, indicating very small sex locus. This simple calculation is only intended to helps provide a rough approximation of the size of the sex locus.
*Samples from (Ouellet-Cauchon et al., 2014). **Sex was recorded in this E. masquinongy population based on the urogenital pores morphology. ***Sex was recorded in N. hubbsi based on the specific coloration of males during the breeding season. NR = phenotypes not recorded. NA = not applicable (sex phenotypes not recorded). Museum collection numbers are as follows: MNHN 2014–2719, MNHN 2014–2720, MNHN 2014–2721, MNHN 2014–2722, and MNHN 2014–2723 for E. cisalpinus and MNHN 2013–1246, MNHN 2013–1245, and MNHN 2013–838 for E. aquitanicus.
The number of markers retained correspond to the number of markers present with depth higher than min. depth in at least one individual.
Data availability
All gene sequences, genomic, Pool-seq and RAD-Seq reads were deposited under the common project number PRJNA634624.
The following dataset was generated:
Pan 2020. Sex determination in the Esociformes. NCBI BioProject. PRJNA634624
The following previously published dataset was used:
Rondeau EB, Minkley DR, Leong JS, Messmer AM, Jantzen JR, von Schalburg KR, Lemon C, Bird NH, Koop BF. 2014. Esox lucius isolate CL-BC-CA-002, whole genome shotgun sequencing project. NCBI Nucleotide. GCA_000721915.2
References
- Alonge M, Soyk S, Ramakrishnan S, Wang X, Goodwin S, Sedlazeck FJ, Lippman ZB, Schatz MC. RaGOO: fast and accurate reference-guided scaffolding of draft genomes. Genome Biology. 2019;20:224. doi: 10.1186/s13059-019-1829-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Research. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amores A, Catchen J, Ferrara A, Fontenot Q, Postlethwait JH. Genome evolution and meiotic maps by massively parallel DNA sequencing: spotted gar, an outgroup for the teleost genome duplication. Genetics. 2011;188:799–808. doi: 10.1534/genetics.111.127324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baroiller JF, D'Cotta H, Saillant E. Environmental effects on fish sex determination and differentiation. Sexual Development. 2009;3:118–135. doi: 10.1159/000223077. [DOI] [PubMed] [Google Scholar]
- Bej DK, Miyoshi K, Hattori RS, Strüssmann CA, Yamamoto Y. A duplicated, truncated amh gene is involved in male sex determination in an old world silverside. G3: Genes, Genomes, Genetics. 2017;117:042697. doi: 10.1534/g3.117.042697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beukeboom LW, Perrin N. The Evolution of Sex Determination. Oxford University Press; 2014. [Google Scholar]
- Blaser O, Grossen C, Neuenschwander S, Perrin N. Sex-chromosome turnovers induced by deleterious mutation load. Evolution. 2013;67:635–645. doi: 10.1111/j.1558-5646.2012.01810.x. [DOI] [PubMed] [Google Scholar]
- Bull JJ, Charnov EL. Changes in the heterogametic mechanism of sex determination. Heredity. 1977;39:1–14. doi: 10.1038/hdy.1977.38. [DOI] [PubMed] [Google Scholar]
- Campbell MA, López JA, Sado T, Miya M. Pike and salmon as sister taxa: detailed intraclade resolution and divergence time estimation of esociformes + salmoniformes based on whole mitochondrial genome sequences. Gene. 2013;530:57–65. doi: 10.1016/j.gene.2013.07.068. [DOI] [PubMed] [Google Scholar]
- Campbell MA, Lopéz JA. Mitochondrial phylogeography of a beringian relict: the endemic freshwater genus of blackfish Dallia (Esociformes): phylogeography of a bberingian endemic. Journal of Fish Biology. 2014;84:523–538. doi: 10.1111/jfb.12314. [DOI] [PubMed] [Google Scholar]
- Catchen J, Hohenlohe PA, Bassham S, Amores A, Cresko WA. Stacks: an analysis tool set for population genomics. Molecular Ecology. 2013;22:3124–3140. doi: 10.1111/mec.12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Zhang G, Shao C, Huang Q, Liu G, Zhang P, Song W, An N, Chalopin D, Volff JN, Hong Y, Li Q, Sha Z, Zhou H, Xie M, Yu Q, Liu Y, Xiang H, Wang N, Wu K, Yang C, Zhou Q, Liao X, Yang L, Hu Q, Zhang J, Meng L, Jin L, Tian Y, Lian J, Yang J, Miao G, Liu S, Liang Z, Yan F, Li Y, Sun B, Zhang H, Zhang J, Zhu Y, Du M, Zhao Y, Schartl M, Tang Q, Wang J. Whole-genome sequence of a flatfish provides insights into ZW sex chromosome evolution and adaptation to a benthic lifestyle. Nature Genetics. 2014;46:253–260. doi: 10.1038/ng.2890. [DOI] [PubMed] [Google Scholar]
- Dabrowski K, Rinchard J, Lin F, Garcia-Abiado M-A, Schmidt D. Induction of gynogenesis in muskellunge with irradiated sperm of yellow perch proves diploid muskellunge male homogamety. Journal of Experimental Zoology. 2000;287:96–105. doi: 10.1002/1097-010X(20000615)287:1<96::AID-JEZ13>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Denys GP, Dettai A, Persat H, Hautecœur M, Keith P. Morphological and molecular evidence of three species of pikes Esox spp. (Actinopterygii, Esocidae) in France, including the description of a new species. Comptes Rendus Biologies. 2014;337:521–534. doi: 10.1016/j.crvi.2014.07.002. [DOI] [PubMed] [Google Scholar]
- di Clemente N, Jamin SP, Lugovskoy A, Carmillo P, Ehrenfels C, Picard JY, Whitty A, Josso N, Pepinsky RB, Cate RL. Processing of anti-mullerian hormone regulates receptor activation by a mechanism distinct from TGF-beta. Molecular Endocrinology. 2010;24:2193–2206. doi: 10.1210/me.2010-0273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earl D, Bradnam K, St John J, Darling A, Lin D, Fass J, Yu HO, Buffalo V, Zerbino DR, Diekhans M, Nguyen N, Ariyaratne PN, Sung WK, Ning Z, Haimel M, Simpson JT, Fonseca NA, Birol İ, Docking TR, Ho IY, Rokhsar DS, Chikhi R, Lavenier D, Chapuis G, Naquin D, Maillet N, Schatz MC, Kelley DR, Phillippy AM, Koren S, Yang SP, Wu W, Chou WC, Srivastava A, Shaw TI, Ruby JG, Skewes-Cox P, Betegon M, Dimon MT, Solovyev V, Seledtsov I, Kosarev P, Vorobyev D, Ramirez-Gonzalez R, Leggett R, MacLean D, Xia F, Luo R, Li Z, Xie Y, Liu B, Gnerre S, MacCallum I, Przybylski D, Ribeiro FJ, Yin S, Sharpe T, Hall G, Kersey PJ, Durbin R, Jackman SD, Chapman JA, Huang X, DeRisi JL, Caccamo M, Li Y, Jaffe DB, Green RE, Haussler D, Korf I, Paten B. Assemblathon 1: a competitive assessment of de novo short read assembly methods. Genome Research. 2011;21:2224–2241. doi: 10.1101/gr.126599.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feron R, Pan Q, Wen M, Imarazene B, Jouanno E, Anderson J, Herpin A, Journot L, Parrinello H, Klopp C, Kottler VA, Roco AS, Du K, Kneitz S, Adolfi M, Wilson CA, McCluskey B, Amores A, Desvignes T, Goetz FW, Takanashi A, Kawaguchi M, Detrich HW, Oliveira M, Nobrega R, Sakamoto T, Nakamoto M, Wargelius A, Ø K, Wang Z, Stöck M, Waterhouse RM, Braasch I, Postlethwait JH, Schartl M, Guiguen Y. RADSex: a computational workflow to study sex determination using restriction Site-Associated DNA sequencing data. bioRxiv. 2020a doi: 10.1101/2020.04.22.054866. [DOI] [PMC free article] [PubMed]
- Feron R, Zahm M, Cabau C, Klopp C, Roques C, Bouchez O, Eché C, Valière S, Donnadieu C, Haffray P, Bestin A, Morvezen R, Acloque H, Euclide PT, Wen M, Jouano E, Schartl M, Postlethwait JH, Schraidt C, Christie MR, Larson WA, Herpin A, Guiguen Y. Characterization of a Y-specific duplication/insertion of the anti-Mullerian hormone type II receptor gene based on a chromosome-scale genome assembly of yellow perch, Perca flavescens. Molecular Ecology Resources. 2020b;20:531–543. doi: 10.1111/1755-0998.13133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J, Sonnhammer EL, Tate J, Punta M. Pfam: the protein families database. Nucleic Acids Research. 2014;42:D222–D230. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gammerdinger WJ, Conte MA, Sandkam BA, Ziegelbecker A, Koblmüller S, Kocher TD. Novel sex chromosomes in 3 cichlid fishes from lake tanganyika. Journal of Heredity. 2018;109:489–500. doi: 10.1093/jhered/esy003. [DOI] [PubMed] [Google Scholar]
- Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, Bairoch A. ExPASy: the proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Research. 2003;31:3784–3788. doi: 10.1093/nar/gkg563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guiguen Y, Bertho S, Herpin A. Sex Determination and Sex Control in Salmonidae. In: Fostier A, editor. Sex Control in Aquaculture. John Wiley & Sons; 2018. pp. 249–280. [DOI] [Google Scholar]
- Hardie DC, Hebert PDN. Genome-size evolution in fishes. Canadian Journal of Fisheries and Aquatic Sciences. 2004;61:1636–1646. doi: 10.1139/f04-106. [DOI] [Google Scholar]
- Hattori RS, Strüssmann CA, Fernandino JI, Somoza GM. Genotypic sex determination in teleosts: insights from the testis-determining amhy gene. General and Comparative Endocrinology. 2013;192:55–59. doi: 10.1016/j.ygcen.2013.03.019. [DOI] [PubMed] [Google Scholar]
- Herrera S, Reyes-Herrera PH, Shank TM. Predicting RAD-seq marker numbers across the eukaryotic tree of life. Genome Biology and Evolution. 2015;7:3207–3225. doi: 10.1093/gbe/evv210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes AL. The evolution of functionally novel proteins after gene duplication. Proceedings. Biological Sciences. 1994;256:119–124. doi: 10.1098/rspb.1994.0058. [DOI] [PubMed] [Google Scholar]
- Jeffries DL, Lavanchy G, Sermier R, Sredl MJ, Miura I, Borzée A, Barrow LN, Canestrelli D, Crochet PA, Dufresnes C, Fu J, Ma WJ, Garcia CM, Ghali K, Nicieza AG, O'Donnell RP, Rodrigues N, Romano A, Martínez-Solano Í, Stepanyan I, Zumbach S, Brelsford A, Perrin N. A rapid rate of sex-chromosome turnover and non-random transitions in true frogs. Nature Communications. 2018;9:4088. doi: 10.1038/s41467-018-06517-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiya T, Kai W, Tasumi S, Oka A, Matsunaga T, Mizuno N, Fujita M, Suetake H, Suzuki S, Hosoya S, Tohari S, Brenner S, Miyadai T, Venkatesh B, Suzuki Y, Kikuchi K. A trans-species missense SNP in Amhr2 is associated with sex determination in the tiger pufferfish, Takifugu rubripes (fugu) PLOS Genetics. 2012;8:e1002798. doi: 10.1371/journal.pgen.1002798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molecular Biology and Evolution. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi K, Hamaguchi S. Novel sex-determining genes in fish and sex chromosome evolution: novel Sex-Determining genes in fish. Developmental Dynamics. 2013;242:339–353. doi: 10.1002/dvdy.23927. [DOI] [PubMed] [Google Scholar]
- Kolmogorov M, Yuan J, Lin Y, Pevzner PA. Assembly of long, error-prone reads using repeat graphs. Nature Biotechnology. 2019;37:540–546. doi: 10.1038/s41587-019-0072-8. [DOI] [PubMed] [Google Scholar]
- Kozielska M, Feldmeyer B, Pen I, Weissing FJ, Beukeboom LW. Are autosomal sex-determining factors of the housefly (Musca Domestica) spreading north? Genetics Research. 2008;90:157–165. doi: 10.1017/S001667230700907X. [DOI] [PubMed] [Google Scholar]
- Kumar S, Stecher G, Suleski M, Hedges SB. TimeTree: a resource for timelines, timetrees, and divergence times. Molecular Biology and Evolution. 2017;34:1812–1819. doi: 10.1093/molbev/msx116. [DOI] [PubMed] [Google Scholar]
- Lartillot N, Brinkmann H, Philippe H. Suppression of long-branch attraction artefacts in the animal phylogeny using a site-heterogeneous model. BMC Evolutionary Biology. 2007;7:S4. doi: 10.1186/1471-2148-7-S1-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lartillot N, Philippe H. A bayesian mixture model for across-site heterogeneities in the amino-acid replacement process. Molecular Biology and Evolution. 2004;21:1095–1109. doi: 10.1093/molbev/msh112. [DOI] [PubMed] [Google Scholar]
- Lartillot N, Philippe H. Computing bayes factors using thermodynamic integration. Systematic Biology. 2006;55:195–207. doi: 10.1080/10635150500433722. [DOI] [PubMed] [Google Scholar]
- Lemey P, Sameli M, Vandamme A. The Phylogenetic Handbook: A Practical Approach to Phylogenetic Analysis and Hypothesis Testing. Cambridge University Press; 2009. [DOI] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup The sequence alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Sun Y, Zhao J, Shi H, Zeng S, Ye K, Jiang D, Zhou L, Sun L, Tao W, Nagahama Y, Kocher TD, Wang D. A tandem duplicate of Anti-Müllerian hormone with a missense SNP on the Y chromosome is essential for male sex determination in nile Tilapia, Oreochromis niloticus. PLOS Genetics. 2015;11:e1005678. doi: 10.1371/journal.pgen.1005678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34:3094–3100. doi: 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marçais G, Kingsford C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 2011;27:764–770. doi: 10.1093/bioinformatics/btr011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall Graves JA. Weird animal genomes and the evolution of vertebrate sex and sex chromosomes. Annual Review of Genetics. 2008;42:565–586. doi: 10.1146/annurev.genet.42.110807.091714. [DOI] [PubMed] [Google Scholar]
- Marshall Graves JA, Peichel CL. Are homologies in vertebrate sex determination due to shared ancestry or to limited options? Genome Biology. 2010;11:205. doi: 10.1186/gb-2010-11-4-205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, von Haeseler A, Lanfear R. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Molecular Biology and Evolution. 2020;37:1530–1534. doi: 10.1093/molbev/msaa015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore AD, Bornberg-Bauer E. The dynamics and evolutionary potential of domain loss and emergence. Molecular Biology and Evolution. 2012;29:787–796. doi: 10.1093/molbev/msr250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muralidhar P, Veller C. Sexual antagonism and the instability of environmental sex determination. Nature Ecology & Evolution. 2018;2:343–351. doi: 10.1038/s41559-017-0427-9. [DOI] [PubMed] [Google Scholar]
- Myosho T, Otake H, Masuyama H, Matsuda M, Kuroki Y, Fujiyama A, Naruse K, Hamaguchi S, Sakaizumi M. Tracing the emergence of a novel sex-determining gene in medaka, Oryzias luzonensis. Genetics. 2012;191:163–170. doi: 10.1534/genetics.111.137497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myosho T, Takehana Y, Hamaguchi S, Sakaizumi M. Turnover of sex chromosomes in Celebensis Group Medaka Fishes. G3: Genes, Genomes, Genetics. 2015;5:2685–2691. doi: 10.1534/g3.115.021543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navara KJ. Mechanisms of Environmental Sex Determination in Fish, Amphibians, and Reptiles. In: Navara K. J, editor. Choosing Sexes: Mechanisms and Adaptive Patterns of Sex Allocation in Vertebrates, Fascinating Life Sciences. Springer International Publishing; 2018. pp. 213–240. [DOI] [Google Scholar]
- Nielsen H. Predicting Secretory Proteins with SignalP. InProtein Function Prediction. Humana Press; 2017. [DOI] [PubMed] [Google Scholar]
- Ouellet-Cauchon G, Normandeau E, Mingelbier M, Bernatchez L. EST-based microsatellites for northern pike (Esox lucius) and cross-amplification across all Esox species. Conservation Genetics Resources. 2014;6:451–454. doi: 10.1007/s12686-013-0123-2. [DOI] [Google Scholar]
- Pan Q, Guiguen Y, Herpin A. Evolution of Sex Determining Genes in Fish Encyclopedia of Reproduction. Laboratoire de Physiologie et Génomique des Poissons; 2017. [Google Scholar]
- Pan Q, Feron R, Yano A, Guyomard R, Jouanno E, Vigouroux E, Wen M, Busnel JM, Bobe J, Concordet JP, Parrinello H, Journot L, Klopp C, Lluch J, Roques C, Postlethwait J, Schartl M, Herpin A, Guiguen Y. Identification of the master sex determining gene in northern pike (Esox lucius) reveals restricted sex chromosome differentiation. PLOS Genetics. 2019;15:e1008013. doi: 10.1371/journal.pgen.1008013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquier J, Cabau C, Nguyen T, Jouanno E, Severac D, Braasch I, Journot L, Pontarotti P, Klopp C, Postlethwait JH, Guiguen Y, Bobe J. Gene evolution and gene expression after whole genome duplication in fish: the PhyloFish database. BMC Genomics. 2016;17:368. doi: 10.1186/s12864-016-2709-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafati N, Chen J, Herpin A, Pettersson ME, Han F, Feng C, Wallerman O, Rubin CJ, Péron S, Cocco A, Larsson M, Trötschel C, Poetsch A, Korsching K, Bönigk W, Körschen HG, Berg F, Folkvord A, Kaupp UB, Schartl M, Andersson L. Reconstruction of the birth of a male sex chromosome present in Atlantic herring. PNAS. 2020;117:24359–24368. doi: 10.1073/pnas.2009925117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, Endo T, Yamahira K, Hamaguchi S, Sakaizumi M. Induction of female-to-male sex reversal by high temperature treatment in medaka, Oryzias latipes. Zoological Science. 2005;22:985–988. doi: 10.2108/zsj.22.985. [DOI] [PubMed] [Google Scholar]
- Sela I, Ashkenazy H, Katoh K, Pupko T. GUIDANCE2: accurate detection of unreliable alignment regions accounting for the uncertainty of multiple parameters. Nucleic Acids Research. 2015;43:W7–W14. doi: 10.1093/nar/gkv318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31:3210–3212. doi: 10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
- Skog A, Vøllestad LA, Stenseth NC, Kasumyan A, Jakobsen KS. Circumpolar phylogeography of the northern pike (Esox lucius) and its relationship to the amur pike (E. reichertii) Frontiers in Zoology. 2014;11:67. doi: 10.1186/s12983-014-0067-8. [DOI] [Google Scholar]
- Solovyev V, Kosarev P, Seledsov I, Vorobyev D. Automatic annotation of eukaryotic genes, pseudogenes and promoters. Genome Biology. 2006;7:S10. doi: 10.1186/gb-2006-7-s1-s10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tree of Sex Consortium. Bachtrog D, Mank JE, Peichel CL, Kirkpatrick M, Otto SP, Ashman TL, Hahn MW, Kitano J, Mayrose I, Ming R, Perrin N, Ross L, Valenzuela N, Vamosi JC. Sex determination: why so many ways of doing it? PLOS Biology. 2014;12:e1001899. doi: 10.1371/journal.pbio.1001899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnquist KN, Larson WA, Farrell JM, Hanchin PA, Kapuscinski KL, Miller LM, Scribner KT, Wilson CC, Sloss BL. Genetic structure of muskellunge in the great lakes region and the effects of supplementation on genetic integrity of wild populations. Journal of Great Lakes Research. 2017;43:1141–1152. doi: 10.1016/j.jglr.2017.09.005. [DOI] [Google Scholar]
- van Doorn GS. Evolutionary Transitions between Sex-Determining Mechanisms: A Review of Theory. Sexual Development. 2014;8:7–19. doi: 10.1159/000357023. [DOI] [PubMed] [Google Scholar]
- van Doorn GS, Kirkpatrick M. Turnover of sex chromosomes induced by sexual conflict. Nature. 2007;449:909–912. doi: 10.1038/nature06178. [DOI] [PubMed] [Google Scholar]
- Vaser R, Sović I, Nagarajan N, Šikić M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Research. 2017;27:737–746. doi: 10.1101/gr.214270.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicoso B. Molecular and evolutionary dynamics of animal sex-chromosome turnover. Nature Ecology & Evolution. 2019;3:1632–1641. doi: 10.1038/s41559-019-1050-8. [DOI] [PubMed] [Google Scholar]
- Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, Cuomo CA, Zeng Q, Wortman J, Young SK, Earl AM. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLOS ONE. 2014;9:e112963. doi: 10.1371/journal.pone.0112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren ML, Burr BM, Tomelleri JR, Echelle AA, Kuhajda BR. Freshwater Fishes of North America: Volume 2: Characidae to Poeciliidae. Johns Hopkins University Press; 2020. [Google Scholar]
- Wilson CA, High SK, McCluskey BM, Amores A, Yan YL, Titus TA, Anderson JL, Batzel P, Carvan MJ, Schartl M, Postlethwait JH. Wild sex in zebrafish: loss of the natural sex determinant in domesticated strains. Genetics. 2014;198:1291–1308. doi: 10.1534/genetics.114.169284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wooller MJ, Gaglioti B, Fulton TL, Lopez A, Shapiro B. Post-glacial dispersal patterns of northern pike inferred from an 8800 year old pike (Esox cf lucius) skull from interior alaska. Quaternary Science Reviews. 2015;120:118–125. doi: 10.1016/j.quascirev.2015.04.027. [DOI] [Google Scholar]
- Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Molecular Biology and Evolution. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- Yano A, Guyomard R, Nicol B, Jouanno E, Quillet E, Klopp C, Cabau C, Bouchez O, Fostier A, Guiguen Y. An immune-related gene evolved into the master sex-determining gene in rainbow trout, Oncorhynchus mykiss. Current Biology. 2012;22:1423–1428. doi: 10.1016/j.cub.2012.05.045. [DOI] [PubMed] [Google Scholar]