

Abstract
Hepatocellular carcinoma (HCC) is a common primary liver cancer with a high incidence and mortality. This study was conducted to identify a long non-coding RNA (lncRNA) signature that may serve as a predictor for HCC prognosis.
RNA-seq data were extracted from The Cancer Genome Atlas database. Differentially expressed genes, lncRNAs, and miRNAs were identified in HCC (n = 374) and control samples (n = 50) and used to screen prognosis-associated lncRNA signatures. The association of the lncRNA signature with HCC prognosis was analyzed and a competitive endogenous RNA regulatory network involving the lncRNA signature was constructed.
A total of 199 mRNAs, 1092 lncRNAs, and 251 miRNAs were differentially expressed between HCC and control samples. Among these lncRNAs, 11 prognosis-associated lncRNAs were used to construct a lncRNA signature. Cox regression analysis showed that patients with higher risk scores of the lncRNA signature were at risk of poor prognosis. Four lncRNAs (including LINC01517, DDX11-AS1, LINC01136, and RP11-20J15.2) and 7 miRNAs (including miR-195, miR-199b, miR-326, miR-424, and let-7c) in the ceRNA network interacted with the upregulated gene E2F2, which was associated with the overall prognosis of patients with HCC.
The 11-lncRNA signature might be useful for predicting the prognosis of patients with HCC.
Keywords: bioinformatics, competitive endogenous RNA, enrichment analysis, hepatocellular carcinoma, risk score
1. Introduction
As the most frequent histologic subtype of primary liver cancer, hepatocellular carcinoma (HCC) is derived from the epithelial or mesenchymal tissues of the liver.[1] HCC is usually induced by virus infection, smoking, aflatoxin, alcohol, obesity, or diabetes.[2] HCC has a high incidence and mortality and leads to approximately 662,000 deaths annually worldwide.[3] Liver transplantation is the most efficient method for treating HCC, but over 70% of patients with advanced HCC are not suitable for transplantation.[4] The tumor recurrence and metastasis rates are higher after transplantation, which results in poor prognosis in patients with HCC.[5] Therefore, prognostic factors for HCC should be explored to improve its therapy.
The prognosis and therapy response of HCC are related to various factors. As reported, the long non-coding RNA (lncRNA) SOX21 antisense divergent transcript 1 (SOX21-AS1) is upregulated in HCC samples, and this upregulation affects tumor progression by silencing p21.[6] The lncRNA TCONS_00027978 has positive correlations with histologic grade, lymph node metastasis, and tumor node metastasis (TNM) stage in patients with HCC, and its low expression serves as an independent adverse prognostic factor for this tumor.[7] Through the miR-128/CD151 pathway, lncRNA small nucleolar RNA host gene 3 (SNHG3) contributes to sorafenib resistance and epithelial-mesenchymal transition (EMT) in patients with HCC.[8] Besides, Ge et al[9] showed that the expression level of the lncRNA HOXA distal transcript antisense RNA (HOTTIP) had a negative correlation with that of miR-192 and miR-204 in HCC tissue specimens, and the miR-192/-204-HOTTIP axis was associated with the prognosis of HCC patients. Gu et al[10] identified lncRNAs significantly related to recurrence-free survival and constructed a 6-lncRNA signature for HCC, which can be applied for accurate diagnosis and targeted therapy of HCC. Sui et al[11] screened the overall survival-associated lncRNAs in HCC and established a 4-lncRNA signature that provided a novel biomarker for prognostic prediction of patients with HCC. These studies have revealed and highlighted the prognostic association of lncRNAs, miRNAs, and genes in HCC. As more changed genes in response to treatments are identified, the molecular mechanism of HCC prognosis will become clearer.
This study aimed to construct a lncRNA signature with a high accuracy in predicting HCC prognosis by using comprehensive analytical approaches. In this study, a lncRNA signature was established and the competitive endogenous RNA (ceRNA) regulatory network involving the above lncRNAs was conducted to reveal the regulatory axes and underlying functions of the key lncRNAs.
2. Materials and methods
2.1. Ethical approval
This study was not conducted on human participants or animals by any of the authors, and ethical approval is not applicable.
2.2. Data source
The RNA-seq data of liver hepatocellular carcinoma (LIHC) (TCGA-LIHC, downloaded on October 18, 2019) and relevant clinical data were extracted from The Cancer Genome Atlas (TCGA, https://cancergenome.nih.gov/) database. There were a total of 424 samples in TCGA, including 374 HCC samples and 50 control samples.
2.3. Differential expression analysis
RNAs with an average expression <1 were excluded. Next, differential expression analysis between HCC samples and control samples was conducted using the edgeR package (http://bioconductor.org/packages/release/bioc/html/edgeR.html)[12] in R. The |log2 fold change (FC)| ≥2 and P-value <.05 were used for screening differentially expressed lncRNAs (DE-lncRNAs) and differentially expressed mRNAs (DE-mRNAs). Meanwhile, P-values <.05 and |log2 FC| ≥1 were defined for selecting differentially expressed miRNAs (DE-miRNAs).
2.4. Identification of the lncRNA signature correlated with prognosis
After collecting the information of patients with HCC with both transcriptome data and clinical prognosis information, 342 samples were selected for subsequent analysis. Next, these samples were randomly divided into the training set and validation set. From the training set, the DE-lncRNAs significantly related to prognosis were analyzed via univariate Cox regression analysis.[13] The threshold was set at a log-rank P-value <.05. Combined with lasso regression analysis,[14] the lncRNA signature correlating with the prognosis of patients with HCC was further screened. Based on the lncRNA signature, the risk score of each sample was calculated. According to the values of the risk scores, the samples in the training set were classified into high-risk (the top 50%) and low-risk (the bottom 50%) groups. Using Kaplan–Meier (KM) survival analysis[15] and receiver operating characteristic (ROC) curve analysis,[16] the prognostic difference between high- and low-risk groups and the predicting power of the lncRNA signature for prognosis were calculated. Moreover, the performance of the lncRNA signature in predicting the outcome of HCC was validated in the validation set.
2.5. Identification of independent clinical prognostic factors
Using univariate Cox regression analysis,[13] the correlations of risk scores and clinical factors (including age, sex, clinical stage, tumor grade, T stage, N stage, and M stage) with prognosis were analyzed. Based on multivariate Cox regression analysis,[13] the independent clinical prognostic factors were further selected. A log-rank P-value <.05 was considered as the cutoff.
2.6. CeRNA regulatory network and enrichment analysis
The Pearson correlation coefficients of the DE-mRNAs and lncRNAs involved in the lncRNA signature were calculated. The values P < .05 and r > 0.4 were defined as the thresholds for screening lncRNA-mRNA coexpression pairs.
The miRNA-lncRNA regulatory pairs in the lncRNA signature were identified using the DIANA-LncBase v2 database (http://carolina.imis.athena-innovation.gr/diana_tools/web/index.php?r=lncbasev2%2Findex-experimental).
Based on the StarBase database (version 3.0, http://starbase.sysu.edu.cn/), the regulatory relationships between the miRNAs in the miRNA-lncRNA regulatory pairs and the mRNAs in the lncRNA-mRNA coexpression pairs were analyzed to identify miRNA-mRNA regulatory pairs.
After integrating the lncRNA-mRNA pairs, miRNA-lncRNA pairs, and miRNA-mRNA pairs, the ceRNA regulatory network was constructed using Cytoscape software (http://www.cytoscape.org/). The Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Ontology (GO) analyses for the mRNAs implicated in the ceRNA regulatory network were conducted using the clusterProfiler package (http://www.bioconductor.org/packages/release/bioc/html/clusterProfiler.html) in R. A false discovery rate <.05 was considered as the threshold for significant enrichment.
2.7. Validation of prognostic association of mRNAs in the ceRNA network
To validate the association of key mRNAs in the final ceRNA network, the correlation of mRNA with overall survival of patients with LIHC was analyzed using the GEPIA online tool (http://gepia.cancer-pku.cn/). A significant correlation was defined as a log-rank P value <.05.
3. Results
3.1. Differential expression analysis
There were 1999 genes (1794 upregulated and 205 downregulated), 1092 lncRNAs (1034 upregulated and 58 downregulated), and 251 miRNAs (229 upregulated and 22 downregulated) that were significantly differentially expressed between the HCC (n = 374) and control samples (n = 50). Volcano plots of the DE-mRNAs, DE-lncRNAs, and DE-miRNAs are shown in Fig. 1.
Figure 1.

Volcano plots showing the results of differential expression analysis: (A) volcano plot of the differentially expressed mRNAs; (B) volcano plot of the differentially expressed lncRNAs; (C) volcano plot of the differentially expressed miRNAs. Red, blue, and gray dots represent upregulated RNAs, downregulated RNAs, and non-differentially expressed RNAs, respectively. FC = fold change.
3.2. Identification of the lncRNA signature correlated with prognosis
Of the 1092 DE-lncRNAs, 210 lncRNAs were significantly related to the prognosis of patients with HCC in the training set. Lasso regression analysis showed that the prognosis-associated lncRNA signature consisted of 11 lncRNAs, including long intergenic non-protein coding RNA 1060 (LINC01060), long intergenic non-protein coding RNA 1136 (LINC01136), eglnine homolog 3 (EGLN3) antisense RNA 1 (EGLN3-AS1), RP11-20J15.2, AC025580.1, Homeobox C (HOXC) antisense RNA 2 (HOXC-AS2), AC114912.1, long intergenic non-protein coding RNA 1517 (LINC01517), AL592043.1, AC089983.1, and DEAD/H box protein 11 (DDX11) antisense RNA 1 (DDX11-AS1). Based on the prognostic coefficients and expression levels of the 11 lncRNAs, the risk scores of samples were calculated as follows: Risk score = 0.026666 × LINC01060 + 0.008425 × LINC01136 + 0.041532 × EGLN3-AS1 + 0.0238 × RP11-20J15.2 + 0.027578 × AC025580.1 + 0.017409 × HOXC-AS2 + 0.054546 × AC114912.1 + 0.017601 × LINC01517 + 0.066435 × AL592043.1 + 0.024386 × AC089983.1 + 0.027224 × DDX11-AS1.
Furthermore, the prognostic difference between the high- and low-risk groups was evaluated using KM and ROC curves. For both the training set (P < .001, the area under the ROC curve [AUC] = 0.896) and the validation set (P < .001, AUC = 0.702), the survival rate of the HCC patients in the high-risk group was lower than patients in the low-risk group (Fig. 2A and B).
Figure 2.

Kaplan–Meier (KM) survival curves and receiver operating characteristic (ROC) curves showing the prognostic difference between high- and low-risk groups divided by the risk scores: (A) KM survival curves and ROC curve for the training set; (B) KM survival curves and ROC curve for the validation set. AUC = area under the receiver operating characteristic curve.
3.3. Identification of independent clinical prognostic factors
The results of univariate Cox regression analysis indicated that the risk score, clinical stage, T stage, and M stage correlated with the prognosis of patients with HCC (Fig. 3A). Multivariate Cox regression analysis suggested that only the risk score was an independent factor for the prognosis of patients with HCC (Fig. 3B).
Figure 3.
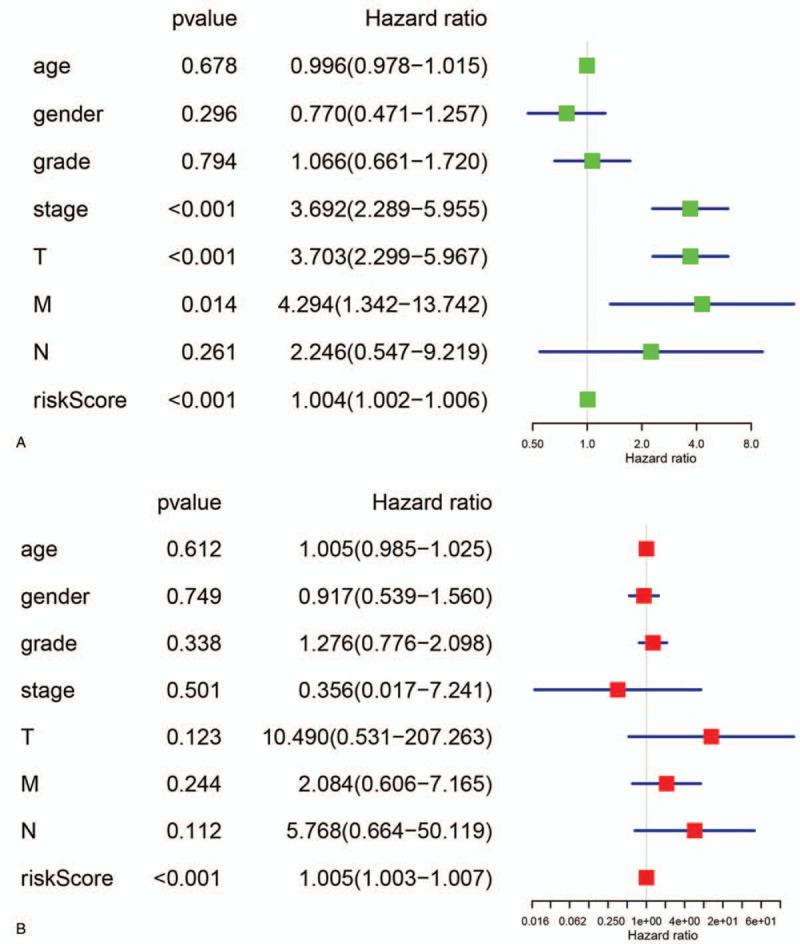
Screening of independent clinical prognostic factors: (A) result of univariate Cox regression analysis; (B) result of multivariate Cox regression analysis.
3.4. CeRNA regulatory network and enrichment analysis
Based on Pearson correlation analysis, 218 lncRNA-mRNA coexpression pairs (involving 11 lncRNAs and 216 mRNAs) were screened out. Combined with the DIANA-LncBase v2 database, 26 miRNA-lncRNA regulatory pairs (involving 10 miRNAs and 6 lncRNAs) were selected. Using the StarBase database, 175 miRNA-mRNA regulatory pairs (involving 7 miRNAs and 85 mRNAs) were obtained. In the ceRNA regulatory network, there were 7 miRNAs, 4 lncRNAs (LINC01517, DDX11-AS1, LINC01136, and RP11-20J15.2), and 85 mRNAs (Fig. 4). In particular, the upregulated gene E2F transcription factor 2 (E2F2) was targeted by 5 miRNAs (let-7c, miR-195, miR-199b, miR-326, and miR-424), which interacted with 4 lncRNAs (LINC01517, DDX11-AS1, LINC01136, and RP11-20J15.2) in the ceRNA regulatory network.
Figure 4.

The competitive endogenous RNA (ceRNA) regulatory network. Diamonds, squares, and circles separately represent long non-coding RNAs (lncRNAs), miRNAs, and mRNAs, respectively. Red and blue note represent upregulation and downregulation, respectively.
GO and KEGG enrichment analyses were performed for the 85 mRNAs in the ceRNA regulatory network. Enrichment analysis showed that these genes were involved in GO biological processes, including chromosome segregation, nuclear division, and DNA packaging, and GO cellular components, including chromosomal region, spindle microtubule, and kinetochore, as well as GO molecular functions, including DNA-dependent ATPase activity, chromatin binding, and tubulin binding (Fig. 5). In addition, these genes, including E2F2, were associated with KEGG pathways, including cell cycle, microRNAs in cancer, non-small-cell lung cancer, and p53 signaling pathway, and are shown in Fig. 6.
Figure 5.

Bubble diagram showing the top 10 Gene Ontology (GO) biological process, GO cellular component, and GO molecular function terms enriched for the mRNAs in the competitive endogenous RNA (ceRNA) regulatory network. The change from red to blue indicates that the adjusted P-value goes from small to large.
Figure 6.

Bubble diagram showing the top 10 Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways enriched for the mRNAs in the competitive endogenous RNA (ceRNA) regulatory network. The change from red to blue indicates that the adjusted P-value goes from small to large.
3.5. Association of E2F2 with overall survival of patients with HCC
Based on TCGA data, we found that patients with HCC with higher expression levels of E2F2 had lower survival percentages compared with patients with lower expression levels (Fig. 7). This result showed that the ceRNA interaction pairs involving E2F2 are of great value for predicting the prognosis of patients with HCC.
Figure 7.

The correlation of E2F2 expression with the overall survival of patients with HCC. Overall survival of patients with LIHC was analyzed using the GEPIA online tool (http://gepia.cancer-pku.cn/). HCC = hepatocellular carcinoma; LIHC = liver hepatocellular carcinoma.
4. Discussion
This study identified that 1999 mRNAs, 1092 lncRNAs, and 251 miRNAs were significantly differentially expressed between HCC and control samples. Integrated bioinformatics analysis showed that the signature of 11 lncRNAs (LINC01060, LINC01136, EGLN3-AS1, RP11-20J15.2, AC025580.1, HOXC-AS2, AC114912.1, LINC01517, AL592043.1, AC089983.1, and DDX11-AS1) was associated with the prognosis of patients with HCC, and may serve as a predictor of prognosis in patients with HCC.
The association of miR-195 with the prognosis, clinical outcome, and recurrence of human cancers has been reported previously.[17–19] It produces antitumor effects by suppressing the migration, invasion, and chemoresistance of cancers.[19,20] The miR-195 is regulated by oncogenic lncRNAs, including plasmacytoma variant translocation 1 (PVT1) and SNHG1. LncRNA DDX11-AS1 has been identified as an oncogenic lncRNA in colorectal cancer.[21,22] Li et al[21] showed that DDX11-AS1 exhibited oncogenic effects by promoting the growth and invasion of HCC cells. DDX11-AS1 is associated with the progression and prognosis of HCC and may be considered a possible target for the treatment of this tumor.[23]DDX11-AS1 inhibits the large tumor suppressor kinase 2 (LATS2) gene.[21] Our present study indicated the potential role of the DDX11-AS1-miR-195-E2F2 axis in HCC development, and its possible involvement in the prognosis of patients with HCC.
Of the miRNAs that have been reported to be associated with HCC development and prognosis, miR-490-3p and miR-490-5p possess antitumor effects.[24,25]MiR-490-3p expression is reduced in HCC, and its targets correlate with the pathways in cancer and possess tumoral functions.[25–27] Decreased let-7c in HCC correlates with poor tissue differentiation, indicating that let-7c may mediate HCC cell differentiation.[28] Our present study identified the upregulated RP11-20J15.2-miR-490/let-7c-E2F2 ceRNA mechanism in HCC. The lncRNA RP11 consists of a cluster of lncRNAs that have been identified as oncogenes in human cancers.[29,30] The identification of RP11-20J15.2-miR-
490/let-7c-E2F2 ceRNA in HCC indicated its potential role in HCC development and outcome.
This study identified a cluster of miRNAs differentially expressed in HCC compared with controls, including miR-195, miR-199b, miR-326, and miR-424. MiR-195 expression was reduced in HCC samples, and miR-195 can exert anti-cancer effects by repressing metadherin (AEG-1).[31]MiR-199b-5p downregulation and N-cadherin upregulation were detected in HCC samples, and miR-199b-5p plays an inhibitory role in EMT and regulates N-cadherin expression in HCC.[32]MiR-326 inhibits the progression of HCC by mediating LIM and SH3 protein 1, and therefore miR-326 may serve as a target for the therapy of patients with HCC.[33] The downregulation of serum miR-424 in patients with HCC was correlated with TNM stage, serum alpha-fetoprotein, and vein invasion, which may be a promising marker for predicting an unfavorable prognosis for HCC.[33] These results indicated that downregulated miR-195, miR-199b, miR-326, and miR-424 and their ceRNA mechanisms might play important roles in the prognosis of HCC.
As indicated in the results, the upregulated E2F2 was involved in several key ceRNAs, including LINC01517–miR-195/miR-199b/miR-326/miR-424E2F2424-E2F2, DDX11-AS1 miR-195/miR-424-E2F2, LINC01136miR-326-E2F2, and RP11-20J15.2-miR-490/let-7c-E2F2 regulatory axes. E2Fs control the cell cycle, and upregulated E2F2 promotes cell proliferation by interacting with p53, Sp1, and cyclin A; its downregulation induces S phase arrest and cell apoptosis.[34,35] High expression of E2F transcription factors is substantially correlated with a poor prognosis for patients with HCC.[36,37] By inhibiting the expression of E2F2 and epithelial cell transforming 2, miR-490-5p overexpression suppresses cell metastasis in HCC.[37] The enrichment analysis in this study revealed that mRNAs, including E2F2, in the ceRNA regulatory networks were implicated in microRNAs in cancer and the p53 signaling pathway. Through the p53-dependent signaling pathway, E2F2 functions in HCC cell metastasis and apoptosis and may be a prognosis-associated biomarker in patients with HCC.[36,37] The present study also showed that higher expression levels of E2F2 in HCC correlated with a poor survival rate. This indicated that LINC01517, DDX11-AS1, LINC01136, and RP11-20J15.2 might also function in the prognosis of patients with HCC by regulating E2F2 and microRNAs in cancer and the p53 signaling pathway.
5. Conclusions
In conclusion, this study confirmed that the signature of 11 lncRNAs might be applied for predicting the prognosis of HCC. These lncRNAs contribute to the development and progression of HCC by regulating miRNAs, including let-7c, miR-195, miR-199b, miR-326, and miR-424, and E2F2, which are associated with the prognosis of patients with HCC. This study provides additional information to clarify the molecular mechanism behind HCC prognosis. The application of this lncRNA signature may improve the accuracy of clinical prognosis assessment for patients with HCC. However, this prognostic 11-lncRNA signature should be validated by experimental studies.
Author contributions
Conceptualization: Anmei Wang, Junhua Lei.
Data curation: Anmei Wang, Junhua Lei.
Formal analysis: Anmei Wang, Junhua Lei.
Investigation: Anmei Wang, Junhua Lei.
Methodology: Anmei Wang.
Resources: Anmei Wang, Junhua Lei.
Software: Anmei Wang, Junhua Lei.
Writing – original draft: Anmei Wang.
Writing – review & editing: Junhua Lei.
Footnotes
Abbreviations: AS = antisense RNA, AUC = area under the ROC curve, ceRNA = competitive endogenous RNA, DDX11 = DEAD/H box protein 11, DE-lncRNAs = differentially expressed lncRNAs, DE-miRNAs = differentially expressed miRNAs, DE-mRNAs = differentially expressed mRNAs, EGLN3 = eglnine homolog 3, EMT = epithelial-mesenchymal transition, FC = fold change, GO = Gene Ontology, HCC = hepatocellular carcinoma, HOTTIP = HOXA distal transcript antisense RNA, HOXC = Homeobox C, KEGG = Kyoto Encyclopedia of Genes and Genomes, KM = Kaplan–Meier, LATS2 = large tumor suppressor kinase 2, LIHC = liver hepatocellular carcinoma, LINC = long intergenic non-protein coding RNA, lncRNA = long non-coding RNA, PVT1 = plasmacytoma variant translocation 1, ROC = receiver operating characteristic, SNHG3 = small nucleolar RNA host gene 3, SOX21-AS1 = SOX21 antisense divergent transcript 1, TCGA = The Cancer Genome Atlas, TNM = tumor node metastasis.
How to cite this article: Wang A, Lei J. Identification of an 11-lncRNA signature with high performance for predicting the prognosis of hepatocellular carcinoma using bioinformatics analysis. Medicine. 2021;100:5(e23749).
The authors declare that they have no competing interests.
Availability of data and material: All data generated or analyzed during this study are included in this published article. The original microarray data are available at The Cancer Genome Atlas (TCGA, https://cancergenome.nih.gov/).
No funding was obtained for this study.
The authors have no conflicts of interest to disclose.
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
The datasets generated during and/or analyzed during the current study are publicly available.
References
- [1].Roth GS, Decaens T. Liver immunotolerance and hepatocellular carcinoma: patho-physiological mechanisms and therapeutic perspectives. Eur J Cancer 2017;87:101–12. [DOI] [PubMed] [Google Scholar]
- [2].Budny A, Kozlowski P, Kaminska M, et al. Epidemiology and risk factors of hepatocellular carcinoma. Pol Merkur Lekarski 2017;43:133–9. [PubMed] [Google Scholar]
- [3].Mcguire S. World Cancer Report 2014. Geneva, Switzerland: World Health Organization, International Agency for Research on Cancer, WHO Press, 2015. Adv Nutr 2016;7:418–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Wang J, Li H, Xia C, et al. Downregulation of CENPK suppresses hepatocellular carcinoma malignant progression through regulating YAP1. Onco Targets Ther 2019;12:869–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Alshahrani AA, Ha SM, Hwang S, et al. Clinical features and surveillance of very late hepatocellular carcinoma recurrence after liver transplantation. Ann Transplant 2018;23:659–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Wei C, Wang H, Xu F, et al. LncRNA SOX21-AS1 is associated with progression of hepatocellular carcinoma and predicts prognosis through epigenetically silencing p21. Biomed Pharmacother 2018;104:137–44. [DOI] [PubMed] [Google Scholar]
- [7].Chen Q, Tian GD, Wang CC. Expression of lncRNA TCONS_00027978 in hepatocellular carcinoma and its influence on prognosis and survival. Eur Rev Med Pharmacol Sci 2017;21:5655–60. [DOI] [PubMed] [Google Scholar]
- [8].Zhang PF, Wang F, Wu J, et al. LncRNA SNHG3 induces EMT and sorafenib resistance by modulating the miR-128/CD151 pathway in hepatocellular carcinoma. J Cell Physiol 2019;234:2788–94. [DOI] [PubMed] [Google Scholar]
- [9].Ge Y, Xiaodan Y, Yiguang J, et al. fMiRNA-192 and miRNA-204 directly suppress lncRNA HOTTIP and interrupt GLS1-mediated glutaminolysis in hepatocellular carcinoma. PLoS Genet 2016;11:e1005726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Gu JX, Zhang X, Miao RC, et al. Six-long non-coding RNA signature predicts recurrence-free survival in hepatocellular carcinoma. World J Gastroenterol 2019;25:220–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Sui J, Miao Y, Han J, et al. Systematic analyses of a novel lncRNA-associated signature as the prognostic biomarker for Hepatocellular Carcinoma. Cancer Med 2018;7:3240–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Robinson MD, Mccarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Biogeosciences 2010;26:139–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Whitehead J. Fitting Cox's regression model to survival data using glim. J R Stat Soc 2018;29:268–75. [Google Scholar]
- [14].Robert FH, Amos CI. Gene set selection via LASSO penalized regression (SLPR). Nucleic Acids Res 2017;45:e114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Lacny S, Wilson T, Clement F, et al. Kaplan-Meier survival analysis overestimates cumulative incidence of health-related events in competing risk settings: a meta-analysis. J Clin Epidemiol 2018;93:25–35. [DOI] [PubMed] [Google Scholar]
- [16].Krupinski EA. Receiver operating characteristic (ROC) analysis. Frontline Learn Res 2017;5:31–42. [Google Scholar]
- [17].Chabre O, Libe R, Assie G, et al. Serum miR-483-5p and miR-195 are predictive of recurrence risk in adrenocortical cancer patients. Endocr Relat Cancer 2013;20:579–94. [DOI] [PubMed] [Google Scholar]
- [18].Cai C, Chen QB, Han ZD, et al. miR-195 inhibits tumor progression by targeting RPS6KB1 in human prostate cancer. Clin Cancer Res 2015;21:4922–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Shen C-J, Cheng Y-M, Wang C-L. LncRNA PVT1 epigenetically silences miR-195 and modulates EMT and chemoresistance in cervical cancer cells. J Drug Target 2017;25:637–44. [DOI] [PubMed] [Google Scholar]
- [20].Zhou Q, Han LR, Zhou YX, et al. MiR-195 suppresses cervical cancer migration and invasion through targeting Smad3. Int J Gynecol Cancer 2016;26:817–24. [DOI] [PubMed] [Google Scholar]
- [21].Li Y, Zhuang W, Huang M, et al. Long noncoding RNA DDX11-AS1 epigenetically represses LATS2 by interacting with EZH2 and DNMT1 in hepatocellular carcinoma. Biochem Biophys Res Commun 2019;514:1051–7. [DOI] [PubMed] [Google Scholar]
- [22].Tian JB, Cao L, Dong GL. Long noncoding RNA DDX11-AS1 induced by YY1 accelerates colorectal cancer progression through targeting miR-873/CLDN7 axis. Eur Rev Med Pharmacol Sci 2019;23:5714–29. [DOI] [PubMed] [Google Scholar]
- [23].Shi M, Zhang XY, Yu H, et al. DDX11-AS1 as potential therapy targets for human hepatocellular carcinoma. Oncotarget 2017;8:44195–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Chen W, Ye L, Wen D, et al. MiR-490-5p inhibits hepatocellular carcinoma cell proliferation, migration and invasion by directly regulating ROBO1. Pathol Oncol Res 2019;25:1–9. [DOI] [PubMed] [Google Scholar]
- [25].Dai W, Dai J, Tang M, et al. lncRNA-SNHG15 accelerates the development of hepatocellular carcinoma by targeting miR-490-3p/histone deacetylase 2 axis. World J Gastroenterol 2019;25:5789–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wang H, Chen W, Jin M, et al. Circslc3a2 functions as an oncogenic factor in hepatocellular carcinoma by sponging mir-490-3p and regulating ppm1f expression. Mol Cancer 2018;17:165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Li M, Zeng J, Pan A, et al. Investigation of miR-490-3p expression in hepatocellular carcinoma based on reverse transcription-polymerase chain reaction (RT-qPCR) and a meta-analysis of 749 cases. Med Sci Monit 2018;24:4914–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Zhu X-M, Wu L-J, Xu J, et al. Let-7c MicroRNA expression and clinical significance in hepatocellular carcinoma. J Int Med Res 2011;39:2323–9. [DOI] [PubMed] [Google Scholar]
- [29].Zhang W, Cai X, Yu J, et al. Exosome-mediated transfer of lncRNA RP11-838N2. 4 promotes erlotinib resistance in non-small cell lung cancer. Int J Oncol 2018;53:527–38. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [30].Huang R, Nie W, Yao K, et al. Depletion of the lncRNA RP11-567G11. 1 inhibits pancreatic cancer progression. Biomed Pharmacother 2019;112:108685. [DOI] [PubMed] [Google Scholar]
- [31].Ai-Hua Z, Shi-Qiang T, Yan Z, et al. Effects of total ginsenosides on the feeding behavior and two enzymes activities of mythimna separata (walker) larvae. Evid Based Complement Alternat Med 2014;2014:451828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Zhou SJ, Liu FY, Zhang AH, et al. MicroRNA-199b-5p attenuates TGF-beta1-induced epithelial-mesenchymal transition in hepatocellular carcinoma. Br J Cancer 2017;117:233–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Fann YW, Lim YA, Cheng YL, et al. Evidence that NF-κB and MAPK signaling promotes NLRP inflammasome activation in neurons following ischemic stroke. Mol Neurobiol 2017;55:1–5. [DOI] [PubMed] [Google Scholar]
- [34].Johnson DG, Schneider-Broussard R. Role of E2F in cell cycle control and cancer. Front Biosci 1998;3:d447–8. [DOI] [PubMed] [Google Scholar]
- [35].Iwasaki T, Tanaka K, Kawano M, et al. Tumor-suppressive microRNA-let-7a inhibits cell proliferation via targeting of E2F2 in osteosarcoma cells. Int J Oncol 2015;46:1543–50. [DOI] [PubMed] [Google Scholar]
- [36].Huang YL, Ning G, Chen LB, et al. Promising diagnostic and prognostic value of E2Fs in human hepatocellular carcinoma. Cancer Manag Res 2019;11:1725–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Fang ZQ, Li MC, Zhang YQ, et al. MiR-490-5p inhibits the metastasis of hepatocellular carcinoma by down-regulating E2F2 and ECT2. J Cell Biochem 2018;119:8317–24. [DOI] [PubMed] [Google Scholar]