

Abstract
Tuberculosis (TB) is one of the leading causes of childhood morbidity and death globally. Lack of rapid, effective non-sputum diagnosis and prediction methods for TB in children are some of the challenges currently faced. In recent years, blood transcriptional profiling has provided a fresh perspective on the diagnosis and predicting the progression of tuberculosis. Meanwhile, combined with bioinformatics analysis can help to identify the differentially expressed genes (DEGs) and functional pathways involved in the different clinical stages of TB. Therefore, this study investigated potential diagnostic markers for use in distinguishing between latent tuberculosis infection (LTBI) and active TB using children's blood transcriptome data.
From the Gene Expression Omnibus database, we downloaded two gene expression profile datasets (GSE39939 and GSE39940) of whole blood-derived RNA sequencing samples, reflecting transcriptional signatures between latent and active tuberculosis in children. GEO2R tool was used to screen for DEGs in LTBI and active TB in children. Database for Annotation, Visualization and Integrated Discovery tools were used to perform Gene Ontology enrichment and Kyoto Encyclopedia of Genes and Genomes pathway analysis. STRING and Cytoscape analyzed the protein-protein interaction network and the top 15 hub genes respectively. Receiver operating characteristics curve was used to estimate the diagnostic value of the hub genes.
A total of 265 DEGs were identified, including 79 upregulated and 186 downregulated DEGs. Further, 15 core genes were picked and enrichment analysis revealed that they were highly correlated with neutrophil activation and degranulation, neutrophil-mediated immunity and in defense response. Among them TLR2, FPR2, MMP9, MPO, CEACAM8, ELANE, FCGR1A, SELP, ARG1, GNG10, HP, LCN2, LTF, ADCY3 had significant discriminatory power between LTBI and active TB, with area under the curves of 0.84, 0.84, 0.84, 0.80, 0.87, 0.78, 0.88, 0.84, 0.86, 0.82, 0.85, 0.85, 0.79, and 0.88 respectively.
Our research provided several genes with high potential to be candidate gene markers for developing non-sputum diagnostic tools for childhood Tuberculosis.
Keywords: active tuberculosis, biomarkers, childhood tuberculosis, differentially expressed genes, latent tuberculosis infection
1. Introduction
Tuberculosis (TB) is a chronic infectious disease caused by mycobacterium tuberculosis (M.tb). Globally, it has the highest mortality rate among single pathogens. When M.tb invades the host, it reproduces in the resident site and causes infection, triggering innate immunity. Various innate immune cells, such as macrophages and dendritic cells, etc. further activate specific T cell immune response by regulating effectors and signaling pathways, thereby killing M.tb and preventing its replication.[1,2] Among them, most of M.tb will be swallowed by macrophages, which could recruit other immune cells to gather organized and form granulomatous lesions to curb the reproduction of M.tb, but the granulation tissue cannot completely eradicate M.tb.[3] At this point, the infection may become stationary or dormant and is considered latent tuberculosis infection (LTBI). Under conditions of failing immune surveillance, approximately 5% to 10% of infected individuals will develop active TB during their lifetime.[4] Due to the diversity of potential states and outcomes, we currently lack reliable methods to identify people who might develop active TB.[5,6] Previous studies have mainly focused on the development of new diagnostic tools for adult tuberculosis, while childhood TB has relatively been neglected. For most childhood cases, the detection rate is about 35% due to lack of effective diagnostic methods especially in early-stage TB and in some cases, the smears and cultures give false positives and negatives.[7,8] One of the unique features of childhood TB is its speedy development into the disease which typically occurs within the first year following infection because of the immature immune system. This is unlike in adults where an M.tb infection could last for decades without worsening.[9] Although extensive preclinical research models have achieved encouraging results in the diagnosis, treatment, and immunological pathogenesis of TB, none of them could perfectly summarize human tuberculosis syndrome.[3,10] Therefore, there is an urgent need to explore potential biomarkers for use in distinguishing between LTBI and active TB in children for early and accurate diagnosis and treatment.
Currently, genome-wide differential expression genes studies, typically host blood transcriptome signatures combined with bioinformatics analysis, have enabled the analysis of RNA expression changes during the progression of childhood TB.[11,12] However, the high rate of false positives in independent microarray analysis has made it difficult to obtain reliable results.[13] Therefore, the current study compared the differentially expressed genes (DEGs) in LTBI and active TB blood in children (aged <15 years) from multiple regions using two gene expression profile data downloaded from Gene Expression Omnibus (GEO). This enabled the identification of potential biomarkers that can distinguish between LTBI from active TB in children hence used in the diagnosis and as potential therapeutic targets.
2. Materials and methods
2.1. Microarray data
The microarray dataset (GSE39939 and GSE39940) were downloaded from the GEO database (https://www.ncbi.nlm.nih.gov/geo/), a public functional genomics data repository. The two gene expression datasets were obtained based on the Illumina GPL10558 platform (Illumina HumanHT-12 V4.0 expression beadchip) which was last updated on August 13, 2018. Unlike in the original study, blood transcriptome of human immunodeficiency virus (HIV) negative children was analyzed and the focus was on the influence of the immune response on LTBI and active TB (sputum culture-positive children). The GSE39939 dataset contained 25 HIV- TB samples and 14 HIV- LTBI samples from Kenya while GSE39940 contained 70 HIV- TB samples and 54 HIV- LTBI samples from South Africa and Malawi.
2.2. Identification of DEGs
GEO2R (http://www.ncbi.nlm.nih.gov/geo/geo2r) was used to screen for DEGs in latent and active tuberculosis in children. GEO2R is an online analysis tool which allows users to compare two or more groups of samples for identification of DEGs across experimental conditions. The adjusted P values (adj. P value) < .01 and |logFC| (fold change) >1 were considered to be statistically significant. Venn diagram web tool (http://bioinformatics.psb.ugent.be/webtools/Venn/) was used to visualize the overlaps and R pheatmap package was used to perform the expression changes of overlapping DEGs.
2.3. GO and KEGG pathway analysis of DEGs
GO functional annotation and KEGG pathway enrichment analysis of DEGs were performed using Database for Annotation, Visualization and Integrated Discovery (DAVID) (https://david.ncifcrf.gov/home.jsp) (version 6.8) tools. DAVID provides a comprehensive set of functional annotation tools which help in understanding the biological meaning behind large list of genes. GO analysis describes DEGs in a standardized way from its biological process (BP), molecular function (MF), and cellular component (CC). KEGG Pathway analysis refers to the metabolic pathway analysis of DEGs which helps to understand the metabolic pathways significantly changed in different disease states and mechanisms. In this study, P value < .05 was considered to be statistically significant. The value of P value was converted to −log10 and the larger the −log10 P value, the greater the statistically significant.
2.4. PPI network construction and modules analysis
STRING database (https://string-db.org/) is an online tool used to predict protein-protein interactions. The DEGs were mapped to STRING to assess the relationships between proteins, and a composite score >0.4 was set to ensure that the interactions had significant statistical significance. Cytoscape software (version 3.6.1) was used to visualize the molecular interaction networks. Molecular Complex Detection (MCODE) in Cytoscape is an app used to screen the most important modules in PPI networks and all the selected parameters were set as default, apart from the MCODE scores >6. The R-package cluster Profiler (version 3.10.1) was used to perform enrichment analyses.
2.5. Hub gene identification and analysis
Cytoscape software identified the 15 top key genes in the PPI networks, with a degree ≥10. R-package cluster Profiler (version 3.10.1) conducted and visualized the functions enrichment and key genes pathways. The gene expression profiles of HIV- LTBI and active TB in the two data sets were downloaded from GEO. Receiver operating characteristics (ROC) curve was used to estimate the hub genes reliability in diagnosis, and to determine the optimal cutoff value (with the highest sensitivity and specificity). The significance level was set to .01. The area under the curve (AUC) was calculated to evaluate the ability of a single candidate gene to predict clinical diagnostic effects. SPSS statistical software version 20.0 was used to perform these analyses.
2.6. Ethical review
All analyses in our study were based on GEO, an international public repository, and therefore ethical approval and informed consent are not required.
3. Results
3.1. Identification of DEGs
DEGs (590 in GSE39939 and 937 in GSE39940) were identified based on the screening conditions: the absolute value of logFC > 1, adj. P value < .01. Comparison of active TB samples with LTBI samples from HIV-negative pediatric patients revealed that a total of 265 genes showed differences in expression in the two datasets, of which 79 were significantly upregulated (logFC > 0) and 186 were downregulated (logFC < 0) genes as shown in the Venn diagram (Fig. 1A and B). We further listed the top 20 up- and down-regulated genes of overlapping DEGs by integrating the analysis results of the two data sets (Fig. 1C, the complete list of 265 genes was shown in supplementary Table 1).
Figure 1.
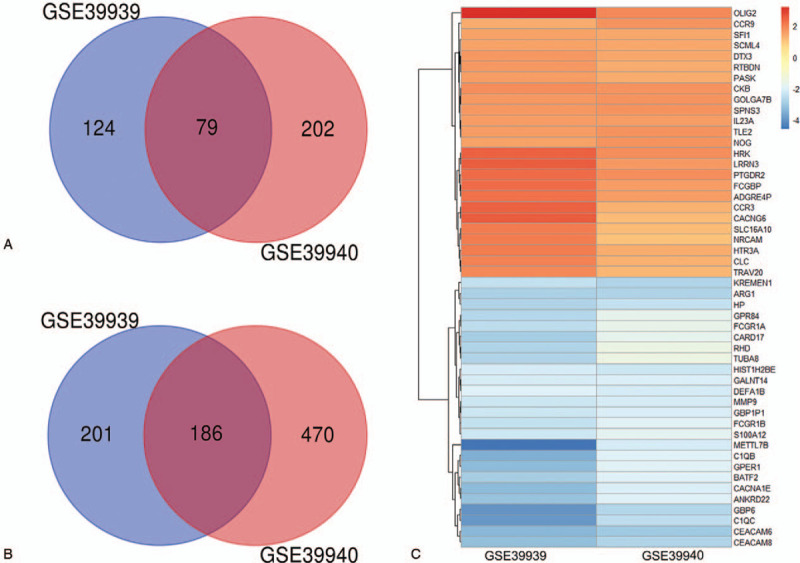
Venn diagram and heat map with two overlapping data sets. (A) Upregulated genes, (B) Downregulated genes, (C) The fold change (logFC) of the top 20 up-and down-regulated DEGs. Each row represents one gene and each column represents one dataset; The color in each rectangle corresponds to the logFC value; Red indicates upregulated genes; Blue indicates downregulated genes.
3.2. KEGG pathway and Enrichment function analysis of DEGs
The screened DEGs were uploaded onto DAVID to assess their biological classification. The results revealed that changes in BP were mainly enriched in innate immune response, antibacterial humoral response, innate immune response in mucosa, inflammatory and immune response (Fig. 2A). Changes in CC were mainly involved in the integral component of the plasma membrane, extracellular exosome, plasma membrane, extracellular space and nucleosome (Fig. 2B). For MF, the DEGs were mainly associated with carbohydrate-binding, receptor activity, protease binding and serine-type endopeptidase activity (Fig. 2C). KEGG pathway analysis showed that there was notably enrichment in complement and coagulation cascades signaling pathway, phagosome signaling pathway, leishmaniasis and staphylococcus aureus infection signaling pathway (Fig. 2D).
Figure 2.
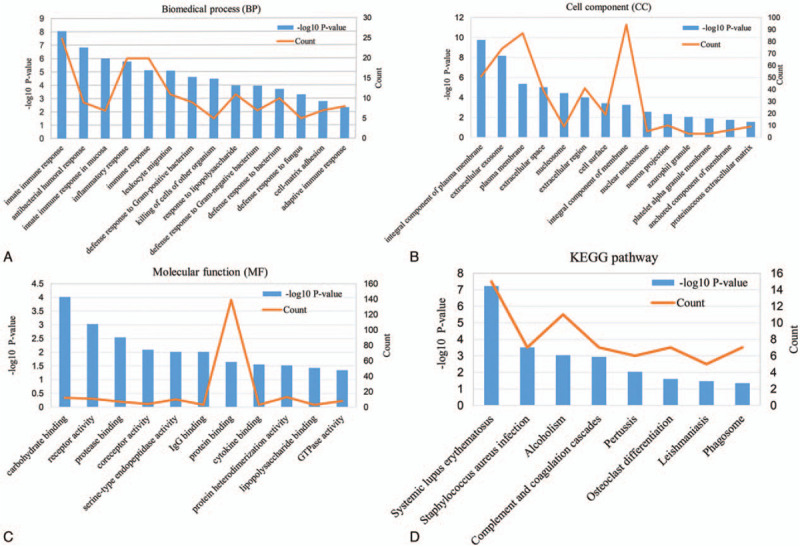
The GO functional annotation and KEGG pathway enrichment analysis of DEGs. (A) Biomedical process (BP), (B) Cell component (CC), (C) Molecular function (MF), (D) KEGG pathway.
3.3. PPI network construction and module analysis
STRING database predicted the PPI network of the DEGs which revealed 257 nodes and 573 edges (Fig. 3). The interactive information was then into the Cytoscape software and the top two modules with high scores were selected via the plug-in MCODE (Fig. 4A and B). R-package cluster Profiler analyzed the enriched modules. The results revealed that the genes were mainly associated with cell chemotaxis, adenylate cyclase-modulating G protein-coupled receptor signaling pathway, second-messenger-mediated signaling, and calcium-mediated signaling in module1. Module 2 was highly connected to neutrophil degranulation and activation, neutrophil activation involved in immune response and defense response to the bacterium (Fig. 4C and D).
Figure 3.
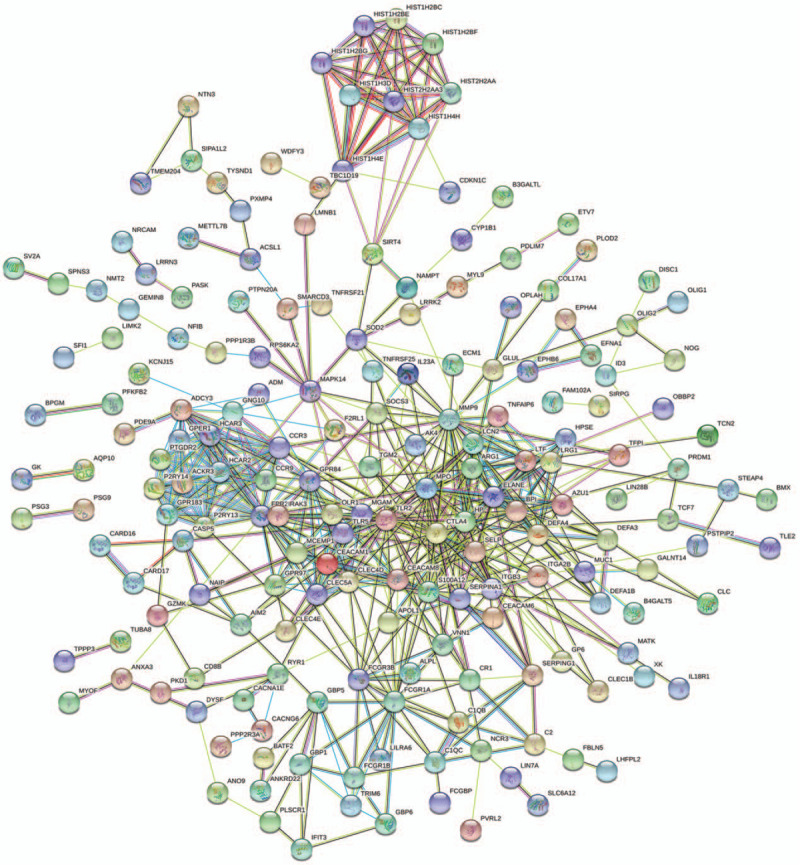
The PPI network of the DEGs from STRING. The PPI network contained a total of 257 nodes and 573 edges. Colored nodes represent the first shell of interactors; White nodes represent the second shell of interactors.
Figure 4.
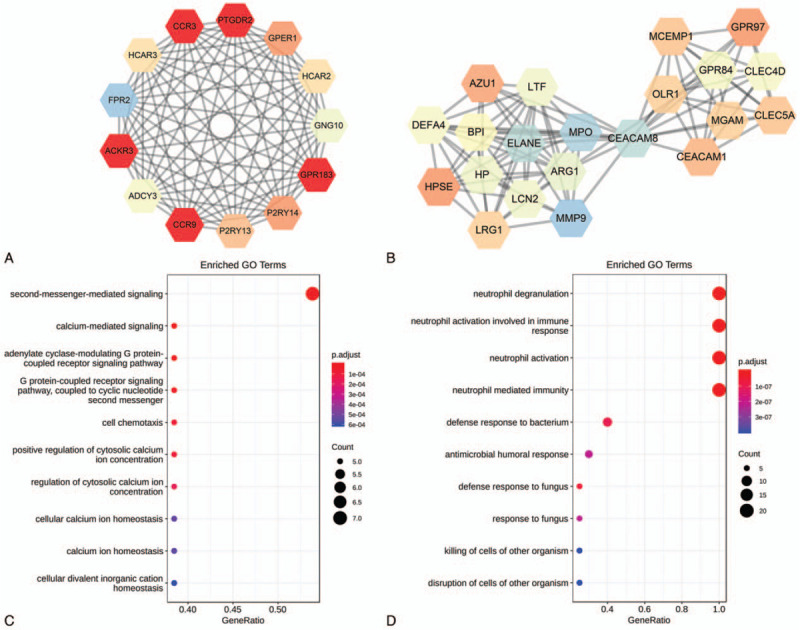
The two most significant modules in the PPI network and GO enrichment analysis. (A) module 1, (B) module 2, (C) The GO enrichment analysis of module 1, (D) The GO enrichment analysis of module 2. Upregulated genes are represented by red nodes; Downregulated genes are represented by colored nodes, and the color of nodes from a bright color to dark color corresponds to genes with low to high connectivity in the PPI network.
3.4. Hub gene identification and analysis
CytoHubba in the Cytoscape software identified the candidate hub nodes in the PPI network, and a subnetwork of the top 15 hub genes with the highest connectivity (Fig. 5A and B) was obtained. These included: TLR2, FPR2, MMP9, MPO, CEACAM8, ELANE, CTLA4, FCGR1A, ASELP, ARG1, GNG10, HP, LCN2, LTF, ADCY3. The names and basic functions of the key genes are shown in Table (Table 1). A total of 15 core genes except TLR2, CTLA4, FCGR1A and SELP were contained in the top two modules, which suggested that these modules might significantly represent the key biological characteristics in the PPI network. This defined the 15 nodes as the major hub genes distinguishing between LTBI and active TB in children. GO enrichment analysis revealed that all the hub genes were associated with neutrophil degranulation and activation of an immune response, response to bacterial molecules and defense response to the bacterium (Fig. 5C). KEGG pathway analysis revealed connection to staphylococcus aureus infection signaling pathway, and phagosome signaling pathway (Fig. 5D).
Figure 5.
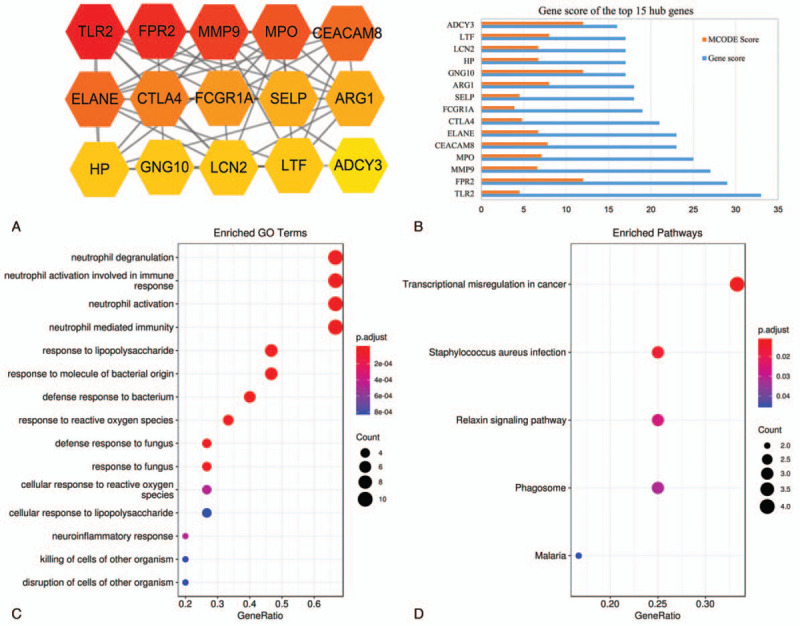
Subnetwork of top 15 hub genes from the PPI network. (A) The top 15 hub genes, (B) MCODE Score and Gene score of the top 15 hub genes, (C) The GO analysis of the top 15 hub genes, (D) The KEGG enrichment analysis of top 15 hub genes.
Table 1.
Top 15 hub genes with a higher degree of connectivity.
| NO. | Gene symbol | Full name | Function |
| 1 | TLR2 | Toll like receptor 2 | Cooperates with LY96 to mediate the innate immune response to bacterial lipoproteins and other microbial cell wall components. |
| 2 | FPR2 | Formyl peptide receptor 2 | Binding of FMLP to the receptor causes activation of neutrophils. |
| 3 | MMP9 | Matrix metallopeptidase 9 | May play an essential role in local proteolysis of the extracellular matrix and in leukocyte migration. |
| 4 | MPO | Myeloperoxidase | Part of the host defense system of polymorphonuclear leukocytes. It is responsible for microbicidal activity against a wide range of organisms. |
| 5 | CEACAM8 | Carcinoembryonic antigen related cell adhesion molecule 8 | Belongs to the immunoglobulin superfamily; CEA family. |
| 6 | ELANE | Elastase, neutrophil expressed | Neutrophil elastase; Modifies the functions of natural killer cells, monocytes and granulocytes. |
| 7 | CTLA4 | Cytotoxic T-lymphocyte associated protein 4 | Inhibitory receptor acting as a major negative regulator of T-cell responses. |
| 8 | FCGR1A | Fc fragment of IgG receptor 1A | High affinity receptor for the Fc region of immunoglobulins gamma. |
| 9 | SELP | Selectin P | Belongs to the family of cell adhesion molecules. Mediates the interaction of activated endothelial cells or platelets with leukocytes. |
| 10 | ARG1 | Arginase 1 | Key element of the urea cycle, which are vital bioenergy pathways for driving collagen synthesis and cell proliferation. |
| 11 | LTF | Lactotransferrin | Lactoferricin binds to the bacterial surface and is crucial for the bactericidal functions. |
| 12 | HP | Haptoglobin | Haptoglobin captures, and combines with free plasma hemoglobin to allow hepatic recycling of heme iron and to prevent kidney damage. |
| 13 | LCN2 | Lipocalin 2 | Iron-trafficking protein involved in multiple processes such as apoptosis, innate immunity and renal development. |
| 14 | GNG10 | G protein subunit gamma 10 | Guanine nucleotide-binding proteins (G proteins) are involved as a modulator or transducer in various transmembrane signaling systems. |
| 15 | ADCY3 | Adenylate cyclase 3 | Catalyzes the formation of the signaling molecule cAMP in response to G-protein signaling. |
We further validated the predictive value of hub genes identified from this study in an independent cohort of children with LTBI and TB. ROCs analysis showed that 15 hub genes including TLR2, FPR2, MMP9, MPO, CEACAM8, ELANE, CTLA4, FCGR1A, SELP, ARG1, GNG10, HP, LCN2, LTF, ADCY3, are potential biomarkers to distinguish LTBI from active TB, and their corresponding AUCs were 0.84, 0.84, 0.84, 0.80, 0.87, 0.78, 0.26, 0.88, 0.84, 0.86, 0.82, 0.85, 0.85, 0.79, and 0.88 respectively (Fig. 6). In addition, the findings revealed that when single genes such as ADCY3, HP, LCN2, CEACAM8, etc were used as biomarkers there was increased diagnostic efficacy in distinguishing active TB from LTBI in children (Table 2).
Figure 6.
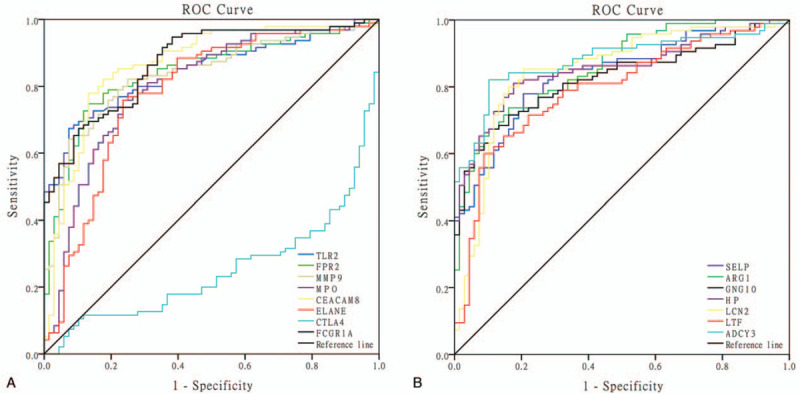
ROCs for discriminating between LTBI and active TB cases using a single hub gene. (A) ROC of the first 8 genes of key genes, (B) ROC of the last 7 genes of the key genes. AUC = area under the curve, ROC = receiver operating characteristics.
Table 2.
Diagnostic value of top 15 hub genes.
| Gene symbol | AUC (95%CI) | P | Sensitivity | Specificity |
| TLR2 | 0.842 (0.783–0.902) | <.001 | 67.40% | 92.60% |
| FPR2 | 0.842 (0.781–0.903) | <.001 | 74.70% | 86.80% |
| MMP9 | 0.837 (0.776–0.899) | <.001 | 82.10% | 75.00% |
| MPO | 0.802 (0.731–0.872) | <.001 | 75.80% | 76.50% |
| CEACAM8 | 0.865 (0.805–0.924) | <.001 | 77.90% | 86.80% |
| ELANE | 0.782 (0.707–0.857) | <.001 | 75.80% | 76.50% |
| CTLA4 | 0.251 (0.176–0.326) | <.001 | 10.30% | 89.70% |
| FCGR1A | 0.872 (0.819–0.925) | <.001 | 67.40% | 89.70% |
| SELP | 0.838 (0.778–0.898) | <.001 | 77.90% | 79.40% |
| ARG1 | 0.860 (0.805–0.914) | <.001 | 73.70% | 83.80% |
| GNG10 | 0.824 (0.761–0.888) | <.001 | 67.40% | 88.20% |
| HP | 0.854 (0.796–0.912) | <.001 | 81.10% | 82.40% |
| LCN2 | 0.849 (0.785–0.912) | <.001 | 85.30% | 79.40% |
| LTF | 0.793 (0.724–0.863) | <.001 | 60.00% | 91.20% |
| ADCY3 | 0.879 (0.825–0.933) | <.001 | 82.10% | 89.70% |
4. Discussion
Tuberculosis is an important disease that threatens children's health. According to the latest WHO report, at least a quarter of the world's population is infected with M.tb, and children with TB account for about 11% of the TB population. However, the number of children with tuberculosis exceeds previous estimates, because the clinical manifestations of children infected with M.tb lack specificity, the bacteria load is relatively small, and the geographical limitations in health care in some areas make it difficult to diagnose TB in children.[14,15] Several limitations have been associated with the use of existing immunological methods, such as tuberculin skin test and interferon-gamma release assays in guiding clinical prevention and treatment of TB, especially in children and immune-compromised individuals.[16,17] In recent years, the analysis of gene expression profiles of adult tuberculosis has improved on people's cognition of the disease pathogenesis. Relatively few studies exist on the use of gene expression profiling to analyze childhood tuberculosis. Therefore, it is crucial to investigate potential biomarkers through gene expression profiling in children with LTBI and active TB to use effective diagnosis and treatment.
This study relied on the use of transcriptional features present in children's whole blood to identify key or potential genes that distinguish between LTBI and active TB. The screening was performed on 2 mRNA microarray datasets from the GEO database where 79 upregulated and 186 downregulated DEGs were obtained. The biological classification of DEGs indicated that they were mainly enriched in innate immune response, antibacterial humoral response, inflammatory and immune response, extracellular exosome, complement and coagulation cascades, and phagosome signaling pathway. Previous studies have shown that these factors play essential roles in TB progression.[18–20] Other studies have shown that exosomes are closely related to the development of LTBI and active TB with the different stages of M.tb infection and as selective packaging of miRNA enters the exosomes.[21,22]
In the PPI network, the first 15 central genes were identified and included TLR2, FPR2, MMP9, MPO, CEACAM8, ELANE, CTLA4, FCGR1A, SELP, ARG1, GNG10, HP, LCN2, LTF, and ADCY3. The results showed that hub genes were involved in neutrophil degranulation, neutrophil activation involved in immunoreaction, response to bacterial molecules, and so forth. KEGG enrichment analysis demonstrated that the hub genes were also highly associated with staphylococcus aureus infection, phagosome, and malaria signaling pathway. Besides, ROC results showed that TLR2, FPR2, MMP9, MPO, CEACAM8, ELANE, FCGR1A, SELP, ARG1, GNG10, HP, LCN2, LTF, ADCY3 had distinguishing ability between LTBI and active TB, suggesting that the state of M.tb infection may be closely related to neutrophil-associated immune response. A recent study on neutrophil-driven IFN-induced blood transcript characteristics by Tobias et al. further supports our view.[23] Therefore, in-depth research on identified DEGs may provide new insights into the progression of LTBI to active TB and their use in diagnosis and treatment of pediatric TB.
TLR2 has been shown to play an important role in pathogen recognition and innate immune activation.[24] It was reported that M.tb suppresses innate immune response by masking the interaction between TLR2 agonists on M.tb and TLR2 on macrophages, allowing it to escape the early detection of the host, thereby delaying the onset of adaptive immune response, which explains the low expression of TLR2 in LTBI in our analysis results.[25,26] The latest research found that TLR2 polymorphisms are important genetic factors related to disease, and can be considered as predictive biomarkers for identifying high-risk individuals with TB.[27] FPR2 is mainly involved in antibacterial host defense and inflammatory response and it is a powerful neutrophil chemokine. Neutrophils in active tuberculosis patients have been reported to flow into the chronic inflammation sites where they release a wide range of inflammatory factors leading to tissue damage.[28] Carmen et al reported that FPR2 can distinguish LTBI from active tuberculosis in cases with non-insulin-dependent diabetes mellitus.[29] Adane et al reported that MMP9, FCGR1A, and LTF had significant identification ability for sputum smear-positive TB cases and household contacts based on the complexity of the infection process and the outcomes of MTB exposure and infection.[30] LTF enhances macrophages and dendritic cells as antigen carriers to activate CD4+T cells and regulate T cell polarization.[31]The Addition of lactoferrin to BCG-infected macrophages can significantly increase the expression of MHC II.[32] Studies on mouse models have shown that susceptibility to TB could be reduced by avoiding iron overload using LTF.[33] MMP9 has been shown to be involved in the pathogenesis of various inflammatory diseases and is highly expressed in both human tuberculosis and mouse tuberculosis models.[34] MMP9 also participates in the molecular mechanism of granuloma formation induced by M.tb infection and promotes macrophage recruitment and tissue remodeling.[35] Recent studies have shown that MMP9 may be used to distinguish LTBI from newly infected cases and it may also be used to prevent the TB epidemic.[36] FCGR1A plays a central role in endocytosis, phagocytosis, etc. TGFB1 is involved in the induction of fibrosis, and excess TGFB1 has been reported in TB-HIV patients where it enhances T cell suppression, the transmission of HIV and TB.[37] Research by Gebremedhin et al proved that the expression profile of FCGR1A could accurately distinguish LTBI and both active TB and non-tuberculosis mycobacterium infection in HIV-positive patients.[37] Moreover, hemodynamic analysis of tuberculosis transcriptome in children's thorax revealed that FCGR1A, FPR1, and MMP9 showed a positive correlation with the extent of the disease, suggesting that these genes may be characteristic markers of different stages of TB.[38] MPO is responsible for the bactericidal activity against various microorganisms. ELANE has been implicated in the protective response against infections, including experimental mycobacterial infections. Kadar et al revealed the plasma level of MPO and ELANE in pulmonary tuberculosis patients were significantly higher than that in LTBI, and these changes were reversed after antituberculosis treatment.[39] In this study, FCGR1A, FPR1, MMP9, MPO, and ELANE showed higher diagnostic efficacy in children TB and this confirmed the reliability of our findings and their potential use in clinical applications.
CTLA4 is an immune checkpoint molecule that prevents immune-driven pathology. It has been reported that the expression of CTLA-4 (on regulatory t cells) is increased in active TB and LTBI compared with healthy subjects. However, the difference in the expression of CTLA4 between LTBI and active TB has not been reported.[40] Unlike other genes, CTLA4 expression is reported to be higher in LTBI than in active TB, and its specificity for identifying TB is higher while the sensitivity is lower, hence there is a need for further research to confirm our results. CEACAM8 is a highly glycosylated protein expressed and released by human granulocytes. Current research on CEACAM8 has focused on cell proliferation, tumor inhibition and survival, but there are no studies on tuberculosis pathogenesis. In this study's PPI network, CEACAM8 showed a higher nodal degree (degree = 23). Granulocytes have been reported to secrete CEACAM8 to reduce respiratory tract inflammation in bacterial infections.[41] However, granulocyte reaction is related to TB pathogenesis and therefore, this may provide a new direction in the study of childhood tuberculosis. SELP controls lymphocyte metastasis to lymph nodes and leukocyte transport to acute inflammation sites. Studies have confirmed that soluble adhesion molecules, especially sE-selectin, sP-selectin, sLCAM-1 are the most sensitive clinical indicators of active TB severity.[42] ARG1 plays a central role in inhibiting tissue damage within granulomas in TB.[43]Additionally, lung injury has been associated with increased serum activity of arginase-1 in patients with TB.[44] These findings suggest that modulation of ARG1 activity is a potential treatment for tuberculosis. Studies have found that GNG10 was involved in human cytomegalovirus infection. ADCY3 has been reported to be an important mediator of energy homeostasis and its deletion mutation leads to severe obesity. However, studies on GNG10 and ADCY3 in TB have not been reported.[45] HP is associated with various infectious and non-infectious diseases such as malaria and tuberculosis. Research has shown that haptoglobin expression increased by nearly twice in patients with latent status to active TB in the malnourished population.[46] LCN2 plays an antibacterial role by combining with iron carriers required for bacterial growth. Other studies have shown that mycobactin-mediated iron uptake is a prerequisite for intracellular mycobacterial growth.[47,48] During M.tb infection, neutrophils inhibit the growth of M.tb by secreting LCN2. Recent studies have indicated that LCN2 promotes the growth of M.tb in the early stage of infection, suggesting that LCN2 is related to different stages of M.tb infection.[49] Currently, limited research exists on the mechanism of action of CEACAM8, GNG10, and ADCY3 in TB.
This study revealed that the key genes identified could be used to distinguish LTBI and active TB and this was reflected in neutrophil activation and defense response to bacterial molecules. But research on whether neutrophils are associated with the host state of M.tb infection is relatively deficient. What excites us is that we found new gene markers CTLA4, CEACAM8, ARG1, GNG10, LCN2, and ADCY3 which are different from the previous researches and therefore, this study proposes a new research direction for future diagnosis and treatment of tuberculosis in children.
In conclusion, our study identified DEGs that can be used to distinguish LTBI from active TB in children through bioinformatics analysis. A total of 265 DEGs were identified, which may play an irreplaceable role in different clinical stages of TB. Our research provided a class of hub genes with high potential to be candidate gene markers for developing non-sputum diagnostic tools for childhood TB. Future experiments are advocated to study the biological functions of the identified DEGs in childhood tuberculosis.
Acknowledgments
We acknowledge the key laboratory of Xinjiang endemic and ethnic diseases.
Author contributions
Conceptualization: Shao Meng, Fang Wu
Data curation: Jie Zhang, Jiangtao Dong, Hui Zhang
Software: Xiaoling Liu, Su Liang
Methodology: Shao Meng, Jie Zhang
Resources: Le Zhang, Chunjun Zhang
Validation: Fang Wu
Supervision: Jiangdong Wu
Writing – original draft: Shao Meng, Wanjiang Zhang
Writing – review & editing: Shao Meng, Wanjiang Zhang
Supplementary Material
Footnotes
Abbreviations: ADCY3 = Adenylate cyclase 3, ARG1 = Arginase 1, AUCs = area under the curves, CEACAM8 = Carcinoembryonic antigen related cell adhesion molecule 8, DAVID = Database for Annotation, Visualization and Integrated Discovery, DEGs = differentially expressed genes, ELANE = Elastase, neutrophil Expressed, FCGR1A = Fc fragment of IgG receptor 1A, FPR2 = Formyl peptide receptor 2, GEO = Gene Expression Omnibus, GNG10 = G protein subunit gamma 10, GO = Gene Ontology, HP = Haptoglobin, KEGG = Kyoto Encyclopedia of Genes and Genomes, LCN2 = Lipocalin 2, LTBI = latent tuberculosis infection, LTF = Lactotransferrin, MMP9 = Matrix metallopeptidase 9, MPO = Myeloperoxidase, PPI = protein-protein interaction, ROC = Receiver operating characteristics, SELP = Selectin P, TB = Tuberculosis, TLR2 = Toll like receptor 2.
How to cite this article: Shao M, Wu F, Zhang J, Dong J, Zhang H, Liu X, Liang S, Wu J, Zhang L, Zhang C, Zhang W. Screening of potential biomarkers for distinguishing between latent and active tuberculosis in children using bioinformatics analysis. Medicine. 2021;100:5(e23207).
MS and FW contributed equally to this work.
This research was supported by grant from National Natural Science Foundation Project (Grant No. U1803127), Key Science and Technology Research Projects in Key Areas of the Corps (Grant No. 2018AB019).
We declare that there are no competing interests.
The datasets analyzed during the current study are available from the corresponding author on reasonable request.
Supplemental digital content is available for this article.
AUC = area under the curve, CI = confidence interval, ROC = receiver-operating characteristics.
References
- [1].Cooper AM. Cell-mediated immune responses in tuberculosis. Annu Rev Immunol 2009;27:393–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Sia JK, Georgieva M, Rengarajan J. Innate immune defenses in human tuberculosis: an overview of the interactions between mycobacterium tuberculosis and innate immune cells. J Immunol Res 2015;2015:747543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Arora G, Misra R, Sajid A. Model systems for pulmonary infectious diseases: paradigms of anthrax and tuberculosis. Curr Top Med Chem 2017;17:2077–99. [DOI] [PubMed] [Google Scholar]
- [4].Van Crevel R, Ottenhoff THM, van der Meer JWM. Innate immunity to Mycobacterium tuberculosis. Clin Microbiol Rev 2002;15:294–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Chai Q, Lu Z, Liu CH. Host defense mechanisms against Mycobacterium tuberculosis. Cell Mol Life Sci 2020;77:1859–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Bucsan AN, Mehra S, Khader SA, et al. The current state of animal models and genomic approaches towards identifying and validating molecular determinants of Mycobacterium tuberculosis infection and tuberculosis disease. Pathog Dis 2019;77:ftz037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Dodd PJ, Gardiner E, Coghlan R, et al. Burden of childhood tuberculosis in 22 high-burden countries: a mathematical modelling study. Lancet Glob Health 2014;2:e453–9. [DOI] [PubMed] [Google Scholar]
- [8].Zar HJ, Connell TG, Nicol M. Diagnosis of pulmonary tuberculosis in children: new advances. Expert Rev Anti Infect Ther 2010;8:277–88. [DOI] [PubMed] [Google Scholar]
- [9].Newton SM, Brent AJ, Anderson S, et al. Paediatric tuberculosis. Lancet Infect Dis 2008;8:498–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Singh AK, Gupta UD. Animal models of tuberculosis: lesson learnt. Indian J Med Res 2018;147:456–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zhuang Z-G, Zhang J-A, Luo H-L, et al. The circular RNA of peripheral blood mononuclear cells: Hsa_circ_0005836 as a new diagnostic biomarker and therapeutic target of active pulmonary tuberculosis. Mol Immunol 2017;90:264–72. [DOI] [PubMed] [Google Scholar]
- [12].O'Shea MK, McShane H. A review of clinical models for the evaluation of human TB vaccines. Hum Vaccin Immunother 2016;12:1177–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Theron G, Venter R, Calligaro G, et al. Xpert MTB/RIF results in patients with previous tuberculosis: can we distinguish true from false positive results? Clin Infect Dis 2016;62:995–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Augustynowicz-Kopeć E, Jagielski T, Kozińska M, et al. Transmission of tuberculosis within family-households. J Infect 2012;64:596–608. [DOI] [PubMed] [Google Scholar]
- [15].Dodd PJ, Yuen CM, Becerra MC, et al. Potential effect of household contact management on childhood tuberculosis: a mathematical modelling study. Lancet Glob Health 2018;6:e1329–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Pai M, Denkinger CM, Kik SV, et al. Gamma interferon release assays for detection of Mycobacterium tuberculosis infection. Clin Microbiol Rev 2014;27:3–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Beshir MR, Zidan AE, El-Saadny HF, et al. Evaluation of the immune response to interferon gamma release assay and tuberculin skin test among BCG vaccinated children in East of Egypt: a cross-sectional study. Medicine 2016;95:e3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Khader SA, Divangahi M, Hanekom W, et al. Targeting innate immunity for tuberculosis vaccination. J Clin Invest 2019;129:3482–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Mehra S, Golden NA, Stuckey K, et al. The Mycobacterium tuberculosis stress response factor SigH is required for bacterial burden as well as immunopathology in primate lungs. J Infect Dis 2012;205:1203–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ernst JD. The immunological life cycle of tuberculosis. Nat Rev Immunol 2012;12:589–91. [DOI] [PubMed] [Google Scholar]
- [21].Hadifar S, Fateh A, Yousefi MH, et al. Exosomes in tuberculosis: Still terra incognita? J Cell Physiol 2019;234:2104–11. [DOI] [PubMed] [Google Scholar]
- [22].Lyu L, Zhang X, Li C, et al. Small RNA profiles of serum exosomes derived from individuals with latent and active tuberculosis. Front Microbiol 2019;10:1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Dallenga T, Repnik U, Corleis B, et al. M. tuberculosis-induced necrosis of infected neutrophils promotes bacterial growth following phagocytosis by macrophages. Cell Host Microbe 2017;22:519–530.e3. [DOI] [PubMed] [Google Scholar]
- [24].Aderem A, Ulevitch RJ. Toll-like receptors in the induction of the innate immune response. Nature 2000;406:782–7. [DOI] [PubMed] [Google Scholar]
- [25].Madan-Lala R, Peixoto KV, Re F, et al. Mycobacterium tuberculosis Hip1 dampens macrophage proinflammatory responses by limiting toll-like receptor 2 activation. Infect Immun 2011;79:4828–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Gopalakrishnan A, Salgame P. Toll-like receptor 2 in host defense against Mycobacterium tuberculosis: to be or not to be-that is the question. Curr Opin Immunol 2016;42:76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Mandala JP, Ahmad S, Pullagurla A, et al. Toll-like receptor 2 polymorphisms and their effect on the immune response to ESAT-6, Pam3CSK4 TLR2 agonist in pulmonary tuberculosis patients and household contacts. Cytokine 2020;126:154897. [DOI] [PubMed] [Google Scholar]
- [28].Alemán M, Beigier-Bompadre M, Borghetti C, et al. Activation of peripheral blood neutrophils from patients with active advanced tuberculosis. Clin Immunol 2001;100:87–95. [DOI] [PubMed] [Google Scholar]
- [29].Serrano CJ, Cuevas-Córdoba B, Macías-Segura N, et al. Transcriptional profiles discriminate patients with pulmonary tuberculosis from non-tuberculous individuals depending on the presence of non-insulin diabetes mellitus. Clin Immunol 2016;162:107–17. [DOI] [PubMed] [Google Scholar]
- [30].Mihret A, Loxton AG, Bekele Y, et al. Combination of gene expression patterns in whole blood discriminate between tuberculosis infection states. BMC Infect Dis 2014;14:257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Actor JK, Hwang S-A, Olsen M, et al. Lactoferrin immunomodulation of DTH response in mice. Int Immunopharmacol 2002;2:475–86. [DOI] [PubMed] [Google Scholar]
- [32].Wilk KM, Hwang S-A, Actor JK. Lactoferrin modulation of antigen-presenting-cell response to BCG infection. Postepy Hig Med Dosw (Online) 2007;61:277–82. [PMC free article] [PubMed] [Google Scholar]
- [33].Schaible UE, Collins HL, Priem F, et al. Correction of the iron overload defect in beta-2-microglobulin knockout mice by lactoferrin abolishes their increased susceptibility to tuberculosis. J Exp Med 2002;196:1507–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Taylor JL, Hattle JM, Dreitz SA, et al. Role for matrix metalloproteinase 9 in granuloma formation during pulmonary mycobacterium tuberculosis infection. Infect Immun 2006;74:6135–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Volkman HE, Pozos TC, Zheng J, et al. Tuberculous granuloma induction via interaction of a bacterial secreted protein with host epithelium. Science 2010;327:466–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Araujo Z, Palacios A, Enciso-Moreno L, et al. Evaluation of the transcriptional immune biomarkers in peripheral blood from Warao indigenous associate with the infection by Mycobacterium tuberculosis. Rev Soc Bras Med Trop 2019;52:e20180516. [DOI] [PubMed] [Google Scholar]
- [37].Gebremicael G, Kassa D, Quinten E, et al. Host gene expression kinetics during treatment of tuberculosis in HIV-coinfected individuals is independent of highly active antiretroviral therapy. J Infect Dis 2018;218:1833–46. [DOI] [PubMed] [Google Scholar]
- [38].Jenum S, Bakken R, Dhanasekaran S, et al. BLR1 and FCGR1A transcripts in peripheral blood associate with the extent of intrathoracic tuberculosis in children and predict treatment outcome. Sci Rep 2016;6:38841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Moideen K, Kumar NP, Nair D, et al. Heightened systemic levels of neutrophil and eosinophil granular proteins in pulmonary tuberculosis and reversal following treatment. Infect Immun 2018;86:e00008–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Wykes MN, Lewin SR. Immune checkpoint blockade in infectious diseases. Nat Rev Immunol 2018;18:91–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Singer BB, Opp L, Heinrich A, et al. Soluble CEACAM8 interacts with CEACAM1 inhibiting TLR2-triggered immune responses. PLoS One 2014;9:e94106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Mukae H, Ashitani J-i, Tokojima M, et al. Elevated levels of circulating adhesion molecules in patients with active pulmonary tuberculosis. Respirology 2003;8:326–31. [DOI] [PubMed] [Google Scholar]
- [43].Duque-Correa MA, Kühl AA, Rodriguez PC, et al. Macrophage arginase-1 controls bacterial growth and pathology in hypoxic tuberculosis granulomas. Proc Natl Acad Sci USA 2014;111:E4024–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Monin L, Griffiths KL, Lam WY, et al. Helminth-induced arginase-1 exacerbates lung inflammation and disease severity in tuberculosis. J Clin Invest 2015;125:4699–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Saeed S, Bonnefond A, Tamanini F, et al. Loss-of-function mutations in ADCY3 cause monogenic severe obesity. Nat Genet 2018;50:175–9. [DOI] [PubMed] [Google Scholar]
- [46].Bapat PR, Satav AR, Husain AA, et al. Differential levels of Alpha-2-macroglobulin, haptoglobin and sero-transferrin as adjunct markers for TB diagnosis and disease progression in the malnourished tribal population of Melghat, India. PLoS One 2015;10:e0133928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Luo M, Fadeev EA, Groves JT. Mycobactin-mediated iron acquisition within macrophages. Nat Chem Biol 2005;1:149–53. [DOI] [PubMed] [Google Scholar]
- [48].Gobin J, Horwitz MA. Exochelins of Mycobacterium tuberculosis remove iron from human iron-binding proteins and donate iron to mycobactins in the M. tuberculosis cell wall. J Exp Med 1996;183:1527–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Dahl SL, Woodworth JS, Lerche CJ, et al. Lipocalin-2 Functions as Inhibitor of Innate Resistance to Mycobacterium tuberculosis. Front Immunol 2018;9:2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.