

Abstract
An 8-month-old male infant patient was referred to our institution (from elsewhere) with a history of fever, convulsions, dystonic posturing, altered sensorium, and loss of motor and mental milestones since past 1 month. Upon admission to our institution, a neuroimaging (magnetic resonance imaging of the brain) revealed frontoparietal atrophy, “bat-wing appearance,” and basal ganglia changes. Carnitine and acylcarnitine profile revealed low total carnitine, very low free carnitine, and low free/acylcarnitine ratio, with normal levels of plasma amino acids. Urine gas chromatography mass spectrometry showed an elevated level of ketones (3-hydroxybutyric acid and acetoacetate) and glutaric acid with the presence of 3-hydroxyglutaric acid, suggestive of glutaric aciduria type 1. Diet modification and pharmacotherapy with riboflavin and carnitine arrested the neurological deterioration in the patient.
Keywords: glutaric aciduria type 1, dystonia, L-carnitine, newborn screening
Introduction
Glutaric aciduria type 1 (GA1) is an autosomal recessive neurometabolic disorder, caused by a deficiency of the enzyme glutaryl CoA dehydrogenase (GCDH), affecting the metabolic pathway of the amino acids lysine, hydroxylysine, and tryptophan; thus leading to accumulation of glutaric acid and other metabolic by-products. 1 2 3 The gene for GCDH is located on chromosome 19p13.2 and many disease-causing mutations have been reported till date. 2 3
GA1-affected infants may develop normally up to 1 to 2 years of life; macrocephaly is a common finding in these infants and it precedes onset of the neurological manifestations. 2 3 Some affected infants may also show subtle neurological symptoms, such as hypotonia, irritability, and feeding problems, during this initial (seemingly) asymptomatic period. 2 The onset of symptoms may occur suddenly after a minor infection or catabolic states. 2 3 The condition is usually heralded by acute encephalopathic findings such as loss of achieved developmental milestones, choreoathetosis, seizures, generalized rigidity, and dystonia. 2 3 Recovery from the attacks usually occur slowly, but some residual neurological abnormalities, especially extrapyramidal symptoms (such as hypotonia/rigidity, choreoathetosis, and dystonia) may persist. 2
We report one such case whose diagnosis got delayed elsewhere (before presentation to our institute), owing to the absence of newborn screening (NBS) for GA1 and, probably, due to paucity of knowledge of the condition among physicians.
Case Report
An 8-month-old male infant patient, Asian Indian in origin, first child born out of a nonconsanguineous marriage, was brought to the pediatric emergency room with 3 days history of fever and three episodes of convulsions 1 month prior, with dystonic posturing since a month and loss of all the achieved motor and mental milestones. The fever was low grade, persisted for 3 days, and had responded to oral paracetamol (15 mg/kg/dose, thrice a day). A month back (with the fever), the child had three episodes of convulsions characterized by tonic posturing of all four limbs with upward deviation of eyes, each seizure lasting for less than a minute, followed by postictal drowsiness persisting for 2 hours. The patient also developed intermittent dystonic posturing, which used to disappear during sleep. During this acute illness, he lost all his achieved milestones. Before this febrile episode, he could hold his neck (achieved at 3 months of age), could roll over (achieved at 5 months of age), had a bidextrous approach (achieved at 5 months of age), and had a social smile with recognition of parents (achieved at 3 and 5 months of age, respectively). The child also had excessive irritability and refusal to accept oral feeds in the past 1 month of the illness. He presented (late) to our hospital on the 30th day of his illness. The birth history was uneventful without any history suggestive of birth asphyxia. There was absence of any significant illnesses in the family.
His weight was 6.3 kg (−2 to −3 standard deviation [SD]), length was 66 cm (−2 to −3 SD), and the head circumference was 44 cm (0 to −1 SD). On examination, he had a modified Glasgow coma scale (GCS) for infants of 10/15 (E2— eye opening to pain, V4—crying to pain, and M4—withdrawal to pain), was afebrile, with a pulse rate of 132/min, with a respiratory rate of 36/min, and blood pressure of 94/62 mm Hg. Central nervous system (CNS) examination revealed the absence of papilledema or retinal hemorrhages, appendicular hypotonia, muscle power of >3/5, normal deep tendon reflexes, and bilateral extensor plantar responses. On pull-to-sit maneuver, there was complete head lag. There was absence of scissoring and absence of signs of meningeal irritation. Remaining systemic examination did not reveal any abnormalities.
The initial laboratory investigations done at admission are enlisted in Table 1 . On day 2 of hospital stay, a serum ammonia level was done, which was normal. The magnetic resonance imaging (MRI) of the brain was performed on day 3 of admission to our hospital, which showed prominent cerebrospinal fluid spaces and Sylvian fissures (“bat-wing appearance”) ( Fig. 1 ). T2/fluid-attenuated inversion recovery (FLAIR) sequences revealed hyperintensities involving bilateral basal ganglia and there was diffusion restriction in the white matter of both cerebral hemispheres on diffusion-weighted imaging. The magnetic resonance (MR) spectroscopy with short echo time showed lipid peak at 0.9 and 1.3 ppm, and glutamate–glutamine (Glx) peak was seen as a shoulder to N-acetyl-aspartate (NAA) at 2.2 and 2.4 ppm. These findings suggested a likely diagnosis of GA1. To confirm our diagnosis, his metabolic work-up was sent. Carnitine and acylcarnitine profile revealed low total carnitine, very low free carnitine, and low free/acylcarnitine ratio, with normal levels of plasma amino acids, suggestive of the possibility of GA1. The urine gas chromatography mass spectrometry showed an elevated level of ketones (3-hydroxybutyric acid and acetoacetate) and glutaric acid with the presence of 3-hydroxyglutaric acid, suggestive of GA1 ( Fig. 2 ).
Table 1. Investigations done in the patient upon admission to our hospital.
| Investigations | Results | Age appropriate reference range |
|---|---|---|
| Hb | 8.3 g/dL | 10.5–14.0 g/dL |
| Leukocyte count | 11.8 × 10 3 cells/mm 3 | 6.0–14.0 × 10 3 cells/mm 3 |
| Platelet count | 280 × 10 3 /mm 3 | 150–400 × 10 3 /mm 3 |
| Serum electrolytes (Na + /K + ) | 140/4.0 mmol/L | 134–144/3.5–6.1 mmol/L |
| BUN/serum creatinine | 5.8/0.48 mg/dL | 5–18/0.22–0.50 mg/dL |
| Serum total protein/albumin | 5.6/3.6 g/dL | 4.6–7.4/1.9–4.9 g/dL |
| AST/ALT | 76/80 U/L | 15–50/5–45 U/L |
| Ionic calcium | 1.26 mmol/L | 1.2–2.8 mmol/L |
| Arterial pH | 7.41 | 7.35–7.45 |
| C-reactive protein | 29.41 mg/L | 0.8–11.2 mg/L |
Abbreviations: ALT, alanine aminotransferase; AST, aspartate transaminase; BUN, blood urea nitrogen; Hb, hemoglobin.
Fig. 1.
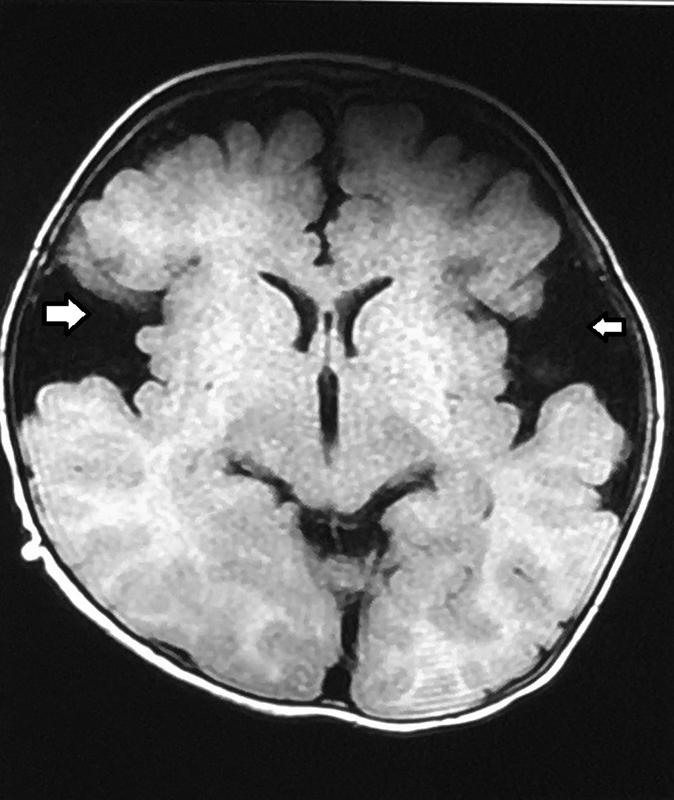
Magnetic resonance image of the brain (T1-weighted, axial section) showing widening of Sylvian fissures, the “bat-wing appearance” (white arrows), characteristic of glutaric aciduria type 1.
Fig. 2.
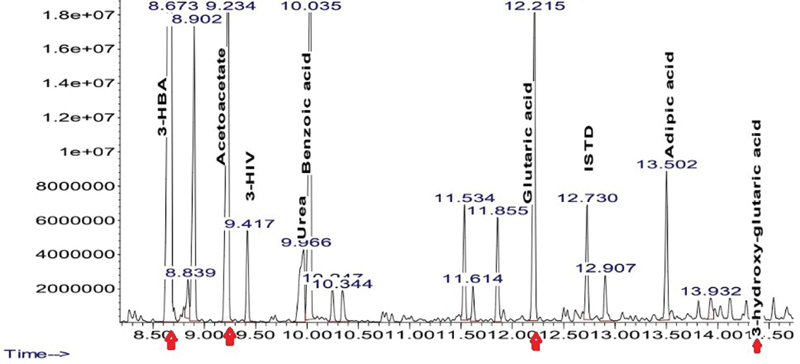
Urine gas chromatography mass spectrometry showing elevated levels of 3-hydroxybutyric acid (3-HBA), acetoacetate, and glutaric acid with the presence of 3-hydroxy-glutaric acid.
Initially (for first 2 days after his indoor admission), the child received supportive treatment in the form of intravenous ceftriaxone (100 mg/kg/d, twice daily), intravenous paracetamol (15 mg/kg/dose, thrice daily), intravenous levetiracetam (20 mg/kg/d, twice daily), and intravenous fluids. The levetiracetam was continued as he was already initiated on the drug before presenting to us. After sending the metabolic work-up (for diagnosing GA1), the child was started on a low-protein diet; initial 4 days, rice kanji with added sugars to provide 120 kcal/kg/d (rice kanji is also known as rice porridge or congee, prepared by boiling rice in water until it softens significantly, being a good source of carbohydrates, more so with added sugars and a poor source of protein, and is helpful in such cases); later a commercial preparation was started. High doses (200 mg/d, orally) of riboflavin and L-carnitine (50 mg/kg/d, thrice daily, orally gradually increased to 100 mg/kg/d, thrice daily before his discharge from the hospital) were given. To control the dystonia, he was started on oral baclofen (10 mg/d, thrice daily, orally). Over the next 6 days, the child showed improvement in the form of mentation (modified GCS for infants improved from 10/15 to 15/15; E4—spontaneous eye opening, V5—cooing and babbling, and M6—spontaneous movements and approach to toys), absence of any repeat episodes of convulsions, and a decrease in the dystonia. With this recovery, the child was discharged on a strict diet plan, with riboflavin, carnitine, levetiracetam, and baclofen after 12 days of indoor admission. After discharge, the parents failed to follow-up, however, as per enquiry on telephone, 5 months after discharge, the child is gaining milestones and there have been no repeat episodes of convulsions; there was continued compliance with the diet and medications advised. The patient can roll over now and there is an improvement in the head control. The remarkable change is that the rapid progression of the neurological disorder has halted and the patient has had no further episodes of vomiting and lethargy during these 5 months after initiating the therapy for GA1.
Discussion and Review of Literature
General Aspects
GA1 is an organic acidemia, associated with a progressive neurological disorder. 1 2 4 By 1 to 2 years of age, the affected individuals generally manifest with recurrent episodes of vomiting and lethargy associated with metabolic acidosis and typical neurological manifestations. 2 However, the age of presentation can vary (presentation as late as 55 years of age has also been reported). 5 The age of clinical onset ranges from early infancy to adulthood and the severity of symptoms may vary from an acute encephalopathy with poor prognosis in early-onset to milder forms in late-onset variants. 3 Birth asphyxia and the metabolic acidosis due to complicated labor may trigger acute decompensation in the neonatal period. 6 The progressive movement disorder, beginning in early childhood, simulates dystonic cerebral palsy but is progressive rather than being static. 1 The initial clinical presentation with encephalopathic features may be mistaken for encephalitis, 3 and this unduly delays the diagnosis and initiation of specific treatment (as happened in our case).
Pathophysiology
GA1 is characterized by bilateral striatal neurodegeneration due to accumulation of glutaric acid and related metabolites leading to vascular dysfunction, abnormal cerebral blood flow, and blood–brain barrier damage. 7 The primary pathological finding of GA1 is the loss of gamma-aminobutyric acid (GABA)—containing neurons and gliosis in the basal ganglia. 1 In our patient, using MRI of the brain, we showed bilateral areas of hyperintensities in the caudate nucleus, bilateral thalamus, and lentiform nucleus, suggestive of basal ganglia necrosis.
Diagnosis
During the acute episodes, mild to moderate metabolic acidosis, ketosis, hypoglycemia, hyperammonemia, and elevations of serum transaminases are seen in some patients. 2 Our patient had only ketosis and mildly elevated liver enzymes; however, ammonia and arterial pH were normal, probably because of the treatment that he had received elsewhere, before presenting to us. High concentrations of glutaric acid are usually found in urine, blood, and cerebrospinal fluid (CSF). 2 3-Hydroxyglutaric acid may also be present in the urine. 2 Plasma amino acids are usually within normal limits. 2 The laboratory findings may be unremarkable between the attacks. 2 Some GA1 patients, termed as high excretors, have large amounts of glutaric and 3-hydroxyglutaric acid in the urine, while others, termed as low excretors, have much less obvious organic aciduria. 4 In any child with progressive dystonia and dyskinesia, activity of the enzyme GCDH should be measured in leukocytes or cultured fibroblasts as urinary glutaric acid may not be elevated. 4 Elevated levels of 3-hydroxyglutaric acid is highly suggestive of GA1 and clinician can reliably start appropriate treatment for GA1; however, detection of a disease-causing mutation confirms the diagnosis. 8 In our patient, the mutation analysis was advised; however, due to financial constraints, it could not be done.
Genotype–Phenotype Correlation
Christensen et al investigated the correlation between genotype and phenotype in 251 patients with GA1, but they could not establish any clear correlation between biochemical phenotype and genotype and the clinical severity. However, they found that patients with a mild mutation on at least one chromosome frequently behave as low excretors and show unusual biochemical findings such as low or normal excretion of glutaric acid and mild or only slightly increased excretion of 3-hydroxyglutaric acid in the urine. However, patients with severe mutations such as R402W or A293T on both alleles have no residual enzyme activity and behave as high excretors. 9 Goodman et al, Mosaeilhy et al, and Kölker et al also did not find any significant correlation between genotype and clinical phenotype. 10 11 12
Treatment
The therapeutic approaches include low-protein diets and diets low in tryptophan and lysine. 2 13 Riboflavin (as flavin adenine dinucleotide), a cofactor of GCDH, was prescribed to enhance residual enzyme activity. 1 2 L-carnitine was used to stimulate the formation and excretion of short-chain acylcarnitine derivatives of glutaric acid. 1 2 14 15 L-carnitine has an important role in GA1, as an antioxidant, anti-inflammatory agent and it also increases the excretion of accumulated toxic metabolites. 15 Glutaric acid is known to inhibit the synthesis of GABA, hence GABA analogs such as baclofen may help in these cases. 2
Neuroimaging (MRI)
MRI is the neuroimaging modality of choice in the assessment of GA1. In severely affected children, bilateral basal ganglia abnormalities are commonly seen, with initial swelling that subsequently progresses to atrophy and necrosis. 16 17 The most notable finding is a “bat-wing appearance” in the frontotemporal area of the brain due to widened Sylvian fissures. 16 This reversible cerebral atrophy is reportedly due to the apoptosis of immature oligodendrocytes. 18 Delay in myelination is a further finding in severely affected infants. 16 17 Other findings are macrocephaly, expansion of subarachnoid spaces. 16 17 The MRI of the brain signal characteristics include T1 hypointensities, T2/FLAIR hyperintensities, and restricted diffusion during acute decompensation. 16 17 MR spectroscopy may reveal decreased NAA/creatine ratio compared with a sex and age-matched control; the presence of the lactate peak reflecting disturbed mitochondrial functions in this disease has rarely been reported. 17 19 In our patient, the MR spectroscopy showed lipid peak at 0.9 and 1.3 ppm with short echo time (demonstrating neuronal necrosis and inflammation), 20 and Glx peak was seen as shoulder to NAA at 2.2 and 2.4 ppm (demonstrating demyelination and neuronal loss), 20 21 latter has been studied for monitoring these patients after initiation of diet restriction. 21
Rare Complications
Life-threatening, severe, acute, subdural hemorrhage with or without retinal hemorrhages have been reported in patients after minor head trauma. 22 It has been hypothesized that microencephalopathic macrocephaly is the predisposing factor. The word “microencephaly” indicates smaller brain size (2 SD below the mean) and “microencephalopathic macrocephaly” indicates relatively smaller brain size for the skull volume, which subsequently leads to expanded CSF spaces and increases the risk of stretching bridging veins and subsequent subdural hemorrhage, which may occur even after minor accidental head trauma. 22 23 In the absence of a history of significant trauma, the occurrence of subdural hemorrhage with retinal hemorrhages may lead clinicians to suspect child abuse especially with undiagnosed GA1. 23 Serrano Russi et al reported three unrelated GA1 patients who presented with malignant CNS tumors (medulloblastoma and glioblastoma) at the ages from 6 to 23 years. 24 Among these three patients, one was diagnosed by NBS but had poor diet compliance, and the other two were diagnosed late. 24 The exact cause of such tumors is not known, but accumulated metabolites such as glutamine, glutaric acid, and 3-hydroxyglutaric acid have been implicated in tumorigenesis. 24 Another potential rare complication of GA1 is rhabdomyolysis. 25 The most obvious precipitating factor for rhabdomyolysis is altered bioenergetics due to the metabolic disorder caused by GCDH deficiency. 25 Investigation of GCDH in mice showed markedly decreased brain and skeletal muscle creatinine phosphokinase activity after administration of lysine. 26 Consumption of mutton (which contains a large amount of lysine) has been reported to cause rhabdomyolysis in these patients. 25 Pierson et al reported subependymal nodules as an uncommon feature of neuroimaging of GA1. 5 Currently, neither the etiology nor the consequence of these nodules is well understood. 5 Kaya Ozcora et al reported infantile stroke in a 9-month-old child with GA1. 27 Badve et al reported a 32-year-old female patient with GA1, who presented with migraine and a bipolar disorder with frontal lobe dysfunction and bilateral pyramidal tract signs. 28 Young-Lin et al reported a 7-month-old male infant with GA1, who presented with flexor spasms, video electroencephalography revealed hypsarrhythmia and the child improved with prednisolone. 29
Newborn Screening and Prenatal Diagnosis
GA1 can be identified by NBS, whereas the prenatal diagnosis of GA1 is feasible by employing enzyme analysis on cultured chorionic villi sample or amniocytes or by metabolite (glutaric acid) screening in amniotic fluid. 6 Molecular analysis on DNA extracted from chorionic villi sample or amniotic fluid is possible only when GCDH gene mutations of the index case have been previously characterized. 6
Cost-effectiveness of NBS
In India, routine NBS for GA1 is not being implemented because of a lack of data on the prevalence of inborn errors of metabolism. 30 It has been proven that extending NBS programs for GA1 represents a highly cost-effective diagnostic strategy. 31 Pfeil et al found that the screening program saves a total of around €30,682 (95% confidence interval: €14,343–€49,176) per 100,000 screened neonates over a 20-year time horizon. 31 Early diagnosis may prevent the progressive brain damage, which, in turn, may reduce the neuromorbidity. 6 Overall, early diagnosis and treatment initiation will be effective in the prevention of complications for patients with GA1, with improved outcomes in areas including oral motor function, ambulatory capability, speech, motor function, and cognition. 32
Follow-up
Biochemical follow-up is needed in these cases, which includes plasma amino acids (especially lysine and tryptophan levels), blood levels of free carnitine, and acylcarnitines. 3 33 Aggressive treatment of the intercurrent infections should be done by cessation (for 24–48 h) or reduction of protein intake to 50% for 24 hours or less, depending on the severity of the illness, while providing a high-energy intake with an extra 10 to 25% of caloric requirements through carbohydrates, lipids, and double dose of carnitine (200 mg/kg/d). 3 33
Conclusion
Physicians caring for such children need to be aware that febrile illnesses often trigger the metabolic presentation in an otherwise well child and the clinical presentation of GA1 to avoid the undue delay in the diagnosis (as happened in our case) and to avoid delay in the initiation of appropriate treatment. It may be a difficult diagnosis to make on a first presentation and if there are other clues such as macrocephaly and preexisting developmental delay or regression of milestones, then a metabolic screen is warranted. A high index of suspicion is necessary for early diagnosis of such metabolic conditions and other inborn errors of metabolism, which will ensure prompt initiation of appropriate treatment and an overall better prognosis.
Acknowledgment
The authors would like to thank Dr. Hemant Deshmukh, Dean, Seth G.S. Medical College and KEM Hospital, for granting permission to submit this manuscript for publication.
Funding Statement
Funding None.
Conflict of Interest None declared.
Authors' Contributions
S.S. and M.S.T. were involved in conceptualizing the manuscript, conducting literature search, and drafting the manuscript. S.S. and M.S.T. are both designated as the first authors of this manuscript. Patient data were collected by S.S. and N.S.; M.A. helped in conducting literature search and revised the manuscript for scientific content. All the authors were involved in clinical management of the patient. M.S.T. will act as the guarantor for the manuscript.
References
- 1.Lipkin P H, Roe C R, Goodman S I, Batshaw M L. A case of glutaric acidemia type I: effect of riboflavin and carnitine. J Pediatr. 1988;112(01):62–65. doi: 10.1016/s0022-3476(88)80123-9. [DOI] [PubMed] [Google Scholar]
- 2.Rezvani I. Philadelphia: Elsevier; 2016. Lysine; p. 676. [Google Scholar]
- 3.Couce M L, López-Suárez O, Bóveda M D. Glutaric aciduria type I: outcome of patients with early- versus late-diagnosis. Eur J Paediatr Neurol. 2013;17(04):383–389. doi: 10.1016/j.ejpn.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 4.Goodman S I, Woontner M. An explanation for metabolite excretion in high- and low-excretor patients with glutaric acidemia type 1. Mol Genet Metab. 2019;127(04):325–326. doi: 10.1016/j.ymgme.2019.07.005. [DOI] [PubMed] [Google Scholar]
- 5.Pierson T M, Nezhad M, Tremblay M A. Adult-onset glutaric aciduria type I presenting with white matter abnormalities and subependymal nodules. Neurogenetics. 2015;16(04):325–328. doi: 10.1007/s10048-015-0456-y. [DOI] [PubMed] [Google Scholar]
- 6.Biasucci G, Morelli N, Natacci F, Mastrangelo M. Early neonatal glutaric aciduria type I hidden by perinatal asphyxia: a case report. Ital J Pediatr. 2018;44(01):8. doi: 10.1186/s13052-018-0450-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Isasi E, Korte N, Abudara V, Attwell D, Olivera-Bravo S. Glutaric acid affects pericyte contractility and migration: possible implications for GA-I pathogenesis. Mol Neurobiol. 2019;56(11):7694–7707. doi: 10.1007/s12035-019-1620-4. [DOI] [PubMed] [Google Scholar]
- 8.Additional individual contributors . Boy N, Mühlhausen C, Maier E M. Proposed recommendations for diagnosing and managing individuals with glutaric aciduria type I: second revision. J Inherit Metab Dis. 2017;40(01):75–101. doi: 10.1007/s10545-016-9999-9. [DOI] [PubMed] [Google Scholar]
- 9.Christensen E, Ribes A, Merinero B, Zschocke J. Correlation of genotype and phenotype in glutaryl-CoA dehydrogenase deficiency. J Inherit Metab Dis. 2004;27(06):861–868. doi: 10.1023/B:BOLI.0000045770.93429.3c. [DOI] [PubMed] [Google Scholar]
- 10.Goodman S I, Stein D E, Schlesinger S. Glutaryl-CoA dehydrogenase mutations in glutaric acidemia (type I): review and report of thirty novel mutations. Hum Mutat. 1998;12(03):141–144. doi: 10.1002/(SICI)1098-1004(1998)12:3<141::AID-HUMU1>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 11.C GPD . Mosaeilhy A, Mohamed M M. Genotype-phenotype correlation in 18 Egyptian patients with glutaric acidemia type I. Metab Brain Dis. 2017;32(05):1417–1426. doi: 10.1007/s11011-017-0006-4. [DOI] [PubMed] [Google Scholar]
- 12.Kölker S, Garbade S F, Greenberg C R. Natural history, outcome, and treatment efficacy in children and adults with glutaryl-CoA dehydrogenase deficiency. Pediatr Res. 2006;59(06):840–847. doi: 10.1203/01.pdr.0000219387.79887.86. [DOI] [PubMed] [Google Scholar]
- 13.Gokmen-Ozel H, MacDonald A, Daly A. Dietary practices in glutaric aciduria type 1 over 16 years. J Hum Nutr Diet. 2012;25(06):514–519. doi: 10.1111/j.1365-277X.2012.01269.x. [DOI] [PubMed] [Google Scholar]
- 14.Flanagan J L, Simmons P A, Vehige J, Willcox M D, Garrett Q. Role of carnitine in disease. Nutr Metab (Lond) 2010;7:30. doi: 10.1186/1743-7075-7-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guerreiro G, Faverzani J, Jacques C ED. Oxidative damage in glutaric aciduria type I patients and the protective effects of l-carnitine treatment. J Cell Biochem. 2018;119(12):10021–10032. doi: 10.1002/jcb.27332. [DOI] [PubMed] [Google Scholar]
- 16.Mohammad S A, Abdelkhalek H S, Ahmed K A, Zaki O K. Glutaric aciduria type 1: neuroimaging features with clinical correlation. Pediatr Radiol. 2015;45(11):1696–1705. doi: 10.1007/s00247-015-3395-8. [DOI] [PubMed] [Google Scholar]
- 17.Desai N K, Runge V M, Crisp D E, Crisp M B, Naul L G. Magnetic resonance imaging of the brain in glutaric acidemia type I: a review of the literature and a report of four new cases with attention to the basal ganglia and imaging technique. Invest Radiol. 2003;38(08):489–496. doi: 10.1097/01.rli.0000080405.62988.f6. [DOI] [PubMed] [Google Scholar]
- 18.Numata-Uematsu Y, Sakamoto O, Kakisaka Y. Reversible brain atrophy in glutaric aciduria type 1. Brain Dev. 2017;39(06):532–535. doi: 10.1016/j.braindev.2017.01.003. [DOI] [PubMed] [Google Scholar]
- 19.Oguz K K, Ozturk A, Cila A. Diffusion-weighted MR imaging and MR spectroscopy in glutaric aciduria type 1. Neuroradiology. 2005;47(03):229–234. doi: 10.1007/s00234-005-1350-3. [DOI] [PubMed] [Google Scholar]
- 20.Verma A, Kumar I, Verma N, Aggarwal P, Ojha R. Magnetic resonance spectroscopy - revisiting the biochemical and molecular milieu of brain tumors. BBA Clin. 2016;5:170–178. doi: 10.1016/j.bbacli.2016.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zinnanti W J, Lazovic J, Housman C. Mechanism of age-dependent susceptibility and novel treatment strategy in glutaric acidemia type I. J Clin Invest. 2007;117(11):3258–3270. doi: 10.1172/JCI31617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ishige M, Fuchigami T, Ogawa E. Severe acute subdural hemorrhages in a patient with glutaric acidemia type 1 under recommended treatment. Pediatr Neurosurg. 2017;52(01):46–50. doi: 10.1159/000448736. [DOI] [PubMed] [Google Scholar]
- 23.Carman K B, Aydogdu S D, Yakut A, Yarar C. Glutaric aciduria type 1 presenting as subdural haematoma. J Paediatr Child Health. 2012;48(08):712. doi: 10.1111/j.1440-1754.2012.02513.x. [DOI] [PubMed] [Google Scholar]
- 24.Serrano Russi A, Donoghue S, Boneh A, Manara R, Burlina A B, Burlina A P. Malignant brain tumors in patients with glutaric aciduria type I. Mol Genet Metab. 2018;125(03):276–280. doi: 10.1016/j.ymgme.2018.08.006. [DOI] [PubMed] [Google Scholar]
- 25.Qian G L, Hong F, Tong F, Fu H D, Liu A M. Recurrent rhabdomyolysis and glutaric aciduria type I: a case report and literature review. World J Pediatr. 2016;12(03):368–371. doi: 10.1007/s12519-016-0042-x. [DOI] [PubMed] [Google Scholar]
- 26.Amaral A U, Cecatto C, Seminotti B.Marked reduction of Na(+), K(+)-ATPase and creatine kinase activities induced by acute lysine administration in glutaryl-CoA dehydrogenase deficient mice Mol Genet Metab 2012107(1-2):81–86. [DOI] [PubMed] [Google Scholar]
- 27.Kaya Ozcora G D, Gokay S, Canpolat M, Kardaş F, Kendirci M, Kumandaş S. Glutaric acidemia type 1: a case of infantile stroke. JIMD Rep. 2018;38:7–12. doi: 10.1007/8904_2017_26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Badve M S, Bhuta S, Mcgill J.Rare presentation of a treatable disorder: glutaric aciduria type 1 N Z Med J 2015128(1409):61–64. [PubMed] [Google Scholar]
- 29.Young-Lin N, Shalev S, Glenn O A. Teaching neuroimages: infant with glutaric aciduria type 1 presenting with infantile spasms and hypsarrhythmia. Neurology. 2013;81(24):e182–e183. doi: 10.1212/01.wnl.0000437291.75075.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kapoor S, Thelma B K. Status of newborn screening and inborn errors of metabolism in India. Indian J Pediatr. 2018;85(12):1110–1117. doi: 10.1007/s12098-018-2681-5. [DOI] [PubMed] [Google Scholar]
- 31.Pfeil J, Listl S, Hoffmann G F, Kölker S, Lindner M, Burgard P. Newborn screening by tandem mass spectrometry for glutaric aciduria type 1: a cost-effectiveness analysis. Orphanet J Rare Dis. 2013;8:167. doi: 10.1186/1750-1172-8-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Viau K, Ernst S L, Vanzo R J, Botto L D, Pasquali M, Longo N. Glutaric acidemia type 1: outcomes before and after expanded newborn screening. Mol Genet Metab. 2012;106(04):430–438. doi: 10.1016/j.ymgme.2012.05.024. [DOI] [PubMed] [Google Scholar]
- 33.Kölker S, Christensen E, Leonard J V. Diagnosis and management of glutaric aciduria type I--revised recommendations. J Inherit Metab Dis. 2011;34(03):677–694. doi: 10.1007/s10545-011-9289-5. [DOI] [PMC free article] [PubMed] [Google Scholar]