

CONSPECTUS:
Total synthesis—the ultimate proving ground for the invention and field-testing of new methods, exploration of disruptive strategies, final structure confirmation, and empowerment of medicinal chemistry on natural products—is one of the oldest and most enduring subfields of organic chemistry. In the early days of this field, its sole emphasis focused on debunking the concept of vitalism, that living organisms could create forms of matter accessible only to them. Emphasis then turned to the use of synthesis to degrade and reconstitute natural products to establish structure and answer questions about biosynthesis. It then evolved to not only an intricate science but also a celebrated form of art. As the field progressed, a more orderly and logical approach emerged that served to standardize the process. These developments even opened up the possibility of computer-aided design using retrosynthetic analysis. Finally, the elevation of this field to even higher levels of sophistication showed that it was feasible to synthesize any natural product, regardless of complexity, in a laboratory. During this remarkable evolution, as has been reviewed elsewhere, many of the principles and methods of organic synthesis were refined and galvanized. In the modern era, students and practitioners are still magnetically attracted to this field due to the excitement of the journey, the exhilaration of creation, and the opportunity to invent solutions to challenges that still persist. Contemporary total synthesis is less concerned with demonstrating a proof of concept or a feasible approach but rather aims for increased efficiency, scalability, and even “ideality.” In general, the molecules of Nature are created biosynthetically with levels of practicality that are still unimaginable using the tools of modern synthesis. Thus, as the community strives to do more with less (i.e., innovation), total synthesis is now focused on a pursuit for simplicity rather than a competition for maximal complexity. In doing so, the practitioner must devise outside-the-box strategies supplemented with forgotten or newly invented methods to reduce step count and increase the overall economy of the approach. The downstream applications of this pursuit not only empower students who often go on to apply these skills in the private sector but also lead to new discoveries that can impact numerous disciplines of societal importance. This account traces some select case studies from our laboratory over the past five years that vividly demonstrate our own motivation for dedicating so much effort to this classic field. In aiming for simplicity, we focus on the elusive goal of achieving ideality, a term that, when taken in the proper context, can serve as a guiding light to point the way to furthering progress in organic synthesis.
Graphical Abstract

INTRODUCTION
The term “ideality” in the context of organic chemistry was introduced in 2010 to pinpoint flaws in a synthetic sequence.2 Inspired by Hendrickson’s seminal studies on efficiency in synthesis it simply states that in order for a synthesis to be ideal, it must only create skeletal bonds of a molecule without recourse to extraneous functional group manipulations, protecting group sequences, or nonstrategic redox fluctuations (concession steps).4 Calculating ideality is rather elementary: the total number of strategic steps is divided by the total step count. Like any singular metric that characterizes a synthesis (yield, step count, atom economy), ideality does not tell the whole story. For example, a two-step synthesis where the first step generates all of the key skeletal bonds followed by a deprotection would only be 50% ideal but clearly superior to a 10-step approach with 80% ideality.5 Similarly, in certain cases it might be valuable to introduce protecting groups or additional steps to aid purification on a large scale. It thus represents an inconvenient truth as even the two-step synthesis mentioned above could be improved. Syntheses are best judged using not only ideality but also by considering step-count,6 atom-economy,7 redox-economy,8 overall yield, convergence,9 number of isolated intermediates,10 and, most importantly, the context. For example, on process scale, although ideality has been used for route scouting,11 various descriptors for overall efficiency that even take into account the quantity of solvent usage are of critical importance (e.g., process mass intensity).12 In radio-chemistry, most of the above variables do not apply as convergency, time, and cost of the radiolabeled element matter most.13 In medicinal or discovery chemistry, the ability to divergently and quickly access multiple vectors of SAR space from a common intermediate is usually more important than overall step count.13 In an academic setting, particularly in natural product synthesis, ideality can be a useful rubric to employ not only at the planning stages and execution, but also in a final analysis when the synthesis is complete. Figure 1 outlines natural product syntheses that our laboratory has completed over the past five years (2016–2020) along with a minimalist retrosynthetic depiction. This account will dive into the motivations for pursuing most of these molecules, what was learned as a consequence, and the lessons of reflecting on the ideality of the final routes (these lessons will be highlighted in bold at the end of each section). The syntheses are graphically illustrated to highlight key reactions and the strategic (blue) or nonstrategic (red) nature of the steps employed. Natural products that leverage two-phase terpene synthesis (phorbol, thapsigargin, and Taxol) will not be covered here as the logic underlying those syntheses has been reviewed elsewhere.14
Figure 1.
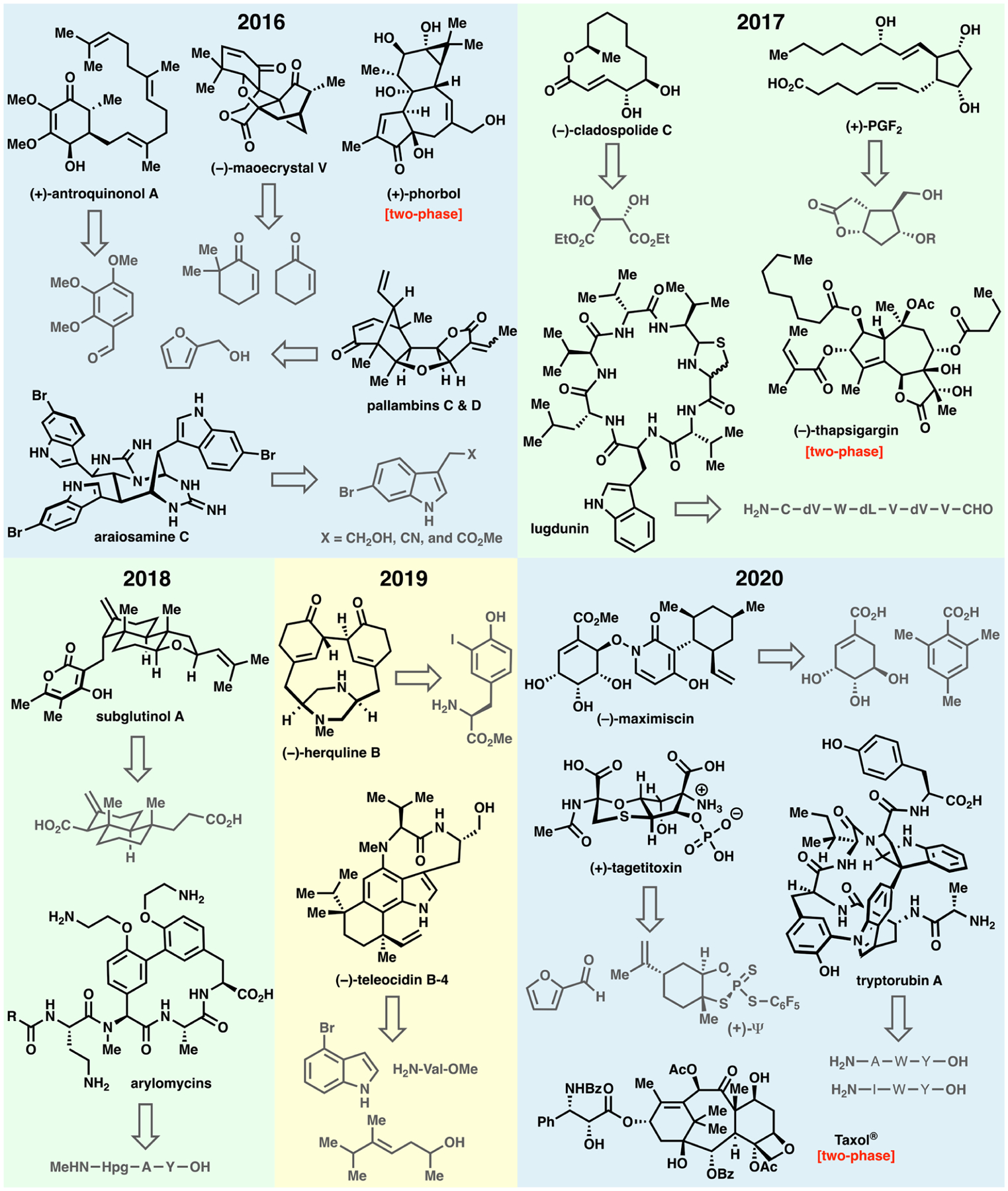
Select molecules synthesized in our laboratory over the past five years.
2016
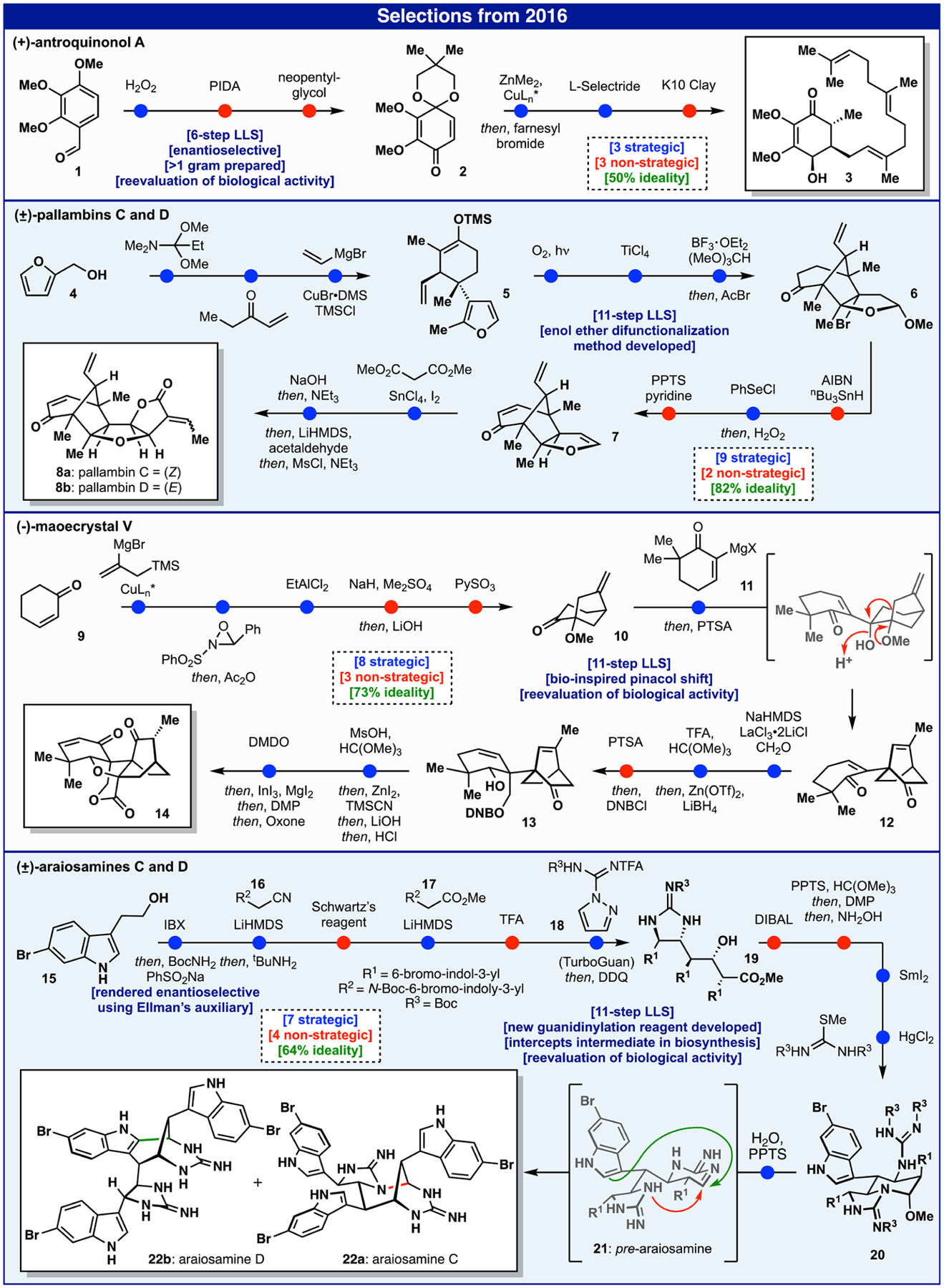
Antroquinonol A (3)15 is a natural product of mixed biosynthetic origin, containing a polyketide-derived quinone portion coupled to a linear diterpene fragment (Figure 2). It was brought to our attention as a target of interest by medicinal chemists at Bristol-Myers-Squibb due to its remarkable anticancer activity.16 In fact, it was granted orphan drug status by the FDA for treatment of pancreatic cancer and acute myeloid leukemia. Multiple phase II studies promoted the use of 3 in a wide range of indications from Alzheimer’s disease and atopic dermatitis to the treatment of COVID-19.17 As the mechanism of action for 3 was (and still is to our knowledge) unknown, a modular and scalable synthetic strategy was devised commencing from inexpensive benzaldehyde derivative 1. The key step involved enantiocontrolled vicinal difunctionalization of quinone 2 by enlisting an organocuprate addition, followed by alkylation with farnesyl bromide. Upon stereocontrolled reduction to install the remaining hydroxyl group, deprotection led to gram quantities of 3. With copious quantities in hand, a barrage of biological assays (in vitro and in vivo) revealed no appreciable cytotoxic activity. It was thus concluded that the enthusiasm associated with initial findings might be due to an impurity in the natural product isolation process rather than 3 itself. The continued use of 3 in clinical trials remains an enigma. Even so, this six-step synthesis achieved all of the goals set forth at the outset in that it provided ample material for extensive biological tests and offered a roadmap for the synthesis of analogs (although this turned out to be unnecessary).However, the 50% ideality points to key problems still remaining in synthesis such as the difficulty in chemoselective manipulation of quinones in their native form.
Figure 2.
Select total syntheses from 2016.
In contrast to the motivations guiding the synthesis of 3, the pursuit of the plant-derived natural products pallambins (8a and 8b) was motivated solely on the basis of structural beauty (Figure 2).18 The retrosynthetic analysis was predicated on the notion that four sequential cyclization/annulation events could consecutively form each ring system. This carefully choreographed sequence, beginning with furfuryl alcohol 4, commenced with a tandem Eschenmoser—Claisen/reduction followed by a Robinson annulation and vicinal difunctionalization to furnish enol ether 5. The furan served as a masked 1,4-dicarbonyl compound that could be strategically unveiled, thus setting the stage for the next cyclization delivering 6. A nonstrategic debromination followed by desaturation and elimination provided furan 7. The final sequence accomplished a formal annulation forging both C—C and C—O bonds of the γ-lactone that could be immediately subjected to an aldol reaction with acetaldehyde to deliver 8a and 8b. While the ideality of this route is high, it is not without warts. For example, the route is linear in nature and one could imagine reducing the step count if a more convergent approach were devised. Additionally, although the use of a furan to shield a 1,4-dicarbonyl group is an often-used tactic19 and does put in place the needed functionality for the ensuing cyclization, it is debatable whether such a sequence is truly strategic. An even more direct approach would involve the cyclization (perhaps through SET oxidation) of the enol ether and furan groups in 5 to deliver a nonbrominated structure analogous to 6, perhaps obviating the need for nonstrategic debromination and elimination steps.
In the case of maoecrystal V (14),20 a popular plant-derived natural product target, the goal was to develop a scalable route to address the unanswered questions surrounding its reported anticancer activity (Figure 2).21 In stark departure from four prior approaches that were all wedded to intuitive Diels—Alder disconnections,22 a convergent approach loosely mirrored on the proposed biosynthesis23 was chosen that, on paper, would dramatically simplify and shorten the route. Thus, fragment 10, derived from cyclohexenone 9 via enantioselective conjugate addition and intramolecular Sakurai cyclization, was merged with enone 11 to generate the key C—C bond linkage, which upon heating with PTSA smoothly afforded key intermediate 12 after a pinacol rearrangement. Although this was a powerfully short entry to the synthesis, extensive optimization was required to install the remaining two carbon atoms, the proper stereochemistry, and the requisite oxidation state found in the natural product. This was accomplished through the following carefully choreographed sequence involving: (1) a La-directed, site-selective, and stereocontrolled aldol with formaldehyde, (2) a Zn-assisted stereoselective ketone reduction, (3) a strategic hemiketal formation followed by stereoselective cyanide addition, and (4) a cascade oxidation sequence to install the enone in 14. Ultimately, with a copious supply of 14 in hand we were able to conclude, as with antroquinonol (vide supra), that the initial enthusiasm associated with the differential cytotoxicity reported for 14 was likely due to an impurity in the initial isolate. Although the ideality of this route was high (73%), it suffered from two protecting group manipulations and an extraneous redox fluctuation. These concession steps were required to set the stage for the fragment union and pinacol shift, and use of a DNB protecting group stemmed from a need to prevent the wrong hemiketal from forming.
Our longstanding interest in the marine-derived pyrroleimidazole alkaloids24 inspired investigation into a conceptually related family of alkaloids bearing a striking set of repeating indole-imidazole subunits called the araiosamines (Figure 2).25 The synthetic challenge posed by these exotic polycyclic architectures was the sole justification for their pursuit in our laboratory. A hypothesis driving our key retrosynthetic disconnections was that their complex structures could be unraveled by invoking a thermodynamic cyclization of a suitably functionalized guanidine-containing indole trimer dubbed prearaiosamine 21.26 While initial studies on ambitious cascade trimerizations of indole-based building blocks failed, a more stepwise approach ultimately proved successful. The construction of the requisite indole trimer 19 could be accomplished using Mannich and aldol reactions with three simple building blocks: an imine derived from 15, nitrile 16, and ester 17. This enabled clean differentiation of the requisite functionality necessary for installing guanidines at key positions. A particularly recalcitrant guanidine installation required the invention of a new reagent for this purpose (18, currently sold by Millipore-Sigma as “TurboGuan”). Subsequent adjustment of the ester oxidation state, setting the remaining amino stereocenter, and appending the second guanidine unit arrived at a functional precursor to pre-araiosamine, 20. Finally, submission of this precursor to PPTS in warm water led to targets 22a and 22b presumably by way of a thermodynamic equilibration. Ironically, biological evaluation of the natural products revealed interesting antibacterial activity despite original reports to the contrary, unlike the seemingly promising isolation studies on 3 and 14. The complexity of the target and its challenging physical properties resulted in a route with only 64% ideality, and while not mentioned above, the stereoselectivity of certain steps was also low (1:1) despite extensive optimization. If trimerization of the simple indole building blocks could be achieved in a controllable fashion, most of the issues of this synthesis could be resolved. This points to unresolved chemical challenges in aldol/Mannich chemistry that Nature has mastered.
2017
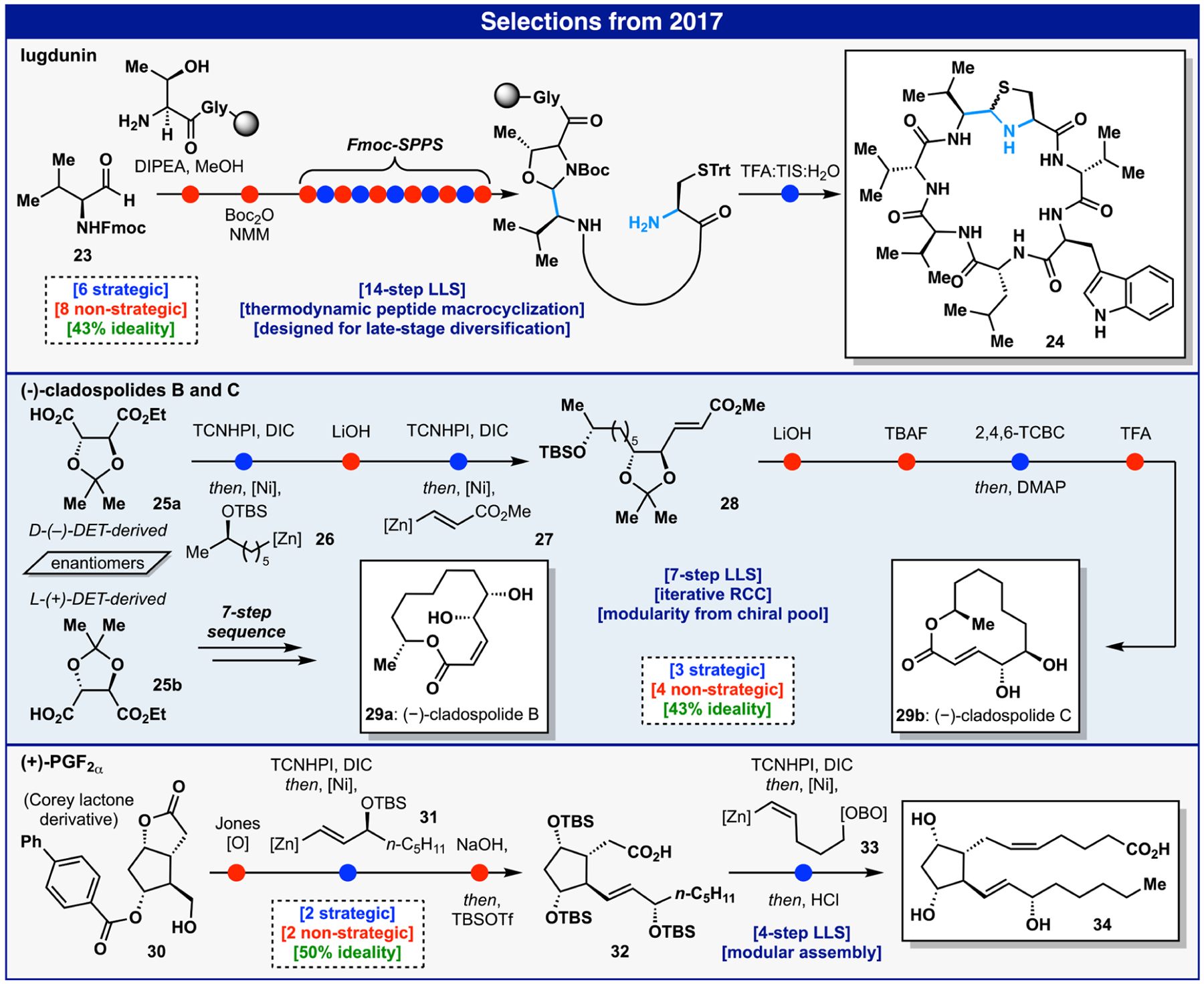
Our interests in large peptidic macrocyclic natural products such as lugdunin27 (24) stemmed from a long-term collaboration with Bristol-Myers-Squibb (Figure 3). Traditionally, macrocyclization strategies to access native cyclic peptides utilize kinetic, irreversible amide bond-forming methods; the premise of this work was to explore a thermodynamic approach. The existence of macrocyclic peptides bearing imines (or derivatives thereof, e.g. 24) inspired an exploration into an imine-based macrocyclization pathway.28 This approach enabled the rapid synthesis of linear amino-aldehyde precursors that upon exposure to a buffered aqueous solution and suitable trap yielded a thermodynamic product via an imine intermediate.27 Studies demonstrated that preorganization of the linear precursors played no role in the final product formation. In other analogues, one could also see striking selectivity for the N-terminus versus adjacent unprotected lysine residues. To demonstrate the value of the approach, a short total synthesis of 24 from 23 was achieved via solid-phase peptide synthesis (SPPS) using a resin-bound hemiaminal followed by tandem cleavage/cyclization. Notably, 24 attracted worldwide interest due to its striking antibacterial activity and origin from the human microbiome.29 Clearly the route benefits from the industrialization of SPPS that makes peptide synthesis so routine. Yet, from an ideality standpoint (43%), one cannot ignore the incessant protecting group manipulations that are required to facilitate such sequences. An interesting challenge for the field, one that has seen some recent progress,30 would be to reduce the reliance on these concessions.
Figure 3.
Select total syntheses from 2017; TCNHPI = tetrachloro-N-hydroxyphthalimide.
The cladospolides 29a and 29b are polyketide-derived natural products that bear a distinct diol motif (Figure 3). Although 1,2-diols traditionally call for a dihydroxylation transform, an alternative strategy was sought. Tartaric acid has historically been used as a diol “cassette” by way of laborious reduction— oxidation-Wittig sequences that often suffer from a lack of geometric control of the olefin product. For example, a prior route to 29 utilized an 18-step sequence where the desired olefin geometry was not controlled, leading to product mixtures and reduced yield.31 Our interest in radical retrosynthesis32 triggered a direct disconnection of the C—CO2H bond in tartaric acid to controllably replace it with a geometrically defined alkene-based nucleophile via decarboxylative alkenylation.33 Sequential use of this cross-coupling on one of the carboxylate moieties, with a decarboxylative alkylation on the other, could allow for a modular approach to a myriad of diol-containing structures. In fact, such an approach accessed cladospolides B 29a and C 29b from the chiral pool derived L- and D-tartrate esters 25b and 25a, respectively. Thus, commercially available tartrate 25a was submitted to two sequential decarboxylative couplings with alkyl zinc 26 and vinyl zinc 27 to afford protected seco-ester 28 and intercepting, in 6 steps (LLS), the prior 14-step route.31 Despite the brevity of this approach, some concession steps were required, namely the use of protecting group manipulations to facilitate the modular nature of this sequence, resulting in a modest ideality score of 43%. Some protecting groups could conceivably be excised (e.g., TBS); however, many of those employed were tactical, and function beyond their ability to block the reactivity of a functional group. For example, the acetonide locks the diol ring conformation to enable diastereoselective coupling, while ester hydrolysis allows for iterative control of the cross-coupling steps.
The prostaglandins, a storied class of molecules, have remained popular targets for the synthetic community.34 PGF2α 34 is among the family’s most notable members and has several important medical applications (Figure 3). The retrosynthetic analysis toward PGF2α, as with 29a and 29b, involved iterative scission of its two side chains: alkyl fragment 31 and alkene 33, tracing back to the commercially available Corey lactone 30.35 This disconnection enabled the modular assembly of PGF2α in four steps with 50% ideality via decarboxylative alkyl and alkenyl cross-couplings, thus leveraging functionality native to 30. While the initial redox manipulation and hydrolysis/TBS protection step are nonideal, they are counterbalanced by the brevity of the route, which carries the potential to access innumerable medicinally relevant analogues of PGF2α via simple cross-coupling. It is worth reiterating that in the syntheses of 29a, 29b, and 34, one is not wedded to the strict rules for controlling olefin geometry in classic reactions (e.g., Wittig olefination) during the retrosynthetic plan since it is programmed by way of cross-coupling. That said, the above routes still illustrate how far we have to go when it comes to controlling chemoselectivity without resorting to protecting group chemistry.
2018
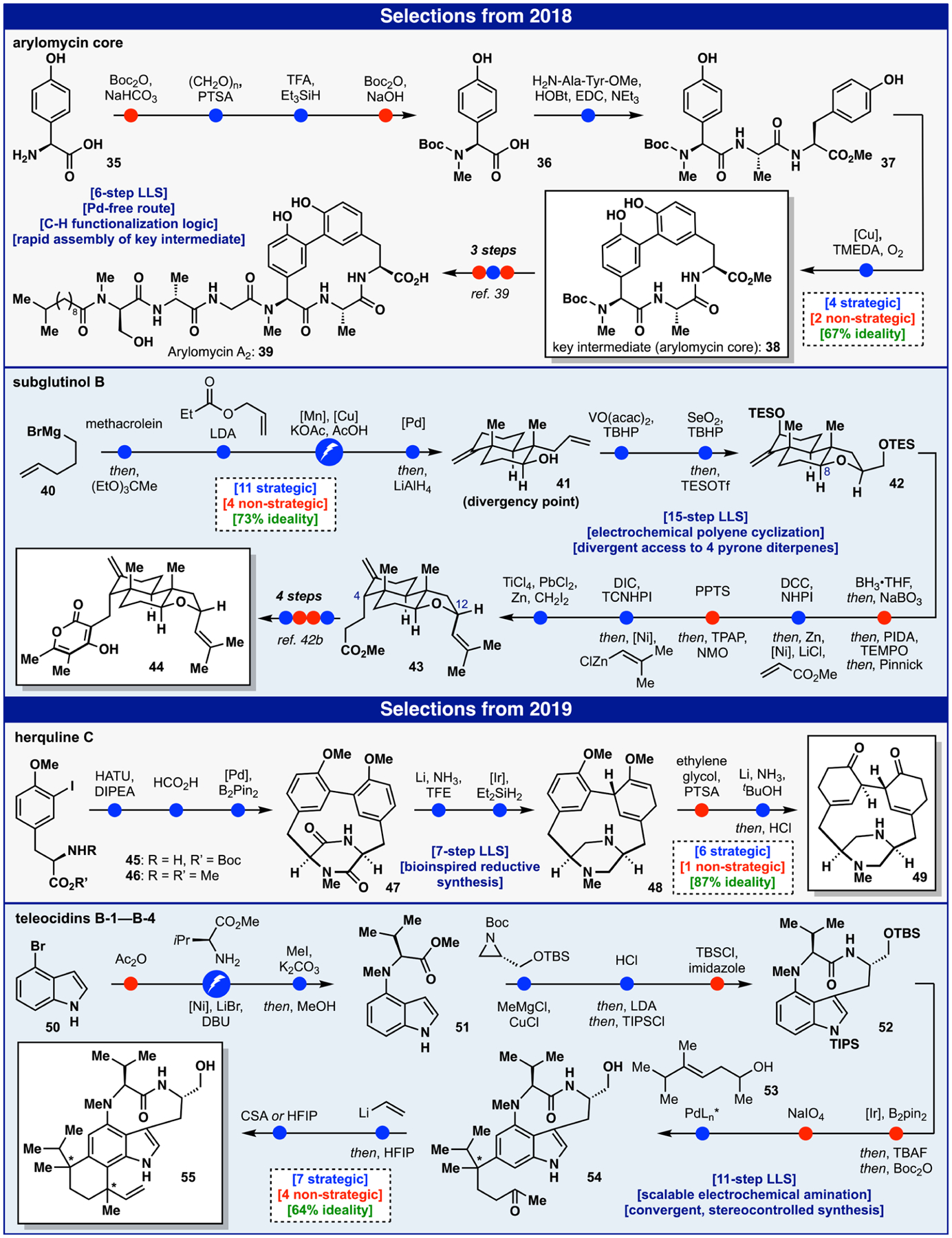
The arylomycins36 are an old class of natural product antibiotics that regained notoriety in 2008 stemming from seminal findings from the Romesberg group37 that served as a basis for further development at Genentech (Figure 4).38 Upon our entry into the area, only one macrocyclization strategy, intramolecular Suzuki coupling,37a,39 was available to forge the key macrocyclic core 38; however, this route was lengthy (14 steps) and posed problems of material throughput (6.4% overall yield, 36% ideality) in the ongoing medicinal chemistry campaigns (RQx and Genentech).
Figure 4.
Select total syntheses from 2018 and 2019.
This key building block not only represented a formal synthesis of 3939 (Figure 1) but was a gateway to a wide variety of analogs. Due to its peptidic nature, our retrosynthetic analysis was predicated on the use of amino acid building blocks and, by mimicking biosynthesis, necessitated the use of C-H functionalization logic to obviate concession steps associated with prefunctionalization of the phenolic building blocks. This allowed a simple unfunctionalized tripeptide 37 to serve as the macrocyclic precursor, which was synthesized through an N-methylation (of tyrosine 35 to form 36) and peptide coupling sequence. Thus, the main hurdle was uncovering conditions best suited for the oxidative macrocyclization of the arylomycin core tripeptide. While nearly all conceivable oxidation methods explored were fruitless, copper-oxo complexes proved viable, and extensive optimization ultimately led to a 60% yield on gram-scale. Development of this route dramatically increased access to supplies of the arylomycins and their analogs for academic research,40 and the technology was ultimately licensed by Genentech for use in their ongoing drug discovery effort. While overall yield was dramatically increased and step count nearly cut in half, the ideality remains at 63%, pointing mainly to the common shortcomings of peptide synthesis as discussed before.
As part of a longstanding collaboration with LEO Pharma to pursue complex natural product synthesis in medicinal chemistry, the subglutinols became targets of interest due to their reported immunosuppressive activity (Figure 4).41 Historically, such structures have been synthesized using purely polar-bond disconnections resulting in long sequences plagued by redox-manipulations and functional group interconversions.42 Our strategy was guided by a desire to access as many members of the family as possible through a divergent approach enlisting a practical and scalable path to a common core followed by methods to controllably access the many substitution patterns and stereochemical configurations.43 Modular, divergent approaches are always ideal in medicinal chemistry, and due to the diversity of methods for cross-coupling of differentially halogenated aromatic precursors, they are often straightforward to plan. Conversely, designing such an approach for a complex, stereochemically ornate natural product is uncommon. We began with the preparation of the key precursor 41 (from 40) that represented the point of divergence, featuring an electrochemical variant of Snider’s radical polycyclization.44 After decarboxylative allylation, epoxidation and reduction could control the C-8 stereocenter in 42 (the opposite configuration was also accessed selectively). Following hydroboration/ oxidation, the C-4 position was primed for use of a recently developed decarboxylative Giese reaction to set the C-4 stereocenter.45 The C-12 center, in turn, could then be stereoselectively installed via a decarboxylative C-C alkenylation en route to 43 as employed in the syntheses of 29 and 34 (Figure 3).33 The alternate stereoisomer could be accessed from the same intermediate by using a conventional Wittig/ metathesis sequence. These radical methods were essential in simplifying this problem to reach not only 44 and three other members of the family but, in principle, a wide variety of analogs in a controlled fashion.42b,46 Radical retrosynthesis, beyond canonical Stork-Curran type cyclizations, clearly offers unusual benefits to address the salient issues of this natural product class. The less than perfect ideality (73%) is reflected in the need for vestigial PG manipulations and redox fluctuations to enable decarboxylative couplings and olefination.
2019
The herquline family of natural products, including herquline C47 (49), has intrigued and confounded the synthetic community since their first isolation by Omura in 1979 (Figure 4).48 Centered on a [6.12.6.6] tetracyclic core, these strained alkaloid natural products possess an unusual reduced dityrosine cyclophane moiety. Successive NADH-mediated reductions are involved in the biosynthesis (from the parent dityrosine macrocycle); we therefore49 sought to recapitulate Nature’s direct and ideal strategy toward the herqulines in our own synthetic effort. Beginning from appropriately substituted iodotyrosines (45 and 46), a straightforward sequence rapidly led to the 12-membered macrocycle 47,50 setting the stage for the key reductive endgame. After testing every conceivable variable from reaction conditions to order of operations, it was ultimately discovered that leveraging a Birch reduction on this highly strained macrocycle was a tractable solution. For the next reduction, Ir-catalyzed, silane mediated conditions were singularly effective in delivering the desired piperazine 48 in 86% yield on gram scale.51 Ketalization of the vinyl methyl ether found in 48 proved pivotal to enable the final Birch reduction. This was presumably a consequence of the newly installed sp3 center that both changed the conformation of the macrocycle and removed a problematic double bond prone to over-reduction. In using biosynthesis to guide the design, we were able to execute a simple and direct synthesis of the herqulines. In fact, apart from the necessary ketalization step, all maneuvers conducted en route to the natural product were strategic (87% ideality). That step is a reminder that methods for chemo- selective arene reduction are still missing from the chemist’s toolbox.
Teleocidins52 are bacterially produced,53 potent inhibitors of protein-kinase-C activation54 exhibiting a hybrid amino-acid/terpene based biosynthetic origin (Figure 4).55 In Nature, these compounds are generated as a mixture of different isomers and reported synthetic routes have similarly led to mixtures of isomers.56 This stems from the challenging problem of long-distance stereochemical relay across molecular architectures. A convergent strategy was devised wherein the indole-containing portion was formed first using chiral-pool derived building blocks and then fused using a catalytic stereoselective method to append the terpene portion. Commencing from 4-bromoindole (50), the valine subunit was appended through electrochemical amination using an amidine-based ligand to access 51.57 The C- 3 tryptophol side chain was established through metalation and chiral aziridine ring opening. After macrolactamization and protection of the primary alcohol to form 52, the stage was set to couple this fragment with a suitable polyunsaturated hydro-carbon after priming the C-6 position for coupling through C- H borylation.58 With the boronic acid in hand after hydrolysis, application of Sigman’s redox relay Heck reaction with homoallylic alcohol 53 cleanly forged the desired quaternary center in 54 with admirable control (6.6-7:1 d.r.).59 Note that both isomers are found in the natural products, and simply changing the stereochemistry of the ligand delivered the opposite selectivity. The redox-economic nature of this powerful C-C bond forming method also prefaced the final ring closure, which was achieved via simple vinyllithium addition/Friedel-to generate a key Crafts alkylation sequence to provide 55. Only modest control could be achieved in this final step as a function of the Brønsted acid employed. Of the 11 steps, 36% of them involved unfortunate protecting group manipulations due to chemo-selectivity/reactivity requirements of the key skeletal bond forming reactions. Finally, the C-H borylation may seem strategic and did indeed simplify the synthesis, but an even more direct approach would have been C-6 palladation followed by Heck to avoid the intermediate C-B bond that is not found in the target.
2020
Tryptorubin A60 (62), isolated by Clardy in 2017,61 is a peptidic macrocycle bearing a remarkable oxidatively fused skeleton wherein one tryptophan is interconnected with neighboring tyrosine and tryptophan residues through C-N and C-C linkages respectively, creating significant strain (Figure 5). Our motivation for pursuing 62 was based purely on chemical curiosity as no bioactivity was reported for this family. The synthesis (starting with iodotyrosine 56) was designed using a convergent strategy wherein an Ullman-derived62 macrocyclic Ala-Trp-Tyr construct (59) could be coupled to a preoxidized Trp (pyrroloindoline)−Tyr (60) using Movassaghi’s Friedel- Crafts arylation63 to generate a key quaternary center. In practice, the natural product could only be obtained by using the indoline 59 rather than the indole 58. After appending the final amino acid (Ile) to provide 61, macrolactamization and reoxidation of the indoline to indole delivered 62 as a single isomer. At the outset, neither our group nor Clardy’s team had any idea that this natural product could possibly exist as a different isomer. In fact, our original synthesis used intermediate 58 following a similar route to deliver an isomer of 62 whose identity was uncovered as the noncanonical atropisomer of 63. For geometric reasons, the indoline oxidation state served to enforce the formation of only one isomer. Given the increased interest in complex macrocycles in drug discovery,64 this kind of isomerism may be important to consider during the planning stages of synthesis and may be more widespread in natural product chemistry than previously appreciated. Had we carried out the initial synthesis with such a plan, this unusual type of isomerism in natural product chemistry may not have been uncovered. From an ideality standpoint this synthesis clearly suffers from the challenges that plague most peptide syntheses: an overreliance on PG chemistry. In addition, the extraneous redox-fluctuation brought about by the indole-indoline-indole sequence clearly points to the difficulty in controlling planar chirality.
Figure 5.
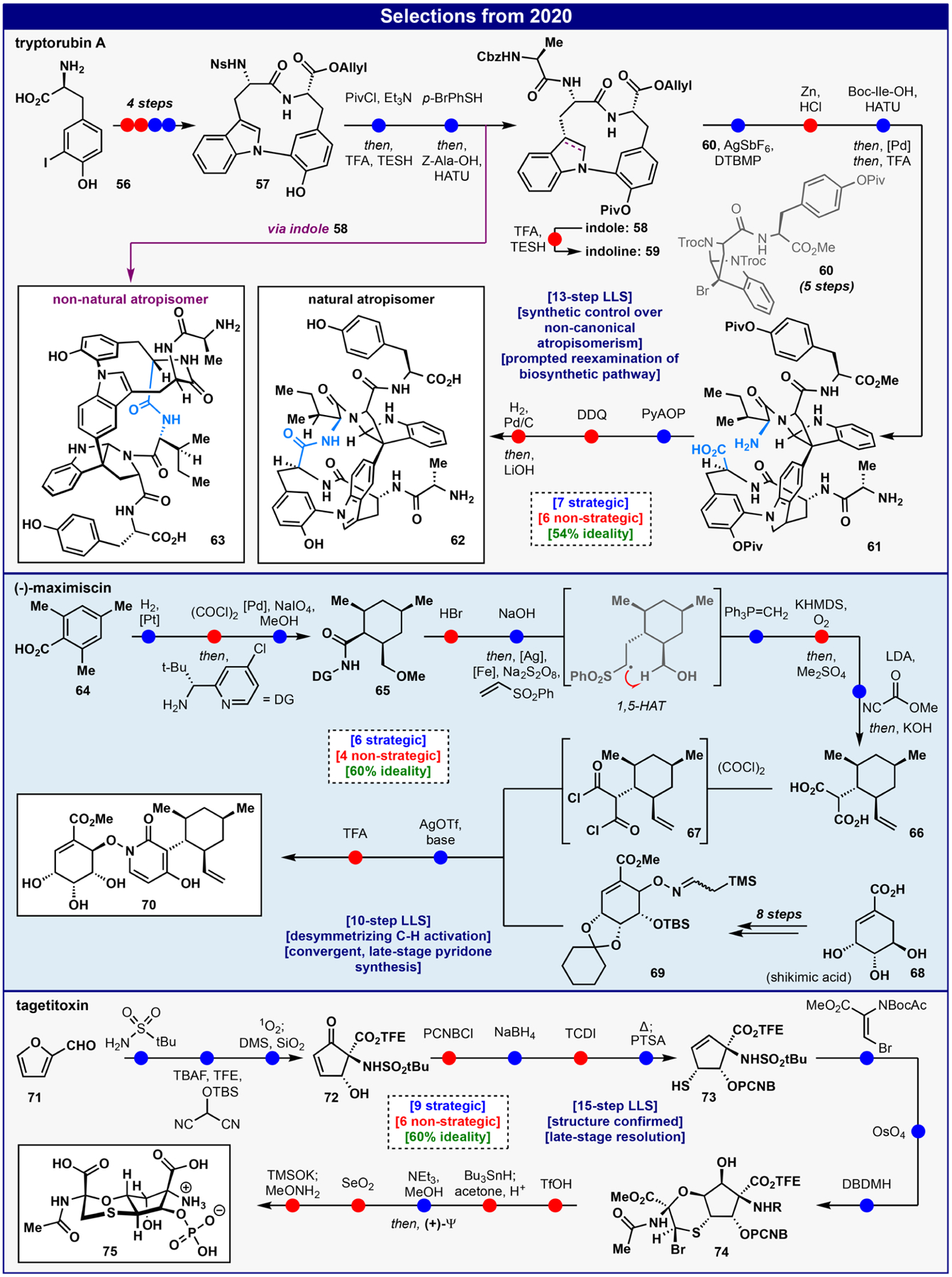
Select total syntheses from 2020.
Maximiscin65 (70) is a natural product of mixed biosynthetic origin that results from the union of three separate metabolic pathways. The molecule exhibits unique structural features, including the peculiar linkage between the N-hydroxypyridone and polyketide-derived fragments that exists as a mixture of interconverting atropisomers (Figure 5).66 This, along with its reported anticancer activity,67 motivated our efforts to pursue its synthesis. A bold late-stage pyridone synthesis was envisaged to cleave the central ring and produce two equally sized fragments 67 and 69. Application of radical retrosynthetic logic and the recognition of hidden symmetry via a desymmetrizing C-H activation allowed this highly convergent approach to begin from mesitylene-derived carboxylic acid 64 and shikimic acid 68. These plans were rapidly put to practice through hydrogenation of 64 leading to a meso carboxylic acid that after a remarkable desymmetrizing C-H activation produced 65 whereby all four stereocenters of the saturated ring were simultaneously defined. Thereafter, directing group cleavage delivered a transient lactone, which was hydrolyzed and directly subjected to a decarboxylative homologation cascade. This carefully orchestrated step, proceeding in >90% yield (gram-scale) utilized a unique Ag/Fe cocatalyst system to both establish the hindered C-C bond with concomitant stereoinversion and effect tandem oxidation of the aldehyde via a 1,5-hydrogen atom transfer (HAT). Subsequently, Wittig olefination, sulfone oxidationalkylation, and acylation with Mander’s reagent followed by in situ hydrolysis led to diacid 66. The pivotal union of both fragments necessitated the use of reaction partners with enhanced reactivity: a trimethylsilyl-substituted oxime ether and a diacyl triflate electrophile generated in situ. This push-pull activation system (“aza-Sakurai”) enabled the construction of the atropisomeric central pyridone ring and provided maximiscin in a 10-step LLS (60% ideality) after deprotection. While concise, common issues relating to C-H activations employing directing groups, a redox fluctuation resulting from a nonideal radical acceptor, and deprotection of a polyol plagued 40% of the steps. This highlights the need for further development of C- H activation methods directed by native functionality that obviate the need for directing group manipulations.
Tagetitoxin68 (75, Figure 5) is a complex alkaloid that has been of intense interest to the scientific community for nearly a century due to its controversial structural assignment (revised 3 times, absolute configuration and optical rotation unknown),69 its collection of remarkable chemical features (fully oxidized cyclopentane core, multiple polar functional groups, more heteroatoms than carbon atoms), and unprecedented bioactivity (novel mechanism of RNA polymerase inhibition).70 Motivated by those compelling attributes, our assembly of the fully oxidized cyclopentane core of tagetitoxin commenced from the commodity chemical furfural (71) via sulfonamide condensation followed by a Mannich-based amino ester synthesis and subsequent oxidative furan rearrangement to furnish 72 in high yield and diastereoselectivity. To install the chiral sulfide functionality, a thio-[3,3]-rearrangement of a suitably substituted allylic alcohol was executed through a sequence involving p-cyanobenzoyl protection, stereoselective Luche reduction, thio-CDI adduct formation, and heating followed by acidic hydrolysis to deliver 73 as a single diastereomer. A thio-Michael addition/catalytic dihydroxylation/bromocyclization sequence afforded the fully substituted bicycle 74. To complete the synthesis of 75, the key phosphate group was installed using the chiral P(V) reagent (+)-Ψ,71 which was singularly successful in forging the key P-O bond and allowed for crystallization and divergent synthesis of each enantiomeric form of 75. The accompanying concession steps were carefully planned deprotections necessitated by the extremely polar nature of 75 along with the oxidative removal (SeO2) of an extra sulfur atom from the Ψ-adduct. Employing an RNA polymerase assay on each enantiomer then allowed for assignment of (+)-75 as the natural isolate. The low ideality of this 15-step route to 75 (60%) is unsurprising given the challenge of manipulating such a polar molecule. This synthesis highlights the need for chemoselective methods that reduce reliance on PG manipulations and strategies for the late-stage incorporation of such functionality in their native form.
CONCLUSION
It is amazingly hard to find a simple solution. A complicated one is relatively easy.
Elon Musk
The progression of organic chemistry can be closely linked to increases in the simplicity and scalability of total syntheses during a given era, just as the age of a tree can be estimated by measuring its growth rings. For example, most skilled practitioners can examine a total synthesis and without knowing when it was published, roughly estimate the decade in which it was reported. New methods and strategies drive the evolution of synthesis to greater levels of ideality, which facilitates an objective and dispassionate analysis of how far we still have to go. This account chronicles the motivations for some of the molecules we have pursued over the past five years along with the sometimes-sobering lessons unearthed by aiming for the ideal synthesis. Several themes emerge from such an analysis that point to future directions in the field, some of which were highlighted above in bold. The syntheses of antroquinonol A,15 pallambins,18 and araiosamines25 show how far the field has to go in terms of site-selective C-C bond formation in molecules with only subtle electronic differences between functional groups. Herqulines,47 tryptorubin A,60 and cladospolides33 remind us of the nonideal, yet often-employed, tactic of controlling selectivity through changes in oxidation state or temporary protection rather than through reagent/catalyst control. Maoecrystal V,20 prostaglandins,33 and subglutinols43 used powerfully simplifying reactions for their rapid construction yet still suffered from superfluous redox changes that set the stage for such maneuvers. Lugdunin,27 arylomycin,36 tryptorubin A, and tagetitoxin68 illuminate the reliance we still have on protecting group chemistry as a result of nonchemoselective reactions. Finally, teleocidins52 and maximiscin65 show the potential of C-H functionalization logic to simplify synthesis, even when those substrates require extra effort to install directing functionality. We also note that several of the syntheses above benefitted from a strategy of employing tandem one-pot sequences of mutually compatible reactions to further streamline routes. This is loosely analogous to biosynthesis where, for example, C-C bond formations, oxidations, and reductions can routinely happen in the same “vessel”. The continuous pursuit of mild and chemoselective methods will make such tandem sequences even more common.
Evaluating syntheses through the lens of ideality can pinpoint areas of methodology and strategy that can have the greatest impact in pushing the field forward. Over the decades to come, as the shortcomings delineated above drive solutions, the syntheses of this era will appear ancient; such an outcome can only happen through the hard work of continuous innovation and creative invention of the organic chemist.
ACKNOWLEDGMENTS
The authors thank Scripps Research, the NIH (postdoctoral fellowship for C.R.P.), Bristol-Myers Squibb (graduate fellowships for D.S.P and K.S.M), and the NSF-GRFP (D.S.P) for their support.
Funding Financial support for the work included in this account was provided by Bristol-Myers-Squibb, LEO Pharmaceuticals, and the NIH/NIGMS (GM118176, GM-097444). Financial support in the form of fellowships for the contributors of the work included in this account was provided by NSF GRFP, NIH, the National Defense Science and Engineering Graduate Fellowship (NDSEG) Program, Bristol-Myers-Squibb, Vividion Therapeutics, Bayer Science, Nankai University, the ACS-MEDI Predoctoral Fellowship Program, the Swiss National Science Foundation, JSPS, the Honjo International Scholarship Program, Fujian Juhong Trade & Business Co., the George E. Hewitt Foundation, the German Research Foundation, the Tsinghua Xuetang Talent Program, and the Fulbright Scholar Program.
Biographies
David S. Peters was born in San Francisco, CA, and received his B.A. in biochemistry from the University of San Diego. He is currently a graduate student in Prof. Baran’s laboratory pursuing the synthesis of antibacterial natural products.
Cody Ross Pitts is from Waterbury, CT. He obtained a B.S. in chemistry with minors in physics and musical theater from Monmouth University (2010), completed his Ph.D. at Johns Hopkins University (2011–2017), and conducted research abroad as an ETH Zürich postdoctoral fellow (2017–2019). Currently, he is an NIH postdoctoral fellow at Scripps Research.
Kyle S. McClymont was born in Ottawa, Canada, and received his B.Sc. and M.Sc. from the University of Ottawa. He then escaped the cold to complete a Ph.D. with Phil S. Baran at Scripps Research in La Jolla and currently works in discovery chemistry at Merck South San Francisco.
Thomas P. Stratton was born in Cranford, New Jersey, and received his B.A. in chemistry from Rutgers University-Newark. He subsequently pursued doctoral studies in the laboratory of Phil S. Baran at Scripps Research, studying alkaloid natural product syntheses. Following completion of his Ph.D., Tom joined the medicinal chemistry department at Gilead Sciences, Inc. He currently resides in San Francisco.
Cheng Bi was born in China and obtained his B.S. in Chemistry from Nankai University in 2018. He then joined Prof. Baran’s laboratory as a graduate student focusing on natural product total synthesis.
Phil S. Baran completed his undergraduate education at New York University in 1997. After earning his Ph.D. at Scripps Research in 2001, he pursued postdoctoral studies at Harvard University until 2003, at which point he returned to TSRI to begin his independent career. He was promoted to the rank of professor in 2008 and is currently the Darlene Shiley Professor of Chemistry.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Newhouse T; Baran PS; Hoffmann RW The economies of synthesis. Chem. Soc. Rev 2009, 38, 3010. [DOI] [PMC free article] [PubMed] [Google Scholar]; The concepts of step, atom, and redox economy are discussed and contextualized with case studies.
- (2).Gaich T; Baran PS Aiming for the Ideal Synthesis. J. Org. Chem 2010, 75, 4657–4673. [DOI] [PubMed] [Google Scholar]; A simple quantification of “ideality” in synthesis is presented alongside a self-evaluation of total syntheses from our laboratory over the seven years prior.
- (3).Kuttruff CA; Eastgate MD; Baran PS Natural product synthesis in the age of scalability. Nat. Prod. Rep 2014, 31, 419–432. [DOI] [PubMed] [Google Scholar]; How the economies of synthesis can be leveraged to access natural products in a scalable fashion is analyzed herein.
- (4).Hendrickson JB Systematic synthesis design. IV. Numerical codification of construction reactions. J. Am. Chem. Soc 1975, 97, 5784–5800. [Google Scholar]
- (5).For a recent example of a 4-step total synthesis see:; Zhuang Z; Herron A; Liu S; Yu J-Q Rapid Construction of Tetralin, Chromane, and Indane Motifs via Cyclative C-H/C-H Coupling: Four-Step Total Synthesis of (±)-Russujaponol F. chemrxiv.org 2020, DOI: 10.26434/chemrxiv.13250294.v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Wender PA; Verma VA; Paxton TJ; Pillow TH Function-Oriented Synthesis, Step Economy, and Drug Design. Acc. Chem. Res 2008, 41, 40–49. [DOI] [PubMed] [Google Scholar]
- (7).Trost B The atom economy-a search for synthetic efficiency. Science 1991, 254, 1471–1477. [DOI] [PubMed] [Google Scholar]
- (8).Burns NZ; Baran PS; Hoffmann RW Redox Economy in Organic Synthesis. Angew. Chem., Int. Ed 2009, 48, 2854–2867. [DOI] [PubMed] [Google Scholar]
- (9).Bertz SH Convergence, molecular complexity, and synthetic analysis. J. Am. Chem. Soc 1982, 104, 5801–5803. [Google Scholar]
- (10).Hayashi Y Pot economy and one-pot synthesis. Chem. Sci 2016, 7, 866–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Roschangar F; Zhou Y; Constable DJC; Colberg J; Dickson DP; Dunn PJ; Eastgate MD; Gallou F; Hayler JD; Koenig SG; Kopach ME; Leahy DK; Mergelsberg I; Scholz U; Smith AG; Henry M; Mulder J; Brandenburg J; Dehli JR; Fandrick DR; Fandrick KR; Gnad-Badouin F; Zerban G; Groll K; Anastas PT; Sheldon RA; Senanayake CH Inspiring process innovation via an improved green manufacturing metric: iGAL. Green Chem 2018, 20, 2206–2211. [Google Scholar]
- (12).(a) Borovika A; Albrecht J; Li J; Wells AS; Briddell C; Dillon BR; Diorazio LJ; Gage JR; Gallou F; Koenig SG; Kopach ME; Leahy DK; Martinez I; Olbrich M; Piper JL; Roschangar F; Sherer EC; Eastgate MD The PMI Predictor app to enable green-by-design chemical synthesis. Nat. Sustain 2019, 2, 1034–1040. [Google Scholar]; (b) Jimenez-Gonzalez C; Ponder CS; Broxterman QB; Manley JB Using the Right Green Yardstick: Why Process Mass Intensity Is Used in the Pharmaceutical Industry To Drive More Sustainable Processes. Org. Process Res. Dev 2011, 15, 912–917. [Google Scholar]
- (13).Ishihara Y; Montero A; Baran PS The Portable Chemist’s Consultant: A Survival Guide for Discovery, Process, and Radiolabeling. Apple Publishing Group, 2013. [Google Scholar]
- (14).Kanda Y; Ishihara Y; Wilde NC; Baran PS Two-Phase Total Synthesis of Taxanes: Tactics and Strategies. J. Org. Chem 2020, 85, 10293–10320. [DOI] [PubMed] [Google Scholar]
- (15).Villaume MT; Sella E; Saul G; Borzilleri RM; Fargnoli J; Johnston KA; Zhang H; Fereshteh MP; Dhar TGM; Baran PS Antroquinonol A: Scalable Synthesis and Preclinical Biology of a Phase 2 Drug Candidate. ACS Cent. Sci 2016, 2, 27–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Lee T-H; Lee C-K; Tsou W-L; Liu S-Y; Kuo M-T; Wen W-C A New Cytotoxic Agent from Solid-State Fermented Mycelium of Antrodia camphorata. Planta Med. 2007, 73, 1412–1415. [DOI] [PubMed] [Google Scholar]
- (17).(a) Clinical Trials. https://goldenbiotech.com/en/antroquinonol-hocena-clinical-trials-information/ (accessed 11/22/ 2020).; (b) ClinicalTrials.gov Identifiers: NCT04523181, NCT03310632, NCT02047344, NCT02047344, NCT02047344, NCT02719028, NCT02719028, NCT04112147, NCT03622463, NCT04110873.
- (18).Martinez LP; Umemiya S; Wengryniuk SE; Baran PS 11-Step Total Synthesis of Pallambins C and D. J. Am. Chem. Soc 2016, 138, 7536–7539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Maezaki N; Gijsen HJM; Sun L-Q; Paquette LA Evaluation of Furan Photooxygenation as a Device for Construction of the Zaragozic Acid (Squalestatin) Core. J. Org. Chem 1996, 61, 6685− 6692. [DOI] [PubMed] [Google Scholar]
- (20).Cernijenko A; Risgaard R; Baran PS 11-Step Total Synthesis of (−)-Maoecrystal V. J. Am. Chem. Soc 2016, 138, 9425–9428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Li S-H; Wang J; Niu X-M; Shen Y-H; Zhang H-J; Sun H-D; Li M-L; Tian Q-E; Lu Y; Cao P; Zheng Q-T Maoecrystal V, Cytotoxic Diterpenoid with a Novel C19 Skeleton from Isodon eriocalyx (Dunn.) Hara. Org. Lett 2004, 6, 4327–4330. [DOI] [PubMed] [Google Scholar]
- (22).(a) Gong J; Lin G; Sun W; Li C-C; Yang Z Total Synthesis of (±) Maoecrystal V. J. Am. Chem. Soc 2010, 132, 16745–16746. [DOI] [PubMed] [Google Scholar]; (b) Lu P; Gu Z; Zakarian A Total Synthesis of Maoecrystal V: Early-Stage C-H Functionalization and Lactone Assembly by Radical Cyclization. J. Am. Chem. Soc 2013, 135, 14552–14555. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Peng F; Danishefsky SJ Total Synthesis of (±)-Maoecrystal V. J. Am. Chem. Soc 2012, 134, 18860–18867. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Zhang W.-b.; Shao W.-b.; Li F.-z.; Gong J.-x.; Yang Z Asymmetric Total Synthesis of (−)-Maoecrystal V. Chem. - Asian J 2015, 10, 1874–1880. [DOI] [PubMed] [Google Scholar]; (e) Zheng C; Dubovyk I; Lazarski KE; Thomson RJ Enantioselective Total Synthesis of (−)-Maoecrystal V. J. Am. Chem. Soc 2014, 136, 17750–17756. [DOI] [PubMed] [Google Scholar]
- (23).Han Q-B; Cheung S; Tai J; Qiao C-F; Song J-Z; Tso T-F; Sun H-D; Xu H-X Maoecrystal Z, a Cytotoxic Diterpene from Isodon eriocalyx with a Unique Skeleton. Org. Lett 2006, 8, 4727–4730. [DOI] [PubMed] [Google Scholar]
- (24).Seiple IB; Su S; Young IS; Nakamura A; Yamaguchi J; Jørgensen L; Rodriguez RA; O’Malley DP; Gaich T; Köck M; Baran PS Enantioselective Total Syntheses of (−)-Palau’amine, (−)-Axinellamines, and (−)-Massadines. J. Am. Chem. Soc 2011, 133, 14710–14726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Tian M; Yan M; Baran PS 11-Step Total Synthesis of Araiosamines. J. Am. Chem. Soc 2016, 138, 14234–14237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Wei X; Henriksen NM; Skalicky JJ; Harper MK; Cheatham TE; Ireland CM; Van Wagoner RM Araiosamines A- D: Tris-bromoindole Cyclic Guanidine Alkaloids from the Marine Sponge Clathria (Thalysias) araiosa. J. Org. Chem 2011, 76, 5515− 5523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Malins LR; deGruyter JN; Robbins KJ; Scola PM; Eastgate MD; Ghadiri MR; Baran PS Peptide Macrocyclization Inspired by Non-Ribosomal Imine Natural Products. J. Am. Chem. Soc 2017, 139, 5233–5241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Becker JE; Moore RE; Moore BS Cloning, sequencing, and biochemical characterization of the nostocyclopeptide biosynthetic gene cluster: molecular basis for imine macrocyclization. Gene 2004, 325, 35–42. [DOI] [PubMed] [Google Scholar]
- (29).Zipperer A; Konnerth MC; Laux C; Berscheid A; Janek D; Weidenmaier C; Burian M; Schilling NA; Slavetinsky C; Marschal M; Willmann M; Kalbacher H; Schittek B; Brötz-Oesterhelt H; Grond S; Peschel A; Krismer B Human commensals producing a novel antibiotic impair pathogen colonization. Nature 2016, 535, 511–516. [DOI] [PubMed] [Google Scholar]
- (30).Kricheldorf HR Polypeptides and 100 Years of Chemistry of α-Amino Acid N-Carboxyanhydrides. Angew. Chem., Int. Ed 2006, 45, 5752–5784. [DOI] [PubMed] [Google Scholar]
- (31).Si D; Sekar NM; Kaliappan KP A flexible and unified strategy for syntheses of cladospolides A, B, C, and iso-cladospolide B. Org. Biomol. Chem 2011, 9, 6988. [DOI] [PubMed] [Google Scholar]
- (32).Smith JM; Harwood SJ; Baran PS Radical Retrosynthesis. Acc. Chem. Res 2018, 51, 1807–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Edwards JT; Merchant RR; McClymont KS; Knouse KW; Qin T; Malins LR; Vokits B; Shaw SA; Bao D-H; Wei F-L; Zhou T; Eastgate MD; Baran PS Decarboxylative alkenylation. Nature 2017, 545, 213–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Das S; Chandrasekhar S; Yadav JS; Grée R Recent Developments in the Synthesis of Prostaglandins and Analogues. Chem. Rev 2007, 107, 3286–3337. [DOI] [PubMed] [Google Scholar]
- (35).Corey EJ; Weinshenker NM; Schaaf TK; Huber W Stereo-controlled synthesis of dl-prostaglandins F2.alpha. and E2. J. Am. Chem. Soc 1969, 91, 5675–5677. [DOI] [PubMed] [Google Scholar]
- (36).Peters DS; Romesberg FE; Baran PS Scalable Access to Arylomycins via C-H Functionalization Logic. J. Am. Chem. Soc 2018, 140, 2072–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).(a) Roberts TC; Smith PA; Cirz RT; Romesberg FE Structural and initial biological analysis of synthetic arylomycin A2. J. Am. Chem. Soc 2007, 129, 15830–15838. [DOI] [PubMed] [Google Scholar]; (b) Smith PA; Roberts TC; Romesberg FE Broad-spectrum antibiotic activity of the arylomycin natural products is masked by natural target mutations. Chem. Biol 2010, 17, 1223–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Smith PA; Koehler MFT; Girgis HS; Yan D; Chen Y; Chen Y; Crawford JJ; Durk MR; Higuchi RI; Kang J; Murray J; Paraselli P; Park S; Phung W; Quinn JG; Roberts TC; Rougé L; Schwarz JB; Skippington E; Wai J; Xu M; Yu Z; Zhang H; Tan M-W; Heise CE Optimized arylomycins are a new class of Gram-negative antibiotics. Nature 2018, 561, 189–194. [DOI] [PubMed] [Google Scholar]
- (39).Dufour J; Neuville L; Zhu J Intramolecular Suzuki-Miyaura Reaction for the Total Synthesis of Signal Peptidase Inhibitors, Arylomycins A2 and B2. Chem. - Eur. J 2010, 16, 10523–10534. [DOI] [PubMed] [Google Scholar]
- (40).Walsh SI; Peters DS; Smith PA; Craney A; Dix MM; Cravatt BF; Romesberg FE The inhibition of protein secretion in Escherichia coli and sub-MIC effects of arylomycin antibiotics. Antimicrob. Agents Chemother 2019, 63, e01253–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Lee JC; Lobkovsky E; Pliam NB; Strobel G; Clardy J Subglutinols A and B: Immunosuppressive compounds from the endophytic fungus Fusarium subglutinans. J. Org. Chem 1995, 60, 7076–7077. [Google Scholar]
- (42).(a) Kikuchi T; Mineta M; Ohtaka J; Matsumoto N; Katoh T Enantioselective Total Synthesis of (−)-Subglutinols A and B: Potential Immunosuppressive Agents Isolated from a Microorganism. Eur. J. Org. Chem 2011, 2011, 5020–5030. [Google Scholar]; (b) Kim H; Baker JB; Lee S-U; Park Y; Bolduc KL; Park H-B; Dickens MG; Lee D-S; Kim Y; Kim SH; Hong J Stereoselective Synthesis and Osteogenic Activity of Subglutinols A and B. J. Am. Chem. Soc 2009, 131, 3192–3194. [DOI] [PubMed] [Google Scholar]
- (43).Merchant RR; Oberg KM; Lin Y; Novak AJE; Felding J; Baran PS Divergent Synthesis of Pyrone Diterpenes via Radical Cross Coupling. J. Am. Chem. Soc 2018, 140, 7462–7465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Snider BB Manganese(III)-Based Oxidative Free-Radical Cyclizations. Chem. Rev 1996, 96, 339–364. [DOI] [PubMed] [Google Scholar]
- (45).Qin T; Malins LR; Edwards JT; Merchant RR; Novak AJ; Zhong JZ; Mills RB; Yan M; Yuan C; Eastgate MD; Baran PS Nickel-Catalyzed Barton Decarboxylation and Giese Reactions: A Practical Take on Classic Transforms. Angew. Chem., Int. Ed 2017, 56, 260–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Zhang F; Danishefsky SJ An Efficient Stereoselective Total Synthesis of dl-Sesquicillin, a Glucocorticoid Antagonist. Angew. Chem., Int. Ed 2002, 41, 1434–1437. [DOI] [PubMed] [Google Scholar]
- (47).He C; Stratton TP; Baran PS Concise Total Synthesis of Herqulines B and C. J. Am. Chem. Soc 2019, 141, 29–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Enomoto Y; Shiomi K; Hayashi M; Masuma R; Kawakubo T; Tomosawa K; Iwai Y; Omura S Herquline B, a New Platelet Aggregation Inhibitor Produced by Penicillium herquei Fg-372. J. Antibiot 1996, 49, 50–53. [DOI] [PubMed] [Google Scholar]
- (49).Yu X; Liu F; Zou Y; Tang M-C; Hang L; Houk KN; Tang Y Biosynthesis of Strained Piperazine Alkaloids: Uncovering the Concise Pathway of Herquline A. J. Am. Chem. Soc 2016, 138, 13529− 13532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Zhu X; McAtee CC; Schindler CS Scalable Synthesis of Mycocyclosin. Org. Lett 2018, 20, 2862–2866. [DOI] [PubMed] [Google Scholar]
- (51).Cheng C; Brookhart M Iridium-Catalyzed Reduction of Secondary Amides to Secondary Amines and Imines by Diethylsilane. J. Am. Chem. Soc 2012, 134, 11304–11307. [DOI] [PubMed] [Google Scholar]
- (52).Nakamura H; Yasui K; Kanda Y; Baran PS 11-Step Total Synthesis of Teleocidins B-1-B-4. J. Am. Chem. Soc 2019, 141, 1494− 1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Takashima M; Sakai H A New Toxic Substance, Teleocidin, Produced by Streptomyces. Bull. Agric. Chem. Soc. Jpn 1960, 24, 647− 655. [Google Scholar]
- (54).Umezawa K; Weinstein IB; Horowitz A; Fujiki H; Matsushima T; Sugimura T Similarity of teleocidin B and phorbol ester tumour promoters in effects on membrane receptors. Nature 1981, 290, 411–413. [DOI] [PubMed] [Google Scholar]
- (55).Awakawa T; Abe I Biosynthesis of the teleocidin-type terpenoid indole alkaloids. Org. Biomol. Chem 2018, 16, 4746–4752. [DOI] [PubMed] [Google Scholar]
- (56).(a) Nakatsuka S.-i.; Masuda T; Goto T Total syntheses of (±)-teleocidin B-3 and B-4. Tetrahedron Lett 1987, 28, 3671–3674. [Google Scholar]; (b) Okabe K; Muratake H; Natsume M Synthesis of teleocidins A, B and their congeners. Part 3. Synthesis of dihydroteleocidin B-4 (dihydroteleocidin B), teleocidin B-3 and teleocidin B-4. Tetrahedron 1991, 47, 8559–8572. [Google Scholar]
- (57).Li C; Kawamata Y; Nakamura H; Vantourout JC; Liu Z; Hou Q; Bao D; Starr JT; Chen J; Yan M; Baran PS Electrochemically Enabled, Nickel-Catalyzed Amination. Angew. Chem., Int. Ed 2017, 56, 13088–13093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Feng Y; Holte D; Zoller J; Umemiya S; Simke LR; Baran PS Total Synthesis of Verruculogen and Fumitremorgin A Enabled by Ligand-Controlled C-H Borylation. J. Am. Chem. Soc 2015, 137, 10160–10163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Mei T-S; Patel HH; Sigman MS Enantioselective construction of remote quaternary stereocentres. Nature 2014, 508, 340–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Reisberg SH; Gao Y; Walker AS; Helfrich EJN; Clardy J; Baran PS Total synthesis reveals atypical atropisomerism in a small-molecule natural product, tryptorubin A. Science 2020, 367, 458− 463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Wyche TP; Ruzzini AC; Schwab L; Currie CR; Clardy J Tryptorubin A: A Polycyclic Peptide from a Fungus-Derived Streptomycete. J. Am. Chem. Soc 2017, 139, 12899–12902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Kunz K; Scholz U; Ganzer D Renaissance of Ullmann and Goldberg Reactions - Progress in Copper Catalyzed C-N-, C-O- and CS-Coupling. Synlett 2003, 2428–2439. [Google Scholar]
- (63).Kim J; Movassaghi M Concise Total Synthesis and Stereochemical Revision of (+)-Naseseazines A and B: Regioselective Arylative Dimerization of Diketopiperazine Alkaloids. J. Am. Chem. Soc 2011, 133, 14940–14943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).(a) Cummings MD; Sekharan S Structure-Based Macrocycle Design in Small-Molecule Drug Discovery and Simple Metrics To Identify Opportunities for Macrocyclization of Small-Molecule Ligands. J. Med. Chem 2019, 62, 6843–6853. [DOI] [PubMed] [Google Scholar]; (b) Driggers EM; Hale SP; Lee J; Terrett NK The exploration of macrocycles for drug discovery — an underexploited structural class. Nat. Rev. Drug Discovery 2008, 7, 608–624. [DOI] [PubMed] [Google Scholar]
- (65).McClymont KS; Wang F-Y; Minakar A; Baran PS Total Synthesis of (−)-Maximiscin. J. Am. Chem. Soc 2020, 142, 8608–8613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Du L; Robles AJ; King JB; Powell DR; Miller AN; Mooberry SL; Cichewicz RH Crowdsourcing Natural Products Discovery to Access Uncharted Dimensions of Fungal Metabolite Diversity. Angew. Chem., Int. Ed 2014, 53, 804–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Robles AJ; Du L; Cichewicz RH; Mooberry SL Maximiscin Induces DNA Damage, Activates DNA Damage Response Pathways, and Has Selective Cytotoxic Activity against a Subtype of Triple-Negative Breast Cancer. J. Nat. Prod 2016, 79, 1822–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).He C; Chu H; Stratton TP; Kossler D; Eberle KJ; Flood DT; Baran PS Total Synthesis of Tagetitoxin. J. Am. Chem. Soc 2020, 142, 13683–13688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Aliev AE; Karu K; Mitchell RE; Porter MJ The structure of tagetitoxin. Org. Biomol. Chem 2016, 14, 238–245. [DOI] [PubMed] [Google Scholar]
- (70).(a) Mathews DE; Durbin RD Tagetitoxin inhibits RNA synthesis directed by RNA polymerases from chloroplasts and Escherichia coli. J. Biol. Chem 1990, 265, 493–498. [PubMed] [Google Scholar]; (b) Vassylyev DG; Svetlov V; Vassylyeva MN; Perederina A; Igarashi N; Matsugaki N; Wakatsuki S; Artsimovitch I Structural basis for transcription inhibition by tagetitoxin. Nat. Struct. Mol. Biol 2005, 12, 1086–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Yuzenkova Y; Roghanian M; Bochkareva A; Zenkin N Tagetitoxin inhibits transcription by stabilizing pretranslocated state of the elongation complex. Nucleic Acids Res. 2013, 41, 9257–9265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Knouse KW; deGruyter JN; Schmidt MA; Zheng B; Vantourout JC; Kingston C; Mercer SE; McDonald IM; Olson RE; Zhu Y; Hang C; Zhu J; Yuan C; Wang Q; Park P; Eastgate MD; Baran PS Unlocking P(V): Reagents for chiral phosphor-othioate synthesis. Science 2018, 361, 1234–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]