

Abstract
The presence of immunosuppressive innate immune cells such as myeloid derived suppressor cells (MDSCs), Ly6C-high monocytes, and tumor-associated macrophages (TAMs) at a tumor can inhibit effector T cell and NK cell function. Immune checkpoint blockade using anti-PD-1 antibody aims to overcome the immune suppressive environment, yet only a fraction of patients responds. Herein, we test the hypothesis that cargo-free PLG nanoparticles administered intravenously can divert circulating immune cells from the tumor microenvironment to enhance the efficacy of anti-PD-1 immunotherapy in the 4T1 mouse model of metastatic triple-negative breast cancer. In vitro studies demonstrate that these nanoparticles decrease the expression of MCP-1 by 5-fold and increase the expression of TNF-α by more than 2-fold upon uptake by innate immune cells. Intravenous administration of particles results in internalization by MDSCs and monocytes, with particles detected in the liver, lung, spleen, and primary tumor. Nanoparticle delivery decreased the abundance of MDSCs in circulation and in the lung, the latter being the primary metastatic site. Combined with anti-PD-1 antibody, nanoparticles significantly slowed tumor growth and resulted in a survival benefit. Gene expression analysis by GSEA indicated inflammatory myeloid cell pathways were downregulated in the lung and upregulated in the spleen and tumor. Upregulation of extrinsic apoptotic pathways was also observed in the primary tumor. Collectively, these results demonstrate that cargo-free PLG nanoparticles can reprogram immune cell responses and alter the tumor microenvironment in vivo to overcome the local immune suppression attributed to myeloid cells and enhance the efficacy of anti-PD-1 therapy.
Keywords: nanoparticles, metastasis, cancer immunotherapy, immune cell reprogramming, immune checkpoint blockade
Introduction
Tumor progression is aided in part by the function of innate immune cells, which can be induced toward an immune suppressive function by tumor-secreted factors [1]. Disease-induced cell types found at the primary tumor or metastatic niche include inflammatory monocytes (CD11b+ Ly6C-hi cells in mice), tumor-associated macrophages (TAMs), and myeloid-derived suppressor cells (MDSCs). Circulating inflammatory monocytes differentiate into tumor-associated macrophages (TAMS) in tissues, and have immunosuppressive phenotypes and induce angiogenesis to aid metastasis [2], [3]. Additionally, myeloid cell recruitment has been shown to be critical for metastatic progression [4]. MDSCs are a class of neutrophil that has been implicated in aiding tumor growth and metastasis, especially to the lung [5], [6]. These cells normally serve to regulate the immune response to pathogens, but in the context of cancer, MDSCs secrete factors, including immunosuppressive cytokines and reactive oxygen species (ROS), to inhibit anti-tumor T cell and NK cell function [7]. These cells contribute to the failure of immune checkpoint blockade or T-cell immunotherapies [8].
The immunosuppression and pro-tumor functions of myeloid cells have been the targets of numerous therapies to alter either their numbers or phenotypes. Small molecule inhibitors have been applied to inhibit cytokine secretion that recruits myeloid cells [9], [10] or T cell function [8], [11], or repolarizing cells such as tumor-associated macrophages [12]. Small molecule inhibitors of specific protein activators of MDSCs and TAMs have been shown to reduce their number and abrogate disease [13], [14]. Depletion of one or more cell types has also been studied in this context [13], [15], [16]. However, the large number of cells, cytokines, and other factors that contribute to the immune suppression limit the efficacy of therapies that target a single cell or protein [17]. In addition, myeloid cell phenotypes within various tissues are distinct [18], which motivates the development of therapies that can elicit a tissue-dependent response.
An emerging approach for modulating innate immune cell response in tissues are nanoparticles administered intravenously that target circulating immune cells. Cargo-free polymer nanoparticles (NPs) have been shown to redirect the trafficking of phagocytic innate immune cells upon, and moderate the disease-induced aberrant behavior of these cells. NPs have highly negative surface charge that result in internalization through scavenger receptors including MARCO [19], and the specificity and biodistribution of polymeric NPs can be tuned with changes to the polymer backbone. As such, internalization of NPs is not limited to targeting ligands [20], and they do not induce systemic responses in multiple tissue as with steroids or NSAIDs. The therapeutic benefit of these NPs has been demonstrated in West Nile virus infection, ischemic reperfusion injury [19], traumatic brain injury [21], as well as in spinal cord injury [22]. These injuries are associated with increased inflammation, which contrasts with cancer that is associated with immune suppression to limit immune cell killing of tumor cells [23].
In this report, we test the hypothesis that cargo-free PLG nanoparticles administered intravenously divert circulating immune cells from the tumor microenvironment or metastatic sites, altering the immune responses and enhancing the efficacy of anti-PD1 immunotherapy. In the orthotopic 4T1 mouse model of metastatic triple negative breast cancer, disease-induced myeloid cells rapidly increase in number systemically with disease progression [24] and metastatic 4T1 cells readily colonize the lung due to the host of factors secreted by aberrant monocytes, macrophages, and MDSCs. These myeloid cells have also been shown to directly contribute to resistance to anti-PD-1 and anti-CTLA-4 in the 4T1 model [13]. The cell types and biodistribution of nanoparticles are analyzed, and the impact of the particles on the cell phenotypes are analyzed both in vitro and in vivo. Particle administration is investigated with respect to the growth of the primary tumor and survival. Finally, the impact of nanoparticles on gene expression within the primary tumor and metastatic site is analyzed. Reprogramming innate immune cell responses within the primary tumor and metastatic tissues represents a new opportunity for this class of nanoparticles that target innate cells in circulation, which has the potential to improve the therapeutic benefit of anti-PD-1 and other immunotherapies.
Materials and Methods
Nanoparticle fabrication
Particles were synthesized as previously described [25], [26]. Briefly, high-molecular weight (acid-terminated, inherent viscosity 0.55–75 dL/g, Lactel) or low-molecular weight (acid-terminated, inherent viscosity 0.15–0.25 dL/g, Lactel) PLG polymer were dissolved at 20% w/v in dichlormethane. The dissolved polymer was added to either 1% w/v (poly(ethylene-alt-maleic anhydride) (PEMA, MW 400,000, Polysciences, Inc) or 2% w/v polyvinylalcohol (PVA, MW 30,000–70,000, Polysciences, Inc) solution and the mixture was sonicated using a Cole-Parmer CPX130 Ultrasonic Processor to form the nanoparticles. The nanoparticle solution was poured immediately into stirring 0.5% w/v PEMA or 0.5% w/v PVA and organic solvent was evaporated overnight. The nanoparticles were then pelleted by centrifugation, the remaining solution was removed, and the nanoparticle pellet was resuspended in DI water. Nanoparticles were lyophilized for storage following three of these washes in DI water. To fabricate fluorescent nanoparticles, acid-terminated 50:50 Poly(DL-Lactide-co-Glycolide)(PLG) polymer (acid-terminated, inherent viscosity 0.55–75 dL/g, Lactel) was first conjugated with cyanine 5.5 amine dye (Lumiprobe) using N-(3-Dimethylaminopropyl)-N’-ethylcarbodiimide hydrochloride (EDC) (Sigma-Aldrich)/N-hydroxysuccinimide (NHS) (Thermo Fisher Scientific) chemistry. Unconjugated polymer and cyanine 5.5-conjugated polymer were then combined at 99:1 weight ratio. Polymer solution was used to make nanoparticles as described above.
Tumor cell culture and inoculation
4T1-luc2-tdTomato cells (Perkin Elmer) were expanded in RPMI 1640 + GlutaMAX (Thermo Fisher Scientific) with 10% FBS for 5 days at 37°C and 5% CO2 prior to inoculation. Cells were removed from culture flasks by incubation with trypsin for 10 minutes at 37°C and resuspended in RPMI 1640. Cells were then pelleted by centrifugation at 300 × g and washed with DPBS, and resuspended at 1 × 107 cells/mL of DPBS. Orthotopic tumor inoculations were performed by injection of 5 × 105 tumor cells resuspended in 50 μL DPBS (Life Technologies) into the fourth right mammary fat pad of 8- to 10-week-old female BALB/c mice (Jackson Laboratory). The cell line was confirmed to be pathogen free and authenticated by short tandem repeat DNA analysis and compared to the ATCC STR profile database (DDC Medical).
Nanoparticle and anti-PD-1 treatment
Nanoparticles were resuspended in DPBS (Life Technologies) at 1 mg in 200 μL and passed through a 35 μm filter mesh prior to intravenous administration via tail vein injection. Control animals received 200 μL of DPBS intravenously. InVivoMab anti-mouse anti-PD-1 (CD279) antibody (clone RMP1-14, Bio X Cell) was diluted in DPBS to a final concentration of 1 mg/mL immediately prior to i.p. injection. For therapeutic efficacy studies, NPs and anti-PD-1 were administered once every 3 days, for a total of 4 doses. For short-term nanoparticle biodistribution studies, a single dose of Cy5.5-NPs at 1mg in 200 μL of PBS was administered prior to organ explant at 12 or 48 hours post-inoculation. For studies of NP accumulation in tissues, Cy5.5-NPs were administered at a dose of 1 mg/200 μL for 6 days to allow for accumulation of NPs and to observe potential NP-induced cell trafficking (n = 4 PBS control, n = 5 NP). Organs were explanted at day 10 post inoculation and innate immune cell distribution was quantified by flow cytometry. For studies of NPs and blood-based immune cell distribution, a single dose of NPs at 1 mg/200 μL was administered and flow cytometry analysis of blood was performed 12 hours post injection.
Tumor size measurement and survival monitoring
Tumors were measured using standard electronic calipers (VWR) while mice were anesthetized with 2% v/v% isoflurane. Tumor volume was calculated as V = 0.5 × L × W2, where L is the length of the longest dimension of the tumor and W is the length of the tumor perpendicular to the longest dimension. Mice were monitored for tumor size and body conditioning to determine survival. Mice were euthanized if any of the following criteria were met: tumor size of > 2cm in any dimension, ulceration of more than 50% of the visible tumor area, partial paralysis due to tumor invasion of hind limb muscle, labored breathing, ascites, lethargy, or visible weight loss.
Ex-vivo fluorescence and bioluminescence imaging
Short-term nanoparticle distribution was measured in explanted whole organs following a single particle injection, with analysis of the organs after 12 or 48 hours using an IVIS Lumina LTE imaging system (Caliper Life Science). Fluorescence signal intensity at 675/700 Ex/Em is reported as photon flux in total photon count/cm2/steradian. Metastatic tumor burden of luciferase-expressing tumor cells in explanted lungs was measured by bioluminescence imaging with the IVIS. Briefly, lungs were incubated in 50 μM d-luciferin (Caliper) at room temperature for 10 minutes prior to imaging in the IVIS. Bioluminescence signal intensity is reported as integrated light flux (photons/sec) as calculated by the Living Image Software (Caliper Life Sciences).
Flow cytometry
Flow cytometry was performed on primary cells obtained from explanted organs. Spleens, lungs, primary tumors, and livers were minced and enzyme digested with Liberase TL or TM (Roche), then filtered through a 70 μm cell strainer (Corning) to obtain a cell suspension. Whole blood was collected by cardiac puncture using a 23 guage needle (BD) attached to a 1 mL BD Luer-Lok syringe and mixed with 10 v/v% 25 mM EDTA (Life Technologies). Red blood cell lysis of whole blood, spleens, and lungs was performed with ACK Lysing Buffer (Life Technologies). Cells were pelleted by centrifugation at 400 × g for 5 minutes and resuspended in MACS buffer. NP+ cells were identified by Cy5.5 fluorescence signal. Nonspecific staining was blocked with anti-CD16/32 (Biolegend) and samples were stained for innate immune markers with anti-mouse CD45 AF700, F4/80 PE-Cy7, Ly6G PacBlue, Ly6C FITC (Biologend), and CD11b BV510 (BD Biosciences). For adaptive immune markers, samples were stained with anti-mouse CD45 AF700, CD4 V500, CD8 FITC, CD19 PacBlue, and CD49b PE-Cy7 (Biolegend). Stained samples were analyzed using the MoFlo Astrios Flow Cytometer or CytoFLEX (Beckman Coulter), and data were processed using FlowJo (BD) with the gating strategies shown in Figure S1.
In vitro nanoparticle uptake and ELISA
Single cell suspensions were isolated from spleens and whole blood of tumor-bearing mice at 14 or 21 days post tumor inoculation as described above. For nanoparticle uptake assays, cells were resuspended in RPMI 1640 media with 10% FBS and seeded in 24-well plates at 1 × 106 cells per well. NPs were resuspended in DPBS at 1 mg/mL and added to the wells. Uptake was characterized following a 30 minute incubation at 37°C by flow cytometry as described above. For cytokine secretion assays, cells were resuspended in RPMI 1640 media and seeded in 96-well plates at 2.5 × 105 cells per well. For the evaluation of cytokine secretion by Gr1+ cells, Gr1+ splenocytes were sorted using the Myeloid-Derived Suppressor Cell Isolation Kit (Miltenyi Biotec), and cells were counted and resuspended in media. NPs were resuspended as described above and added to each well. Following incubation for 18 hours, cells were pelleted at 1,000 × g for 5 minutes and the supernatant was removed and diluted 4x for quantification of cytokine secretion by ELISA, which was performed by the Cancer Center Immunology Core at the University of Michigan.
Gene expression analysis by RNA-seq
Explanted tissues were flash-frozen by submerging them in isopentane on dry ice, and homogenized in TRIzol reagent (Thermo Fisher Scientific). RNA was extracted from each tissue sample with the Direct-zol RNA spin column kit (Zymo Research). Purified RNA concentration was measured by UV spectroscopy using a Nanodrop 2000c (Thermo Scientific) to confirm all samples had concentrations ≥ 10 ng/μL. RNA quality control, QuantSeq 3’ mRNA-seq library prep and quality control, and sequencing were performed by the Advanced Genomics Core at the University of Michigan. Samples were sequenced using the Illumina NextSeq 550 sequencer, and sequences were aligned to Gene Symbols. Raw sequencing counts were normalized and differential gene expression was calculated in R and using DESeq2 [27] [28]. For pathway analysis, mouse gene symbols for counts obtained through RNA-seq were first converted to human gene symbols using the biomaRt package in R. Gene Set Enrichment Analysis (GSEA) was performed on DESeq2-normalized counts and the corresponding human gene symbols for a total of 5,944 MsigDB gene sets (Hallmark, Reactome, PID, and Gene Ontology (GO)). A positive normalized enrichment score (NES) indicates enrichment in the treated cohort compared with PBS control. In comparisons between the two treatment groups, a positive NES indicates enrichment in the combination (NP + anti-PD-1) treatment group compared with the nanoparticle (NP) treatment group.
Statistical analysis
One-way or two-way ANOVA and Tukey’s multiple comparisons tests were performed on groups with more than 2 conditions. Two-tailed unpaired t test was used for single comparisons between two conditions. Median survival and survival curves were analyzed with the log-rank (Mantel-Cox) test for statistical significance. Following normalization of RNA-seq gene expression data with DESeq2, genes with adjusted p-value < 0.1 were considered differentially expressed. Statistical analyses were performed using Prism 8 (GraphPad Software) and R. A p-value < 0.05 was considered to be statistically significant and all values are expressed in mean ± standard deviation (SD). Error bars on plotted data represent standard error of the mean (SEM).
Results
Nanoparticle internalization by innate immune cells
PLG nanoparticle internalization by tumor-induced immune cells was initially investigated in vitro. By varying the surfactant type and PLG molecular weight, three fluorescently-labeled PLG NP formulations were synthesized (Fig 1A): PEMA-coated low-molecular weight (PEMA-Low), PEMA-coated high-molecular weight (PEMA-High), and PVA-coated high-molecular weight (PVA-High). These NPs were incubated with blood leukocytes from tumor-bearing mice at 5 μg/mL and 50 μg/mL NP concentration (Fig 1B, S2). Because PEMA-High NPs exhibited the maximal internalization at both concentrations, subsequent studies were performed with PEMA-High NPs. Myeloid-derived suppressor cells (MDSCs, CD11b+/Ly6Clo/−/Ly6G+) and other myeloid cells (CD11b+/Ly6C−/Ly6G−) showed increased Cy5.5-NP internalization at 50 μg/mL relative to 5 μg/mL (Fig 1C), whereas the increased concentration had no effect on the number of Cy5.5-NP+ monocytes as a percentage of total cells. As MDSCs comprise a large majority of immune cells in circulation, internalization was also quantified as the percentage of Cy5.5-NP+ cells relative to the number of single cells for each cell subtype (Fig 1D). At 5 μg/mL, nearly all monocytes are NP+ (89.4 ± 5.4%) while a lower percentage of MDSCs and other myeloid cells have lower relative internalization (23.7 ± 10.4% and 72.3 ± 14.5%, respectively). When the NP+ cell numbers are increased at 50 μg/mL, nearly all cells are Cy5.5-NP+ in all three cell populations (99.3 ± 0.5% of MDSCs, 99.9 ± 0.1% of monocytes, and 99.6 ± 0.2% of other myeloid cells). Taken together, these data demonstrate that monocytes more readily internalize NPs than other myeloid subtypes at low concentrations, yet nearly all MDSCs and other myeloid cells have internalized NPs.
Figure 1.
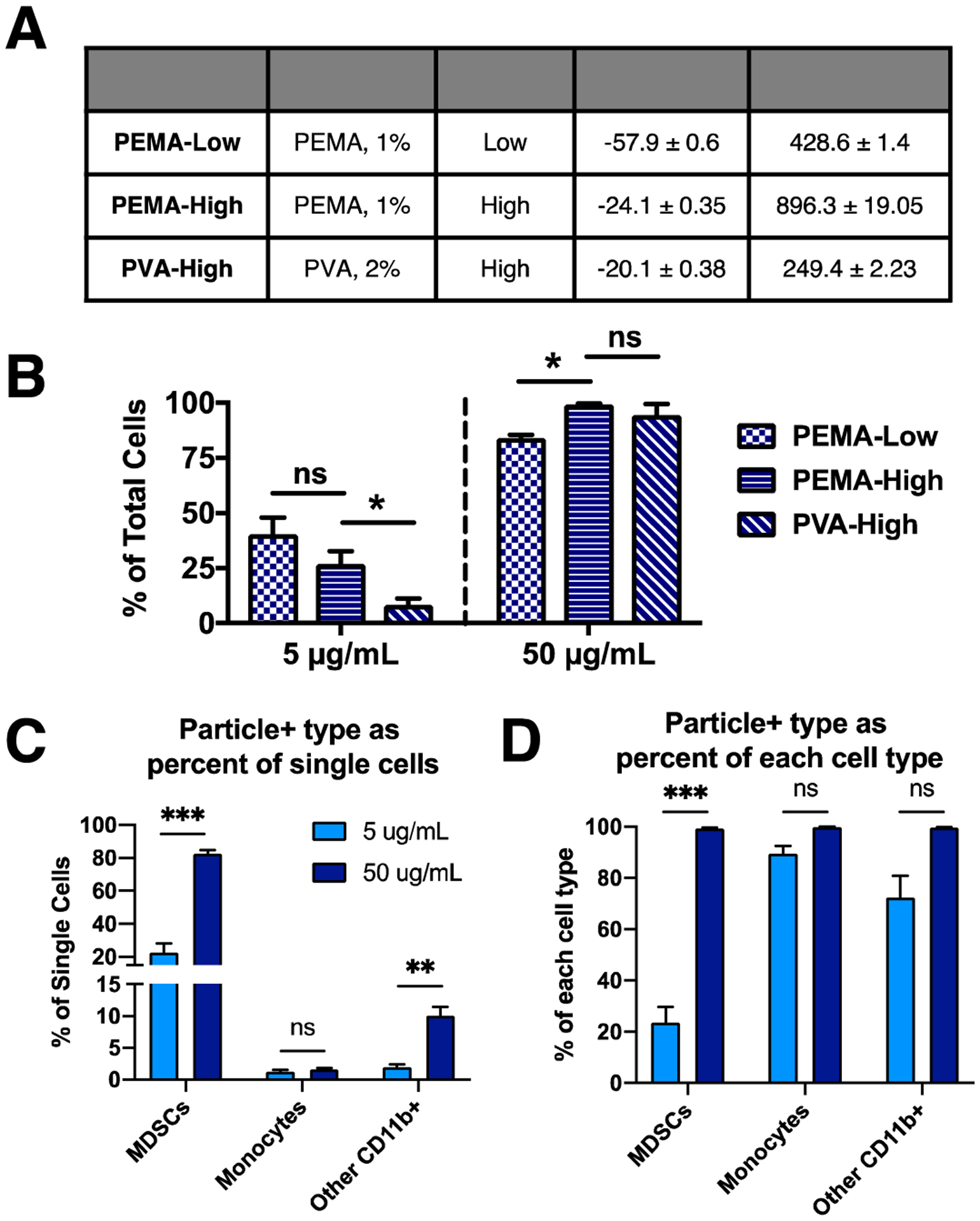
Cy5.5-NPs are internalized by innate immune cells and are distributed in disease-relevant tissues. (A) Three NP formulations with combinations of surfactant and PLG molecular weight were tested for internalization in vitro. (B) Blood leukocytes from 4T1 tumor-bearing mice at 21 days post inoculation were incubated in vitro with each formulation at 5 μg/mL and 50 μg/mL, and internalization was quantified. PEMA-High NPs exhibited high internalization at both concentrations, and subsequent studies were performed with the PEMA-High NPs. Internalization was quantified (C and D) for myeloid cell subtypes (C) as a percentage of all cells and (D) as a percentage of each cell subtype. CD11b+/Ly6Clo/−/Ly6G+ cells (MDSCs) and CD11b+/Ly6C−/Ly6G− cells (other myeloid cells) showed increased internalization with increase in NP concentration, while monocytes did not. These data indicate that monocytes more readily internalize NPs compared to MDSCs and other myeloid cells. A 2-way ANOVA with Tukey’s multiple comparisons test was performed (A and B), bars show mean ± SEM for n = 3 biological replicates per condition, where ** p < 0.01, *** p < 0.001, ns, not significant.
The biodistribution and in vivo internalization by myeloid cell subsets was next studied following intravenous injection of Cy5.5-NPs. Assessment of whole-organ fluorescence (Figs 2A, 2B, S3A) indicated that Cy5.5-NPs accumulated primarily in the liver and spleen, and Cy5.5-NPs were detected in these organs through 48 hours post injection. NPs accumulated in the lung at 12 hours, yet the majority was cleared from the lung by 48 hours. Relatively low levels of Cy5.5-NPs were detected in the tumor. The cell types within these organs that were associated with NPs was quantified by flow cytometry and each NP+ cell type was shown as a percentage of all NP+ cells within each organ (Figs 2B, S3B). Monocytes comprised the largest population of NP+ myeloid cells in all organs (37.6 ± 7.9%, 46.3 ± 7.9%, 26.6 ± 4.3%, 7.6 ± 2.4% in tumor, lung, spleen, and liver, respectively) compared to MDSCs (15.6 ± 5.3%, 34.8 ± 10.0%, 7.8 ± 2.0%, 3.6 ± 0.7% in tumor, lung, spleen, and liver, respectively) and macrophages (19.5 ± 7.7%, 3.3 ± 0.5%, 3.5 ± 0.8%, 4.0 ± 0.9% in tumor, lung, spleen, and liver, respectively).
Figure 2.
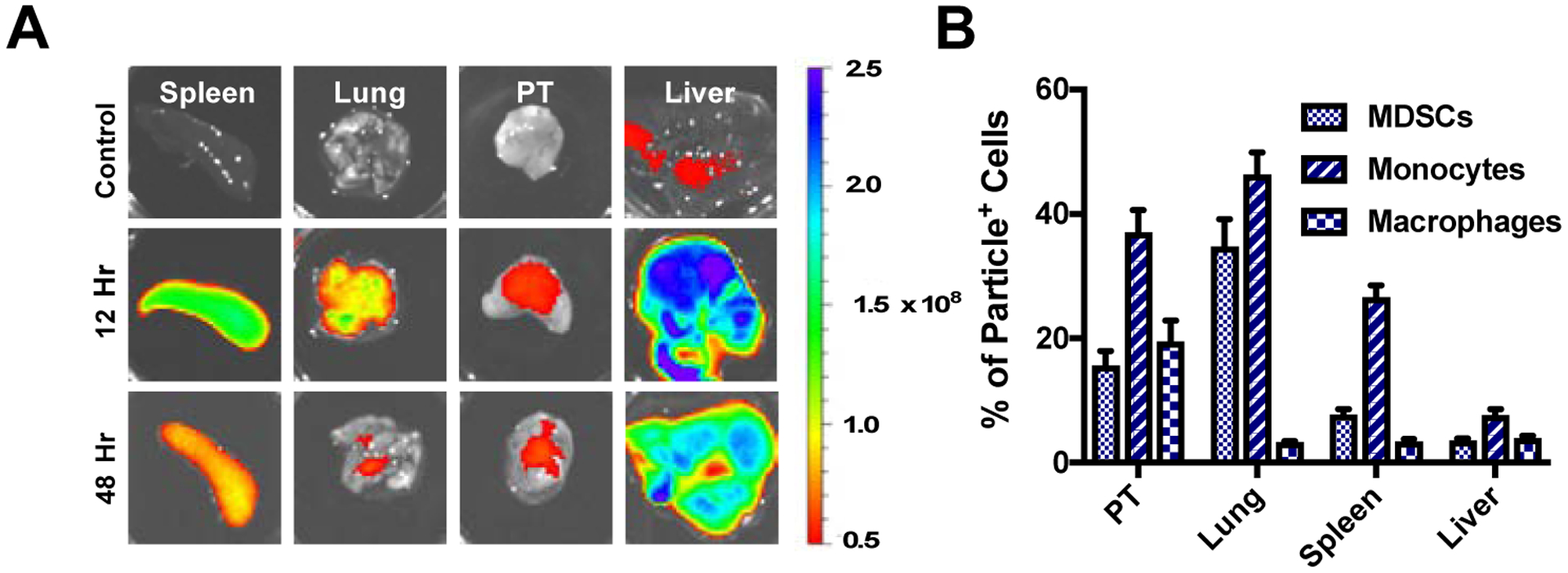
In vivo distribution of Cy5.5-NPs. (A) Ex vivo imaging following i.v. injection of 1 mg of Cy5.5-NPs shows they primarily accumulate in spleen and liver, and are detectable for more than 48 hours post injection. (B) Quantification of particle internalization by myeloid cell subsets within tissues as a percentage of total NP+ cells for a given tissue.
Nanoparticle administration alters immune cell distribution in blood and organs
We next investigated whether the intravenously delivered NPs would influence the distribution of innate immune cells in circulation and at the primary tumor or metastatic sites (i.e., lung). The analysis of blood 12 hours following a single dose of NPs revealed that the proportion of MDSCs decreased from 82.5 ± 2.8% to 63.5 ± 13.9% (Fig 3A). No significant change was observed in the percentage of macrophages (CD11b+/F4/80+), monocytes, or dendritic cells (DCs, CD11c+) in the blood with NP administration (Fig 3A). The accumulation of cells within the primary tumor and metastatic sites was analyzed following 6 consecutive days of NP administration. The quantity of MDSCs as a percentage of all single cells in the lung (Fig 3B) decreased with NP administration (30.0 ± 3.7% for PBS vs. 21.1 ± 6.2% for NP), consistent with a decrease observed in the blood (Fig 3A). No significant differences in myeloid cell proportions were observed in the spleen or primary tumor (Fig 3C and 3D).
Figure 3.
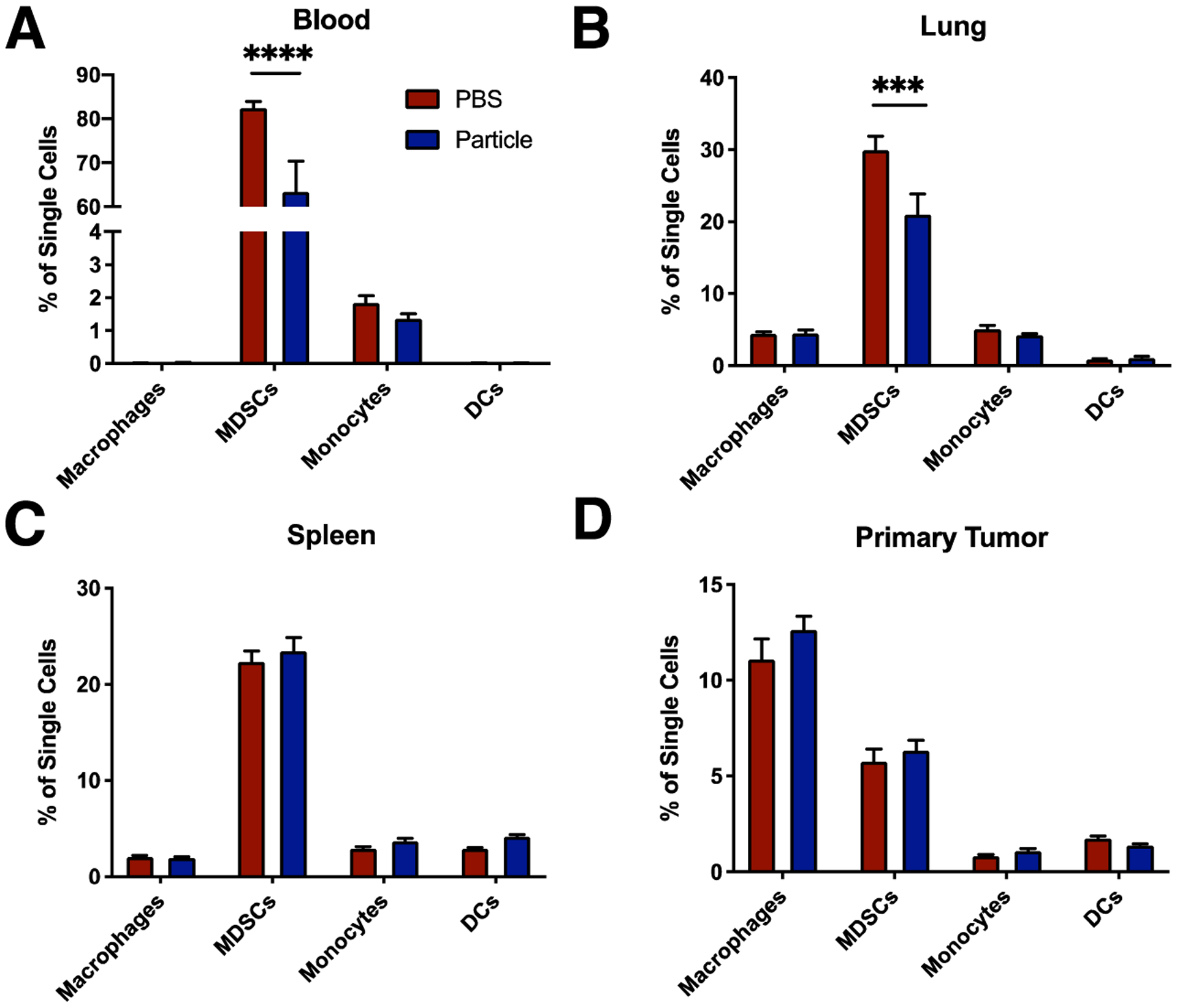
In vivo administration of cargo-free NPs reduced the proportion of MDSCs in circulation and at metastatic organs. (A) Tumor-bearing mice at were administered i.v. 1 mg of NPs in 200 μL of PBS or the equivalent volume of PBS only (n = 4 per group), and innate immune cells were quantified in the blood 12 hours post injection by flow cytometry. (B–D) NPs were administered at a dose of 1 mg/200 μL for 6 days to allow for accumulation and uptake of NPs (n = 4 PBS control, n = 5 NP). Flow cytometry quantification of immune cells in the lung (B), spleen (C), and primary tumor (D) was performed on day 10 post inoculation. Decrease in MDSCs observed in the blood (A) and lung (C) with NP administration. A 2-way ANOVA with Tukey’s multiple comparisons test was performed (A–D), bars show mean ± SEM, where *** p < 0.001, and **** p < 0.0001.
Synergistic therapeutic effect observed in nanoparticles combined with anti-PD-1
The therapeutic efficacy of NPs against 4T1 tumor growth and metastasis was next investigated. Mice were inoculated with orthotopic 4T1 tumors and placed in one of four treatment groups: 1) PBS control, 2) anti-PD-1 antibody only, 3) NPs only, and 4) NPs + anti-PD-1 (Fig. 4A). Average tumor size was decreased for combination NPs + anti-PD-1 (V = 1240 ± 298 mm3, volume increase of 16.0 ± 6.7 compared to initial tumor volume) compared to PBS control (V = 1940 ± 431 mm3; volume increase by 28.4 ± 12.4; p = 0.038), but was not decreased for either monotherapy (V = 1630 ± 578 mm3, volume increase by 23.2 ± 8.3 for anti-PD-1; V = 1690 ± 575 mm3, volume increase by 21.5 ± 5.4 for NPs) (Fig 4B). Survival, based on body condition and tumor appearance, was monitored after day 22 post inoculation (Fig 4C). Median survival was 24 days for the PBS and anti-PD-1 groups, 25 days for NPs alone, and 28 days for NPs + anti-PD-1 combination treatment. A survival benefit was observed for the combination treatment cohort compared to cohorts treated with PBS (p = 0.001), and compared to cohorts treated with either anti-PD-1 or NP monotherapy (p = 0.015 and p = 0.030, respectively). Taken together, these data indicate an additive or synergistic therapeutic effect by combining NPs and anti-PD-1 antibody.
Figure 4.
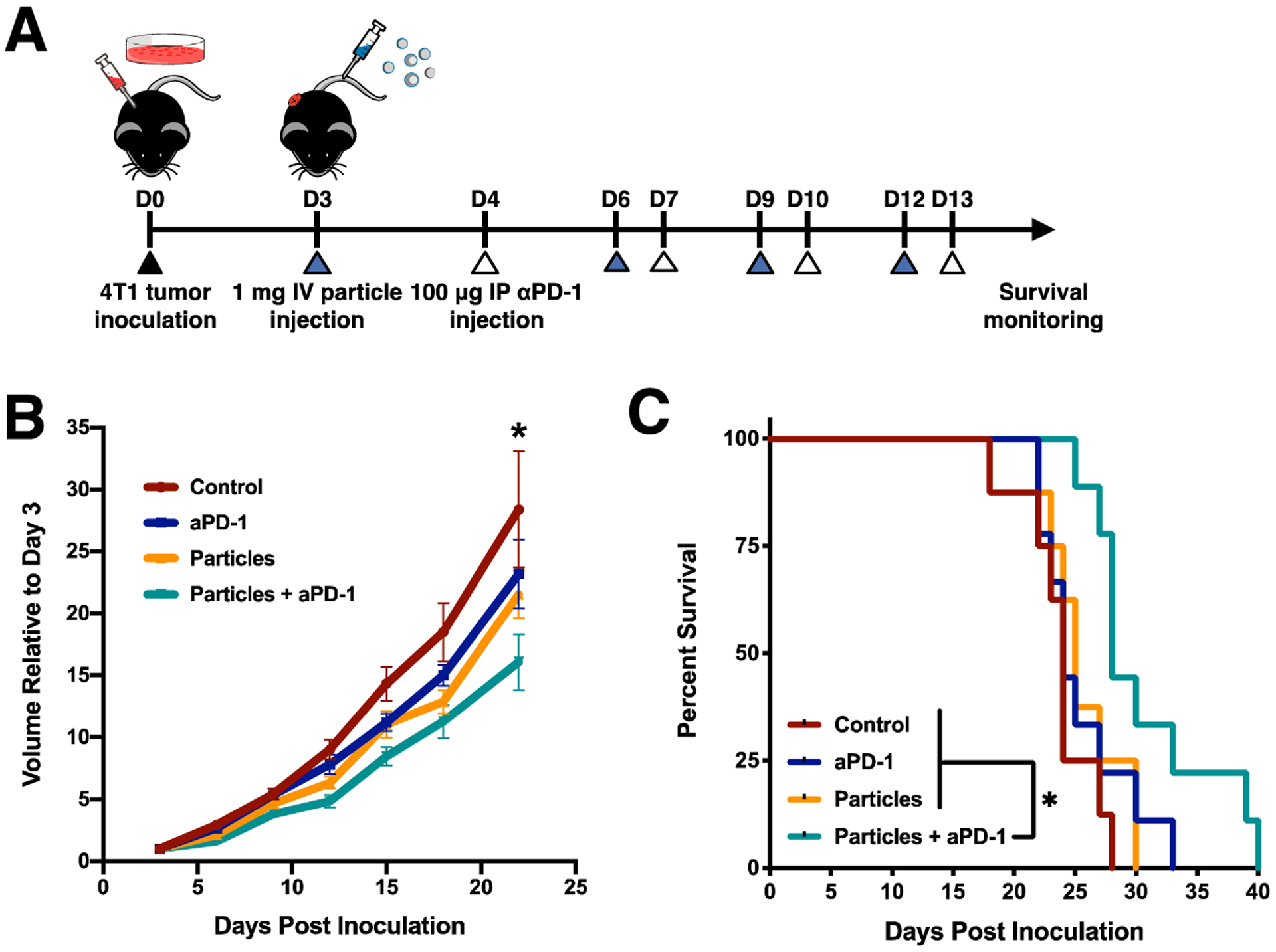
NPs + anti-PD-1 combination therapy delays 4T1 tumor growth and reduces metastasis. 4T1-bearing mice were treated with PBS (control), anti-PD-1 (aPD-1), NPs (particles), or combination therapy (particles + aPD-1). (A) Schematic of disease model and treatment. (B) Tumor volumes, and (C) survival curves are shown for the four treatment conditions (control, n = 7; aPD-1, n = 9; particles, n = 8, particles + aPD-1, n = 9), indicating a therapeutic benefit for particles + aPD-1 but not for either monotherapy compared to control. Two-way ANOVA with Tukey’s multiple comparisons tests were performed. Bars show mean ± SEM where * p < 0.05 compared to control (B), and where * p < 0.05 compared to particles + aPD-1 (C).
Nanoparticle internalization results in upregulation of inflammatory pathways
The mechanism by which NP administration may reduce tumor growth and metastasis was investigated in vitro by analyzing secretion of multiple cytokines associated with tumor progression following nanoparticle treatment. Mice were first treated with anti-PD-1 antibody post inoculation (Fig 5A), and splenocytes were then isolated from healthy and tumor-bearing mice (with and without anti-PD-1 treatment) at day 14 post inoculation. Splenocytes from tumor-bearing (Tumor+) mice secreted elevated amounts of the cytokine interleukin 6 (IL-6), granulocyte-macrophage colony-stimulating factor (GM-CSF), monocyte chemoattractant protein 1 (MCP-1), and tumor necrosis factor alpha (TNF-α), but not transforming growth factor beta (TGF-β) relative to those of Tumor− mice (Figs 5B–F). Treatment of these cells with NPs significantly decreased secretion of MCP-1 (Fig 5D) and increased TNF- α secretion (Fig 5E). This effect is amplified with the addition of anti-PD-1 (Tumor+ αPD-1+). No alteration in cytokine expression was observed for NP treatment for production of IL-6 (Fig 5B) or TGF-β (Fig 5F) in tumor-bearing mice with or without anti-PD-1. Increased granulocyte-macrophage colony-stimulating factor (GM-CSF) production was observed with NP treatment only in the Tumor+ αPD-1+ group (Fig 5C). Notably, NP treatment increased TNF-α production by more than 2-fold in tumor-bearing mice both with and without anti-PD-1 (Fig 5E). The production of MCP-1 is also greatly increased in tumor-bearing mice, yet there is a nearly 5-fold decrease in secreted MCP-1 with NP treatment in both the Tumor+ and Tumor+ αPD-1+ groups. We sought to identify the source of the produced cytokines, and thus Gr1+ cells were sorted by MACS and cytokine analysis was performed. Gr1+ cells were the focus as they are the predominant cell type in the disease model. These studies indicate an increased expression of TNF-α by the Gr1+ cells (Fig S4), consistent with the analysis of single cell data sets. However, the increase in MCP-1 was not observed for the Gr1+ cells, indicating an alternative cell type. These data indicate that NPs can alter the production of inflammatory cytokines and that anti-PD-1 contributes to additional changes in cytokine levels.
Figure 5.
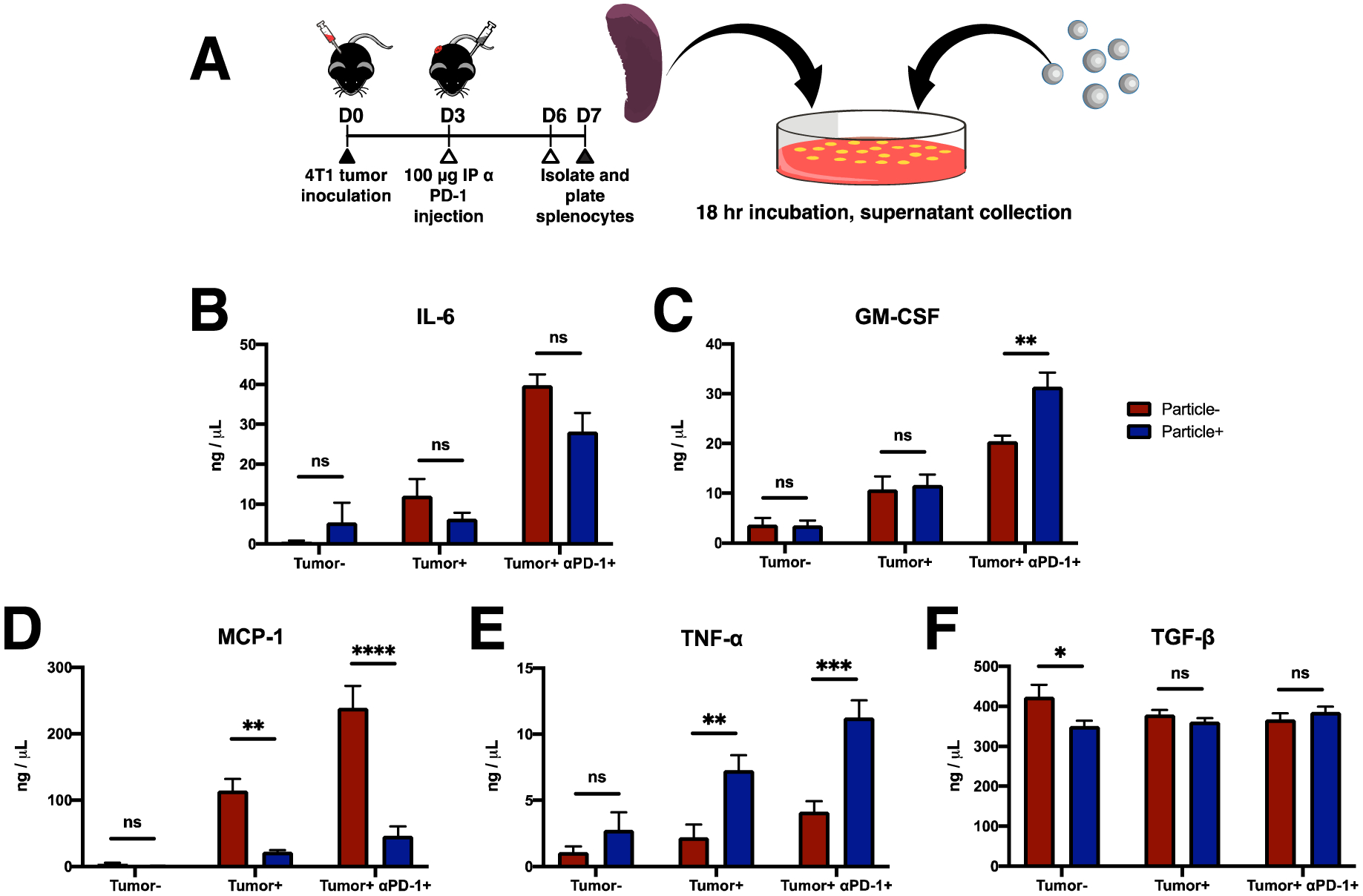
Reprogramming of inflammatory response by NP treatment in vitro. (A) Schematic and timeline of the study. Splenocytes from tumor-bearing mice that were treated with or without aPD-1 and incubated with NPs. (B–F) Cytokine secretion as measured by ELISA for splenocytes isolated from healthy (tumor−), tumor-bearing (tumor+), and in vivo aPD-1-treated (tumor+ aPD-1+) mice. Splenocytes were treated in vitro with (particle+) or without (particle−) NPs. Notably, there was a large decrease in the production of the MDSC-recruiting chemokine MCP-1 (D) and an increase in the production of proinflammatory TNF-α (C). 2-way ANOVA with Tukey’s multiple comparisons tests were performed. Bars show mean ± SEM where * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001 compared to particle− control (n = 4 per condition).
Tissue-specific deactivation of disease-relevant pathways in vivo
Gene expression within the primary tumor, spleen, and the lung, the primary metastatic site was analyzed at day 14 post inoculation to assess the impact of NP and anti-PD-1 treatment on these tissues. Gene expression at the spleen was analyzed to provide a relative measure of systemic immune changes, which could then be compared to the tissue specific responses within the primary tumor and lung. Differential gene expression was evaluated for each tissue compared to the PBS control group (Table 1, Figs 6A–C). In addition, pathway changes were investigated using Gene Set Enrichment Analysis (GSEA) for a total of 5,944 gene sets were examined (Fig S5). Furthermore, we subcategorized the pathway changes into those that would be supportive of tumor progression versus those that would inhibit tumor progression (Fig S6). The spleen and tumor had a substantially larger number of pathways that were predicted to have upregulated activity relative to those that would be downregulated, though the number of pathways that would be predicted to be supportive of tumor progression relative to those that would inhibit tumor progression were more similar. Gene sets that showed the greatest enrichment (positive or negative) for immune cells in all data sets were obtained and condensed into a list of 14 disease-relevant pathways (Fig 6D, E). Pathways with normalized enrichment score (NES) > 1.3 or NES < −1.3 were considered differentially expressed compared to untreated control. Relative to no treatment, the spleen and lung had a similar NES for 9 of the 14 pathways, with the differences associated with innate and adaptive immune cell responses. NP treatment resulted in downregulation of pathways associated with pro-tumor innate cells. The impact of NPs was distinct within the lung, the primary metastatic site, relative to the spleen or primary tumor. In the lung, the number of pathways predicted to have downregulated activity was substantially larger than the number predicted to be upregulated, which also associated with an increased in the activity of pathways associated with anti-tumor activity relative to pro-tumor activity. For the 14 common pathways, only 4 were similar between the lung and the spleen or primary tumor.
Table 1.
Summary of in vivo gene expression changes with treatment compared to PBS. A total of 14,472 genes and 5,944 pathways were analyzed. Note that pro-tumor indicates upregulation of pro-tumor pathways or downregulation of anti-tumor pathways. Similarly, the column anti-tumor indicates upregulation of anti-tumor pathways or downregulation of pro-tumor pathways. The listing of pathways is available in Fig S6.
| Upreg. Genes | Downreg. Genes | Upreg. Pathways | Downreg. Pathways | Pro-Tumor | Anti-Tumor | ||
|---|---|---|---|---|---|---|---|
| NP | Spleen | 243 | 119 | 327 | 264 | 124 | 105 |
| Tumor | 2 | 2 | 353 | 71 | 82 | 75 | |
| Lung | 15 | 20 | 221 | 623 | 96 | 128 | |
| NP+αPD-1 | Spleen | 28 | 62 | 612 | 165 | 107 | 97 |
| Tumor | 21 | 13 | 411 | 110 | 125 | 102 | |
| Lung | 340 | 240 | 84 | 934 | 87 | 148 |
Figure 6.

Tissue-specific in vivo reprogramming by NP and aPD-1 treatment. (A–C) Gene expression by RNAseq was performed on spleen, tumor, and lung tissue from mice treated with PBS, NPs, or NPs + aPD-1 (n = 3 per condition), and differential gene expression between the control and NP groups or the control and NP + aPD-1 groups were analyzed. Phenotypic differences were analyzed by Gene Set Enrichment Analysis. (D and E) Pathway enrichment for NP and NP and aPD-1 treated mice as shown by normalized enrichment score (NES) compared with PBS for the given gene sets. (F) Pathway enrichment for NP + aPD-1 compared with NP alone was also analyzed. Pathways with NES > 1.3 or NES < −1.3 were considered differentially expressed. Changes in the tumor largely reflected systemic changes as seen in the spleen, with positive enrichment of immune cell function, whereas the lung showed negative enrichment in many of these pathways.
The RNAseq data was also analyzed by comparing the NP + anti-PD-1 condition to NP alone in order to more effectively isolate the impact of the additional anti-PD-1 treatment. A list of 15 disease-relevant pathways were identified (Fig 6F). As before, the spleen and primary tumor had many similarities, and the lung was the most distinct with most pathways having a negative NES. With the primary tumor, an increased expression was observed for many pathways associated with activation of the immune system for both adaptive and innate immune cells, which is consistent with the decreased tumor growth that was observed and also the expected effects of treatment with anti-PD-1. Within the lung, most of these 15 pathways had a negative NES. Based on the differential response of the lung relative to the primary tumor and spleen, we also analyzed the cell types present within the lung at day 14 for differences in cell types for the conditions (Fig S7). For NP treatment with and without anti-PD-1, the number of neutrophils, macrophages, T cells, B cells, and NK cells was similar, suggesting that the RNAseq results indicate a differential phenotype and do not result from the accumulation of differential cell types. Analysis of the tumor cell numbers within the lung indicate an accumulation on the order of 100 cells within the lung, indicate of early stage metastasis, that was not significantly different between conditions (Fig S8).
Discussion
In this study, cargo-free PLG NPs were investigated for systemic delivery and therapeutic efficacy in the 4T1 mouse model of metastatic breast cancer. Innate immune cells, such as MDSCs, inflammatory monocytes, and TAMs have been revealed to support pro-tumor functions at the primary tumor and metastatic niche. The cells have been widely studied for their potential as biomarkers for early diagnostics of cancer and metastasis [29]–[33]. Furthermore, the therapeutic benefit of immunotherapies such as immune checkpoint blockade (ICB) and CAR T-cells, which primarily aim to improve the adaptive anti-tumor immune response, are limited by the presence of these immunosuppressive innate cells [13], [15], [16]. As such, targeting of these innate immune cells has emerged as a potential cancer immunotherapy. The data presented here demonstrate that NPs delivered systemically alter the phenotype of the pro-tumor innate immune cells at the primary tumor, resulting in a therapeutic benefit when combined with anti-PD-1 that is targeting the adaptive immune cells.
Previous studies have targeted innate immune cells with antibodies or small molecules against cell surface receptors or their ligands. MDSC depletion with antibodies either against Gr1 or Ly6G has been reported to have a therapeutic benefit in multiple cancer models [34], [35]. In the 4T1 model, diversion or depletion of MDSCs has resulted in a survival benefit following resection of the primary tumor [24], [36]. Inhibiting MDSC activation resulted in total elimination of tumors in mice treated with combination anti-PD-1 and anti-CTLA-4 [13]. Alternatively, small molecule inhibitors of myeloid cell recruitment and activation have also proven to be effective for targeting specific myeloid cells and improving the efficacy of immuno-, chemo-, and radio-therapies. However, there have been several reports of adverse effects of existing therapies. Stopping CCL2 inhibition has been shown to accelerate breast cancer metastasis [37]. CSF1R inhibition delays tumor growth [14], yet also promoted metastasis by diminishing the amount of IL-15 and reducing the number of NK cells [38]. The NPs reported here do not deplete the immune cell as can occur with antibodies, and do not contain an active pharmaceutical ingredient. No adverse effects were observed for NP treated animals in these studies, suggesting that NPs may provide an opportunity to enhance outcomes and improve safety by avoiding off target effects. These NPs are based on a formulation that completed both Phase I and Phase II clinical trials in celiac disease (NCTG03486990, NCT03738475), supporting the safety of these particles for translation. In addition, human MDSCs cannot be targeted with the same antibodies due to differences between mouse and human surface proteins [5], [39], [40]. NPs may provide an opportunity to modify human MDSCs based on a similar functionality between human and mice.
Administration of cargo-free PLG NPs intravenously in the cancer model leads to their subsequent association with monocytes, MDSCs, and other myeloid cells (Fig 2B), which results in a reduction in the percentage of myeloid cells present in the blood and lung (Fig 3). These NPs have been previously employed in animal models of trauma and autoimmune disease, and have been able to reduce inflammation that is associated with loss of function [22], [41]. Intravenous administration leads to NP accumulation primarily in the spleen and liver, and to a lesser extent in the lung for the first 48 hours post injection. Previous reports have suggested that intravenous NP administration diverted inflammatory cells to the spleen [19], [22], while others have observed granulocyte trafficking to the liver in several inflammatory disease models [42]. While NP administration reduced levels of MDSCs within the blood and lung, the primary tumor had stable levels of MDSCs, which were the lowest relative to other tissues at approximately 5% of single cells. This relatively low abundance would suggest a low level of MDSC recruitment. Furthermore, the lifetime of MDSCs can be up to 4 times longer in tumors relative to blood [43], which is also consistent with a low level of recruitment and may explain why the nanoparticles that divert a fraction of the circulating cells did not substantially impact the abundance of MDSCs within the primary tumor.
Herein, the NPs were applied to a model of cancer progression, which is typically associated with immune suppression to enable the tumor cells to escape destruction by the immune system. The NPs can target the innate cells and the ICB can primarily target the adaptive immune cell responses. A greater percentage of Ly6C-hi monocytes was observed to internalize NPs relative to MDSCs and other myeloid cells, though no significant decrease in monocytes was observed in tissues. These monocytes are thought to be precursors to TAMs [2], and previous studies found decreased metastatic seeding in the lung when monocyte recruitment was inhibited [44]. The lower level of association with MDSCs is consistent with a decreased capacity for phagocytosis [45], yet confirms previous findings that MDSCs can still be targeted through particle internalization [46]. The difference in NP uptake may also be explained in part by physicochemical properties of the NPs. Preferential internalization by specific cell types was reported to be a function of the surfactant used for NP manufacturing [47] or polymer properties [41]. In addition, nanoparticle size and surface charge influence internalization by myeloid cells [48], and selectivity can be achieved without targeting moieties [49], [50].
Reduced primary tumor growth was observed in the 4T1 model with combination NP and anti-PD-1 treatment (Fig 4). Interestingly, the NP administration was applied for only 12 days, yet administration impacted tumor growth over longer time frames. NP internalization induces phenotypic changes in the myeloid cells, with the response dependent upon the status of the immune system. PLG nanoparticles have been applied to reduce inflammation in spinal cord injury, yet the NPs appear to induce inflammatory pathways both in vitro and in vivo in the 4T1 model (Fig 5). In vitro studies of NP internalization indicated upregulation of two immunostimulatory cytokines TNF-α and GM-CSF. These cytokines have relatively nuanced roles in cancer progression. Neutralization of systemic GM-CSF with “cytokine sponges” has been reported to enhance the efficacy of anti-PD-1 treatment [51]. Conversely, combinations of GM-CSF, IL-6, and TNF-α have also been shown to inhibit human T cell activation in vitro, though solid tumors that contained MDSCs were found to consistently downregulate GM-CSF [39]. NP treatment in vitro also resulted in a decrease in the production of MCP-1 (CCL2), which is a critical cytokine associated with MDSC function [44]. Reviewing single cell data from the 4T1 model, macrophages and monocytes appear to be the predominant cell type expressing MCP-1 [52]. Signaling within the tumor microenvironment is complex, and the survival advantage provided by these particles warrant further investigation in the mechanisms of action.
NP treatment led to substantial changes in the gene expression systemically and within the primary tumor, which may contribute to the delayed tumor growth and the survival benefit. The percentage of MDSCs within the primary tumor was not significantly reduced with NP treatment, yet the phenotype of the immune cells was altered both in vivo (Fig 6) and in vitro (Fig S4), consistent with a report that MDSC alteration unrelated to recruitment can have a significant impact in the 4T1 model [53]. MDSCs are increasingly being appreciated for their ability to modulate and contribute to tumor development, as they secrete a variety of cytokines that are associated with pro- and anti-inflammatory phenotypes. The influence of these cytokines on cancer progression is complex. ARG-1, inducible NO synthase, reactive oxygen species, S100A8/9, and PD-L1 are generally immunosuppressive in their ability to inhibit DC, NK and T cell activity, while S100A8/9, TGF-β, IL-10 promote the function of Tregs and TAMs, which are present at a metastatic niche [54]–[57]. TNF-α is implicated in tumor progression in a variety of contexts, and has been observed to contribute to MDSC necroptosis [58]–[60]. Due, in part, to their role in cancer progression, MDSCs have become a target of biomaterial strategies to modulate multiple stages of tumorigenesis [61]–[64]. MDSCs share a considerable number of phenotypic and functional roles with neutrophils [65], [66], which have been reported to exhibit altered trafficking, phenotype, and cytokine secretion following various size-, charge-, and shape-dependent biomaterial interactions [67]–[70]. The mechanisms by which these highly-negatively charged NPs specifically influence MDSC function have not been fully elucidated, yet are likely to involve multiple complex interactions that modulate the cell phenotype. As seen in Figure 6, NP delivery alters a number of innate immune cell processes such as myeloid cell differentiation, cytokine secretion, and signaling that can impact multiple aspects of the adaptive immune response. Addition of anti-PD-1 along with NP administration provided an opportunity to target both the innate and adaptive immune suppression. The addition of anti-PD-1 with NP administration further enhanced pathways associated with myeloid cell function and also enhanced a number of pathways associated with T cell and natural killer cell responses. Anti-PD-1 led to downregulated negative regulation of T cell proliferation, which is consistent with findings that PD-1 downregulates T cell proliferation [71]. The impact of the NP and anti-PD-1 within the lung was distinct, which may be due to the relatively small numbers of tumor cells in the organs at the time points analyzed.
In conclusion, we report that PLG NPs delivered systemically to target circulating immune cells improve the efficacy of ICB. The NPs are well-tolerated and the absence of an active pharmaceutical ingredient may reduce off target effects. The NPs reprogram innate immune cell phenotypes by altering their trafficking and expression of key cytokines, which altered the microenvironment of the primary tumor. Modulating the innate cell responses by the NPs combined with ICB therapy that targets the adaptive responses contributes to an improved therapeutic outcome. PLG NPs that target circulating innate immune cells may provide novel therapeutic strategies in cancer, used either alone or to complement current or emerging T-cell-targeted immunotherapies.
Supplementary Material
Acknowledgments:
Funding for this research was provided by the National Institutes of Health R01 CA214384, R01 CA243916 and R01 AI148076. We are grateful to the scientists at Cour Pharmaceutical that aided in the experimental design.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests: L.D.S. consults and has financial interests in Cour Pharmaceutical Development Company, Inc.
References
- [1].Kitamura T, Qian B-Z, and Pollard JW, “Immune cell promotion of metastasis,” Nat. Rev. Immunol, vol. 15, no. 2, pp. 73–86, 2015, doi: 10.1038/nri3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Qian B et al. , “A distinct macrophage population mediates metastatic breast cancer cell extravasation, establishment and growth,” PLoS One, vol. 4, no. 8, 2009, doi: 10.1371/journal.pone.0006562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Singhal S et al. , “Human tumor-associated monocytes/macrophages and their regulation of T cell responses in early-stage lung cancer,” Sci. Transl. Med, vol. 11, no. 479, 2019, doi: 10.1126/scitranslmed.aat1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Psaila B and Lyden D, “The metastatic niche: adapting the foreign soil,” Nat. Rev. Cancer, vol. 9, pp. 285–293, 2009, doi: 10.1038/nrc2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Gabrilovich DI and Nagaraj S, “Myeloid-derived-supressor cells as regulators of th eimmune system,” Nat. Rev. Immunol, vol. 9, no. 3, pp. 162–174, 2009, doi: 10.1038/nri2506.Myeloid-derived-suppressor. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Wculek SK and Malanchi I, “Neutrophils support lung colonization of metastasis-initiating breast cancer cells.,” Nature, vol. advance on, pp. 1–21, 2015, doi: 10.1038/nature16140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Sceneay J et al. , “Primary tumor hypoxia recruits CD11b+/Ly6Cmed/ Ly6G+ immune suppressor cells and compromises NK cell cytotoxicity in the premetastatic niche,” Cancer Res, vol. 72, no. 16, pp. 3906–3911, 2012, doi: 10.1158/0008-5472.CAN-11-3873. [DOI] [PubMed] [Google Scholar]
- [8].Sun L et al. , “Inhibiting myeloid-derived suppressor cell trafficking enhances T cell immunotherapy,” JCI Insight, vol. 4, no. 7, 2019, doi: 10.1172/jci.insight.126853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Gazzaniga S et al. , “Targeting tumor-associated macrophages and inhibition of MCP-1 reduce angiogenesis and tumor growth in a human melanoma xenograft,” J. Invest. Dermatol, vol. 127, no. 8, pp. 2031–2041, 2007, doi: 10.1038/sj.jid.5700827. [DOI] [PubMed] [Google Scholar]
- [10].Gil-Bernabé AM et al. , “Recruitment of monocytes/macrophages by tissue factor-mediated coagulation is essential for metastatic cell survival and premetastatic niche establishment in mice,” Blood, vol. 119, no. 13, pp. 3164–3175, 2012, doi: 10.1182/blood-2011-08-376426.The. [DOI] [PubMed] [Google Scholar]
- [11].Colligan SH, Tzetzo SL, and Abrams SI, “Myeloid-driven mechanisms as barriers to antitumor CD8+ T cell activity,” Mol. Immunol, vol. 118, no. September 2019, pp. 165–173, 2020, doi: 10.1016/j.molimm.2019.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].He W, Kapate N, Shields CW, and Mitragotri S, “Drug delivery to macrophages: A review of targeting drugs and drug carriers to macrophages for inflammatory diseases,” Adv. Drug Deliv. Rev, 2019, doi: 10.1016/j.addr.2019.12.001. [DOI] [PubMed] [Google Scholar]
- [13].Kim K et al. , “Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells.,” Proc. Natl. Acad. Sci. U. S. A, vol. 111, no. 32, pp. 11774–9, 2014, doi: 10.1073/pnas.1410626111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Strachan DC et al. , “CSF1R inhibition delays cervical and mammary tumor growth in murine models by attenuating the turnover of tumor-associated macrophages and enhancing infiltration by CD8+ T cells,” Oncoimmunology, vol. 2, no. 12, 2013, doi: 10.4161/onci.26968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Nakamura K and Smyth MJ, “Myeloid immunosuppression and immune checkpoints in the tumor microenvironment,” Cell. Mol. Immunol, no. September, pp. 1–12, 2019, doi: 10.1038/s41423-019-0306-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Rashidian M et al. , “Immuno-PET identifies the myeloid compartment as a key contributor to the outcome of the antitumor response under PD-1 blockade,” Proc. Natl. Acad. Sci. U. S. A, vol. 116, no. 34, pp. 16971–16980, 2019, doi: 10.1073/pnas.1905005116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lechner MG et al. , “Immunogenicity of Murine Solid Tumor Models as a Defining Feature of In Vivo Behavior and Response to Immunotherapy,” J. Immunother, vol. 36, no. 9, pp. 477–489, 2013, doi: 10.1097/01.cji.0000436722.46675.4a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Movahedi K et al. , “Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes,” Cancer Res, vol. 70, no. 14, pp. 5728–5739, 2010, doi: 10.1158/0008-5472.CAN-09-4672. [DOI] [PubMed] [Google Scholar]
- [19].Getts DR et al. , “Therapeutic Inflammatory Monocyte Modulation Using Immune-Modifying Microparticles,” Sci. Transl. Med, vol. 6, no. 219, p. 219ra7, January 2014, doi: 10.1126/scitranslmed.3007563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Le QV, Yang G, Wu Y, Jang HW, Shokouhimehr M, and Oh YK, “Nanomaterials for modulating innate immune cells in cancer immunotherapy,” Asian J. Pharm. Sci, vol. 14, no. 1, pp. 16–29, 2019, doi: 10.1016/j.ajps.2018.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Sharma S et al. , “Intravenous Immunomodulatory Nanoparticle Treatment for Traumatic Brain Injury,” Ann. Neurol, vol. 87, no. 3, pp. 442–455, 2020, doi: 10.1002/ana.25675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Park J et al. , “Intravascular innate immune cells reprogrammed via intravenous nanoparticles to promote functional recovery after spinal cord injury,” Proc. Natl. Acad. Sci. U. S. A, vol. 116, no. 30, pp. 14947–14954, 2019, doi: 10.1073/pnas.1820276116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Pyzer AR, Cole L, Rosenblatt J, and Avigan DE, “Myeloid-derived suppressor cells as effectors of immune suppression in cancer,” International Journal of Cancer. 2016, doi: 10.1002/ijc.30232. [DOI] [PubMed] [Google Scholar]
- [24].Rao SS et al. , “Enhanced survival with implantable scaffolds that capture metastatic breast cancer cells in vivo,” Cancer Res, vol. 76, no. 18, pp. 5209–5218, 2016, doi: 10.1158/0008-5472.CAN-15-2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hunter Z et al. , “A biodegradable nanoparticle platform for the induction of antigen-specific immune tolerance for treatment of autoimmune disease,” ACS Nano, 2014, doi: 10.1021/nn405033r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Smarr CB et al. , “Biodegradable antigen-associated PLG nanoparticles tolerize Th2-mediated allergic airway inflammation pre- and postsensitization,” Proc. Natl. Acad. Sci. U. S. A, 2016, doi: 10.1073/pnas.1505782113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Love MI, Huber W, and Anders S, “Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2,” Genome Biol, vol. 15, no. 12, pp. 1–21, 2014, doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Zhu A, Ibrahim JG, and Love MI, “Heavy-Tailed prior distributions for sequence count data: Removing the noise and preserving large differences,” Bioinformatics, vol. 35, no. 12, pp. 2084–2092, 2019, doi: 10.1093/bioinformatics/bty895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Angell TE et al. , “Circulating myeloid-derived suppressor cells predict differentiated thyroid cancer diagnosis and extent,” Thyroid, 2016, doi: 10.1089/thy.2015.0289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Feng AL et al. , “CD16+ monocytes in breast cancer patients: Expanded by monocyte chemoattractant protein-1 and may be useful for early diagnosis,” Clin. Exp. Immunol, vol. 164, no. 1, pp. 57–65, 2011, doi: 10.1111/j.1365-2249.2011.04321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Mukhtar RA, Nseyo O, Campbell MJ, and Esserman LJ, “Tumor-associated macrophages in breast cancer as potential biomarkers for new treatments and diagnostics,” Expert Rev. Mol. Diagn, vol. 11, no. 1, pp. 91–100, 2011, doi: 10.1586/erm.10.97. [DOI] [PubMed] [Google Scholar]
- [32].Hamm A et al. , “Tumour-educated circulating monocytes are powerful candidate biomarkers for diagnosis and disease follow-up of colorectal cancer,” Gut, vol. 65, no. 6, pp. 990–1000, 2016, doi: 10.1136/gutjnl-2014-308988. [DOI] [PubMed] [Google Scholar]
- [33].Schauer D et al. , “Intermediate Monocytes but Not TIE2-Expressing Monocytes Are a Sensitive Diagnostic Indicator for Colorectal Cancer,” PLoS One, vol. 7, no. 9, pp. 1–10, 2012, doi: 10.1371/journal.pone.0044450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Daley JM, Thomay AA, Connolly MD, Reichner JS, and Albina JE, “Use of Ly6G-specific monoclonal antibody to deplete neutrophils in mice,” J. Leukoc. Biol, vol. 83, no. 1, pp. 64–70, 2008, doi: 10.1189/jlb.0407247. [DOI] [PubMed] [Google Scholar]
- [35].Srivastava MK et al. , “Myeloid suppressor cell depletion augments antitumor activity in lung cancer,” PLoS One, vol. 7, no. 7, 2012, doi: 10.1371/journal.pone.0040677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Bosiljcic M et al. , “Targeting myeloid-derived suppressor cells in combination with primary mammary tumor resection reduces metastatic growth in the lungs,” Breast Cancer Res, vol. 21, no. 1, pp. 1–16, 2019, doi: 10.1186/s13058-019-1189-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Bonapace L et al. , “Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis,” Nature, vol. 515, no. 7525, pp. 130–133, 2014, doi: 10.1038/nature13862. [DOI] [PubMed] [Google Scholar]
- [38].Beffinger M et al. , “CSF1R-dependent myeloid cells are required for NK-mediated control of metastasis,” JCI insight, vol. 3, no. 10, 2018, doi: 10.1172/jci.insight.97792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Lechner MG, Liebertz DJ, and Epstein AL, “Characterization of Cytokine-Induced Myeloid-Derived Suppressor Cells from Normal Human Peripheral Blood Mononuclear Cells,” J. Immunol, vol. 185, no. 4, pp. 2273–2284, 2010, doi: 10.4049/jimmunol.1000901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Perez C et al. , “Immunogenomic identification and characterization of granulocytic myeloid derived suppressor cells in multiple myeloma.,” Blood, 2020, doi: 10.1182/blood.2019004537. [DOI] [PubMed] [Google Scholar]
- [41].Saito E et al. , “Designing drug-free biodegradable nanoparticles to modulate inflammatory monocytes and neutrophils for ameliorating inflammation,” J. Control. Release, vol. 300, no. February, pp. 185–196, 2019, doi: 10.1016/j.jconrel.2019.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Fromen CA et al. , “Neutrophil-Particle Interactions in Blood Circulation Drive Particle Clearance and Alter Neutrophil Responses in Acute Inflammation,” ACS Nano, vol. 11, no. 11, pp. 10797–10807, 2017, doi: 10.1021/acsnano.7b03190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Sawanobori Y et al. , “Chemokine-mediated rapid turnover of myeloid-derived suppressor cells in tumor-bearing mice,” Blood, 2008, doi: 10.1182/blood-2008-01-136895. [DOI] [PubMed] [Google Scholar]
- [44].Qian B-Z et al. , “CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis.,” Nature, vol. 475, no. 7355, pp. 222–5, 2011, doi: 10.1038/nature10138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Bronte V et al. , “Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards,” Nat. Commun, vol. 7, p. 12150, 2016, doi: 10.1038/ncomms12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Wu C et al. , “Repolarization of myeloid derived suppressor cells via magnetic nanoparticles to promote radiotherapy for glioma treatment,” Nanomedicine Nanotechnology, Biol. Med, vol. 16, no. xxxx, pp. 126–137, 2019, doi: 10.1016/j.nano.2018.11.015. [DOI] [PubMed] [Google Scholar]
- [47].Casey LM et al. , “Cargo-less nanoparticles program innate immune cell responses to toll-like receptor activation,” Biomaterials, vol. 218, no. July, p. 119333, 2019, doi: 10.1016/j.biomaterials.2019.119333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Jeon S, Clavadetscher J, Lee DK, Chankeshwara SV, Bradley M, and Cho WS, “Surface charge-dependent cellular uptake of polystyrene nanoparticles,” Nanomaterials, vol. 8, no. 12, pp. 1–11, 2018, doi: 10.3390/NANO8121028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Pinton L et al. , “Targeting of immunosuppressive myeloid cells from glioblastoma patients by modulation of size and surface charge of lipid nanocapsules,” J. Nanobiotechnology, vol. 18, no. 1, pp. 1–12, 2020, doi: 10.1186/s12951-020-00589-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Smith BR et al. , “Selective uptake of single-walled carbon nanotubes by circulating monocytes for enhanced tumour delivery,” Nat. Nanotechnol, vol. 9, no. 6, pp. 481–487, 2014, doi: 10.1038/nnano.2014.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Li S et al. , “Pseudoneutrophil Cytokine Sponges Disrupt Myeloid Expansion and Tumor Trafficking to Improve Cancer Immunotherapy.,” Nano Lett, pp. 2–11, 2019, doi: 10.1021/acs.nanolett.9b03753. [DOI] [PubMed] [Google Scholar]
- [52].Oakes RS et al. , “Metastatic Conditioning of Myeloid Cells at a Subcutaneous Synthetic Niche Reflects Disease Progression and Predicts Therapeutic Outcomes,” Cancer Res, vol. 80, no. 3, pp. 602–612, February 2020, doi: 10.1158/0008-5472.CAN-19-1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Xiao P et al. , “Interleukin 33 in tumor microenvironment is crucial for the accumulation and function of myeloid-derived suppressor cells,” Oncoimmunology, 2016, doi: 10.1080/2162402X.2015.1063772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Ostrand-Rosenberg S, Beury DW, Parker KH, and Horn LA, “Survival of the fittest: how myeloid-derived suppressor cells survive in the inhospitable tumor microenvironment,” Cancer Immunol. Immunother, no. 0123456789, 2019, doi: 10.1007/s00262-019-02388-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Umansky V, Blattner C, Gebhardt C, and Utikal J, “The role of myeloid-derived suppressor cells (MDSC) in cancer progression,” Vaccines. 2016, doi: 10.3390/vaccines4040036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Atretkhany KSN and Drutskaya MS, “Myeloid-derived suppressor cells and proinflammatory cytokines as targets for cancer therapy,” Biochemistry (Moscow). 2016, doi: 10.1134/S0006297916110055. [DOI] [PubMed] [Google Scholar]
- [57].Liu Y and Cao X, “Characteristics and Significance of the Pre-metastatic Niche,” Cancer Cell, vol. 30, no. 5, pp. 668–681, 2016, doi: 10.1016/j.ccell.2016.09.011. [DOI] [PubMed] [Google Scholar]
- [58].Montfort A, Colacios C, Levade T, Andrieu-Abadie N, Meyer N, and Ségui B, “The TNF paradox in cancer progression and immunotherapy,” Frontiers in Immunology. 2019, doi: 10.3389/fimmu.2019.01818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Smith AD et al. , “Autocrine IL6-Mediated Activation of the STAT3-DNMT Axis Silences the TNFα-RIP1 Necroptosis Pathway to Sustain Survival and Accumulation of Myeloid-Derived Suppressor Cells,” Cancer Res, 2020, doi: 10.1158/0008-5472.CAN-19-3670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Moriwaki K, Bertin J, Gough PJ, Orlowski GM, and Chan FKM, “Differential roles of RIPK1 and RIPK3 in TNF-induced necroptosis and chemotherapeutic agent-induced cell death,” Cell Death Dis, 2015, doi: 10.1038/cddis.2015.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Wilkerson A, Kim J, Huang AY, and Zhang M, “Nanoparticle Systems Modulating Myeloid-Derived Suppressor Cells for Cancer Immunotherapy,” Curr. Top. Med. Chem, 2017, doi: 10.2174/1568026617666161122121412. [DOI] [PubMed] [Google Scholar]
- [62].Sasso MS et al. , “Low dose gemcitabine-loaded lipid nanocapsules target monocytic myeloid-derived suppressor cells and potentiate cancer immunotherapy,” Biomaterials, 2016, doi: 10.1016/j.biomaterials.2016.04.010. [DOI] [PubMed] [Google Scholar]
- [63].Yu GT et al. , “Myeloid-Derived Suppressor Cell Membrane-Coated Magnetic Nanoparticles for Cancer Theranostics by Inducing Macrophage Polarization and Synergizing Immunogenic Cell Death,” Adv. Funct. Mater, 2018, doi: 10.1002/adfm.201801389. [DOI] [Google Scholar]
- [64].Sau S, Alsaab HO, Bhise K, Alzhrani R, Nabil G, and Iyer AK, “Multifunctional nanoparticles for cancer immunotherapy: A groundbreaking approach for reprogramming malfunctioned tumor environment,” Journal of Controlled Release. 2018, doi: 10.1016/j.jconrel.2018.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Zhou J, Nefedova Y, Lei A, and Gabrilovich D, “Neutrophils and PMN-MDSC: Their biological role and interaction with stromal cells,” Seminars in Immunology. 2018, doi: 10.1016/j.smim.2017.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Gabrilovich DI, “Myeloid-derived suppressor cells,” Cancer Immunol. Res, 2017, doi: 10.1158/2326-6066.CIR-16-0297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Veiseh O et al. , “Size- and shape-dependent foreign body immune response to materials implanted in rodents and non-human primates,” Nat. Mater, 2015, doi: 10.1038/nmat4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Zhao Z, Ukidve A, Kim J, and Mitragotri S, “Targeting Strategies for Tissue-Specific Drug Delivery,” Cell. 2020, doi: 10.1016/j.cell.2020.02.001. [DOI] [PubMed] [Google Scholar]
- [69].Goncalves DM, de Liz R, and Girard D, “Activation of neutrophils by nanoparticles.,” TheScientificWorldJournal. 2011, doi: 10.1100/2011/768350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Dobrovolskaia MA, Shurin M, and Shvedova AA, “Current understanding of interactions between nanoparticles and the immune system,” Toxicol. Appl. Pharmacol, 2016, doi: 10.1016/j.taap.2015.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Patsoukis N, Brown J, Petkova V, Liu F, Li L, and Boussiotis VA, “Selective Effects of PD-1 on Akt and Ras Pathways Regulate Molecular Components of the Cell Cycle and Inhibit T Cell Proliferation,” Sci. Signal, vol. 5, no. 230, pp. ra46–ra46, June 2012, doi: 10.1126/scisignal.2002796. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.