

Abstract
In the intestine, IgA antibody-secreting B cells (IgA-ASCs) and helper T cells coordinate to maintain local homeostasis while their dysregulation could lead to development of intestinal inflammatory diseases. However, mechanisms underlying the coordinated localization and function of the B and T cells into the intestine, particularly the colon, are poorly understood. We herein report the first evidence that the gut-homing chemokine receptor CCR10+ IgA-ASCs form conjugates with helper T cells, preferentially regulatory T cells, at their differentiation sites of gut-associated lymphoid organs for their coordinated co-localization into the colon to promote local homeostasis. In CCR10-knockout mice, defective migration of IgA-ASCs also resulted in defective T cell migration and homeostasis, and development of inflammatory symptoms in the colon. Antigen-specific interaction of CCR10+ IgA-ASCs and T cells is crucial for their homeostatic establishment in the colon. On the other hand, in IgA-knockout mice, preferential expansion of CCR10+ IgG1-ASCs with regulatory functions compensated for CCR10+ IgA-ASCs to help maintain colonic homeostasis. The preferential expansion of specific subclasses of CCR10+ IgG-ASCs with regulatory functions was also found in asymptomatic IgA-deficient patients. These findings suggest coordinated cell migration as a novel mechanism underlying localization and function of B and T cells in colonic homeostatic regulation.
Keywords: IgA-antibody-secreting cells (IgA-ASC), helper T (Th) cells, regulatory T (Treg) cells, CCR10, coordinated cell migration, colonic immune homeostasis
INTRODUCTION
The intestine is constantly exposed to commensal bacteria and other food-born antigens. Properly coordinated B and T cell responses are crucial for maintenance of intestinal homeostasis, while their dysregulation lead to increased incidences of intestinal inflammatory diseases and various other disorders.
IgA-ASCs are a major component of the B cell response involved in intestinal homeostasis. Predominantly generated in the gut-associated lymphoid tissue (GALT) such as Peyer’s patches (PP) and cecal patches (CP) from naïve B cells, isotype-switched IgA+ plasmablast cells acquire gut-homing properties and migrate into the lamina propria (LP) of intestines, where they mature into IgA antibody-secreting plasma cells 1, 2, 3, 4. Mature IgA-ASCs produce large amounts of IgA antibodies that are transported across intestinal epithelia and released into intestinal lumen, where they help maintain homeostasis of commensal microbiota and neutralize food-borne pathogens and toxins 5. IgA-ASCs also express molecules with immune regulatory functions such as PD-L1 and can produce cytokines such as TNFα and TGFβ, suggesting that they have additional functions in intestinal homeostatic regulation besides production of IgA 6, 7, 8.
In coordination with IgA-ASCs, CD4+ αβT cells are a major component of T cell responses in intestinal homeostasis. Under homeostatic conditions, there is preferential activation of CD4+ FOXP3+ regulatory T (Treg) cells, which promote homeostatic maintenance through regulation of various other types of immune cells 9, 10. Like IgA-ASCs, T cells are activated in the GALT and migrate into intestinal LP to participate in homeostatic regulation. However, mechanisms directing localization of T cells and IgA-ASCs into the intestine, particularly the colon, seem different. CCR10 is a major chemokine receptor upregulated on IgA+ plasmablast cells generated in the GALT for their migration into the colon, which highly expresses CCL28, the mucosa-specific ligand of CCR10 11, 12, 13, 14. CCR10 is predominantly expressed by intestinal IgA+ plasmablasts and plasma cells but not B cells 11, 12, 15. We previously reported that CCR10 is important for migration of IgA-ASCs into the colon and colonic IgA responses 15. In contrast, CCR10 is not expressed on colonic T cells 11. Few molecules are identified to direct migration of T cells into the colon. GPR15 is an important homing molecule for migration of Treg cells into the colon in mice 16. However, in humans, GPR15 is expressed mainly on pathogenic effector T cells of the colon 17. CCR6 is another homing molecule reportedly involved in migration of T cells to the colon under inflammatory conditions 18. Mechanisms regulating coordinated migration of B and T cells into the colon remain poorly understood.
In this report, we provide the first evidence that gut-homing CCR10+ IgA-ASCs form conjugates with T cells to help their migration into the colon for homeostatic regulation. Furthermore, in IgA-deficient conditions, specific subclasses of CCR10+ IgG-ASCs with regulatory functions preferentially expand to substitute for CCR10+ IgA-ASCs to help maintain colonic homeostasis. These findings shed insight into mechanisms coordinating migration and function of IgA-ASCs and T cells in the colon.
RESULTS
Dysregulated T cell homeostasis associated with increased inflammatory symptoms in colons of CCR10−/− mice
We previously generated a strain of CCR10-KO/EGFP-KI mice in which the CCR10 coding sequence was replaced with a DNA fragment coding for EGFP that could be used to report the CCR10 expression 15, 19. Compared to wild-type or heterozygous CCR10-KO/EGFP-KI (CCR10+/EGFP, or CCR10+/− for simplicity) littermates, homozygous CCR10-KO/EGFP-KI (CCR10EGFP/EGFP, or CCR10−/−) mice had significantly shortened colons at 2-3 months of age, which became more evident at 9-12 months of age (Fig. 1A, Supplementary Fig. 1A). CCR10−/− mice also had elevated levels of transcripts of IL-17A and IL-1α in colons compared to CCR10+/− mice (Fig. 1B). Associated with this, CD4+ T cells isolated from the colonic LP of CCR10−/− mice expressed higher levels of IL-17A and IFNγ but lower levels of IL-10 compared to CCR10+/− controls (Fig. 1C). While no obvious lesions were observed in colons of untreated CCR10−/− mice (Supplementary Fig. 1B), significantly higher percentages of CCR10−/− mice had fecal occult bleeding and died after feeding with dextran sulphate sodium (DSS) compared to CCR10+/− controls (Fig. 1D-E). DSS-fed CCR10−/− mice also had higher expression of TNFα, IL-17A and IL-22 in the colon than same-treated CCR10+/− mice (Fig. 1F). These results reveal that colons of CCR10−/− mice have dysregulated T cell homeostasis with an increased inflammatory status and are prone to over-active inflammation after tissue damage.
Figure 1.
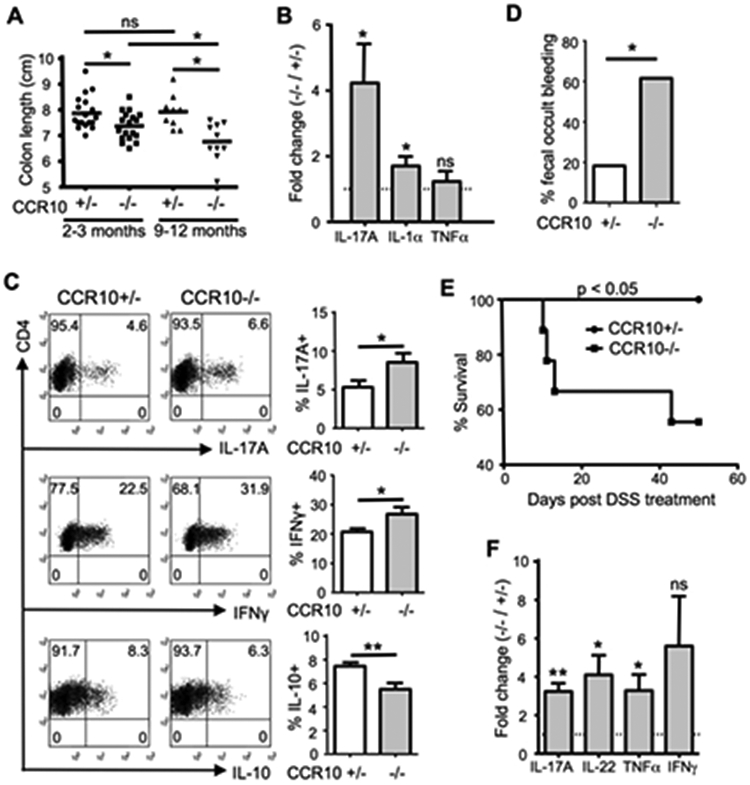
CCR10−/− mice developed colonic inflammation associated with dysregulated T cells. (A) Lengths of colons of CCR10−/− and CCR10+/− mice at different ages. One dot represents one mouse. (B) Ratios of expression levels of different cytokine transcripts (determined by qRT-PCR) in colons of CCR10−/− over those of CCR10+/− mice at 9-12 months of age (1 indicating no difference). N=8 mice of each genotype. (C) Analysis of IL-17A, IFNγ and IL-10 expression in gated CD45+CD3+CD4+ T cells of the colonic LP of CCR10+/− and CCR10−/− mice. N≥9 mice of each genotype for IL-17A, N=4 mice of each genotype for IFNγ and N=15 of each genotype for IL-10 analyses. (D) Percentages of CCR10+/− and CCR10−/− mice with detectable fecal occult bleeding 3 days after starting the DSS treatment. 11 CCR10+/− and 13 CCR10−/− mice were analyzed. (E) Survival rates of CCR10+/− and CCR10−/− mice after starting the DSS treatment. 10 CCR10+/− and 9 CCR10−/− mice were analyzed. (F) Ratios of expression levels of indicated cytokine transcripts in colons of CCR10−/− over those of CCR10+/− mice 7 days after starting DSS treatment. 5 mice of each genotype were analyzed. *p<0.05, **p<0.01, ***p<0.001, ns: no significant difference (applied to all figures).
A high frequency of T cells, particularly Treg cells, form conjugates with CCR10+ IgA-ASCs in colon
Since IgA-ASCs were reportedly the only type of colonic immune cells that express CCR10 11, impaired localization and function of IgA-ASCs in the colon were likely the cause of the colonic T cell dysregulation and increased inflammatory status in CCR10−/− mice. Surprisingly, however, a sizable fraction of colonic LP (but not intraepithelial) T cells were EGFP(CCR10)+ in CCR10+/EGFP reporter mice (Fig. 2A, Supplementary Fig. 2A). The colonic LP EGFP(CCR10)+ T cells were predominantly composed of CD4+ αβT cells (Supplementary Fig. 2A). Among them, FOXP3+CD4+ Treg cells contained relatively higher percentages of EGFP(CCR10)+ cells than FOXP3−CD4+ or CD8+ T cells (Fig. 2B-C, Supplementary Fig. 2A). Compared to CCR10+/− mice, CCR10−/− mice had the preferentially higher reduction of colonic EGFP+ T cells, particularly Treg cells (Fig. 2B and D). EGFP+ cells in CCR10−/− mice are “CCR10-wannabe cells” that are supposed to express CCR10 but could not because both of their CCR10 coding sequences are replaced by the knocked-in EGFP coding sequences 19. These results indicate that CCR10-knockout leads to impaired presence of EGFP(CCR10)+ T cells, particularly Treg cells, in colons.
Figure 2.

CCR10+IgA+ cells preferentially conjugate with Treg cells in the colon. (A) Flow cytometric detection of EGFP(CCR10) on gated colonic LP CD45+CD3+CD4+ T cells in CCR10+/− mice. Gray areas are the staining of wild-type colonic T cells as negative controls for EGFP. Representative of more than 30 mice. (B) Representative flow cytometric analysis of gated colonic LP CD45+CD3+CD4+ T cells of CCR10+/− and CCR10−/− mice for EGFP(CCR10) and FOXP3. The CCR10+/+ sample is used as a negative control for EGFP. (C) Comparison of percentages of different subsets of colonic LP T cells in CCR10+/− mice that were positive for EGFP(CCR10). N≥7 mice of each genotype were analyzed. (D) Comparison of percentages of total and EGFP(CCR10)+FOXP3+ Treg, and EGFP(CCR10)+FOXP3−CD4+ T cells of the colonic LP of CCR10−/− vs. CCR10+/− mice. N≥7 mice of each genotype were analyzed. (E) Flow cytometric analysis of gated EGFP(CCR10)+CD3−, EGFP(CCR10)+CD3+ and EGFP(CCR10)−CD3+ CD45+ cells of the colons of CCR10+/− mice for CD3 and surface IgA. Representative of more than 10 experiments. (F) Imaging flow cytometric detection of conjugates of EGFP(CCR10)+ IgA-ASCs and EGFP(CCR10)−CD3+ T cells of the colon. Representatives of 30 conjugates. (G) Immunofluorescent microscopic analysis of interaction of IgA-ASCs and FOXP3+ Treg cells in colonic sections. A DAPI-only image is shown besides the image with both DAP1 and antibody staining signals. Note that the IgA+ cell and CD3+Foxp3+ cell indicated by arrows in the inlet have nucleuses based on the DAPI signal. The graph on the right summarizes percentages of FOXP3+ Treg cells and FOXP3− T cells that interact with IgA-ASCs based on the immunofluorescent microscopic analysis. One dot represents one mouse. The line links the data of the same mouse. Total 1196 FOXP3(EGFP)− and 65 FOXP3(EGFP)+ CD3+ T cells of 5 mice were analyzed.
We then analyzed EGFP(CCR10)+ colonic T cells in more detail to gain potential clues underlying their impairment in the colon of CCR10−/− mice. All EGFP(CCR10)+ colonic T cells were of the large size (Supplementary Fig. 2B-C) and did not express CD62L or CCR7 (Supplementary Fig. 2D), suggesting that they were activated T cells. The EGFP(CCR10)+ colonic T cells stained positive for IgA (Fig. 2E), suggesting that they might be conjugates of activated T cells and CCR10+ IgA-ASCs. Imaging flow cytometric analysis confirmed that “EGFP(CCR10)+” colonic T cells were indeed conjugates of EGFP(CCR10)− T cells and EGFP(CCR10)+ IgA-ASCs and that colonic T cells themselves did not express CCR10(EGFP−) (Fig. 2F).
To more precisely assess the frequency of interaction of IgA-ASCs and T cells within the colon in situ, we further performed immunofluorescent microscopic imaging analysis of colonic sections co-stained for IgA-ASCs and T cells. Markedly, within the colonic LP, about 20% of FOXP3− T cells and 40% of FOXP3+ Treg cells conjugated with IgA-ASCs (Fig. 2G). These results reveal a high frequency of interaction of T cells, particularly Treg cells, with IgA-ASCs in the colonic LP.
CCR10-knockout impaired co-migration of IgA-ASCs and their conjugated T cells into colon
IgA-ASCs express MHCII and co-stimulatory/inhibitory molecules such as CD80 and PD-L1 for their interaction with CD4+ T cells 20. They also uniquely express CD28 21. These molecules were expressed similarly on colonic EGFP+ IgA-ASCs of CCR10−/− and CCR10+/− mice (Supplementary Fig. 3A), suggesting that CCR10-knockout would not affect their interaction with T cells. Considering that CCR10+ IgA-ASCs and T cells likely start to form conjugates during their differentiation stages in the GALT 22, we tested whether impaired migration of CCR10−/− IgA-ASCs from the GALT into the colon resulted in impaired migration of their conjugated T cells. Indeed, even higher percentages of EGFP(CCR10)+IgA+ cells conjugated with T cells in the PP, CP and mesenteric lymph node (mLN) compared to the colonic LP (Fig. 3A). As in the colon, Treg cells preferentially interacted with CCR10+ IgA-ASCs in the PP and CP compared to CD4+ or CD8+ effector T cells (Fig. 3B). In addition, treatment of mice with FTY720, a drug that sequestered IgA-ASCs in lymphoid organs of the PP, CP and mLN 23, resulted in the increased accumulation of EGFP(CCR10)+ IgA-ASCs in the PP and CP but their reduction in the mLN and colonic LP (Supplementary Fig. 3B), supporting the notion that CCR10+ IgA-ASCs were generated in the PP and CP and migrated through the mLN to reach the colonic LP. Therefore, in absence of CCR10, there would be impaired migration of IgA-ASCs and their conjugated T cells.
Figure 3.
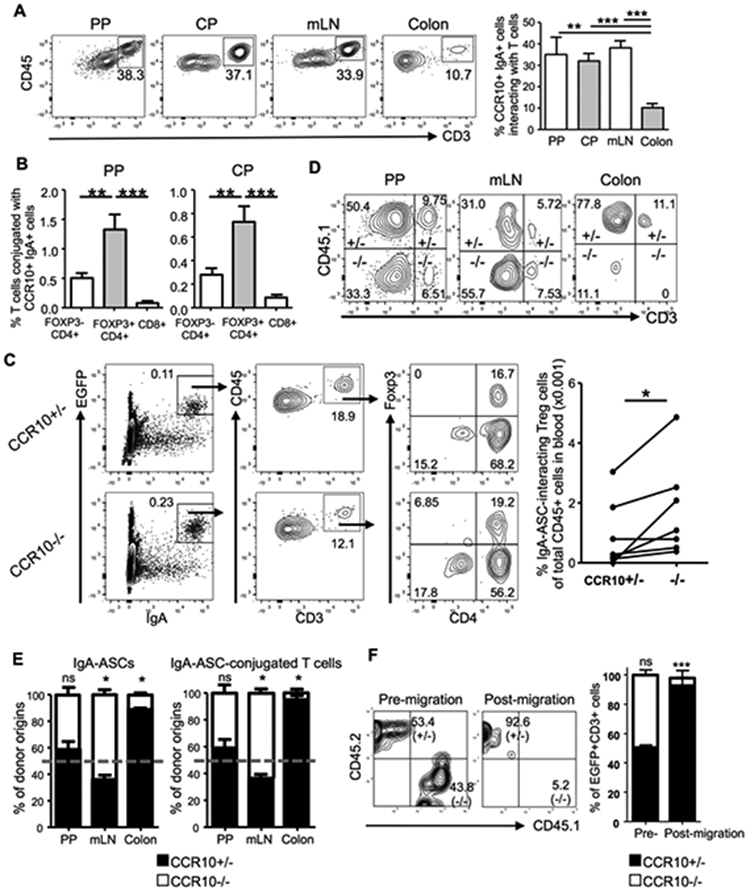
CCR10 mediates co-migration of IgA-ASCs and conjugating T cells into the colon. (A) Analysis of gated EGFP(CCR10)+CD45+intracellular IgA+ cells of the PP, CP, mLN and colons of CCR10+/EGFP mice to detect interacting CD3+ T cells. The bar graph on the right shows average percentages of CCR10(EGFP)+IgA+ cells that interact with T cells. 6 mice for PP and CP, 10 for mLN and 8 for colon were analyzed in at least 3 independent experiments. (B) Comparison of percentages of different subsets of T cells in the PP and CP of CCR10+/− mice that form conjugates with EGFP(CCR10)+ IgA-ASCs. 6 mice were analyzed. (C) Analysis of gated blood CD45+ cells of CCR10+/− and CCR10−/− mice for detection of interacting CCR10(EGFP)+intracellular IgA+ ASCs and T cells. Average percentages of Treg cells that conjugate with EGFP(CCR10)+ IgA+ cells of total CD45+ cells in the blood are presented in the bar graph on the right. One dot represents one mouse. Pairs of mice analyzed in parallel in same experiments are linked by lines. (D) Representative flow cytometric analysis of gated colonic EGFP+ intracellular IgA+ cells for their CCR10+/− (CD45.1+) versus CCR10−/− (CD45.1−) donor B cell origins and their interacting CD3+ T cells in the PP, mLN and colonic LP in μMT recipient mice two weeks after transfer. (E) Relative percentages of contribution of CCR10−/− (−/−) versus CCR10+/− (+/−) donor B cells to total donor-derived EGFP+ IgA-ASCs (left panel) and their interacting T cells (right panel) in PP, mLN and colon of μMT recipients two weeks after transfer. The relative percentages of contribution of CCR10+/− versus CCR10−/− donor B cells to IgA-ASCs were normalized on ratios of CCR10+/− versus CCR10−/− donor B cells in spleens of the recipients. The dashed line indicates the 50% of relative contribution from each donor origin if CCR10+/− and CCR10−/− donor cells contribute equally. Total 8 recipient mice were analyzed. (F) Flow cytometric analysis of 1:1 mixture of gated intestinal EGFP+CD3+ IgA-ASC/T cell conjugates of CCR10+/− and CCR10−/− mice (pre-migration) and cells recovered in bottom of a transwell after migration towards CCL28 in an in vitro migration assay (post-migration). Cells of 3 mice of each genotype were analyzed.
We then searched for evidence of impaired migration of T cells conjugated with IgA-ASCs in CCR10−/− mice. Since IgA-ASCs enter the colonic LP through blood, we tested whether we could find conjugates of IgA-ASCs and T cells in the blood. Indeed, a significant percentage of EGFP(CCR10)+IgA+ cells of the blood conjugated with T cells, with relative enrichment of Treg cells, in CCR10+/− mice (Fig. 3C). In addition, compared to CCR10+/− mice, CCR10−/− mice had increased percentages of EGFP(CCR10)+IgA+ cells and their conjugates with T cells in blood (Fig. 3C), consistent with the notion that the impaired migration of CCR10−/− IgA-ASCs and their T cell conjugates to the colon (and other mucosal sites) resulted in their increased accumulation in blood. To directly demonstrate that impaired migration of CCR10−/− IgA-ASCs resulted in the impaired migration of their conjugated T cells, we co-transferred similar numbers of naïve splenic B cells of CCR10+/− (CD45.1/2) and CCR10−/− (CD45.2/2) mice into B cell-deficient μMT mice. In this transfer scheme, only donor-derived cells could express EGFP, which was used to identify IgA+ cells of all donor origins while CD45.1 was used to distinguish EGFP(CCR10)+IgA+ cells of CCR10+/− (CD45.1+) versus CCR10−/− (CD45.1−) donor origins (Fig. 3D). Two weeks after transfer, we analyzed generation of EGFP(CCR10)+ IgA-ASCs of the donor B cells and their formation of conjugates with T cells of the host origin in the GALT and their localization into the colon. EGFP(CCR10)+ IgA-ASCs were generated equally from CCR10+/− and CCR10−/− donor B cell origins in the PP and they also equally formed conjugates with T cells of recipients (Fig. 3D-E, Supplementary Fig. 3C). However, there were drastically reduced percentages of EGFP(CCR10)+ IgA-ASCs of CCR10−/− donor B cell origin and their conjugated T cells in the colon compared to corresponding populations of CCR10+/− donor B cell origin (Fig. 3D-E). Reciprocally, there was increased accumulation of EGFP(CCR10)+ IgA-ASCs of CCR10−/− donor B cell origin and their conjugated T cells in the mLN compared to corresponding populations of CCR10+/− donor B cell origin (Fig. 3D-E). Together, these results suggest that CCR10+ IgA-ASCs form conjugates with T cells in the PP during their differentiation phases and help migration of the conjugated T cells into the colon. CCR10−/− IgA-ASCs and their conjugated T cells could not efficiently migrate into the colon due to loss of CCR10-mediated migration to chemo-attraction of colon-expressed ligands.
We performed an in vitro co-migration assay in which a mixture of similar numbers of intestinal EGFP(CCR10)+ ASC/T cell conjugates of CCR10+/− and CCR10−/− mice were assessed for their migration towards CCL28 in a transwell. The expression of CD45.2 and CD45.1 distinguished the cells of CCR10+/− and CCR10−/− origins respectively (Fig. 3F). Markedly, of all EGFP(CCR10)+ ASC/T cell conjugates that migrated towards CCL28, more than 90% were of CCR10+/− origin (CD45.2+) and the rest were mostly of CCR10−/− (CD45.1+) origin (Fig. 3F). There were almost no CD45.2+CD45.1+ EGFP(CCR10)+ ASC/T cell conjugates in the post-migration cell population, suggesting no formation of new ASC/T cell conjugates during the migration process (Fig. 3F). These results indicate that the intestinal T cells conjugated with CCR10-expressing IgA-ASCs migrated towards CCL28 while T cells conjugated with CCR10-knockout IgA-ASCs did not, demonstrating that CCR10 could mediate co-migration of CCR10+ IgA-ASC and their T cell conjugates to chemoattraction of its colon-derived ligand.
MHCII-dependent antigen-specific interaction of IgA-ASCs and T cells is crucial for their homeostatic establishment in the colon
Coordinated co-migration of CCR10+ IgA-ASCs and T cells into the colon suggests that the process is important for their proper establishment in the colon. Since MHCII is involved in antigen-specific interaction between IgA-ASCs and CD4+ T cells, we tested whether MHCII was involved in formation of conjugates of IgA-ASCs and T cells and/or their establishment in the colon. To test this, we co-transferred similar numbers of MHCII+/+CCR10+/− (CD45.1/2) and MHCII−/−CCR10+/− (CD45.2/2) B cells into μMT mice. Two weeks after transfer, we analyzed IgA-ASCs of the donor B cells and their formation of conjugates with T cells in the PP and localization into the colon. All donor B cells carried a CCR10-KO/EGFP-KI (CCR10+/−) allele for purpose of identifying donor-derived CCR10+ IgA-ASCs with EGFP. CD45.1 was used to distinguish EGFP(CCR10)+IgA+ cells of MHCII+/+CCR10+/− (CD45.1+) versus MHCII−/−CCR10+/− (CD45.1−) donor origins (Fig. 4A). There were similar percentages of CCR10(EGFP)+ IgA-ASCs of MHCII+/+CCR10+/− and MHCII−/−CCR10+/− donor B cells in the PP, suggesting that B cells could differentiate into gut-homing IgA-ASCs in a T cell antigen-independent fashion (Fig. 4A-B). There was also no difference in the percentage of T cells conjugating with CCR10(EGFP)+ IgA-ASCs of MHCII−/−CCR10+/− and MHCII+/+CCR10+/− donor B cells in the PP and mLN (Fig. 4A-B). Strikingly, however, there were drastically reduced percentages of CCR10(EGFP)+ IgA-ASCs of MHCII−/−CCR10+/− donor B cells and their T cell conjugates in the colon compared to corresponding populations of the MHCII+/+CCR10+/− donor B cells (Fig. 4A-B), suggesting that antigen-specific interaction is crucial for establishment of co-migrated IgA-ASCs and T cells in the colon.
Figure 4.
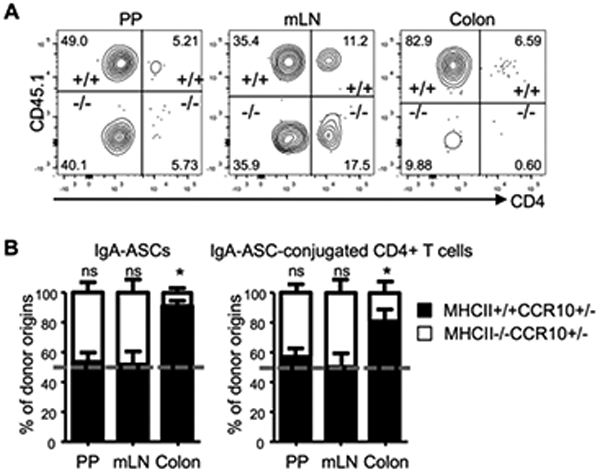
MHCII expressed by CCR10+ IgA-ASCs is not important for their formation of conjugates with T cells but is crucial for their establishment in the colon. (A) Representative flow cytometric analysis of gated EGFP+ intracellular IgA+ cells of the PP, mLN and colons for their MHCII+/+CCR10+/− (+/+) versus MHCII−/−CCR10+/− (−/−) donor B cell origin and interacting CD4+ T cells in μMT recipient mice two weeks after transfer. Inclusion of one CCR10-KO/EGFP-KI allele (CCR10+/−) in the donor B cells was to report the CCR10 expression with EGFP. (B) Relative percentages of CCR10+ IgA-ASCs of MHCII+/+CCR10+/− versus MHCII−/−CCR10+/− donor B cell origin (left panel) and interacting CD4+ T cells (right panel) in the PP, mLN and colons of μMT recipients two weeks after transfer. Relative percentages of contribution of MHCII+/+CCR10+/− versus MHCII−/−CCR10+/− donor B cells to IgA-ASCs were normalized on ratios of MHCII+/+CCR10+/− versus MHCII−/−CCR10+/− donor B cells in spleens of recipients. The dashed line indicates the 50% of relative contribution from each donor origin if MCHII+/+CCR10+/− and MHCII−/−CCR10+/− donor cells contribute equally. 7 recipient mice were analyzed.
CCR10+ IgG1-ASCs are the major isotype of ASCs substituting for CCR10+ IgA-ASCs in the colons of IgA−/− mice
While CCR10 was almost exclusively expressed on all IgA-ASCs in the colon, IgA-knockout (IgA−/−) mice, unlike CCR10−/− mice, did not spontaneously develop colonic inflammation 24, 25. Similarly, most IgA-deficient humans did not have overt intestinal inflammation 26, 27, 28. IgM antibodies have been suggested to compensate for lost functions of IgA in IgA-deficient conditions since IgM is the only antibody isotype besides IgA that could be transported through intestinal epithelia for release into intestinal lumen 3. We therefore assessed what isotypes of antibody-producing cells substituted for CCR10+ IgA-ASCs in the colons of IgA−/−CCR10+/− mice. IgA−/−CCR10+/− mice carried a CCR10-KO/EGFP-KI (CCR10+/−) allele for purpose of reporting the CCR10 expression with EGFP. Compared to IgA+/+CCR10+/− mice, IgA−/−CCR10+/− mice had only mildly reduced percentages of CCR10(EGFP)+ cells in the colon (Fig. 5A), consistent with the notion that other isotypes of antibody-producing cells increased to compensate for loss of IgA-ASCs. Colonic EGFP(CCR10)+ cells of IgA−/−CCR10+/− mice expressed same surface markers of MHCII, PD-L1 and CD28 as colonic EGFP(CCR10)+ IgA-ASCs of IgA+/+CCR10+/− mice (Supplementary Fig. 4A), and also formed conjugates with T cells, preferentially Treg cells (Fig. 5B). Surprisingly, intracellular staining analysis for isotypes of antibodies found that only 20% of colonic EGFP(CCR10)+ cells of IgA−/−CCR10+/− mice stained positive for IgM while most of them (~70%) stained positive for IgG1 (Fig. 5C, Supplementary Fig. 4B). As a result, there was a highly increased number of EGFP(CCR10)+ IgG1+ cells in the colon of IgA−/−CCR10+/− mice compare to IgA+/+CCR10+/− mice (Fig. 5D). The EGFP(CCR10)+ IgG1-ASCs also expressed the membrane form of IgG1 (Fig. 5E). Consistent with predominance of CCR10(EGFP)+ IgG1-ASCs in the colon, increased percentages of colonic EGFP(CCR10)− immune cells, including macrophages, of IgA−/−CCR10+/− mice stained positive for IgG1 (Fig. 5F-G, Supplementary Fig. 4C), likely because increased amounts of secreted IgG1 antibodies bound to phagocytes that express Fc receptors for IgG1. Together, these results suggest that CCR10+ IgG1-ASCs predominantly substitute for CCR10+ IgA-ASCs in the colons of IgA−/− mice to regulate local homeostasis.
Figure 5.
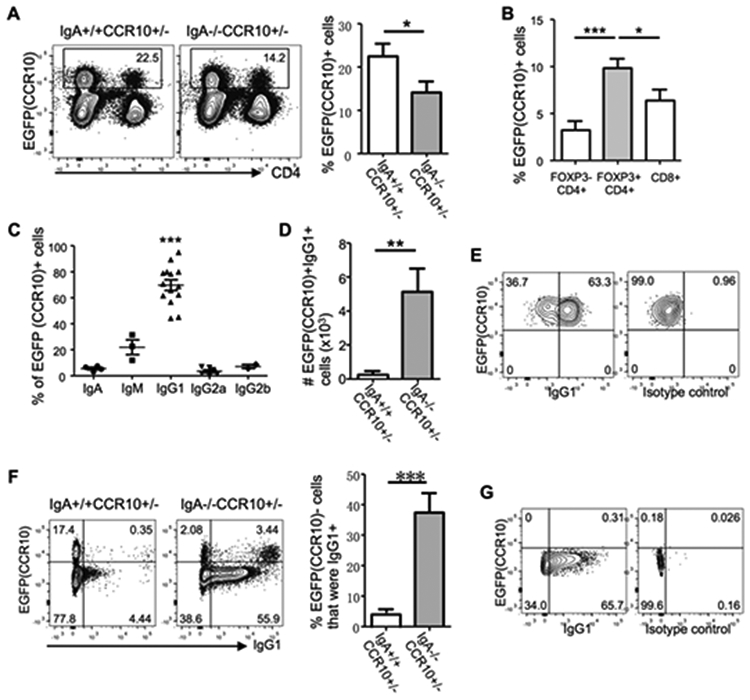
CCR10+ IgG1-ASCs are the major isotype of ASCs that substitute for CCR10+ IgA-ASCs in the colons of IgA-knockout mice. All mice carried a CCR10-KO/EGFP-KI allele (CCR10+/−) for reporting the CCR10 expression with EGFP. (A) Flow cytometric analysis of gated colonic CD45+ immune cells for EGFP(CCR10)+ plasma cells and their interacting CD4+ T cells in IgA+/+CCR10+/− and IgA−/−CCR10+/− mice. The bar graph on the right shows average percentages of EGFP(CCR10)+ cells of total CD45+ immune cells in the colon of IgA+/+CCR10+/− and IgA−/−CCR10+/− mice. N = 6 mice of each genotype. (B) Average percentages of different subsets of colonic T cells that form conjugates with EGFP(CCR10)+ plasma cells in IgA−/−CCR10+/− mice. 6 mice were analyzed. (C) Average percentages of EGFP(CCR10)+ colonic plasma cells that express indicated antibody isotypes in IgA−/−CCR10+/− mice. One dot represents one mouse. (D) Comparison of the number of EGFP(CCR10)+IgG1+ cells in the colon of IgA+/+CCR10+/− and IgA−/−CCR10+/− mice. 5 mice of each genotype were analyzed. (E) Flow cytometric analysis of gated CD45+EGFP(CCR10)+ colonic plasma cells for their surface expression of IgG1 in IgA−/− CCR10+/− mice. Repeated twice. (F) Flow cytometric analysis of gated colonic CD45+ immune cells for intracellular IgG1 in EGFP+ and EGFP− cells in IgA−/−CCR10+/− and IgA+/+CCR10+/− mice. The bar graph on the right shows percentages of EGFP(CCR10)− IgG1+ cells of total EGFP(CCR10)- immune cells in the colon of IgA+/+CCR10+/− and IgA−/−CCR10+/− mice. 5 mice of each genotype were analyzed. (G) Flow cytometric analysis of gated CD45+F4/80+ cells of IgA−/−CCR10+/− mice for intracellular IgG1. Representative of 3 independent experiments.
IgG1-ASCs and IgG2-ASCs are the major isotypes of plasma cells substituting for IgA-ASCs in the colons of IgA-deficient human patients
Preferential expansion of CCR10+ IgG1-ASCs in the colons of IgA−/− mice prompted us to assess isotypes of antibody-producing cells that substituted for IgA-ASCs in the colons of asymptomatic IgA-deficient human individuals. In colonic sections of most IgA-deficient individuals (12/14), there were significantly more IgG+ plasma cells than IgM+ plasma cells (Fig. 6A-B). Further analysis revealed that there were about equal numbers of IgG1+ and IgG2+ plasma cells but few IgG3+ or IgG4+ plasma cells in colonic sections of IgA-deficient individuals (Fig. 6C-D). Colonic plasma cells of IgA-deficient individuals stained positive for CCR10 (Fig. 6E). These results suggest that CCR10+ IgG1/IgG2-ASCs predominantly substitute for CCR10+ IgA-ASCs in the colons of most asymptomatic IgA-deficient individuals.
Figure 6.

IgG1-ASCs and IgG2-ASCs are the major isotypes of plasma cells substituting for IgA-ASCs in IgA-deficient human patients. (A) Representative microscopic images of colonic sections of IgA-sufficient and IgA-deficient patients immunohistochemically stained for IgA+, IgM+ and IgG+ plasma cells. The sections were H&E counter-stained. (B) Numbers of IgA+, IgM+ and IgG+ plasma cells per representative high-power imaging field (HPF) of colonic sections of IgA-sufficient and IgA-deficient patients. One dot represents one patient sample. (C) Representative microscopic images of colonic sections of IgA-deficient patients immunohistochemically stained for IgG1+, IgG2+, IgG3+ and IgG4+ plasma cells. The sections were H&E counter-stained. (D) Numbers of IgG1+, IgG2+, IgG3+ and IgG4+ plasma cells per representative high-power imaging field of colonic sections of IgA-deficient patients. One dot represents one patient sample. (E) Representative microscopic images of colonic sections of IgA-sufficient and IgA-deficient patients immunohistochemically stained for CCR10+ plasma cells. The sections were H&E counter-stained.
DISSCUSSION
In the intestine, IgA-ASCs and T cells, particularly Treg cells, could form a loop to support each other for homeostatic maintenance 29, 30. FOXP3+ Treg cells could help differentiation of IgA-ASCs in the PP 31, and promote survival and antibody production of IgA-ASCs in the intestinal LP 32. IgA-ASCs are suggested to help Treg cell differentiation by secreting TGFβ and retinoic acid to induce the FOXP3 expression 7. However, the in vivo evidence of IgA-ASCs in regulation of T cells is scarce up to now. We here provided the evidence suggesting that CCR10+ IgA-ASCs form conjugates with helper T cells, preferentially Treg cells, during their induction phases in the GALT to coordinate their co-migration into the colon to promote local homeostasis.
Our finding that IgA-ASC/T cell conjugates migrate together into the colon suggests a potentially important mechanism coordinating IgA and T cell responses in intestinal homeostatic regulation. That is, while T cells themselves do not express the colon-homing chemokine receptor CCR10, their conjugation with CCR10+ IgA-ASCs allows them to migrate together with IgA-ASCs for proper localization into the colon. In addition, the co-migration/localization of T cells and IgA-ASCs in the colon might also help their maintenance and function in the colon. Likely, interacting IgA-ASCs and T cells would have specificities to same antigens and their co-localiztion in the colon would help their coordinated responses to the antigens. Furthermore, cross-interaction of colocalized IgA-ASCs and T cells would allow them help each other more efficiently for their survival and maintenance in the colon. These findings provide a physical basis to the notion that IgA-ASCs and T cells, particularly Treg cells, form a loop to for homeostatic maintenance 29, 30. How interacting CCR10+ IgA-ASCs and T cells coordinately respond to the antigen stimulation in colonic homeostatic regulation needs further investigation.
While single cell migration has long been a paradigm in regulation of immune responses, our findings suggest that coordinated co-migration of immune cells of different functions might be required for proper regulation of immune responses in certain settings. Although further confirmation and dissection of molecular and cellular components involved in the coordinated B and T cell migration are needed, coordinated cell migration (or collective cell migration) has been demonstrated as an essential process underlying many aspects of biologic and pathologic functions, from embryonic development, wound healing to cancer metastasis 33, 34, 35. A recent study reported that neutrophils form clusters with circulating tumor cells in blood to help their metastasis, implicating immune cells in coordinated migration with cancer cells 36. Considering that a balanced response of the complex immune system is crucial in health, the coordinated cell migration might be an essential process in regulating immune homeostasis that is worth further investigation.
Our studies also suggest that in absence of IgA-ASCs, specific isotypes of IgG-ASCs could substitute to help the colonic immune homeostasis. IgA-deficiency is a common primary immune-deficient disorder in humans. IgA-deficient individuals have increased incidences of celiac disease and other complications of immune dysregulation 37. However, most IgA-deficient individuals and mice do not display obvious symptoms, suggesting that compensatory mechanisms exist to help maintain immune homeostasis. IgM is suggested to substitute for IgA in IgA-deficient conditions since IgM is the only isotype of antibody besides IgA that could be transported through intestinal epithelium and released into intestinal lumen 3. However, we found that CCR10+ IgG1-ASCs, but not IgM-ASCs, are the dominant isotype of substituting ASCs in the colons of IgA-knockout mice. Compared to the other IgG isotypes, IgG1 preferentially binds to FcγRIIb, an inhibitory Fc receptor that transduces regulatory signals 38, 39. Therefore, while IgG1 could not be secreted into intestinal lumen, it could opsonize invading bacteria for destruction by phagocytes within the tissue without inducing detrimental inflammation. Similarly, IgG1- and IgG2-ASCs are major isotypes of substituting ASCs in the colons of asymptomatic IgA-deficient human individuals. Since these individuals do not have intestinal inflammatory symptoms, the IgG1- and IgG2-ASCs likely help maintain intestinal homeostasis in absence of IgA. Like IgG1 in mice, IgG2 of humans is primarily involved in immune regulation, since it has the lowest activating-to-inhibitory ratio among all IgG subclasses 40. In contrast to the predominance of IgG1- and IgG2-ASCs in asymptomatic IgA-deficient individuals, IgM-ASCs were reported to dominate in the gut of symptomatic IgA-deficient individuals 41, 42, 43. These results suggest that IgA-deficient individuals with properly compensatory isotypes of ASCs (i.e., IgG1/IgG2-ASCs) would have better maintenance of intestinal homeostasis. Consistent with this notion, individuals with simultaneous deficiency of IgA and IgG2 have higher incidences of intestinal diseases than those with combinatory deficiency of IgA and other antibody isotypes 44. Further studies are needed to establish how differential expansion of specific isotypes of ASCs affects the intestinal homeostasis versus inflammation in IgA-deficient conditions.
MATERIALS AND METHODS
Mice
CCR10-knockout/EGFP-knockin (CCR10-KO/EGFP-KI) mice on C57BL/6 background (CD45.2/2) were described 19. MHCII-knockout, μMT and FOXP3-EGFP reporter mice were purchased from Jackson lab. IgA-knockout mice were obtained from the Mutant Mouse Regional Resource Centers (MMRRC) 24,45. A CCR10-KO/EGFP-KI allele (CCR10+/−) was introduced to MHCII-knockout and IgA-knockout mice for purpose of reporting the CCR10 expression with EGFP by proper crossing. All mice were on C57BL/6 (CD45.2+) background unless indicated otherwise. Mice were used at 8-12 weeks of age unless indicated otherwise. Mouse experiments were approved by the Institutional Animal Care and Use Committees (IACUC) of The Pennsylvania State University and University of Texas Health Science Center at San Antonio.
Human samples
Archives at Department of Pathology, Montefiore Medical Center/Albert Einstein College of Medicine were searched for colon biopsy specimens from patients with selective IgA deficiency using Clinical Looking Glass@. Selective IgA deficiency was defined as serum IgA level < 7 mg/dL with normal or increased levels of serum IgG (≥ 844 mg/dL) and IgM (≥ 50 mg/dL) in individuals older than 4 years of age. A total of 14 colon biopsy specimens from 11 patients were identified. The clinical information of the patients was summarized in Supplementary Table 1. Five colon biopsy specimens from age- and gender-matched IgA-sufficient patients were used as control. This retrospective study was approved by the Institutional Review Board of Albert Einstein College of Medicine.
Antibodies and chemicals
Antibodies and chemical reagents are listed in Supplementary Tables 2 and 3.
Cell isolation
Isolation of lymphocytes were performed similarly as we previously described 15. Briefly, for isolation of lymphocytes from colonic LP, colons were flushed with cold HBSS containing 15 mM HEPES (CMF/HEPES), opened longitudinally, cut into 5-mm segments and washed 4-5 times with cold CMF/HEPES. Colon segment cuts were then incubated for 15 min at 37°C with shaking at 200 rpm in HBSS containing 15 mM HEPES, 5 mM EDTA and 10% FBS (Equitech-Bio, Kerrville, TX), followed by intense vortex to remove the epithelium and intraepithelial lymphocytes. This step was repeated 3-4 times until no more epithelium shedding occurred. The remaining pieces were washed with RPMI medium (Mediatech, Manassas, VA) containing 10% FBS, minced, and then digested for 60 min at 37°C with shaking at 200 rpm in RPMI medium containing 5% FBS, 1.5 mg/mL collagenase (Worthington, Lakewood, NJ). To isolate lymphocytes from Peyer’s patches and cecal patches, the patches were dissected out and then minced and digested in RPMI medium containing 0.5 mg/mL collagenase for 30 min at 37°C with shaking at 200 rpm. Dissociated cells from the digestion were washed once with PBS, resuspended in 3 mL of 80% Percoll (GE Healthcare), underlaid with 4 mL of 40% Percoll, and centrifuged for 20 min at 850g at room temperature. Lymphocytes were recovered from the interphase of the Percoll gradient, washed twice, and resuspended in FACS buffer (PBS containing 3% FBS and 0.05% sodium azide) or RPMI medium. Spleen and mesenteric lymph node cells were prepared by pressing the tissues through cell strainers using the end of a sterile plunger of a 5-ml syringe. Peripheral blood lymphocytes were isolated by gradient centrifugation using Lympholyte-Mammal (CEDARLANE Laboratories, Burlington, NC).
Cell staining and flow cytometric analysis
For antibody staining of surface markers, cells were incubated with fluorescently labeled antibodies for 30 min at 4°C. For antibody staining of intracellular Foxp3, IgA and other different isotypes of antibodies, cells were first stained for cell surface markers, then fixed with 4% paraformaldehyde (PFA), permeabilized with the Foxp3/Transcription Factor Staining Buffer Set (eBioscience), and stained with antibodies against the intracellular molecules. For the intracellular staining of cytokines, cells were first stimulated for 2–4 h in culture with PMA (phorbol 12-myristate 13-acetate) and ionomycin in the presence of brefeldin A and then were stained as for the intracellular staining of the other intracellular molecules. Cells stained with a single antibody were used for the calibration experiments. Flow cytometric analyses were performed on FC500 (Beckman Coulter) or BD Fortessa LSRII (BD Biosciences, San Jose, CA). Imaging flow cytometric analysis was performed on Amnis FlowSight (Luminex). Data were analyzed with FlowJo software (BD Biosciences).
DSS-induced colitis
Mice were treated for 7 days with 3% DSS in drinking water. Fecal occult bleeding was detected with Coloscreen (Helena Laboratories, Beaumont, Texas) at day 3 after starting the DSS treatment. Treated mice were monitored for bodyweight change. A drop of bodyweight more than 30% was counted as death. Colons were collected at day 7 after starting the DSS treatment and used for the H&E staining and qRT-PCR analyses.
FTY720 treatment
Mice were provided drinking water containing FTY720 (10 μg/mL) for 14-21 days.
B cell transfer into μMT mice
Naïve B cells were purified from spleens using B cell isolation kit (Miltenyi Biotec). Twenty million B cells of CD45.1/2 CCR10+/− and CD45.2/2 CCR10−/− or CD45.2/2 MHCII−/−CCR10+/− mixtures at the 1:1 ratio were intravenously injected into μMT mice. Two weeks after transfer, mice were euthanized for analysis of donor-derived EGFP(CCR10)+IgA+ cells and their formation of T cell conjugates in the PP, mLN and colonic LP.
In vitro migration assay
The experiment was performed similarly as we previously described 46. Briefly, colonic LP EGFP+CD3+ conjugates of CCR10+/− (CD45.2+) and CCR10−/− (CD45.1+) mice were mixed at the 1:1 ratio, suspended in RPMI-1640/10% FBS, immediately placed into the upper chamber of a Transwell with 5-μm pore filters (Costar, Corning, NY), and incubated with CCL28 in the bottom chamber for 4 hours. Cells migrating into the bottom chamber were collected and analyzed by flow cytometry for EGFP+CD3+ conjugates of CCR10+/− and CCR10−/− origins. The ratio of EGFP+CD3+ conjugates of CCR10+/− and CCR10−/− origins migrating into the bottom chamber was quantified based on their different expression of CD45.2 and CD45.1, compared to the ratio of the mixture before migration. In this study, average 19% (19%±3%) of CCR10+/− EGFP+CD3+ cells migrated into the bottom chamber in the presence of CCL28.
Real-time quantitative RT-PCR
The experiment was performed as we described 46. Sequences of primers used are listed in Supplementary Table 4.
H&E staining and pathological scoring of colonic sections
The paraffin embedding, sectioning and H&E staining of colons were performed by the Animal Diagnostic Laboratory of Pennsylvania State University. Colitic lesions were scored in four categories (inflammation, degeneration of epithelium, edema, and thickening of epithelium) based on following criteria: 0= within normal limits, 1= minimal, 2= mild, 3= moderate, and 4= extensive. Sums of scores in the four categories are pathological scores.
Immunofluorescent staining and microscopic analysis of colonic sections for detection of interaction of IgA-ASCs and Treg cells
Colons of FOXP3-EGFP reporter mice were fixed in 4% paraformaldehyde, frozen in O.C.T. medium, cut at the 3-μm thickness on a cryostat, and placed on glass slides. The sections were stained with Alexa Flour 568-conjugated anti-mouse CD3ε (17A2) and APC-conjugated anti-mouse IgA (11-44-2), and counterstained with DAPI. Stained sections were imaged with an Olympus Fluoview 10i microscope. Images were analyzed with the Fiji software.
Immunohistochemical staining of human colonic sections
The antigen retrieval and immunohistochemical staining of human colonic sections were performed as previously described 47.
Statistical analyses
Statistical significance was determined by unpaired student T tests (dot plots or bar graphs) or paired student T tests (in the figures where lines linked the data of paired samples). The data is presented as means ± standard errors (SEM). Time to death post DSS treatment was estimated using Kaplan-Meier estimates. Survival curves between groups were compared using log-rank tests. P < 0.05 is considered significant.
Supplementary Material
ACKNOWLEDGEMENTS
Research reported in this publication was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under Award Numbers AR064831 and Pennsylvania State University institutional support (to N.X) and Montefiore Medical Center Pathology Research Funding (to S.H). The content is solely the responsibility of the authors and does not necessarily represent official views of the funding agencies. We thank the Clinical Immunology lab of Montefiore Medical Center for excellent technical support for IHC staining.
Footnotes
Disclosure: All authors declare no conflict of interest.
REFERENCES
- 1.Cerutti A The regulation of IgA class switching. Nat Rev Immunol 8, 421–434 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mora JR & von Andrian UH Differentiation and homing of IgA-secreting cells. Mucosal Immunol 1, 96–109 (2008). [DOI] [PubMed] [Google Scholar]
- 3.Macpherson AJ, Geuking MB & McCoy KD Homeland security: IgA immunity at the frontiers of the body. Trends Immunol 33, 160–167 (2012). [DOI] [PubMed] [Google Scholar]
- 4.Xiong N & Hu S Regulation of intestinal IgA responses. Cell Mol Life Sci 72, 2645–2655 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brandtzaeg P Secretory IgA: Designed for Anti-Microbial Defense. Front Immunol 4, 222 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fritz JH et al. Acquisition of a multifunctional IgA+ plasma cell phenotype in the gut. Nature 481, 199–203 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim MS & Kim TS IgA+ plasma cells in murine intestinal lamina propria as a positive regulator of Treg differentiation. J Leukoc Biol 95, 461–469 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Gommerman JL, Rojas OL & Fritz JH Re-thinking the functions of IgA(+) plasma cells. Gut Microbes 5, 652–662 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shale M, Schiering C & Powrie F CD4(+) T-cell subsets in intestinal inflammation. Immunol Rev 252, 164–182 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Josefowicz SZ, Lu LF & Rudensky AY Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol 30, 531–564 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kunkel EJ et al. CCR10 expression is a common feature of circulating and mucosal epithelial tissue IgA Ab-secreting cells. J Clin Invest 111, 1001–1010 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lazarus NH et al. A common mucosal chemokine (mucosae-associated epithelial chemokine/CCL28) selectively attracts IgA plasmablasts. J Immunol 170, 3799–3805 (2003). [DOI] [PubMed] [Google Scholar]
- 13.Pan J et al. A novel chemokine ligand for CCR10 and CCR3 expressed by epithelial cells in mucosal tissues. J Immunol 165, 2943–2949 (2000). [DOI] [PubMed] [Google Scholar]
- 14.Wang W et al. Identification of a novel chemokine (CCL28), which binds CCR10 (GPR2). J Biol Chem 275, 22313–22323 (2000). [DOI] [PubMed] [Google Scholar]
- 15.Hu S, Yang K, Yang J, Li M & Xiong N Critical roles of chemokine receptor CCR10 in regulating memory IgA responses in intestines. Proc Natl Acad Sci U S A 108, E1035–1044 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim SV et al. GPR15-mediated homing controls immune homeostasis in the large intestine mucosa. Science 340, 1456–1459 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nguyen LP et al. Role and species-specific expression of colon T cell homing receptor GPR15 in colitis. Nat Immunol 16, 207–213 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang C, Kang SG, Lee J, Sun Z & Kim CH The roles of CCR6 in migration of Th17 cells and regulation of effector T-cell balance in the gut. Mucosal Immunol 2, 173–183 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jin Y, Xia M, Sun A, Saylor CM & Xiong N CCR10 is important for the development of skin-specific gammadeltaT cells by regulating their migration and location. J Immunol 185, 5723–5731 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Doi T et al. IgA plasma cells express the negative regulatory co-stimulatory molecule programmed cell death 1 ligand and have a potential tolerogenic role in the intestine. Biochem Biophys Res Commun 425, 918–923 (2012). [DOI] [PubMed] [Google Scholar]
- 21.Rozanski CH et al. Sustained antibody responses depend on CD28 function in bone marrow-resident plasma cells. J Exp Med 208, 1435–1446 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tsuji M et al. Preferential generation of follicular B helper T cells from Foxp3+ T cells in gut Peyer's patches. Science 323, 1488–1492 (2009). [DOI] [PubMed] [Google Scholar]
- 23.Gohda M et al. Sphingosine 1-phosphate regulates the egress of IgA plasmablasts from Peyer's patches for intestinal IgA responses. J Immunol 180, 5335–5343 (2008). [DOI] [PubMed] [Google Scholar]
- 24.Harriman GR et al. Targeted deletion of the IgA constant region in mice leads to IgA deficiency with alterations in expression of other Ig isotypes. J Immunol 162, 2521–2529 (1999). [PubMed] [Google Scholar]
- 25.Macpherson AJ & McCoy KD Independence Day for IgA. Immunity 43, 416–418 (2015). [DOI] [PubMed] [Google Scholar]
- 26.Suzuki K et al. Aberrant expansion of segmented filamentous bacteria in IgA-deficient gut. Proc Natl Acad Sci U S A 101, 1981–1986 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khounlotham M et al. Compromised intestinal epithelial barrier induces adaptive immune compensation that protects from colitis. Immunity 37, 563–573 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moon C et al. Vertically transmitted faecal IgA levels determine extra-chromosomal phenotypic variation. Nature 521, 90–93 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Feng T, Elson CO & Cong Y Treg cell-IgA axis in maintenance of host immune homeostasis with microbiota. Int Immunopharmacol 11, 589–592 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang L et al. T regulatory cells and B cells cooperate to form a regulatory loop that maintains gut homeostasis and suppresses dextran sulfate sodium-induced colitis. Mucosal Immunol 8, 1297–1312 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kawamoto S et al. Foxp3(+) T cells regulate immunoglobulin a selection and facilitate diversification of bacterial species responsible for immune homeostasis. Immunity 41, 152–165 (2014). [DOI] [PubMed] [Google Scholar]
- 32.Cong Y, Feng T, Fujihashi K, Schoeb TR & Elson CO A dominant, coordinated T regulatory cell-IgA response to the intestinal microbiota. Proc Natl Acad Sci U S A 106, 19256–19261 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Friedl P & Gilmour D Collective cell migration in morphogenesis, regeneration and cancer. Nat Rev Mol Cell Biol 10, 445–457 (2009). [DOI] [PubMed] [Google Scholar]
- 34.Shellard A & Mayor R Supracellular migration - beyond collective cell migration. J Cell Sci 132 (2019). [DOI] [PubMed] [Google Scholar]
- 35.Labernadie A et al. A mechanically active heterotypic E-cadherin/N-cadherin adhesion enables fibroblasts to drive cancer cell invasion. Nat Cell Biol 19, 224–237 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Szczerba BM et al. Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature 566, 553–557 (2019). [DOI] [PubMed] [Google Scholar]
- 37.Yazdani R, Azizi G, Abolhassani H & Aghamohammadi A Selective IgA Deficiency: Epidemiology, Pathogenesis, Clinical Phenotype, Diagnosis, Prognosis and Management. Scand J Immunol 85, 3–12 (2017). [DOI] [PubMed] [Google Scholar]
- 38.Nimmerjahn F & Ravetch JV Divergent immunoglobulin g subclass activity through selective Fc receptor binding. Science 310, 1510–1512 (2005). [DOI] [PubMed] [Google Scholar]
- 39.Ravetch JV & Bolland S IgG Fc receptors. Annu Rev Immunol 19, 275–290 (2001). [DOI] [PubMed] [Google Scholar]
- 40.Bruhns P Properties of mouse and human IgG receptors and their contribution to disease models. Blood 119, 5640–5649 (2012). [DOI] [PubMed] [Google Scholar]
- 41.Crabbe PA & Heremans JF Lack of gamma A-immunoglobulin in serum of patients with steatorrhoea. Gut 7, 119–127 (1966). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Savilahti E IgA deficiency in children. Immunoglobulin-containing cells in the intestinal mucosa, immunoglobulins in secretions and serum IgA levels. Clin Exp Immunol 13, 395–406 (1973). [PMC free article] [PubMed] [Google Scholar]
- 43.Brandtzaeg P et al. The human secretory immune system shows striking heterogeneity with regard to involvement of J chain-positive IgD immunocytes. J Immunol 122, 503–510 (1979). [PubMed] [Google Scholar]
- 44.Oxelius VA et al. IgG subclasses in selective IgA deficiency: importance of IgG2-IgA deficiency. N Engl J Med 304, 1476–1477 (1981). [DOI] [PubMed] [Google Scholar]
- 45.Mbawuike IN et al. Mucosal immunity to influenza without IgA: an IgA knockout mouse model. J Immunol 162, 2530–2537 (1999). [PubMed] [Google Scholar]
- 46.Hu S & Xiong N Programmed downregulation of CCR6 is important for establishment of epidermal gammadeltaT cells by regulating their thymic egress and epidermal location. J Immunol 190, 3267–3275 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wolgast LR et al. Spectrin isoforms: differential expression in normal hematopoiesis and alterations in neoplastic bone marrow disorders. Am J Clin Pathol 136, 300–308 (2011). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.