

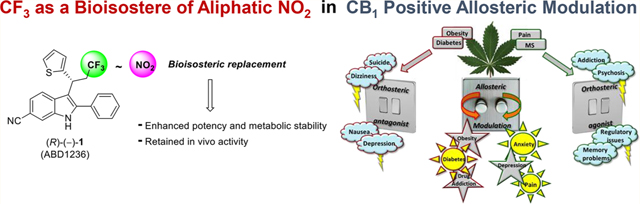
Abstract
The first generation of CB1 positive allosteric modulators (e.g., ZCZ011) featured a 3-nitroalkyl-2-phenyl-indole structure. Although a small number of drugs include the nitro group, it is generally not regarded as being “drug-like”, and this is particularly true for aliphatic nitro groups. There are very few case studies where an appropriate bioisostere replaced a nitro group that had a direct role in binding. This may be indicative of the difficulty of replicating its binding interactions. Herein, we report the design and synthesis of ligands targeting the allosteric binding site on the CB1 cannabinoid receptor, in which a CF3 group successfully replaced the aliphatic NO2. In general, the CF3-bearing compounds were more potent than their NO2 equivalents and also showed improved in vitro metabolic stability. The CF3 analogue (1) with the best balance of properties was selected for further pharmacological evaluation. Pilot in vivo studies showed that (±)-1 has similar activity to (±)-ZCZ011, with both showing promising efficacy in a mouse model of neuropathic pain.
Graphical Abstract
INTRODUCTION
The endocannabinoid system plays a major role in modulating neurotransmitter release in the central nervous system, and thus its modulation elicits a broad range of physiological effects. Compounds activating cannabinoid receptors represent a promising therapeutic opportunity for the treatment of many disorders, including depression, anxiety, and pain. However, global activation of the cannabinoid CB1 receptor by direct agonism leads to psychoactive side effects, physical dependence, and abuse liability. There are both an opportunity and a need to provide safer and more effective ways of harnessing the therapeutic benefits of CB1 receptor activation. Recent reviews have highlighted the therapeutic potential of targeting the allosteric binding site on the CB1 receptor using positive allosteric modulators (PAMs),1,2 which have the advantage of enhancing the actions of endocannabinoids directly at the orthosteric receptors expressed within spinal and brain pain processing centers. Thus, PAMs are predicted to be free of the psychoactive side effects associated with direct CB1 agonists that globally activate these receptors. The impressive efficacy of small molecule tool CB1 PAMs in models of neuropathic pain and post-traumatic stress disorder, without any evidence of addiction or psychotropic side effects, supports this hypothesis.
The activity of the first CB1 PAM - AZ-4,3 also known as F-0874 and now commonly referred to as GAT2115 (Figure 1), was reported in a conference poster by researchers at AstraZeneca Montreal,3 having been first synthesized by Noland and Lange in 1959.6 The structure has since been modified to improve potency (ZCZ011,7 5 in Table 1), and the enantiomers of AZ-4 have been separated (GAT228/229),5 but no substantial development or comprehensive structure activity relationships have been reported. The primary problematic issue with these compounds is the presence of an aliphatic nitro group. Despite its presence in a small number of major drugs, including nifedipine, chloramphenicol, ranitidine, and metronidazole, most academic and industrial medicinal chemists do not regard a nitro group as being “drug-like”. However, surprisingly little published evidence exists to support this blanket exclusion of the nitro group from the drug-like space. Justification for this exclusion may be based upon accepted wisdom, derived from personal experience (e.g., see Muegge and co-workers,8 Beck and co-workers,9 and discussions on internet forums, including the well-regarded “In the Pipeline”10,11), or based on a well-established (if only potential) toxic liability associated solely with the aromatic nitro and its propensity for formation of an aryl nitrenium ion, which can bind to DNA.12 In additional examples of this general dismissal of the nitro group from the drug-like space, the nitro was one of the groups eliminated prior to the studies leading to Baell’s identification of the range of pan-assay interference compounds;13 likewise, any structures containing a nitro are explicitly excluded by Rankovic and Morphy14 as being acceptable for use in fragment-based lead discovery.
Figure 1.

Molecular structures of AZ-4 (GAT211) and ZCZ-011 (5).
Table 1.
Enhancement of β-Arrestin Recruitment Induced by the CB1 Agonist CP 55,940 and Metabolic Stability Data from Human, Rat, and Mouse Liver Microsomal Stability Studies (HLM, RLM, and MLM)
 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| comp | R1 | R2 | R3 | R4 | PAM activitya: >50% enhancement | EC50 |
Emax |
t½(min)e |
||||
| PAMb | Agoc | PAMd | Agod | HLM | RLM | MLM | ||||||
| 2 | 5-Me | 2-thienyl | Ph | CH2NO2 | * | |||||||
| 3 | 5-CN | 2-thienyl | Ph | CH2NO2 | NA | 49.4 | 8.2 | |||||
| 4 | 5-NO2 | 2-thienyl | Ph | CH2NO2 | NA | |||||||
| 5 | 6-Me | 2-thienyl | Ph | CH2NO2 | ** | 283 ± 140 | 777 ± 363 | 106 ± 18 | 55 ± 24 | 41.2 ± 3.1 | 6.4 ± 1.2 | 10.4 ± 2.1 |
| 6 | 6-Cl | 2-thienyl | Ph | CH2NO2 | ** | 462 ± 363 | 1530 ± 1128 | 154 ± 10 | 82 ± 48 | 32.7 | 13.8 | |
| 7 | 6-CF3 | 2-thienyl | Ph | CH2NO2 | ** | 284 ± 211 | 974 ± 780 | 163 ± 17 | 106 ± 47 | 30.7 | 15.6 | |
| (±)-8 | 6-CN | 2-thienyl | Ph | CH2NO2 | * | |||||||
| (+)-8 | 6-CN | 2-thienyl | Ph | CH2NO2 | * | 863 ± 211 | >10000 | 51 ± 18## | 9 ± 4# | |||
| (-)-8 | 6-CN | 2-thienyl | Ph | CH2NO2 | * | 664 ± 215 | >10000 | 117 ± 19## | 21 ± 6# | |||
| 9 | 6-Cl | 2-thienyl | 4-OMe—C6H4− | CH2NO2 | NA | |||||||
| 10 | 6-Cl | 2-thienyl | 4-NMe2−C6H4− | CH2NO2 | NA | |||||||
| 11 | 6-Cl | 2-thienyl | 4-CN−C6H4− | CH2NO2 | NA | |||||||
| 12 | 6-Cl | 2-thienyl | 4-CO2Me−C6H4− | CH2NO2 | NA | |||||||
| 13 | 6-Cl | 2-thienyl | 4-SO2Me−C6H4− | CH2NO2 | NA | |||||||
| 14 | 6-Cl | 2-thienyl | 4-COOH−C6H4− | CH2NO2 | NA | |||||||
| 15 | 6-Cl | 2-thienyl | 4-Ac−C6H4− | CH2NO2 | NA | |||||||
| 16 | 6-Me | 2-thienyl | 4-C1−C6H4− | CH2NO2 | NA | 24.2 | 9.8 | |||||
| 17 | 6-Cl | 2-thienyl | 4-C1−C6H4− | CH2NO2 | NA | |||||||
| 18 | 6-Cl | 2-thienyl | 4-F−C6H4− | CH2NO2 | NA | |||||||
| 19 | 6-Me | 2-thienyl | 4-F−C6H4− | CH2NO2 | NA | |||||||
| 20 | 6-Cl | 2-thienyl | 3-OMe−C6H4− | CH2NO2 | NA | |||||||
| 21 | 6-Cl | 2-thienyl | 3-CN−C6H4− | CH2NO2 | NA | |||||||
| 22 | 6-Me | 2-thienyl | cyclopropyl | CH2NO2 | * | |||||||
| 23 | 6-Me | 2-thienyl | cyclopentyl | CH2NO2 | * | |||||||
| 24 | 6-Me | 2-thienyl | cyclohexyl | CH2NO2 | NA | |||||||
| 25 | 5-CN | 2-thienyl | cyclohexyl | CH2NO2 | NA | 21.4 | 3.8 | |||||
| (±)-26 | Ph | Ph | CH2CO2H | NA | 62.7 | |||||||
| (+)-26 | Ph | Ph | CH2CO2H | NA | ||||||||
| (−)-26 | Ph | Ph | CH2CO2H | NA | ||||||||
| 27 | Ph | Ph | CH2OH | NA | ||||||||
| 28 | Ph | Ph | CH2CN | NA | 42.7 | 14.5 | ||||||
| 29 | Ph | Ph | CH(CN)2 | NA | 26.2 | 14.8 | ||||||
| 30 | 6-Me | Ph | Ph | CH2CN | * | 45.7 | 18.7 | |||||
| 31 | 6-Cl | Ph | Ph | CH2CN | * | 1113 ± 401 | 2354 ± 601 | 100 ± 36 | 24 ± 7 | 43.2 | 22.5 | |
| 32 | 6-Me | Ph | Ph | CH(CN)2 | * | 39.3 | 12.4 | |||||
| 33 | 6-Me | 2-thienyl | Ph | CH(CN)2 | * | 31.2 | 4.9 | |||||
| 34 | 6-Cl | 2-thienyl | Ph | CH(CN)2 | NA | 29.6 | 13.5 | |||||
| 35 | Ph | Ph | COCF3 | NA | 51.1 | 41.4 | ||||||
| 36 | Ph | Ph | CHOHCF3 | NA | 39.4 | 18.2 | ||||||
| 37 | Ph | Ph | CH2CF3 | * | 85.2 | 40.7 | ||||||
| 38 | 6-Me | Ph | Ph | COCF3 | ** | 267 | 24.8 | |||||
| 39 | 6-Me | Ph | Ph | CH2CF3 | ** | 87.2 | 28.2 | |||||
| 40 | 6-Me | cyclopropylmethyl | Ph | CH2CF3 | * | 74.4 | 19.8 | |||||
| 41 | 6-Me | 2-thienyl | Ph | CH2CF3 | ** | 108 | 17.5 | |||||
| 42 | 6-Me | 2-furyl | Ph | CH2CF3 | * | 59.1 | 10.3 | |||||
| 43 | 6-Cl | 2-furyl | Ph | CH2CF3 | ** | 63.9 | 46.3 | |||||
| 44 | 6-Cl | 2-thienyl | Ph | CH2CF3 | *** | 157 | 54.3 | |||||
| 45 | 6-Cl | Ph | Ph | CH2CF3 | 98.6 | 78.0 | ||||||
| 46 | 6-CF3 | Ph | Ph | CH2CF3 | *** | |||||||
| 47 | 6-CF3 | 2-thienyl | Ph | CH2CF3 | ** | 322 ± 252 | 801 ± 554 | 203 ± 54 | 95 ± 15 | 49.7 | 36.7 | |
| 48 | 5-Me | Ph | Ph | CH2CF3 | NA | |||||||
| 49 | 5-Me | 2-thienyl | Ph | CH2CF3 | NA | |||||||
| (±)-50 | 6-CO2Me | 2-thienyl | Ph | CH2CF3 | *** | |||||||
| (+)-50 | 6-CO2Me | 2-thienyl | Ph | CH2CF3 | *** | 160 ± 147‡‡ | 682 ± 211‡ | 88 ± 28## | 26 ± 5## | |||
| (−)-50 | 6-CO2Me | 2-thienyl | Ph | CH2CF3 | *** | 113 ± 62‡ | 416 ± 167‡ | 156 ± 41## | 51 ± 11## | |||
| 51 | 6-CO2H | 2-thienyl | Ph | CH2CF3 | NA | |||||||
| 52 | 6-CH2OH | 2-thienyl | Ph | CH2CF3 | NA | |||||||
| 53 | 6-NHAc | 2-thienyl | Ph | CH2CF3 | NA | |||||||
| (±)-1 | 6-CN | 2-thienyl | Ph | CH2CF3 | *** | 177 ± 66 | 647 ± 270 | 100 ± 25 | 30 ± 14 | |||
| (S)-(+)-1 | 6-CN | 2-thienyl | Ph | CH2CF3 | ** | 342 ± 142‡ | 1206 ± 345‡ | 79 ± 32# | 17 ± 4 | |||
| (R)-(−)-1 | 6-CN | 2-thienyl | Ph | CH2CF3 | ** | 363 ± 169‡‡ | 828 ± 310‡‡ | 125 ± 29# | 25 ± 6 | 72.3 ± 30.5 | 34.9 ± 7.4 | 26.5 ± 1.5 |
| 54 | 6-Ac | 2-thienyl | Ph | CH2CF3 | *** | 158 ± 60 | 741 ± 362 | 121 ± 23 | 39 ± 9 | 51.2 | 12.2 | |
| 55 | 6-NH2 | 2-thienyl | Ph | CH2CF3 | NA | |||||||
| 56 | 6-NHSO2Me | 2-thienyl | Ph | CH2CF3 | NA | |||||||
| 57 | 6-CF3 | 2-thienyl | cyclopentyl | CH2CF3 | ** | 301 ± 66 | 755 ± 237 | 162 ± 47 | 68 ± 14 | 47.1 | 44.4 | |
Pilot data were generated as described previously7 and represent data from a single experiment.
at 1000 nM;
at 500 nM;
at 200 nM.
Selected compounds were evaluated by Eurofins DiscoverX in three independent experiments, using the PathHunter β-arrestin assay. Positive allosteric modulation was assessed using EC20 CP 55,940. The EC50 value represents the compound concentration at which 50% of the maximal achievable EC20 CP 55,940-induced β-arrestin recruitment is seen (± standard deviation) for each specific compound.
Agonism was evaluated in the same assay, and EC50 and Emax values were generated as described for positive allosteric modulation using the compound alone in the absence of the agonist.
Emax for each compound is the maximum EC20 CP 55,940-induced β-arrestin recruitment relative to the maximum achievable with CP 55,940.
Human, rat, and mouse liver microsomal stability studies were conducted by Cyprotex Ltd. Half-life values represent data from a single experiment (five time points), except where ± standard deviation is given (three independent experiments).
P < 0.005;
P < 0.05, between enantiomers (Student’s t test).
P < 0.005;
P < 0.05, between IC50 values of the PAM and agonist (two-tailed t test). NA = not active (defined as <50% enhancement in CP 55,940-induced β-arrestin recruitment at 1 μM).
While little information exists on the toxicological issues associated with aliphatic nitro groups, the widely held opinions described above usually require its removal or replacement early in the development of a bioactive compound. However, there are only a few case studies in which a nitro group has been successfully replaced by an appropriate bioisostere,9 which may be indicative of the difficulty of replicating its binding interactions. Nitro groups may act as hydrogen bond acceptors and may also stack with other groups, including ketones and alcohols.15 Additionally, Alston and co-workers16 reported that the nitroalkyl group can serve as a one- and two-electron reductant, oxidant, ambident nucleophile, electrophile, ligand, and leaving group in reactions with enzymes and other biomolecules and suggested that “the richness of its reactivity may have hindered the application of the nitroalkyl group in medicinal chemistry.” Much more commonly, nitro groups are initially present in bioactive compounds as aromatic substituents where their electron-withdrawing properties may polarize the ring, giving optimal interaction with electron-rich moieties within the biological target. In such cases, simple replacement with other strongly electron-withdrawing substituents, for example, halogens or haloalkyl groups, may be possible.
Because of this strong electron-withdrawing nature, the presence of a nitro group also facilitates many chemical reactions; thus, it is often found in screening collections (including the collection from which AZ-4 was originally identified). The reaction of an indole with nitrostyrene has been widely reported in the literature, and there are a considerable range of methods available for this simple 1,4-conjugate addition reaction.17–24 There are literature precedents for the nitro group to be replaced with a carboxylic acid in this series of compounds, which is achieved by initial reaction of an indole with the highly electron-deficient diethylbenzylidene malonate.21,22 The limited similarity of nitro group binding to carboxylic acid binding in spite of the near-perfect isosterism has been discussed by Kelly and Kim.25 A small range of derivatives can be accessed from the carboxylic acid (for example, CH2OH, CO2H, CO2Et, CONHNH2, CONH2, and CONHMe, Figure 2; see the Supporting Information); however, these were inactive as CB1 PAMs.
Figure 2.

Inactive AZ-4 derivatives.
Likewise, simple derivatives based on reduction of the nitro to an amine (Figure 2), with or without subsequent substitution, gave inactive compounds (see the Supporting Information).
Modeling studies on GAT22826 indicate that the nitro group may form polar interactions with a serine, a methionine, and a cysteine residue in the absence of the orthosteric agonist (CP 55,940). However, this case appears to apply to an orthosteric agonist. Although not stated as a conclusion by the authors, the modeling suggests that in the presence of an agonist, GAT228 moves deeper into the receptor; accordingly, the nitro group may be too far from these binding groups to retain a strong interaction. Thus, it remains uncertain that specific binding interactions provided by the nitro group are required for its activity as a PAM. However, the lack of activity shown by any of the compounds in Figure 2 suggests that such interactions will be necessary for PAM activity.
Herein, we report that CF3 offers a viable bioisosteric replacement for an aliphatic nitro group, demonstrating the removal of the “non-drug-like” nitro group from the most commonly studied and utilized CB1 PAMs, to achieve compounds with improved potency and metabolic stability.
RESULTS AND DISCUSSION
Chemistry.
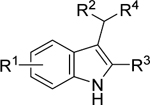
A library of indole derivatives 1–57 (Table 1) was synthesized using a variety of methods. Nitro-indole derivatives 2–25 were obtained by InBr3-catalyzed Michael addition of 2-(2-nitrovinyl)-aryl/heteroaryl derivatives to the corresponding indoles, upon microwave heating (see Supporting Information, Scheme SI-1 for details). Dicyano derivatives 29 and 32–34 having R2 = Ph and R4 = CH(CN)2 were obtained by addition of lithium indolyl cuprate(I) to benzylidene malononitriles (Supporting Information, Scheme SI-4). Compounds having R2 = Ph and R4 = CH2CN were obtained either via Fischer indole syntheses (28 and 30) or via InBr3-promoted addition of phenyl oxirane to an indole followed by one-carbon homologation (31). A similar strategy followed by one-carbon homologation using TMS-CF3 and Barton–McCombie dehydroxylation was used to access the CF3-indoles 37 and 39 (Supporting Information, Scheme SI-5). The cyclopropyl CF3-indole 40 was also prepared using a multistep process involving TMS-CF3 homologation and Barton–McCombie dehydroxylation (Supporting Information, Scheme SI-7). However, the most versatile method for efficiently preparing a wide range of CF3 indoles is shown in Scheme 1. The starting indole was iodinated at the 3-position with NIS to be then subjected to Sonogashira coupling to give 79. Terminal alkyne trifluoromethylation with in situ generated ″Cu-CF3″ under aerobic conditions gave CF3 alkynes 80,27 which were transformed into iodoalkenes 81 with lithium iodide under mildly acidic aonditions,28 which were then subjected to Stille coupling with the corresponding aryl stannanes leading to N-MOM-protected indoles 82. The latter were first deprotected with HCl and then reduced with Et3SiH in the presence of TFA to afford the target CF3 indoles 1, 41–50, and 57.
Scheme 1. Synthesis of CF3-PAMs via Stille Coupling/Ionic Hydride Reduction Strategya.

aReagents and conditions: (a) NIS, 0 °C, CH2Cl2; (b) TMSCCH, PdCl2(PPh3)2, CuI, Et3N, 23 °C, DMF; (c) TBAF, 0 °C, THF; (d) TMSCF3, CuI, TMEDA, K2CO3, air, 23 °C, DMF; (e) LiI, HOAc, CH2Cl2, 0–23 °C; (f) R2SnBu3, PdCl2(MeCN)2, P(o-Tol)3, CuI, 55 °C, DMF; (g) 6 M HCl, 60 °C, THF then Et3SiH, TFA, 0–23 °C, CH2Cl2.
Further late-stage functional group manipulations, shown in Scheme 2, expanded the library of CF3-indole derivatives to include compounds 52–56 and 83–89.
Scheme 2. Late-Stage Functional Group Interconversion Strategya.

aReagents and conditions: (a) Super-Hydride, THF; (b) SEMCl; (c) HNMe(OMe)-HCl, iPrMgCl, THF, 0 °C; (d) MeLi, THF, −78 °C; (e) NH2OH-HCl; (f) cyanuric chloride, DMF, 23 °C; (g) N2H4 hydrate, EtOH, 100 °C; (h) MeSO2Cl, Et3N, 23 °C; (i) HFaq-TFA.
All the compounds above were obtained in the racemic form, but pure enantiomers could be obtained for a number of derivatives, such as 1, 8, 26, and 50 using preparative HPLC separation on a chiral column (see the Supporting Information for details). Compound (+)-1 was crystallized from EtOAc/hexane solution, and its structure was determined by X-ray crystallography,29 which showed that the stereogenic centers C9 and C31 had (S) configurations (Figure 3). Note that as a consequence of the change of priority of the substituents, (R)-(−)-1 and (S)-(−)-GAT229 have different descriptors but share the same stereocenter’s topicity.
Figure 3.

X-ray crystal structure of (S)-(+)-1.
In Vitro Pharmacology.
Modification of the ZCZ011 βstructure indicated that either a 6-Me (5), 6-Cl (6), 6-CF3 (7), or 6-CN (8) substitution led to moderately potent and efficacious CB1 PAMs as determined by enhancement of β-arrestin recruitment, induced by the CB1 agonist CP 55,940 at its EC20 in the PathHunter β-arrestin assay (Eurofins DiscoverX), by a variation of the methods described previously.7 The results are shown in Table 1. Substitutions on the 5-position of the indole gave compounds with poor potency [5-Me (2), 5-CN (3), 5-NO2 (4)]. However, results from the same assay demonstrated that, alongside their PAM activity, most of these compounds were also allosteric agonists, giving receptor activation in the absence of the orthosteric ligand. This compares with previous reports5 that the enantiomers of AZ-4 (GAT228 and GAT229) show considerable differences in their properties, with the (+) enantiomer being an allosteric agonist and the (−) being a pure PAM. Separation of the enantiomers of (±)-8 (6-CN) confirmed that (−)-8 was the more potent PAM, but contrary to GAT228/GAT229,5 it did not indicate that (+)-8 was an agonist: both (−)-8 and (+)-8 showed enhancement of CP55,940-induced β-arrestin recruitment (Emax = 117 and 51% for the (−) and (+) enantiomers, respectively, p < 0.005 between enantiomers) with EC50 values that were not significantly different (664 and 863 nM, for the (−) and (+) enantiomers, respectively). However, both showed very low agonism (activation in the absence of CP 55,940) (Emax = 21 and 6% for the (−) and (+) enantiomers, respectively) with very poor potency (EC50 > 10 μM) making them both effectively pure PAMs (Table 1). Replacement of the 2-phenyl group (R3) with different aryl or cycloalkyl groups as in 9–25 was not tolerated. Other researchers30 have noted that in the absence of any (R1) substitutions on the indole, 4-fluoro, 4-chloro, or 3-chloro-4-fluoro substitution on the 2-phenyl (R2) appeared to be beneficial for potency in a cAMP assay; however, no examples featuring both an R1 and R2 substitution were shown.
In addition to the potential toxicological concerns, the methylene group adjacent to the nitro group was expected to be acidic and potentially a metabolic liability, contributing to the short half-life. ZCZ011 (5) was shown to be highly unstable in the rat and mouse liver microsomal assays (t½ ≤ 10 min), with moderate stability in human liver microsomes (t½ = 41 min) (Cyprotex Ltd.). No significant increase in stability was seen with the 6-Cl (6) and 6-CF3 (7) derivatives or with the placement of electron-withdrawing 4-Cl on the 2-phenyl group (16) (see Table 1). We therefore concluded that further development of the nitro-bearing compounds was unlikely to lead to “drug-like” compounds (suitable either for preclinical development or as tool compounds for in vivo target validation studies), and set about developing the synthetic methods required to permit replacement of the nitro group with a range of bioisosteres.
Modeling software programs (FieldStere and FieldAlign, products available from Cresset Biomolecular at the time) were used to suggest moieties that might mimic the electronic properties of the nitro group, with the primary feature modeled identified, but synthetic methods were generally lacking. Various five-membered heterocycles were investigated (Figure 4); however, none of these were active, and the syntheses were not straightforward. Thus, this line of investigation was terminated because of unproductivity.
Figure 4.

Inactive heterocyclic analogues of ZCZ011 investigated in this work (see the Supporting Information: Het-1–Het-4).
The cyano group is sometimes proposed as a bioisostere for a non-aromatic nitro group31 although perhaps the most widely known example of this is in fact the use of nitro as a bioisostere of a cyano group, as seen in the development of ranitidine from cimetidine.32 This bioisosterism was confirmed by the (albeit modest) potency of compounds 30 and 32, which featured replacement of the CH2NO2 with either a CH2CN or a CH(CN)2 moiety. Compound 31 showed trends for somewhat reduced potency and efficacy as compared to its nearest nitro analogue (not significant), and showed similarly poor metabolic stability in rat liver microsomes, possibly again related to the acidity of the proton adjacent to the CN group(s).
Studies aimed at replacement of the stereogenic carbon atom with an amine led to the preparation of a range of compounds bearing a −COCF3 substituent, some of which (such as compounds 58 and 59, Figure 5) showed a modest degree of enhancement (∼50% at 1 μM).
Figure 5.

Trifluoroacetamide derivatives tested in this work.
It was assumed that these compounds would be highly susceptible to hydrolysis and they were not regarded as development candidates or even appropriate for use as tool compounds. However, these results suggested that the −CF3 group might be a suitable bioisostere. Consequently, we set about developing synthetic methodologies to allow access to target compounds.
The unsubstituted derivatives (35, 36, and 37) featuring COCF3, CH(OH)CF3, CH2CF3, respectively, showed a small degree of enhancement (30–50%) at 1 μM (see Table 1). Incorporation of the 6-methyl group from ZCZ011 (compounds 38 and 39) gave a further increase in potency and efficacy (50–100% enhancement at 300 nM). Compounds showed good stability in human liver microsomes and potential improvement in rat liver microsomes (RLMs) compared to their nitro analogues (see Table 1).
We prepared a range of CF3 derivatives (40–57) with the aim of SAR elucidation and further improvements in potency and metabolic stability. Several compounds showed excellent potency and efficacy, with enhancement of over 100% at <200 nM and HLM/RLM t1/2 > 50 min; however, results from these compounds tended to be variable due to solubility issues not seen with the nitro derivatives. In order to address these solubility issues, we prepared analogues with a range of polar substituents on the indole. In parallel, we used the nitro series of compounds as a model system for rapid preparation of the analogues bearing substituents on the 2-phenyl group. Unfortunately, these studies showed an unforgiving SAR (see the Supporting Information for observations on the SAR) in that none of the substitutions on the 2-phenyl were tolerated and none of 6-CO2H (51), 6-CH2OH (52), 6-NHAc (53), 6-NH2 (55), or 6-NHSO2Me (56) gave active compounds. The 6-Ac derivative (54) was potent and efficacious but showed poor stability in the RLM assay (t1/2 = 12 min). Results are shown in Table 1.
Aside from those compounds with solubility issues, the most potent derivative was 6-CO2Me (50) for which the enantiomers were separated. As expected from GAT228/GAT2295 and our own observations from nitro compound 8, as shown in Table 1, the (−) enantiomer was significantly more efficacious (p < 0.005) than the (+) enantiomer both as a PAM (Emax = 156 ± 41 vs 88 ± 28%, for the (−) and (+) enantiomers, respectively) and as an agonist (Emax = 51 ± 11 vs 26 ± 5%, for the (−) and (+) enantiomers, respectively). Both the (−) and (+) enantiomers were more potent (p < 0.05) as PAMs than as agonists (EC50 = 113 ± 62 and 160 ± 147 nM as PAMs vs EC50 = 416 ± 167 and 682 ± 211 nM for the (−) and (+) enantiomers, respectively as agonists). However, as an ester, compound 50 was assumed to be unsuitable for further evaluation due to metabolic instability and, unfortunately, that the likely metabolite (the 6-CO2H analogue 51) was inactive.
The compound with the best balance of properties was the 6-CN analogue (1). As with the 6-CO2Me analogue 50, both the (−) and (+) enantiomers were significantly (p < 0.05) more efficacious as PAMs (Emax = 125 ± 29 and 79 ± 32% for the (−) and (+) enantiomers, respectively) than as agonists (Emax = 25 ± 6 and 17 ± 4% for the (−) and (+) enantiomers, respectively), with the (−) enantiomer significantly more efficacious than the (+) enantiomer (p < 0.05) as a PAM. Likewise, both the (−) and (+) enantiomers were more potent (p < 0.05) as PAMs than agonists (EC50 = 363 ± 169 and 342 ± 142 nM as PAMs and EC50 = 828 ± 310 and 1206 ± 345 nM as agonists for the (−) and (+) enantiomers, respectively). The 6-CN derivative (−)-1 was considerably more stable than ZCZ011, with HLM/RLM/MLM t½ of 72.3 ± 30.5 (N.S. in comparison with ZCZ011), 34.9 ± 7.4 (p < 0.005 to ZCZ011), and 26.5 ± 1.5 min (p < 0.005 to ZCZ011), respectively, compared to 41.2 ± 3.1, 6.4 ± 1.2, and 10.4 ± 2.1 min, respectively, for ZCZ011. Results are shown in Table 1.
Thus, compound (−)-1 was selected for further pharmacological evaluation. The enantiomers of CF3 derivative 1 and the enantiomers of its nitro analogue 8 were compared to ZCZ011 in several assays, to assess further the bioisosterism of the −CF3 and −NO2 groups.
A similar pattern for compound potency, but not efficacy, was seen in the DiscoverX Hithunter cAMP assay (Eurofins DiscoverX): CF3-bearing compounds 1 showed a trend for greater potency, than their NO2 equivalents 8, and the (−) enantiomers also showed a trend for greater potency than their (+) analogues. The results are shown in Table 2. However, contrary to the situation seen with β-arrestin recruitment, the compounds possessed similar efficacy, with all compounds acting as both PAMs and full agonists, completely inhibiting forskolin-stimulated cAMP production. The significance of this apparent difference between the effects on β-arrestin and cAMP is unclear. However, it must be noted that as neither ZCZ011 or (±)-1 showed any activity in the tetrad or psychoactive effects (see the Supporting Information), agonism with regard to cAMP signaling does not appear to translate to agonism at CB1 in vivo.
Table 2.
Effects of Compounds on cAMP Generation, Either Alone (Agonist) or Induced by EC20 CP 55,940 (PAM) in the Presence of EC80 Forskolin in the DiscoverX Hithunter cAMP Assay Using a Variation of the Methodology Described Previously7,a
| compound | cAMP EC50 (nM) |
cAMP Emax (%) |
|||
|---|---|---|---|---|---|
| PAM | agonist | PAM | agonist | ||
| ZCZ011 | 18 ± 1 | 32 ± 10 | 98 ± 0 | 94 ± 4 | |
| (+)-8 | 181 ± 95 | 251 ± 75 | 102 ± 5 | 96 ± 2 | |
| (−)-8 | 36 ± 10 | 46 ± 7 | 99 ± 2 | 97 ± 2 | |
| (S)-(+)-1 | 40 ± 17 | 76 ± 8 | 96 ± 3 | 89 ± 6 | |
| (R)-(−)-1 | 17 ± 5 | 29 ± 6 | 90 ± 1 | 84 ± 8 | |
Data is the result of three independent experiments.
Binding studies were also conducted (Eurofins Cerep) using a variant of the methodology described previously7 and demonstrated that like ZCZ011, 1 μM (+)-1 and (−)-1 showed significant enhancement in binding of the CB1 agonist, [3H]CP 55,940. At this concentration, no enhancement was noted with either (+)-8 and (−)-8. The results are shown in Table 3.
Table 3.
Effect of 1 μM ZCZ011, (+)-8, (−)-8, (+)-(S)-1, or (R)-(−)-1 on [3H]CP 55,940 Binding Using a Variation of the Methodology Described Previously7,a
| compound | % enhancement of [3H]CP 55,940 binding |
|---|---|
| ZCZ011 | 127.1 ± 13.3 |
| (+)-8 | 99.0 ± 10.1 |
| (−)-8 | 108.4 ± 8.3 |
| (S)-(+)-1 | 144.4 ± 3.5 |
| (R)-(−)-1 | 131.4 ± 4.3 |
Data is the result of three independent experiments.
In Vivo Efficacy.
Using the methodology previously described for the racemic ZCZ011,7 (±)-1 was evaluated in several CB1 receptor-sensitive in vivo models and was shown to possess a similar activity profile as ZCZ011.
(±)-1 (40 mg/kg) did not elicit pharmacological effects in the tetrad assay when administered alone. (±)-1 did not affect either the distance traveled or immobility time during assessment of locomotor behavior, did not produce antinociception in the tail withdrawal test, and did not produce hypothermia or cause immobility in the catalepsy bar test (see the Supporting Information). Thus, even at a high dose, (±)-1 did not elicit in vivo effects indicative of CB1 receptor orthosteric agonism. In contrast, (±)-1 (40 mg/kg) augmented a subset of pharmacological effects of the CB1 receptor orthosteric agonist CP 55,940. Importantly, (±)-1 produced significant leftward shifts in the dose–response relationships of CP 55,940 in producing antinociception in the tail withdrawal test (see the Supporting Information) and thus was further evaluated for its potential in the mouse chronic constrictive injury of the sciatic nerve (CCI) model of neuropathic pain.
As shown in Figure 6, mice displayed profound mechanical allodynia 7 days post-CCI surgery in both the ipsilateral (panel A) and contralateral (panel B) paws. At 40 mg/kg (the same dose at which ZCZ011 produces anti-allodynic effects7), (±)-1 significantly reversed CCI-induced mechanical allodynia in a time-dependent manner in the ipsilateral [Ftime(6, 168) = 6.411; Fdrug(3, 28) = 101.5; P < 0.0001; Finteraction(18, 168) = 7.394; P < 0.0001, Figure 6A] and contralateral [Ftime(6, 168) = 14.81; Fdrug(3, 28) = 182.2; Finteraction(18, 168) = 4.901; P < 0.0001, Figure 6B] paws. In particular, (±)-1 produced a significant anti-allodynic effect 0.5 h post-administration and achieved maximal effectiveness by 1 h.
Figure 6.

Acute administration of 40 mg/kg (±)-1 time-dependently reverses CCI-induced mechanical sensitivity in ipsilateral and contralateral paws. Data are expressed as mean ± SEM (n = 8/group). ****p < 0.0001, *p < 0.05 vs sham + vehicle; ###p < 0.001 ##p < 0.01, #p < 0.05 vs CCI + vehicle. pre-sx (pre-surgery); post-sx (post-surgery).
CONCLUSIONS
In conclusion, we have demonstrated that −CF3 serves as a viable bioisosteric replacement for an aliphatic nitro group. Thus, we have shown that it is possible to remove the “nondrug-like” nitro group from the most commonly studied CB1 PAMs to give compounds with improved potency and metabolic stability, which retain the activity profile of the parent nitro compound. These findings offer major assistance in the development of a preclinical candidate suitable for evaluation of the therapeutic potential of CB1 PAMs in a range of unmet medical needs. We hypothesize that the CF3 group might be a bioisosteric replacement for a nitro group also in other classes of bioactive compounds, especially when the nitro group is not involved in highly stabilizing hydrogen bonds with the receptor as the CF3 group is known to be a weak hydrogen bond acceptor.
EXPERIMENTAL SECTION
General Methods.
All reactions were carried out under a nitrogen atmosphere, with dry solvents under anhydrous conditions unless mentioned otherwise.
Solvents and Reagents.
Anhydrous tetrahydrofuran (THF), diethyl ether (Et2O), methylene chloride (CH2Cl2), and toluene were purchased from commercial suppliers. Reagents were purchased at the highest commercial quality, used without further purification, and handled in accordance with COSHH regulations.
Chromatography.
Flash chromatography (FC) was carried out on a silica gel (Merck Silica gel Si 60, 40–63 μm). Thin-layer chromatography (TLC) was carried out on glass-based 0.25 mm Merck silica gel plates (60F-254), which were developed with UV irradiation (254 nm and 365 nm), an aqueous solution of KMnO4 and an ethanolic solution of ammonium molybdate, and heat as developing agents.
1H NMR Spectra.
1H NMR spectra were recorded at 400 MHz on a Bruker ADVANCE III 400 instrument. Chemical shifts (δH) are given in parts per million (ppm) as referenced to the appropriate residual solvent peak.
13C NMR Spectra.
13C NMR spectra were recorded at 100.6 MHz on a Bruker ADVANCE III 400 instrument.
19F NMR Spectra.
19F NMR spectra were recorded at 376 MHz on a Bruker ADVANCE III 400 instrument without 1H decoupling. Chemical shifts (δC) are given in parts per million (ppm) as referenced to CFCl3 as 0 ppm. All NMR spectra were recorded at 298 K unless otherwise stated. Chemical shifts (δC) are given in parts per million (ppm) as referenced to CHCl3. The following abbreviations were used to explain the multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, quint = quintet, m = multiplet, br = broad.
Purity Analysis.
Purities of all final compounds were determined by reverse-phase HPLC-MS using an Agilent 1200 series chromatograph system coupled with an Agilent G6120 signal quadrupole detector equipped with an electrospray ionization source and detection in positive mode.
HPLC Conditions for Purity Analysis.
The HPLC conditions are as follows: Phenomenex Luna C18(2) 100 Å column, 4.6 × 250 mm, 5 μm; mobile phase: 75% MeOH/25% H2O with 0.1% formic acid; flow rate: 1 mL/min. All tested compounds were found to be at least of 96% purity.
Chiral Separation.
Semipreparative enantiomeric separations were carried out using an Agilent Technologies 1200 Series HPLC system equipped with a diode-array detector (DAD) and a normal-phase ChiralPak IA (10.0 × 250 mm, 5 μm) chiral column from Daicel Chemical Industries Ltd.
Optical Rotations.
Optical rotations were measured on an AA-65 angular scale automatic polarimeter from Optical Activity Ltd. with a 1 dm cell at the sodium D line.
The following procedures for the preparation of CF3-PAMs 1, 41 and 52–56 are representative examples:
2-Phenyl-3-(3,3,3-trifluoro-1-(thiophen-2-yl)propyl)-1H-indole-6-carbonitrile (1).
To a solution of compound 82l (147 mg, 0.335 mmol) in THF (5 mL) was added 6 M HCl. (0.5 mL). The mixture was stirred at 60 °C for 16 h prior to quenching with a saturated solution of NaHCO3 (10 mL) and diluted with CH2Cl2 (10 mL). The layers were separated, and the aqueous layer was extracted with CH2Cl2 (4 × 5 mL). The combined organic layers were dried over Na2SO4 and concentrated in vacuo. The residue was passed through a short plug of silica gel (CH2Cl2) and concentrated in vacuo prior to use.
To a solution of the crude product in CH2Cl2 (5 mL) were added Et3SiH (233 mg, 320 μL, 2.0 mmol) and trifluoroacetic acid (193 mg, 130 μL, 1.689 mmol) at 0 °C. The resulting red solution was stirred at 23 °C for 16 h before it was quenched with a saturated solution of NaHCO3 (10 mL) and diluted with CH2Cl2 (10 mL). The layers were separated, and the aqueous layer was extracted with CH2Cl2 (4 × 5 mL). The combined organic layers were dried over Na2SO4 and concentrated in vacuo. The residue was purified by flash column chromatography (silica gel, hexanes/EtOAc 10:1) to give compound 1 (102 mg, 77% for two steps) as a pale yellow solid. 1H NMR (400 MHz, CDCl3) δ = 8.58 (s, 1H), 7.77 (dd, J = 1.3, 0.6 Hz, 1H), 7.61– 7.46 (m, 6H), 7.34 (dd, J = 8.3, 1.4 Hz, 1H), 7.22 (dd, J = 5.1, 0.8 Hz, 1H), 6.98 (dd, J = 5.1, 3.6 Hz, 1H), 6.93 (dt, J = 3.6, 1.2 Hz, 1H), 4.96 (dd, J = 9.5, 4.8 Hz, 1H), 3.35–2.93 (m, 2H); 13C NMR (101 MHz, CDCl3) δ = 146.9, 140.2, 134.9, 131.2, 129.8, 129.3, 129.1, 128.8, 127.0, 126.0 (q, J = 277.9 Hz), 124.5, 124.2, 123.0, 120.7, 120.5, 116.0, 113.6, 104.6, 39.2 (q, J = 37.5 Hz), 31.8 (q, J = 3.1 Hz); 19F NMR (376 MHz, CDCl3) δ = −64.34 (t, J = 10.2 Hz, 3F). m/z (ESI) 397.1 [M + H+].
6-Methyl-2-phenyl-3-(3,3,3-trifluoro-1-(thiophen-2-yl)propyl)-1H-indole (41).
Using the method analogous to preparing compound 1, Et3SiH (176 mg, 242 μL, 1.51 mmol), compound 82a (108 mg, 0.25 mmol), and trifluoroacetic acid (144 mg, 97 μL, 1.26 mmol) were employed. Purification by flash column chromatography (silica gel, hexanes/EtOAc 10:1) gave compound 41 (64 mg, 66% for 2 steps) as a white solid. 1H NMR (400 MHz, CDCl3) δ = 7.95 (s, 1H), 7.62–7.36 (m, 6H), 7.23 (s, 1H), 7.20 (dd, J = 4.2, 2.4 Hz, 1H), 7.04–6.91 (m, 3H), 4.97 (dd, J = 9.2, 4.8 Hz, 1H), 3.30–3.16 (m, 1H), 3.16–3.00 (m, 1H), 2.53 (s, 3H); 13C NMR (101 MHz, CDCl3) δ = 148.0, 136.7, 135.4, 132.6, 132.3, 128.9 128.7, 128.3, 126.8, 126.3 (q, J = 278.1 Hz), 124.5, 124.2, 124.1, 121.7, 119.9, 112.8, 111.3, 39.6 (q, J = 27.1 Hz), 32.1 (q, J = 3.2 Hz), 21.7; 19F NMR (376 MHz, CDCl3) δ = −64.42 (t, J = 10.7 Hz, 3F). m/z (ESI) 386.1 [M + H+].
(2-Phenyl-3-(3,3,3-trifluoro-1-(thiophen-2-yl)propyl)-1H-indol-6-yl)methanol (52).
To a solution of compound 50 (15 mg, 0.035 mmol) in THF (2 mL) was added a solution of Super-Hydride (105 μL, 0.105 mmol, 1.0 M in THF) at 0 °C. The resulting mixture was stirred at 0 °C and allowed to warm to 23 °C for 16 h before it was quenched with a saturated solution of NH4Cl (10 mL) and diluted with CH2Cl2 (10 mL). The layers were separated, and the aqueous layer was extracted with CH2Cl (4 × 5 mL). The combined organic layers were dried over Na2SO4 and concentrated in vacuo. The residue was purified by flash column chromatography (silica gel, hexanes/EtOAc 5:1 → 1:1) to give compound 52 (13 mg, 94%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ = 8.13 (s, 1H), 7.69–7.35 (m, 7H), 7.19 (dd, J = 4.7, 1.5 Hz, 1H), 7.12 (d, J = 8.1 Hz, 1H), 7.02–6.83 (m, 2H), 4.95 (dd, J = 9.3, 4.8 Hz, 1H), 4.81 (s, 2H); 19F NMR (376 MHz, CDCl3) δ = −64.54 (t, J = 10.6 Hz, 3F). m/z (ESI) 424.1 [M + Na+].
N-(2-Phenyl-3-(3,3,3-trifluoro-1-(thiophen-2-yl)propyl)-1H-indol-6-yl)acetamide (53).
To a solution of compound 87 (15 mg, 0.027 mmol) in acetonitrile (2 mL) in a PTFE container was added an aqueous solution of HF (1 mL, 48 wt %) at 0 °C. The resulting solution was allowed to warm to 23 °C and stirred for 16 h before it was quenched with a saturated solution of NaHCO3 (20 mL). The mixture was stirred at 23 °C for 10 min before EtOAc (20 mL) was added. The layers were separated, and the organic layer was extracted with H2O (2 × 10 mL). The combined organic layers were dried over Na2SO4 and concentrated in vacuo.
The residue was redissolved in MeOH (2 mL), and K2CO3 was added at 0 °C. The mixture was stirred at 0 °C for 30 min prior to quenching with a saturated solution of NH4Cl and diluted with EtOAc (30 mL). The layers were separated, and the organic layer was extracted with H2O (2 × 10 mL). The combined organic layers were dried over Na2SO4 and concentrated in vacuo. The residue was purified by flash column chromatography (silica gel, hexanes/EtOAc 3:1) to give compound 53 (9 mg, 78% for 2 steps) as a pale yellow solid. 1H NMR (400 MHz, CDCl3) δ = 8.56 (brs, 1H), 8.14 (d, J = 1.5 Hz, 1H), 7.57–7.45 (m, 4H), 7.45–7.36 (m, 2H), 7.31 (s, 1H), 7.18 (dd, J = 5.0, 1.1 Hz, 1H), 6.97–6.89 (m, 2H), 6.79 (dd, J = 8.5, 1.8 Hz, 1H), 4.92 (dd, J = 8.9, 4.9 Hz, 1H), 3.22–2.93 (m, 2H), 2.17 (s, 3H); 13C NMR (101 MHz, CDCl3) δ = 168.3, 148.2, 142.3, 137.8, 134.2, 132.9, 129.0, 128.6, 128.2, 126.9, 126.4 (q, J = 278.5 Hz), 124.3, 121.1, 120.4, 113.1, 110.9, 97.0, 39.1 (q, J = 26.8 Hz), 32.2 (q, J = 2.8 Hz), 24.9; 19F NMR (376 MHz, CDCl3) δ = −64.42 (t, J = 10.4 Hz, 3F). m/z (ESI) 429.1 [M + H+].
1-(2-Phenyl-3-(3,3,3-trifluoro-1-(thiophen-2-yl)propyl)-1H-indol-6-yl)ethan-1-one (54).
Using the method analogous to preparing compound 53, compound 85 (22 mg, 0.04 mmol) was employed. Purification by flash column chromatography (silica gel, hexanes/EtOAc 3:1) to give compound 54 (12 mg, 72% for two steps) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ = 8.59 (s, 1H), 8.13 (s, 1H), 7.74 (dd, J = 8.4, 1.3 Hz, 1H), 7.59–7.42 (m, 6H), 7.20 (d, J = 4.8 Hz, 1H), 7.02–6.86 (m, 2H), 4.96 (dd, J = 9.5, 4.6 Hz, 1H), 3.18–3.03 (m, 2H), 2.67 (s, 3H); 13C NMR (101 MHz, CDCl3) δ = 198.2, 147.3, 140.1, 135.7, 131.7, 131.6, 130.4, 129.1, 128.7, 126.9, 126.2 (q, J = 276.2 Hz), 124.4, 124.2, 120.4, 119.7, 113.3, 112.2, 39.4 (q, J = 27.1 Hz), 31.9 (q, J = 2.8 Hz), 26.8; 19F NMR (376 MHz, CDCl3) δ = −64.39 (t, J = 10.4 Hz, 3F). m/z (ESI) 414.1 [M + H+].
2-Phenyl-3-(3,3,3-trifluoro-1-(thiophen-2-yl)propyl)-1H-indol-6-amine (55).
Using the method analogous to preparing compound 53, compound 88 (30 mg, 0.058 mmol) was employed. Purification by flash column chromatography (silica gel, hexanes/EtOAc 1:1) gave compound 55 (18 mg, 80% for two steps) as a pale yellow solid. 1H NMR (400 MHz, CDCl3) δ = 7.86 (brs, 1H), 7.53– 7.42 (m, 4H), 7.41 (dd, J = 5.2, 3.6 Hz, 1H), 7.31 (s, 1H), 7.20–7.13 (m, 1H), 6.94 (d, J = 3.6 Hz, 2H), 6.72 (d, J = 1.8 Hz, 1H), 6.56 (dd, J = 8.4, 2.0 Hz, 1H), 4.91 (dd, J = 8.8, 5.1 Hz, 1H), 3.28–2.55 (m + brs, 4H); 13C NMR (101 MHz, CDCl3) δ = 148.0, 142.2, 137.6, 134.0, 132.7, 128.9, 128.5, 128.0, 126.7, 126.3 (q, J = 277.1 Hz), 124.1, 120.9, 120.2, 112.9, 110.7, 96.8, 39.6 (q, J = 26.8 Hz), 32.0 (q, J = 3.2 Hz); 19F NMR (376 MHz, CDCl3) δ = −64.45 (t, J = 10.6 Hz, 3F). m/z (ESI) 387.1 [M + H+].
N-(2-Phenyl-3-(3,3,3-trifluoro-1-(thiophen-2-yl)propyl)-1H-indol-6-yl)methanesulfonamide (56).
Using the method analogous to preparing compound 53, compound 89 (32 mg, 0.054 mmol) was employed. Purification by flash column chromatography (silica gel, hexanes/EtOAc 1:1) gave compound 56 (20 mg, 81% for 2 steps) as a pale yellow solid. 1H NMR (400 MHz, CDCl3) δ = 8.29 (brs, 1H), 7.64–7.40 (m, 7H), 7.19 (dd, J = 5.0, 1.2 Hz, 1H), 7.02–6.91 (m, 2H), 6.89 (dd, J = 8.5, 2.0 Hz, 1H), 6.67 (brs, 1H), 4.94 (dd, J = 9.1, 4.9 Hz, 1H), 3.22–3.01 (m, 2H), 3.00 (s, 3H); 13C NMR (101 MHz, CDCl3) δ = 147.5, 137.1, 136.5, 132.0, 131.1, 129.0, 128.7, 126.8, 126.3 (q, J = 276.8 Hz), 125.0, 124.3, 124.1, 120.9, 115.3, 112.8, 105.7, 39.4 (q, J = 27.2 Hz), 39.0, 31.9 (q, J = 3.1 Hz); 19F NMR (376 MHz, CDCl3) δ = −64.38 (t, J = 10.4 Hz, 3F). m/z (ESI) 464.1 [M + H+].
3-Ethynyl-1-(methoxymethyl)-6-methyl-2-phenyl-1H-indole (79a).
To a solution of 1-(methoxymethyl)-6-methyl-2-phenyl1H-indole (575 mg, 2.29 mmol) in CH2Cl2 (10 mL) was added N-iodosuccinimide (525 mg, 2.33 mmol) at 0 °C. The reaction mixture was stirred for 3 h before it was quenched with a saturated solution of Na2SO3 (5 mL) and diluted with H2O (10 mL). The layers were separated, and the aqueous layer was extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were dried over Na2SO4 and concentrated in vacuo to give the corresponding 3-iodoindole, which was used without further purification.
To a solution of the crude 3-iodoindole in DMF (5 mL) were added trimethylsilylacetylene (337 mg, 475 μL, 3.43 mmol), PdCl2(PPh3)2 (80 mg, 0.114 mmol), CuI (26 mg, 0.137 mmol), and Et3N (695 mg, 957 μL, 6.87 mmol) at 23 °C. The reaction mixture was stirred for 6 h at 23 °C before it was quenched with a saturated solution of NH4Cl (10 mL) and diluted with EtOAc (50 mL). The layers were separated, and the organic layer was extracted with H2O (3 × 10 mL). The combined organic layers were dried over Na2SO4 and concentrated in vacuo. The residue was passed through a short plug of silica gel (CH2Cl2) and concentrated in vacuo prior to use.
To a solution of the crude product was added a solution of tetra-n-butylammonium fluoride (2.8 mL, 2.8 mmol, 1.0 M in THF) at 0 °C. The reaction mixture was stirred for 2 h at 23 °C before it was quenched with a saturated solution of NH4Cl (10 mL) and diluted with EtOAc (50 mL). The layers were separated, and the organic layer was extracted with H2O (3 × 10 mL). The combined organic layers were dried over Na2SO4 and concentrated in vacuo. The residue was purified by flash column chromatography (silica gel, hexanes/EtOAc 5:1) to give compound 79a (566 mg, 90% for three steps). 1H NMR (400 MHz, CDCl3) δ = 7.76–7.66 (m, 1H), 7.59–7.44 (m, 5H), 7.40–7.33 (m, 1H), 7.15 (d, J = 8.0 Hz, 1H), 5.39 (s, 2H), 3.28 (s, 3H), 3.18 (s, 1H), 2.56 (s, 3H); 13C NMR (101 MHz, CDCl3) δ = 144.4, 137.1, 133.6, 131.2, 130.5, 130.3, 128.8, 128.5, 127.2, 123.4, 119.7, 110.5, 79.7, 78.00, 74.9, 56.0, 22.0. m/z (ESI) 276.1 [M + H+].
3-Ethynyl-1-(methoxymethyl)-2-phenyl-1H-indole-6-carbonitrile (79g).
Using the method analogous to preparing compound 79a, compound 94d (700 mg, 2.67 mmol), N-iodosuccinimide (616 mg, 2.74 mmol), trimethylsilylacetylene (394 mg, 555 μL, 4.01 mmol), PdCl2(PPh3)2 (94 mg, 0.134 mmol), CuI (25 mg, 0.131 mmol), Et3N (813 mg, 1.12 mL, 8.03 mmol), and tetra-n-butylammonium fluoride (3.40 mL, 3.40 mmol, 1.0 M in THF) were used. Purification by flash column chromatography (silica gel, hexanes/EtOAc 5:1) gave compound 79g (596 mg, 78% for three steps) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ = 7.88 (d, J = 0.5 Hz, 1H), 7.85 (dd, J = 8.2, 0.5 Hz, 1H), 7.72 (d, J = 1.8 Hz, 1H), 7.70 (d, J = 1.4 Hz, 1H), 7.61–7.49 (m, 4H), 5.41 (s, 2H), 3.30 (s, 3H), 3.19 (s, 1H); 13C NMR (101 MHz, CDCl3) δ = 147.8, 135.5, 132.5, 130.2, 129.7, 129.1, 129.0, 128.8, 124.6, 120.9, 120.2, 115.5, 106.1, 80.9, 76.2, 75.3, 56.3. m/z (ESI) 309.1 [M + Na+].
1-(Methoxymethyl)-6-methyl-2-phenyl-3-(3,3,3-trifluoroprop-1-yn-1-yl)-1H-indole (80a).
Using a method described by Blanchard et al.,27 to a solution of N,N,N′,N′-tetramethylethylenediamine (324 mg, 418 μL, 2.79 mmol) in DMF (5 mL) were added CuI (531 mg, 2.79 mmol) and K2CO3 (771 mg, 5.58 mmol) at 23 °C. The resulting blue mixture was stirred vigorously at 23 °C under air (1 atm) for 20 min before trimethyl(trifluoromethyl)silane (529 mg, 550 μL, 3.72 mmol) was added, and the resulting mixture was stirred for additional 15 min under air. After this, the deep green mixture was cooled to 0 °C prior to adding a mixture of compound 79a (512 mg, 1.86 mmol) and trimethyl(trifluoromethyl)silane (529 mg, 550 μL, 3.72 mmol) in DMF (2 mL). The reaction mixture was stirred at 0 °C for 30 min and allowed to warm to 23 °C for 6 h before it was quenched with a saturated solution of NH4Cl (10 mL) and diluted with EtOAc (50 mL). The layers were separated, and the organic layer was extracted with H2O (3 × 10 mL). The combined organic layers were dried over Na2SO4 and concentrated in vacuo. The residue was purified by flash column chromatography (silica gel, hexanes/EtOAc 4:1) to give compound 80a (556 mg, 87%) as a pale yellow oil. 1H NMR (400 MHz, CDCl3) δ = 7.75–7.64 (m, 3H), 7.64–7.47 (m, 3H), 7.39 (s, 1H), 7.19 (d, J = 8.0 Hz, 1H), 5.40 (s, 2H), 3.32 (s, 3H), 2.58 (s, 3H); 13C NMR (101 MHz, CDCl3) δ = 147.0 (q, J = 1.7 Hz), 137.1, 134.3, 130.1, 129.50, 129.47, 128.8, 126.5, 124.1, 119.4, 115.5 (q, J = 256.2 Hz), 110.9, 93.6 (q, J = 2.5 Hz), 83.3 (q, J = 6.2 Hz), 78.7 (q, J = 52.2 Hz), 75.0, 56.1, 22.0; 19F NMR (376 MHz, CDCl3) δ = −48.59 (s, 3F). m/z (ESI) 344.1 [M + H+].
1-(Methoxymethyl)-2-phenyl-3-(3,3,3-trifluoroprop-1-yn-1yl)-1H-indole-6-carbonitrile (80g).
Using the method analogous to preparing compound 80a, compound 79g (300 mg, 1.05 mmol), N,N,N′,N′-tetramethylethylenediamine (185 mg, 255 μL, 1.57 mmol), CuI (300 mg, 1.58 mmol), K2CO3 (435 mg, 3.15 mmol), and trimethyl(trifluoromethyl)silane (596 mg, 620 μL, 4.19 mmol) were used. Purification by flash column chromatography (silica gel, hexanes/EtOAc 6:1) gave compound 80g (352 mg, 95%) as a pale yellow oil. 1H NMR (400 MHz, CDCl3) δ = 7.92 (s, 1H), 7.83 (d, J = 8.2 Hz, 1H), 7.72–7.65 (m, 2H), 7.64–7.59 (m, 3H), 7.56 (dd, J = 8.2, 1.2 Hz, 1H), 5.44 (s, 2H), 3.33 (s, 3H); 13;C NMR (101 MHz, CDCl3) δ = 150.5 (q, J = 1.7 Hz), 135.6, 131.8, 130.4, 130.0, 129.0, 128.2, 125.3, 120.7, 119.7, 115.9, 115.1 (q, J = 256.4 Hz), 106.9, 94.5 (q, J = 2.2 Hz), 81.1 (q, J = 6.2 Hz), 79.5 (q, J = 52.4 Hz), 75.4, 56.4; 19F NMR (376 MHz, CDCl3) δ = −49.13 (s, 3F). m/z (ESI) 355.1 [M + H+].
(E)-1-(Methoxymethyl)-6-methyl-2-phenyl-3-(3,3,3-trifluoro-1-iodoprop-1-en-1-yl)-1H-indole (81a).
Using a modified method described by Zhong et al.,28 to a solution of compound 80a (420 mg, 1.22 mmol) in CH2Cl2 (4 mL) were added LiI (180 mg, 1.35 mmol) and HOAc (1 mL) at 0 °C. The resulting mixture was stirred at 23 °C for 16 h before it was quenched with a saturated solution of NaHCO3 (10 mL) and diluted with CH2Cl2 (10 mL). The layers were separated, and the aqueous layer was extracted with CH2Cl2 (2 × 10 mL). The combined organic layers were dried over Na2SO4 and concentrated in vacuo. The residue was purified by flash column chromatography (silica gel, hexanes/EtOAc 10:1) to give compound 81a (558 mg, 97%) as a pale yellow solid. 1H NMR (400 MHz, CDCl3) δ = 7.65–7.48 (m, 6H), 7.38 (s, 1H), 7.24–7.14 (m, 1H), 6.71 (q, J = 7.1 Hz, 1H), 5.38 (s, 2H), 3.25 (s, 3H), 2.58 (s, 3H); 13C NMR (101 MHz, CDCl3) δ = 137.5, 137.0, 133.6, 132.0 (q, J = 33.9 Hz), 130.4, 130.2, 129.0, 128.6, 124.0, 123.3, 121.3 (q, J = 274.4 Hz), 119.4, 115.8, 110.6, 104.4 (q, J = 6.2 Hz), 74.8, 55.9, 22.0; 19F NMR (376 MHz, CDCl3) δ = −60.80 (d, J = 7.1 Hz, 3F). m/z (ESI) 472.0 [M + H+].
(E)-1-(Methoxymethyl)-2-phenyl-3-(3,3,3-trifluoro-1-iodoprop-1-en-1-yl)-1H-indole-6-carbonitrile (81g).
Using the method analogous to preparing compound 81a, compound 80g (330 mg, 0.931 mmol) and LiI (150 mg, 1.12 mmol) were used. The residue was passed through a short plug of silica gel (CH2Cl2) and concentrated in vacuo to give compound 81g, which was used without further purification.
(E)-1-(Methoxymethyl)-6-methyl-2-phenyl-3-(3,3,3-trifluoro-1-(thiophen-2-yl)prop-1-en-1-yl)-1H-indole (82a).
To a solution of compound 81a (146 mg, 0.31 mmol) in DMF (5 mL) were added tri-n-butyl(thiophen-2-yl)stannane (175 mg, 0.47 mmol), P(o-Tol)3 (14 mg, 0.046 mmol), PdCl2(MeCN)2 (7.8 mg, 0.03 mmol), and CuI (9 mg, 0.046 mmol) at 23 °C. The resulting mixture was stirred at 23 °C for 20 min before it was heated to 55 °C for 3 h. After this, the reaction mixture was quenched with a saturated solution of NH4Cl (10 mL) and diluted with Et2O (50 mL). The layers were separated, and the organic layer was extracted with H2O (3 × 10 mL). The combined organic layers were dried over Na2SO4 and concentrated in vacuo. The residue was purified by flash column chromatography (silica gel, hexanes/EtOAc 10:1) to give compound 82a (120 mg, 90%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ = 7.50–7.31 (m, 7H), 7.27 (d, J = 5.0 Hz, 1H), 7.06 (d, J = 8.1 Hz, 1H), 6.98 (d, J = 3.4 Hz, 1H), 6.91 (dd, J = 4.9, 3.8 Hz, 1H), 6.31 (q, J = 8.0 Hz, 1H), 5.46 (s, 2H), 3.24 (s, 3H), 2.57 (s, 3H); 13C NMR (101 MHz, CDCl3) δ = 144.3, 138.4 (q, J = 5.5 Hz), 137.3, 132.9, 130.8, 130.3, 128.4, 128.3, 128.1, 127.7, 127.4, 126.2, 123.1 (q, J = 270.6 Hz), 123.0, 119.4, 115.1 (q, J = 33.4 Hz),111.1, 110.5, 74.8, 55.6, 22.0; 19F NMR (376 MHz, CDCl3) δ = −58.23 (d, J = 8.0 Hz, 3F). m/z (ESI) 428.1 [M + H+].
(E)-1-(Methoxymethyl)-2-phenyl-3-(3,3,3-trifluoro-1-(thiophen-2-yl)prop-1-en-1-yl)-1H-indole-6-carbonitrile (82l).
Using the method analogous to preparing compound 82a, compound 81g (449 mg, 0.931 mmol), tri-n-butyl(thiophen-2-yl)stannane (522 mg, 1.40 mmol), P(o-Tol)3 (43 mg, 0.141 mmol), PdCl2(MeCN)2 (24 mg, 0.093 mmol), and CuI (18 mg, 0.095 mmol) were used. Purification by flash column chromatography (silica gel, hexanes/EtOAc 5:1) gave compound 82l (334 mg, 82%) as a pale yellow oil. 1H NMR (400 MHz, CDCl3) δ = 7.93 (s, 1H), 7.48 (d, J = 8.3 Hz, 1H), 7.45–7.32 (m, 6H), 7.31–7.23 (m, 1H), 6.93–6.83 (m, 2H), 6.30 (q, J = 8.2 Hz, 1H), 5.47 (s, 2H), 3.26 (s, 3H); 13C NMR (101 MHz, CDCl3) δ = 143.5, 142.6, 137.1 (q, J = 6.0 Hz), 135. 6, 131.1, 130.1, 129.5, 129.3, 128.5, 128.2, 127.89, 127.86, 124.2, 123.3 (q, J = 268.2 Hz), 121.5, 120.3, 115.9 (q, J = 34.2 Hz), 115.7, 111.4, 105.3, 75.1, 56.0; 19F NMR (376 MHz, CDCl3) δ = −58.18 (d, J = 8.2 Hz, 3F). m/z (ESI) 439.1 [M + H+].
Methyl-2-phenyl-3-(3,3,3-trifluoro-1-(thiophen-2-yl)propyl)-1-((2-(trimethylsilyl)-ethoxy)methyl)-1H-indole-6-carboxylate (83).
To a solution of compound 50 (540 mg, 1.257 mmol) in THF (3 mL) was added NaH (75 mg, 1.886 mmol, 60% in mineral oil) at 0 °C. After stirring at 0 °C for 30 min, 2-(trimethylsilyl)ethoxymethyl chloride (SEM-Cl, 282 mg, 300 μL, 1.51 mmol, 90% technical grade) was added. The resulting reaction mixture was stirred at 0 °C and allowed to warm to 23 °C for 3 h before it was quenched with a saturated solution of NH4Cl (5 mL) and diluted with EtOAc (50 mL). The layers were separated, and the organic layer was extracted with H2O (4 × 10 mL). The combined organic layers were dried over Na2SO4 and concentrated in vacuo. The residue was purified by flash column chromatography (silica gel, hexanes/EtOAc 5:1) to give compound 83 (680 mg, 97%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ = 8.33 (s, 1H), 7.85 (dd, J = 8.4, 1.3 Hz, 1H), 7.59–7.39 (m, 6H), 7.16 (d, J = 5.0 Hz, 1H), 6.93 (dd, J = 5.1, 3.6 Hz, 1H), 6.86 (d, J = 3.5 Hz, 1H), 5.41 (s, 2H), 4.65 (dd, J = 9.9, 4.3 Hz, 1H), 3.97 (s, 3H), 3.34 (ABq, J = 7.6 Hz, 2H), 3.26–3.08 (m, 1H), 3.08–2.92 (m, 1H), 0.80 (ABq, J = 7.6 Hz, 2H), −0.07 (s, 9H); 13C NMR (101 MHz, CDCl3) δ = 167.9, 147.3, 142.0, 136.7, 130.8, 130.2, 129.6, 129.3, 128.6, 126.7, 126.1 (q, J = 277.1 Hz), 124.2, 124.0, 123.8, 121.5, 119.4, 114.7, 113.1, 94.3, 72.9, 65.8, 52.0, 39.0 (q, J = 27.5 Hz), 32.1 (q, J = 2.6 Hz), 17.8, −1.5; 19F NMR (376 MHz, CDCl3) δ = −64.20 (t, J = 10.4 Hz, 3F). m/z (ESI) 560.2 [M + H+].
N-Methoxy-N-methyl-2-phenyl-3-(3,3,3-trifluoro-1-(thiophen-2-yl)propyl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indole-6-carboxamide (84).
Using a modified method described by Williams et al.,33 to a solution of compound 83 (408 mg, 0.729 mmol) and N,O-dimethylhydroxylamine hydrochloride (108 mg, 1.11 mmol) in THF (3 mL) was added a solution of isopropylmagnesium chloride (1.10 mL, 2.19 mmol, 2.0 M in THF) at 0 °C. After stirring at 0 °C for 30 min, the solution was allowed to warm to 23 °C and stirred for additional 3 h before it was quenched with a saturated solution of NH4Cl (5 mL) and diluted with EtOAc (50 mL). The layers were separated, and the organic layer was extracted with H2O (4 × 10 mL). The combined organic layers were dried over Na2SO4 and concentrated in vacuo. The residue was purified by flash column chromatography (silica gel, hexanes/EtOAc 3:1) to give compound 84 (326 mg, 76%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ = 8.00 (s, 1H), 7.61–7.39 (m, 7H), 7.16 (dd, J = 5.1, 0.9 Hz, 1H), 6.92 (dd, J = 5.1, 3.6 Hz, 1H), 6.87 (dt, J = 3.5, 1.2 Hz, 1H), 5.38 (ABq, J = 12.7 Hz, 2H), 4.64 (dd, J = 9.7, 4.3 Hz, 1H), 3.65 (s, 3H), 3.42 (s, 3H), 3.33 (ddd, J = 9.5, 6.2, 1.9 Hz, 2H), 3.25–3.09 (m, 1H), 3.12– 2.89 (m, 1H), 0.80 (ddd, J = 9.5, 6.2, 1.9 Hz, 2H), −0.07 (s, 9H); 13C NMR (101 MHz, CDCl3) δ = 170.7, 147.4, 140.9, 136.4, 130.9, 130.4, 129.2, 128.6, 127.9, 127.8, 126.7, 126.2 (q, J = 278.5 Hz), 124.2, 123.8, 120.7, 119.3, 114.5, 111.8, 73.0, 65.7, 61.0, 39.0 (q, J = 27.2 Hz), 34.4, 32.1 (q, J = 2.8 Hz), 17.8, −1.5; 19F NMR (376 MHz, CDCl3) δ = −64.19 (t, J = 10.3 Hz, 3F). m/z (ESI) 589.2 [M + H+].
1-(2-Phenyl-3-(3,3,3-trifluoro-1-(thiophen-2-yl)propyl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indol-6-yl)ethan-1-one (85).
To a solution of compound 84 (408 mg, 0.54 mmol) in THF (3 mL) was added a solution of methyllithium (860 μL, 0.860 mmol, 1.6 M in Et2O) at −78 °C. After stirring at −78 °C for 30 min, the solution was allowed to warm to 0 °C and stirred for additional 3 h before it was quenched with a saturated solution of NH4Cl (5 mL) and 0.1 M HCl solution (5 mL). The mixture was stirred at 23 °C for 10 min before EtOAc (50 mL) was added. The layers were separated, and the organic layer was extracted with H2O (4 × 10 mL). The combined organic layers were dried over Na2SO4 and concentrated in vacuo. The residue was purified by flash column chromatography (silica gel, hexanes/EtOAc 5:1) to give compound 85 (252 mg, 86%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ = 8.26 (s, 1H), 7.80 (dd, J = 8.4, 1.4 Hz, 1H), 7.64–7.41 (m, 6H), 7.17 (dd, J = 5.1, 1.0 Hz, 1H), 6.94 (dd, J = 5.1, 3.6 Hz, 1H), 6.90–6.84 (m, 1H), 5.44 (s, 2H), 4.66 (dd, J = 10.0, 4.2 Hz, 1H), 3.41–3.30 (m, 2H), 3.25–3.10 (m, 1H), 3.09–2.95 (m, 1H), 2.71 (s, 3H), 0.85–0.74 (m, 2H), −0.06 (s, 9H); 13C NMR (101 MHz, CDCl3) δ = 198.0, 147.3, 142.5, 136.8, 131.7, 130.8, 130.1, 129.8, 129.4, 128.6, 126.8, 126.1 (q, J = 277.4 Hz), 124.3, 123.8, 120.8, 119.5, 114.8, 111.8, 73.0, 65.9, 38.9 (q, J = 27.6 Hz), 32.1 (q, J = 3.3 Hz), 26.82, 17.78, −1.49; 19F NMR (376 MHz, CDCl3) δ = −64.14 (t, J = 10.3 Hz, 3F). m/z (ESI) 544.2 [M + H+].
(E)/(Z)-1-(2-Phenyl-3-(3,3,3-trifluoro-1-(thiophen-2-yl)propyl)-1-((2-(trimethyl-silyl)ethoxy)methyl)-1H-indol-6-yl)ethan-1-one Oxime (86).
To a solution of compound 85 (220 mg, 0.405 mmol) in MeOH (5 mL) were added hydroxylamine hydrochloride (34 mg, 0.489 mmol) and sodium acetate (50 mg, 0.61 mmol). The resulting mixture was heated to 70 °C for 4 h before it was cooled to 23 °C, quenched with a saturated solution of NH4Cl (10 mL), and then diluted with CH2Cl2 (10 mL). The layers were separated, and the aqueous layer was extracted with CH2Cl2 (4 × 5 mL). The combined organic layers were dried over Na2SO4 and concentrated in vacuo to give oxime 86 as a mixture of (E) and (Z) isomers. Oxime 86 was used without further purification.
N-(2-Phenyl-3-(3,3,3-trifluoro-1-(thiophen-2-yl)propyl)-1((2-(trimethylsilyl)-ethoxy)methyl)-1H-indol-6-yl)acetamide (87).
Using a modified method described by Giacomelli et al.,34 2,4,6-trichloro-[1,3,5]triazine (cyanuric chloride, 32 mg, 0.445 mmol) was added to DMF (1 mL) at 23 °C, and the solution was stirred for 30 min before a solution of oxime 86 (226 mg, 0.404 mmol) in DMF (1 mL) was added. The resulting mixture was stirred for 4 h at 23 °C prior to quenching with a saturated solution of NaHCO3 (10 mL) and diluted with EtOAc (50 mL). The layers were separated, and the organic layer was extracted with H2O (4 × 10 mL). The combined organic layers were dried over Na2SO4 and concentrated in vacuo. The residue was purified by flash column chromatography (silica gel, hexanes/EtOAc 1:1) to give compound 87 (208 mg, 92% for two steps) as a pale yellow oil. 1H NMR (400 MHz, CDCl3) δ = 8.01 (d, J = 1.0 Hz, 1H), 7.59 (s, 1H, NH), 7.55–7.42 (m, 5H), 7.40 (d, J = 8.5 Hz, 1H), 7.14 (d, J = 5.0 Hz, 1H), 7.05 (dd, J = 8.5, 1.5 Hz, 1H), 6.96–6.88 (m, 1H), 6.86 (d, J = 3.4 Hz, 1H), 5.31 (s, 2H), 4.62 (dd, J = 9.5, 4.3 Hz, 1H), 3.32 (ddd, J = 9.7, 5.2, 3.0 Hz, 2H), 3.21–3.07 (m, 1H), 3.07–2.91 (m, 1H), 2.19 (s, 3H), 0.82–0.73 (m, 2H), −0.07 (s, 9H); 13C NMR (101 MHz, CDCl3) δ = 168.4, 147.8, 138.9, 137.5, 133.1, 131.0, 130.7, 128.9, 128.5, 126.7, 126.2 (q, J = 279.0 Hz), 124.1, 123.7, 123.0, 120.0, 114.3, 114.0, 103.1, 72.8, 65.6, 39.0 (q, J = 27.1 Hz), 32.1 (q, J = 2.7 Hz), 24.6, 17.8, −1.5; 19F NMR (376 MHz, CDCl3) δ = −64.19 (t, J = 10.3 Hz, 3F). m/z (ESI) 559.2 [M + H+].
2-Phenyl-3-(3,3,3-trifluoro-1-(thiophen-2-yl)propyl)-1-((2(trimethylsilyl)ethoxy)-methyl)-1H-indol-6-amine (88).
To a solution of compound 87 (106 mg, 0.19 mmol) in EtOH (2 mL) in a sealable tube was added hydrazine hydrate (50 μL, 1.03 mmol). The tube was sealed and heated to 100 °C for 16 h before the reaction mixture was quenched with 0.1 M HCl (1 mL) and diluted with EtOAc (50 mL). The layers were separated, and the organic layer was washed with a saturated solution of NaHCO3 (2 × 10 mL) and H2O (2 × 10 mL). The combined organic layers were dried over Na2SO4 and concentrated in vacuo to give compound 32 as a pale yellow solid. Compound 88 was used without further purification.
N-(2-Phenyl-3-(3,3,3-trifluoro-1-(thiophen-2-yl)propyl)-1((2-(trimethylsilyl)-ethoxy)methyl)-1H-indol-6-yl)methanesulfonamide (89).
To a solution of compound 88 (37 mg, 0.072 mmol) in CH2Cl2 (2 mL) was added pyridine (10 μL, 0.124 mmol) at 0 °C followed by methanesulfonyl chloride (6 μL, 0.078 mmol). The resulting mixture was stirred at 0 °C before it was quenched by a saturated solution of NaHCO3 (5 mL) and diluted with EtOAc (20 mL). The layers were separated, and the organic layer was extracted with H2O (2 × 10 mL). The combined organic layers were dried over Na2SO4 and concentrated in vacuo to give compound 89 as a yellow oil. The residue was used without further purification.
CCI Mouse Model of Neuropathic Pain.
Subjects consisted of male C57BL/6J mice (6–8 weeks old, body mass of 27–32 g) obtained from the Virginia Commonwealth University Transgenic Mouse Core (Richmond, Virginia). Mice were group-housed (four per cage) for at least 1 week before the beginning of experiments on a 12/12 light/dark cycle (lights on at 0600 h), with an ambient temperature of 20–22 °C and humidity of 55–60%. Standard rodent chow and water were available ad libitum. Animal experiments were conducted in accordance with the guidelines of the Institutional Animal Care and Use Committee of Virginia Commonwealth University and followed the NIH Guidelines for the Care and Use of Laboratory Animals. (±)-1 was dissolved in ethanol (5% of total volume), ALKAMULS-620 (Sanofi-Aventis, Bridgewater, NJ) (5% of total volume), and saline (0.9% NaCl) (90% of total volume) and administered intraperitoneally in a volume of 10 mL/kg.
The CCI model of neuropathic pain was used to assess the anti-allodynic effects of (±)-1. Surgical procedure for chronic constriction of the sciatic nerve was performed as previously described,35 but modified for the mouse.36 In brief, the mice were anesthetized with isoflurane (5 vol % induction followed by 2.0% in oxygen), and the right hind leg was shaved and cleaned with betadine and ethanol. Using aseptic procedures, the sciatic nerve was carefully isolated and loosely ligated using three segments of 5–0 chromic gut sutures (Ethicon, Somerville, NJ, USA). Sham surgery was identical to CCI surgery, but the nerve was neither ligated nor manipulated. The overlying muscle was sutured closed with (1) 4–0 sterile silk suture (Ethicon), and animals recovered from anesthesia within approximately 5 min. Mice were randomly assigned into the CCI or sham surgical groups.
Behavioral Assessment of Mechanical Allodynia.
Following 15 or 20 min of habituation to the testing environment, von Frey filaments were used to determine baseline paw withdrawal responses as previously described.37 Mice were unrestrained and were singly placed under an inverted wire mesh basket to allow for unrestricted air flow, and the mechanical allodynia was assessed with von Frey filaments (North Coast Medical, Morgan Hill, CA) applied randomly to the left and right plantar surfaces of the hind paw. Lifting, licking, or shaking the paw in response to three stimulations was coded as a positive response. Basal von Frey paw withdrawal responses were assessed prior to CCI or sham surgery (pre-sx) and again on postsurgery day 7 prior to drug administration (post-sx). Following the assessment of von Frey thresholds on day 7, CCI and sham mice were assigned to different treatment groups and were given a singular i.p. injection of vehicle or (±)-1 (40 mg/kg). Each mouse was tested for paw withdrawal thresholds 0.5, 1, 2, 4, and 8 h after the injections.
Statistical Analysis.
For all in vivo data, statistical analyses were performed using the computer program GraphPad Prism version 6.0 (GraphPad Software, Inc., San Diego, CA). Data were analyzed by two-way analysis of variance (ANOVA) with drug treatment as a between subject factor and time as a within subject factor. A P value of <0.05 was considered statistically significant. Following significant ANOVAs, the Bonferroni post hoc test was used to ascertain further differences for both ipsilateral and contralateral paws. All data are expressed as the mean ± SEM.
PathHunter β-Arrestin Assay.
Beta-arrestin recruitment studies were conducted by Eurofins DiscoverX using the PathHunter β-arrestin assay. Briefly, this assay monitors the activation of CB1 using enzyme fragment complementation, with β-galactosidase as the functional reporter. When the CB1 receptor is activated and β-arrestin is recruited to the receptor, enzyme complementation occurs, restoring β-galactosidase activity, effecting substrate transformation. Product formation is detected by chemiluminescence. PathHunter cell lines were expanded from freezer stocks according to standard procedures. Cells were seeded in a total volume of 20 μL into white walled, 384-well microplates and incubated at 37 °C for the appropriate time prior to testing. Cells were pre-incubated with the sample followed by agonist induction at the EC20 concentration of CP 55,940 (2 nM). Intermediate dilution of sample stocks was performed to generate a 5× sample in assay buffer. The 5× sample (5 μL) was added to cells and incubated at 37 °C or room temperature for 30 min. The vehicle concentration was 1%. A 6× EC20 agonist (5 μL) in assay buffer was added to the cells and incubated at 37 °C or room temperature for 90 or 180 min. The assay signal was generated through a single addition of 12.5 or 15 μL (50% v/v) of PathHunter detection reagent cocktail followed by a 1 h incubation at room temperature. Microplates were read following signal generation with a PerkinElmer EnVision instrument for chemiluminescence signal detection. Compound activity was analyzed using the CBIS data analysis suite (ChemInnovation, CA). Percentage modulation was calculated using the following formula: %modulation =100% × ((mean RLU of test sample – mean RLU of EC20 control)/(mean RLU of MAX control ligand – mean RLU of EC20 control)).
Hit Hunter cAMP Assay.
cAMP generation studies were conducted by Eurofins DiscoverX as described previously.7 Briefly, cells were pre-incubated with the sample followed by agonist induction at the EC20 concentration (0.09 nM CP 55,940). Media were aspirated from cells and replaced with 10 μL of 1:1 HBSS/10 mM Hepes/cAMP XS+ Ab reagent. Intermediate dilution of sample stocks was performed to generate a 4× sample in assay buffer. The 4× (5 μL) compound was added to the cells and incubated at room temperature or 37 °C for 30 min. The 4× EC20 agonist (5 μL) was added to the cells and incubated at room temperature or 37 °C for 30 or 60 min. EC80 forskolin (20 μM) was included. After appropriate compound incubation, the assay signal was generated through incubation with 20 μL of cAMP XS + ED/CL lysis cocktail for 1 h followed by incubation with 20 μL of cAMP XS + EA reagent for 3 h at room temperature. Microplates were read following signal generation with a PerkinElmer EnVision instrument for chemiluminescence signal detection. Compound activity was analyzed using the CBIS data analysis suite (ChemInnovation, CA). Percentage modulation is calculated using the following formula: %modulation =100% × (1 − (mean RLU of test sample – mean RLU of MAX control)/(mean RLU of EC20 control – mean RLU of MAX control)).
Pharmacokinetics Assays.
Routine in vitro stability studies were conducted by Cyprotex Ltd. (Macclesfield, U.K.).
Supplementary Material
ACKNOWLEDGMENTS
We gratefully thank Signal Pharma and the Canadian Institutes of Health Research Proof of Principle grants PPP-125784 and PP2-139101 for financial support and fellowship (C.C.T.), NIH grants R01DA039942 and P30DA033934 and VCU School of Pharmacy start-up funds (A.H.L.). We thank the EPSRC National Crystallography Service (University of Southampton) for the X-ray data collection. We are grateful to Dr. Monica Sani (CNR-ICRM, Milan, Italy) and Mr. Massimo Frigerio (Politecnico di Milano, Italy) for the synthesis of two tetrazole-substituted indoles (Het-1 and Het-2).
ABBREVIATIONS USED
- HLM
human liver microsomes
- RLM
rat liver microsomes
- MLM
mouse liver microsomes
- SEM
standard error of mean
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.9b00252.
Molecular formula strings of target compounds (CSV) Experimental procedures, characterization of all intermediates and target compounds, and copies of NMR spectra of compounds 1 and 39–57 (PDF)
R.A.R., M.Z., and I.R.G. are co-founders of Signal Pharma Ltd., which owns the I.P. of some of the molecules described in this article.
The authors declare the following competing financial interest(s): R.A.R., M.Z. and I.R.G. are co-founders of Signal Pharma Limited, which owns the I.P. of some of the molecules described in this article.
REFERENCES
- (1).Dopart R; Lu D; Lichtman AH; Kendall DA Allosteric Modulators of Cannabinoid Receptor 1: Developing Compounds for Improved Specificity. Drug Metab. Rev 2018, 50, 3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Alaverdashvili M; Laprairie RB The Future of Type 1 Cannabinoid Receptor Allosteric Ligands. Drug Metab. Rev 2018, 50, 14–25. [DOI] [PubMed] [Google Scholar]
- (3).Adam L; Rihakova L; Lapointe S; St-Onge S; Labrecque J; Payza K. Positive Allosteric Modulators of CB1 Receptors. In 17th Annual Symposium on the Cannabinoids; Poster 86; Saint-Sauveur, Québec, Canada Abstract available from http://icrs.co/SYMPOSIUM.2007/2007.ICRS.Program.and.Abstracts.pdf, 2007. Accessed on March 26 2019, 2007. [Google Scholar]
- (4).Kerr JR; Trembleau L; Storey JMD; Wardell JL; Harrison WTA Crystal Structures of Four Indole Derivatives as Possible Cannabinoid Allosteric Antagonists. Acta Crystallogr., Sect. E: Crystallogr. Commun 2015, 71, 654–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Laprairie RB; Kulkarni PM; Deschamps JR; Kelly MEM; Janero DR; Cascio MG; Stevenson LA; Pertwee RG; Kenakin TP; Denovan-Wright EM; Thakur GA Enantiospecific Allosteric Modulation of Cannabinoid 1 Receptor. ACS Chem. Neurosci. 2017, 8, 1188–1203. [DOI] [PubMed] [Google Scholar]
- (6).Noland WE; Lange RF The Nitroethylation of Indoles. III.1‑3 A Synthetic Route to Substituted Tryptamines. J. Am. Chem. Soc 1959, 81, 1203–1209. [Google Scholar]
- (7).Ignatowska-Jankowska BM; Baillie GL; Kinsey S; Crowe M; Ghosh S; Owens RA; Damaj IM; Poklis J; Wiley JL; Zanda M; Zanato C; Greig IR; Lichtman AH; Ross RA A Cannabinoid CB1 Receptor-Positive Allosteric Modulator Reduces Neuropathic Pain in the Mouse with No Psychoactive Effects. Neuropsychopharmacology 2015, 40, 2948–2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Muegge I; Heald SL; Brittelli D. Simple Selection Criteria for Drug-like Chemical Matter. J. Med. Chem 2001, 44, 1841–1846. [DOI] [PubMed] [Google Scholar]
- (9).Beck DE; Abdelmalak M; Lv W; Reddy PVN; Tender GS; O’Neill E; Agama K; Marchand C; Pommier Y; Cushman M. Discovery of Potent Indenoisoquinoline Topoisomerase I Poisons Lacking the 3-Nitro Toxicophore. J. Med. Chem 2015, 58, 3997–4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Lowe D. I Want A New Nitro. In the Pipeline, https://blogs.sciencemag.org/pipeline/archives/2007/03/29/i_want_a_new_nitro. 2007, Accessed Mar 26, 2019.
- (11).Lowe D. Ligands From Nothing. In the Pipeline, https://blogs.sciencemag.org/pipeline/archives/2013/09/12/ligands_from_nothing, 2013. Accessed Mar 26, 2019.
- (12).Kazius J; McGuire R; Bursi R. Derivation and Validation of Toxicophores for Mutagenicity Prediction. J. Med. Chem 2005, 48, 312–320. [DOI] [PubMed] [Google Scholar]
- (13).Baell JB; Holloway GA New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J. Med. Chem 2010, 53, 2719–2740. [DOI] [PubMed] [Google Scholar]
- (14).Albert JS Chapter 4 - Fragment-Based Lead Discovery In Lead Generation Approaches in Drug Discovery; Rankovic Z., Morphy R., Eds.; John Wiley & Sons, Ltd: Hoboken, New Jersey, USA, 2010; pp 105–139. [Google Scholar]
- (15).Meanwell NA Synopsis of Some Recent Tactical Application of Bioisosteres in Drug Design. J. Med. Chem 2011, 54, 2529–2591. [DOI] [PubMed] [Google Scholar]
- (16).Alston TA; Porter DJT; Bright HJ The Bioorganic Chemistry of the Nitroalkyl Group. Bioorg. Chem 1985, 13, 375–403. [Google Scholar]
- (17).Bandini M; Melchiorre P; Melloni A; Umani-Ronchi A. A Practical Indium Tribromide Catalysed Addition of Indoles to Nitroalkenes in Aqueous Media. Synthesis 2002, 2002, 1110–1114. [Google Scholar]
- (18).Suresh Babu K; Rama Subba Rao V; Sunitha P; Sivaram Babu S; Madhusudana Rao J. Mild and Efficient Michael Addition of Activated Olefins to Indoles Using TBAB as a Catalyst: Synthesis of 3-Substituted Indoles. Synth. Commun 2008, 38, 1784–1791. [Google Scholar]
- (19).Gu Y; Barrault J; Jérôme F. Glycerol as An Efficient Promoting Medium for Organic Reactions. Adv. Synth. Catal 2008, 350, 2007–2012. [Google Scholar]
- (20).Habib PM; Kavala V; Kuo C-W; Yao C-F Catalyst-Free Aqueous-Mediated Conjugative Addition of Indoles to β-Nitrostyrenes. Tetrahedron Lett. 2008, 49, 7005–7007. [Google Scholar]
- (21).Kumar VP; Sridhar R; Srinivas B; Narender M; Rao KR Friedel–Crafts Alkylation of Indoles with Nitroolefins in the Presence of β-Cyclodextrin in Water under Neutral Conditions. Can. J. Chem 2008, 86, 907–911. [Google Scholar]
- (22).Kuo C-W; Wang C-C; Fang H-L; Raju BR; Kavala V; Habib PM; Yao C-F An Efficient Method for the N-Bromosuccinimide Catalyzed Synthesis of Indolyl-Nitroalkanes. Molecules 2009, 14, 3952–3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Praveen C; Karthikeyan K; Perumal PT Efficient Synthesis of 3-Substituted Indoles through a Domino Gold(I) Chloride Catalyzed Cycloisomerization/C3-Functionalization of 2-(Alkynyl)-Anilines. Tetrahedron 2009, 65, 9244–9255. [Google Scholar]
- (24).Habib PM; Kavala V; Kuo C-W; Raihan MJ; Yao C-F Catalyst Free Conjugate Addition of Indoles and Pyrroles to Nitro Alkenes under Solvent Free Condition (SFC): An Effective Greener Route to Access 3-(2-Nitro-1-Phenylethyl)-1H-Indole and 2-(2-Nitro-1-Phenylethyl)-1H-Pyrrole Derivatives. Tetrahedron 2010, 66, 7050–7056. [Google Scholar]
- (25).Kelly TR; Kim MH Relative Binding Affinity of Carboxylate and Its Isosteres: Nitro, Phosphate, Phosphonate, Sulfonate, and .Delta.-Lactone. J. Am. Chem. Soc 1994, 116, 7072–7080. [Google Scholar]
- (26).Saleh N; Hucke O; Kramer G; Schmidt E; Montel F; Lipinski R; Ferger B; Clark T; Hildebrand PW; Tautermann CS Multiple Binding Sites Contribute to the Mechanism of Mixed Agonistic and Positive Allosteric Modulators of the Cannabinoid CB1 Receptor. Angew. Chem., Int. Ed 2018, 57, 2580–2585. [DOI] [PubMed] [Google Scholar]
- (27).Tresse C; Guissart C; Schweizer S; Bouhoute Y; Chany AC; Goddard M-L; Blanchard N; Evano G. Practical Methods for the Synthesis of Trifluoromethylated Alkynes: Oxidative Trifluoromethylation of Copper Acetylides and Alkynes. Adv. Synth. Catal 2014, 356, 2051–2060. [Google Scholar]
- (28).Zhang X-G; Chen M-W; Zhong P; Hu M-L Regio- and Stereo-Specific Preparation of (E)-1-Aryl-3,3,3-Trifluoro-1-IodoPropenes and Their Palladium-Catalyzed Reaction with Terminal Alkynes. J. Fluorine Chem 2008, 129, 335–342. [Google Scholar]
- (29).CCDC 1879918 Contains the Supplementary Crystallographic Data for This Paper. These Data Can Be Obtained Free of Charge via http://Www.Ccdc.Cam.Ac.Uk/Data_request/CifWww.Ccdc.Cam.Ac.Uk/Data_request/Cif, or by e-Mailing Data_request@ccdc.Cam.Ac.Uk, or by Contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44 1223 336033.
- (30).Thakur G; Kulkarni P. Allosteric Modulators of Cb1 Cannabinoid Receptors. WO/2013/103967, July 12, 2013. [Google Scholar]
- (31).Tang H; de Jesus RK; Walsh SP; Zhu Y; Yan Y; Priest BT; Swensen AM; Alonso-Galicia M; Felix JP; Brochu RM; Bailey T; Thomas-Fowlkes B; Zhou X; Pai L-Y; Hampton C; Hernandez M; Owens K; Roy S; Kaczorowski GJ; Yang L; Garcia ML; Pasternak A. Discovery of a Novel Sub-Class of ROMK Channel Inhibitors Typified by 5-(2-(4-(2-(4-(1H-Tetrazol-1-Yl)Phenyl)Acetyl)Piperazin-1-Yl)Ethyl)Isobenzofuran-1(3H)-One. Bioorg. Med. Chem. Lett 2013, 23, 5829–5832. [DOI] [PubMed] [Google Scholar]
- (32).Patrick G. An Introduction to Medicinal Chemistry, 6th ed.; Oxford University Press: Oxford, UK, 2017. [Google Scholar]
- (33).Williams JM; Jobson RB; Yasuda N; Marchesini G; Dolling U-H; Grabowski EJJ A New General Method for Preparation of N-Methoxy-N-Methylamides. Application in Direct Conversion of an Ester to a Ketone. Tetrahedron Lett. 1995, 36, 5461–5464. [Google Scholar]
- (34).De Luca L; Giacomelli G; Porcheddu A. Beckmann Rearrangement of Oximes under Very Mild Conditions. J. Org. Chem 2002, 67, 6272–6274. [DOI] [PubMed] [Google Scholar]
- (35).Bennett GJ; Xie Y-K A Peripheral Mononeuropathy in Rat That Produces Disorders of Pain Sensation like Those Seen in Man. Pain 1988, 33, 87–107. [DOI] [PubMed] [Google Scholar]
- (36).Ignatowska-Jankowska B; Wilkerson JL; Mustafa M; Abdullah R; Niphakis M; Wiley JL; Cravatt BF; Lichtman AH Selective Monoacylglycerol Lipase Inhibitors: Antinociceptive versus Cannabimimetic Effects in Mice. J. Pharmacol. Exp. Ther 2015, 353, 424–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Murphy PG; Ramer MS; Borthwick L; Gauldie J; Richardson PM; Bisby MA Endogenous Interleukin-6 Contributes to Hypersensitivity to Cutaneous Stimuli and Changes in Neuropeptides Associated with Chronic Nerve Constriction in Mice. Eur. J. Neurosci 1999, 11, 2243–2253. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.