

Abstract
In previous studies, we showed that the topical application of dibenzo[a,l]pyrene (DB[a,l]P), also known as dibenzo[def,p]chrysene, to the oral cavity of mice induced oral squamous cell carcinoma. We also showed that dA and dG adducts likely account for most of the mutagenic activity of DB[a,l]P in the oral tissues in vivo. Here we report for the first time that the oral treatment of lacI mice with a combination of tobacco smoke carcinogens, DB[a,l]P and N′-nitrosonornicotine (NNN), induces a higher fraction of mutations than expected from a simple sum of their induced individual mutation fractions, and a change in the mutational profile compared with that expected from the sum of the individual agents. The mutational profile of the combination of agents resembled that of the P53 gene in human head and neck cancers more than that of either of the individual agents, in that the percentage of the major class of mutations (GC > AT transitions) is similar to that seen in the P53 gene. A preliminary study was performed to understand the origin of the unexpected mutagenesis observations by measuring specific DNA adducts produced by both NNN and DB[a,l]P in human oral leukoplakia cells. No significant differences in the expected and observed major adduct levels from either agent were observed between individual or combined treatments, suggesting that additional adducts are important in mutagenesis induced by the mixture. Taken together, the above observations support the use of this animal model not only to investigate tobacco smoke-induced oral cancer but also to study chemoprevention.
INTRODUCTION
Cancer of the oral cavity and pharynx in the U.S. has an annual incidence of 53 000, with almost 11 000 annual deaths from this disease.1 It has a 5 year survival rate of ~65%1 and treatment is often highly disfiguring and may seriously interfere with eating, speaking, and quality of life.2 In general, this disease has a devastating outcome. Tobacco smoking increases the risk for oral cancer several fold and synergizes with excess alcohol consumption.3–5 There are several experimental animal models for oral cancer, but most utilize synthetic carcinogens that are not found in tobacco smoke.6 We have established a mouse model for oral carcinogenesis and mutagenesis in the tongue and other pooled oral tissues using the topical application into the mouse oral cavity of the polycyclic aromatic hydrocarbon environmental pollutant and tobacco smoke constituent dibenzo[a,l]pyrene (DB[a,l]P), (also known as dibenzo[def,p]chrysene) or its ultimate carcinogen, (±)-anti-11,12-dihydroxy-13,14-epoxy-11,12,13,14-tetrahydrodibenzo[a,l]pyrene, (DBPDE).7,8 These compounds induce mutations and oral squamous cell carcinoma (OSCC) in the mouse oral cavity; histologically, >90% of oral cancers in humans are OSCC.7,8 However, it may be optimistic to believe that the induction of OSCC following animal exposure to a single tobacco carcinogen is the ideal model to fully understand the molecular basis of disease progression. Thus to better model tobacco-smoke-induced oral carcinogenesis, we treated mice with (DB[a,l]P) in combination with the oral carcinogen N′-nitrosonornicotine (NNN) (Figure 1). NNN is found in tobacco and tobacco smoke and is carcinogenic in the rat oral cavity.9,10 It is also mutagenic in the mouse oral cavity.11 Hecht et al. developed a useful model to study tobacco carcinogenesis and to assess the efficacy of chemopreventive agents in mice using the combination of the tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butamone (NNK) and benzo[a]pyrene (BaP) as inducers of lung tumors.12,13 Encouraged by these previous studies, it seems appropriate to utilize the combination of DB[a,l]P and NNN as a model for tobacco smoke-induced oral cancer. Here we report for the first time that the oral treatment of lacI mice with the combination DB[a,l]P and NNN induces a higher fraction of mutations than expected from a simple sum of their individual mutation inductions and a change in the mutational profile over the expected mutation profile of the sum of the individual agents. To provide some insights that can account for the observed mutation fraction and profile, we performed a preliminary investigation aimed at determining the effects of NNN and DB[a,l]P on the levels of DNA adducts in human oral leukoplakia cells.
Figure 1.
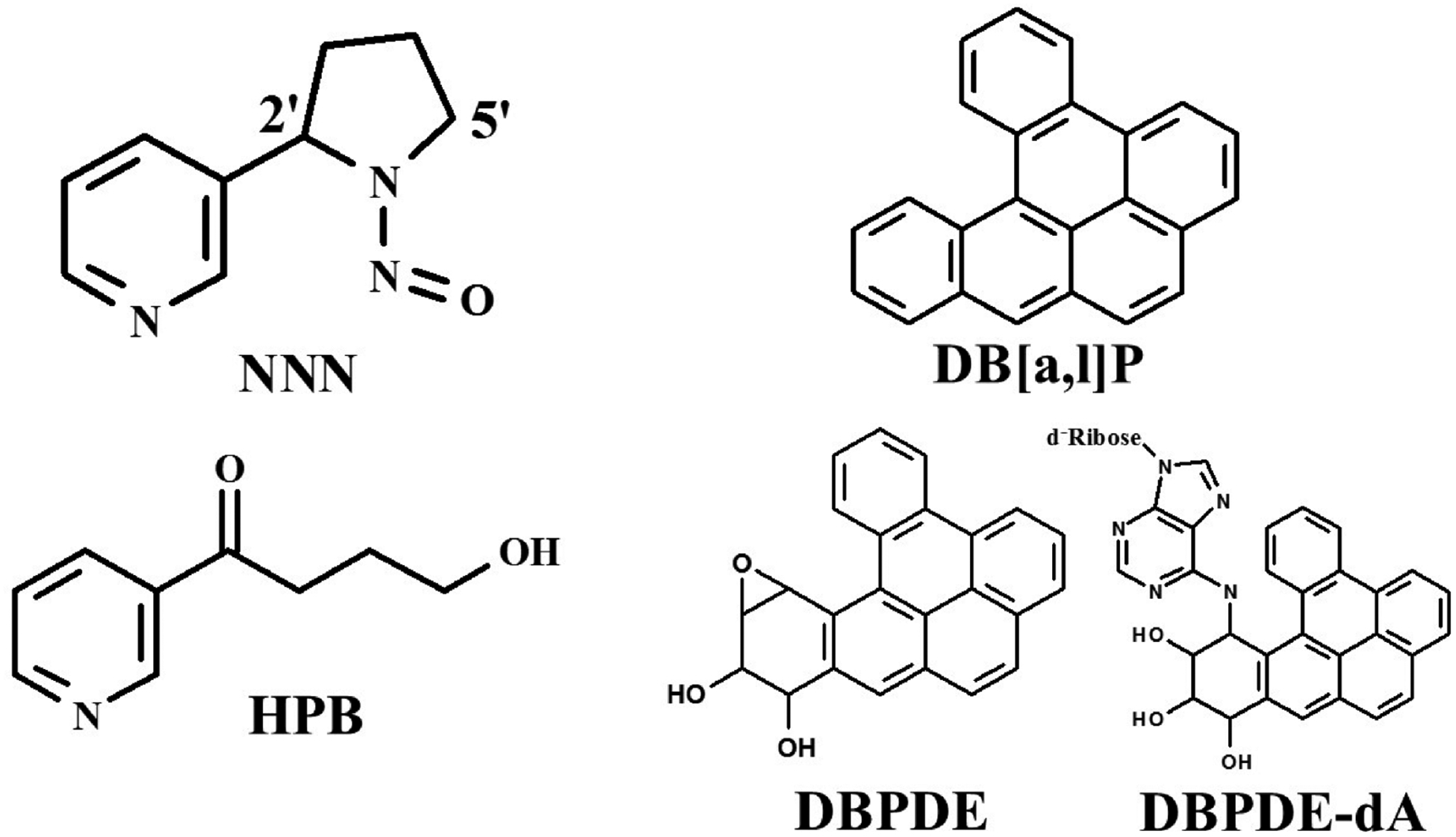
Structures of NNN, DB[a,l]P, DB[a,l]P-diolepoxide (DBPDE), DBPDE-dA adduct, and HPB.
MATERIALS AND METHODS
Chemicals and Medium.
DB[a,l]P, NNN, and DBPDE were prepared as previously described.7,14,15 4-Hydroxy-1-(3-pyridyl)-1-butanone (HPB) and its deuterated analog [3,3,4,4-d4]-HPB as well as [13C215N]guanine were obtained commercially (Toronto Research Chemical, Toronto, ON, Canada). Protease K and RNase A were purchased from Sigma Chemical. Keratinocyte growth medium (KGM) was obtained from Lonza Bioscience.
Cell Line and Culture Conditions.
MSK-Leuk1 cells were established from a premalignant leukoplakic lesion adjacent to a squamous cell carcinoma of the tongue.16,17 The cells were obtained from Dr. Peter Sacks, who is an emeritus faculty member in the same department as J.B.G. The cells were authenticated by Genetica DNA Laboratories (Burlington, NC) using short tandem repeat DNA profiling. Sequencing studies indicated that a GC > AT transition in exon 8 in one allele of p53, resulting in a Glu to Lys mutation in codon 286, was present in the MSK-Leuk1 cells.16,17 This cell line was routinely maintained in KGM grown to 70% confluence and trypsinized with a 0.125% trypsin–2 mmol/L ethylenediaminetetraacetic acid (EDTA) solution before passage.
Treatment of MSK Cells with NNN, DB[a,l]P, and NNN + DB[a,l]P.
Cells were grown to ~50% confluence and treated with (a) 2 mM NNN, (b) 1 μM DB[a,l]P, or (c) a combination of 2 mM NNN and 1 μM DB[a,l]P. 24 h later, cells were harvested and DNA was isolated.
Analysis of DNA Adducts.
DNA was isolated from cells using the Qiagen DNA easy kit, as described by the manufacturer. The concentration of DNA was determined using a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE). The method used for the analysis of the DBPDE-dA adducts by LC/MS/MS is identical to our previously published methods.18 The method used for the analysis of NNN–DNA adducts by assessing levels of HPB-releasing compound is identical to that reported in the literature.19,20
Analysis of DB[a,l]PDE-N(6)-dA.
The major deoxyadenosine adduct was analyzed by LC/MS/MS based on our published procedure.18 Prior to enzymatic hydrolysis, 150 pg of [15N5]-(−)-anti-trans-DB[a,l]PDE-N(6)-dA was added as an internal standard to ~40 μg DNA. Subsequently, DNA samples were hydrolyzed by DNase I (0.2 mg/mg DNA), snake venom phosphodiesterase (0.08 unit/mg DNA), and alkaline phosphatase (2 units/mg DNA). An aliquot of the hydrolysate was taken for dA analysis by high-performance liquid chromatography (HPLC). The remaining mixture was partially purified by solid phase extraction (SPE) (Oasis HLB column, 1 cm3, Waters). Then, the analysis was performed on an API 3200 LC/MS/MS triple quadrupole mass spectrometer interfaced with an Agilent 1200 series HPLC. The MS/MS transitions of m/z 604→ m/z 335 and m/z 609→ m/z 335 were monitored for targeted adducts and the internal standard, respectively, using the multiple reaction monitoring (MRM) mode.
Analysis of HPB.
The formation of HPB-releasing DNA adducts has been reported after the metabolic activation of NNN in human oral cells.19,20 HPB was released from DNA by acid hydrolysis and analyzed by HPLC/MS/MS according to a reported method.20 The internal standards, [3,3,4,4-d4]-HPB and [13C215N]guanine were added to DNA samples prior to acid hydrolysis. After SPE purification (HyperSep Hypercarb cartridges), the hydrolyzed DNA samples were analyzed on an API 4000 QTrap LC/MS/MS mass spectrometer interfaced with an Agilent 1100 series HPLC through monitoring the MRM transitions of [3,3,4,4d4]-HPB, m/z 170 → 106, and HPB, m/z 166 → 106. For the analysis of guanine, DNA samples were analyzed by an API 3200 LC/MS/MS triple quadrupole mass spectrometer interfaced with an Agilent 1200 series HPLC to monitor the MRM transitions of [13C215N]guanine, m/z 155 → 138, and guanine, m/z 152 → 135.
Mice and Treatments.
Female Big Blue C57BL6 mice between 10 and 12 weeks of age at the start of the study were kindly provided by Robert Young, MilliporeSigma, BioReliance Toxicology Testing Services (Rockville, MD). Groups of 10 mice were treated with (a) DB[a,l]P 0.16 μmol, three times/week, (b) NNN 8.46 μmol, two times/week, or (c) the two agents on alternate days. All mice were treated for 5 weeks and then euthanized 4 weeks after the last dose (Figure 2). The total doses of these agents were based on our previous studies where they induced mutagenesis or carcinogenesis, except the individual doses of DB[a,l]P were increased and administered over a shorter time period to reduce the length of the experiment.7,11
Figure 2.
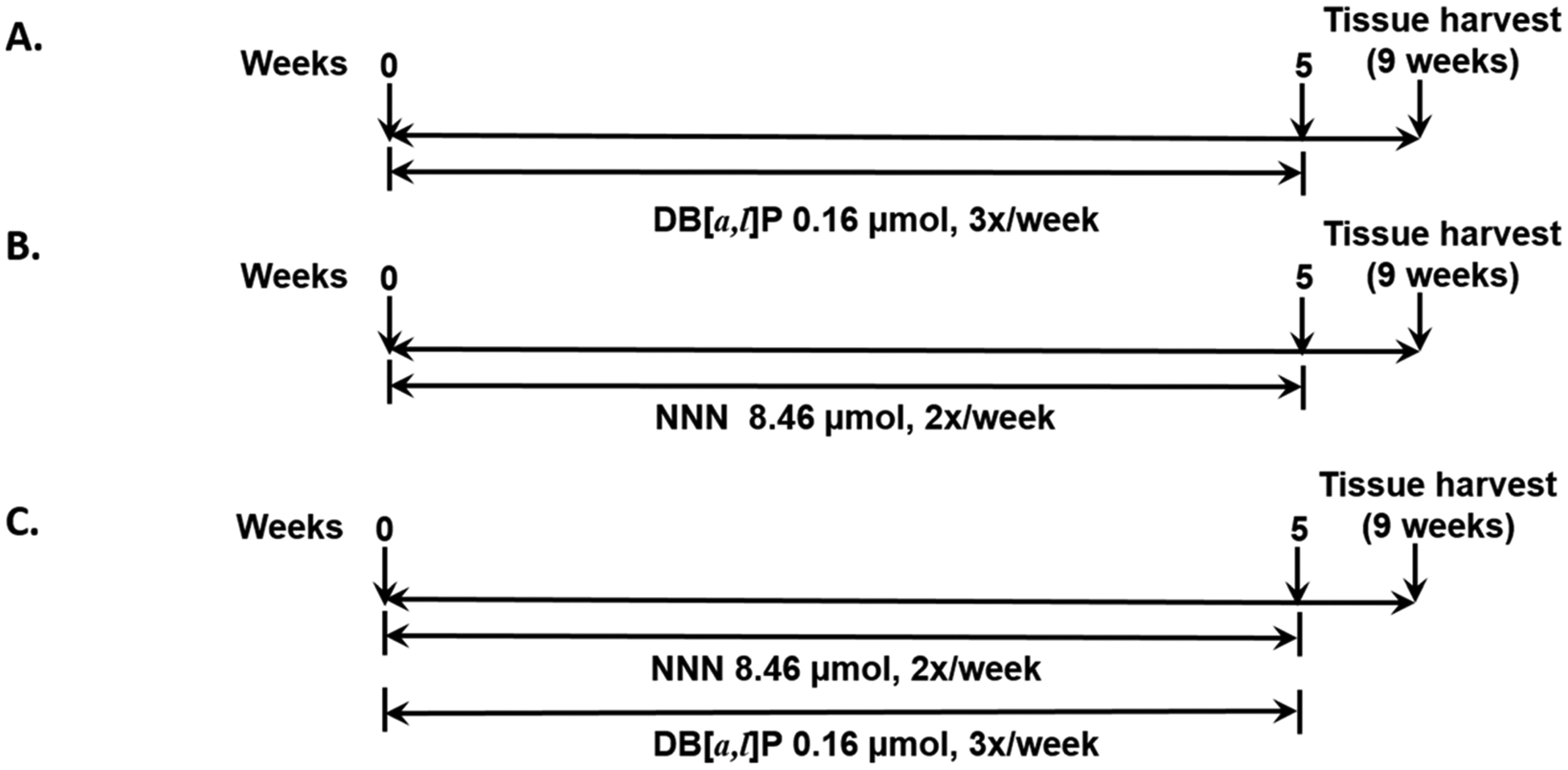
Experimental protocol to compare the mutagenesis induced by (A) DB[a,l]P, (B) NNN, and (C) DB[a,l]P + NNN in oral tissues of lacI mice (not to scale).
Mutagenesis Assay.
The lacI mice contain a lambda shuttle vector that includes the bacterial lacI locus and also the cII gene, which is the reporter gene for the mutagenesis studies. This assay detects mutations at the cII locus.21 The cII protein is a positive regulator of gene transcription that controls the decision between lytic or lysogenic development pathways in phage-infected cells. In appropriate E. coli (E. coli 1250) host cells, under specified conditions (25 °C), only mutants give rise to phage plaques, whereas at 37 °C, all infected cells give rise to plaques, providing a phage titer.21,22 The ratio of mutant to nonmutant plaques is the measure of mutagenesis, the mutant fraction (MF). DNA isolation was performed as previously described using an ammonium acetate precipitation method.7 Phage packaging was carried out using a homemade packaging extract prepared from bacterial strains supplied by Dr. Peter Glazer (Yale University School of Medicine, New Haven, CT), and the positive selection cII mutation assay was performed as previously described.23 At least three packaging reactions were carried out for each DNA sample. The historical spontaneous background MF was subtracted from the MF from each group.
Amplification and Sequencing.
Mutant plaques were randomly selected from all animals in each group and cored from the Petri dishes. The agar plug was mixed with 100 μL of phage buffer. Ten microliters of the buffer was then spread on a selective plate to confirm the mutant phenotype and purify mutant phages. Fifty-four mutant plaques/group of mice were then randomly selected for sequencing. Amplification and sequencing were performed as previously described24 with updated purification and sequencing. In brief, the purified mutant plaques were subjected to amplification using a Terra Direct polymerase chain reaction (PCR) kit, followed by ExoSAP-IT cleanup. Sequencing of the cII gene was carried out by Sanger sequencing using an Applied Biosystems 3730XL DNA analyzer. Sequencing and amplification were performed by the University of Arizona Genomics Core. The primers used for sequencing and amplification were cII forward primer 5′-CCACACCTATGGTGTATG-3′ and cII reverse primer 5′-CCTCTGCCGAAGTTGAGTAT-3′.
RESULTS
Mutagenesis.
Individually, NNN and DB[a,l]P both significantly increased mutagenesis in the mouse tongues over background levels, and the combination of DB[a,l]P and NNN induced an unexpectedly high MF (Figure 3). To determine whether the MF in the group treated with NNN and DB[a,l]P was statistically different from that expected from the sum of the mean MFs of NNN and DB[a,l]P, we compared the confidence band around the mean of the DB[a,l]P and NNN groups with the sum of the means of the MFs of NNN and DB[a,l]P using the pooled estimate of the standard deviation. The mean for the NNN group and the DB[a,l]P group is 12.2, and the 95% confidence limits are (10.3, 14.1). The best estimates of the independent (additive) effects of NNN and DB[a,l]P groups are 2.7 + 5.5, or 8.2. This sum is outside the 95% confidence limit of the NNN + DB[a,l]P group, and so this group is significantly greater than the expected independent contributions of NNN and DB[a,l]P. The exact p value for that t test is 0.004.
Figure 3.
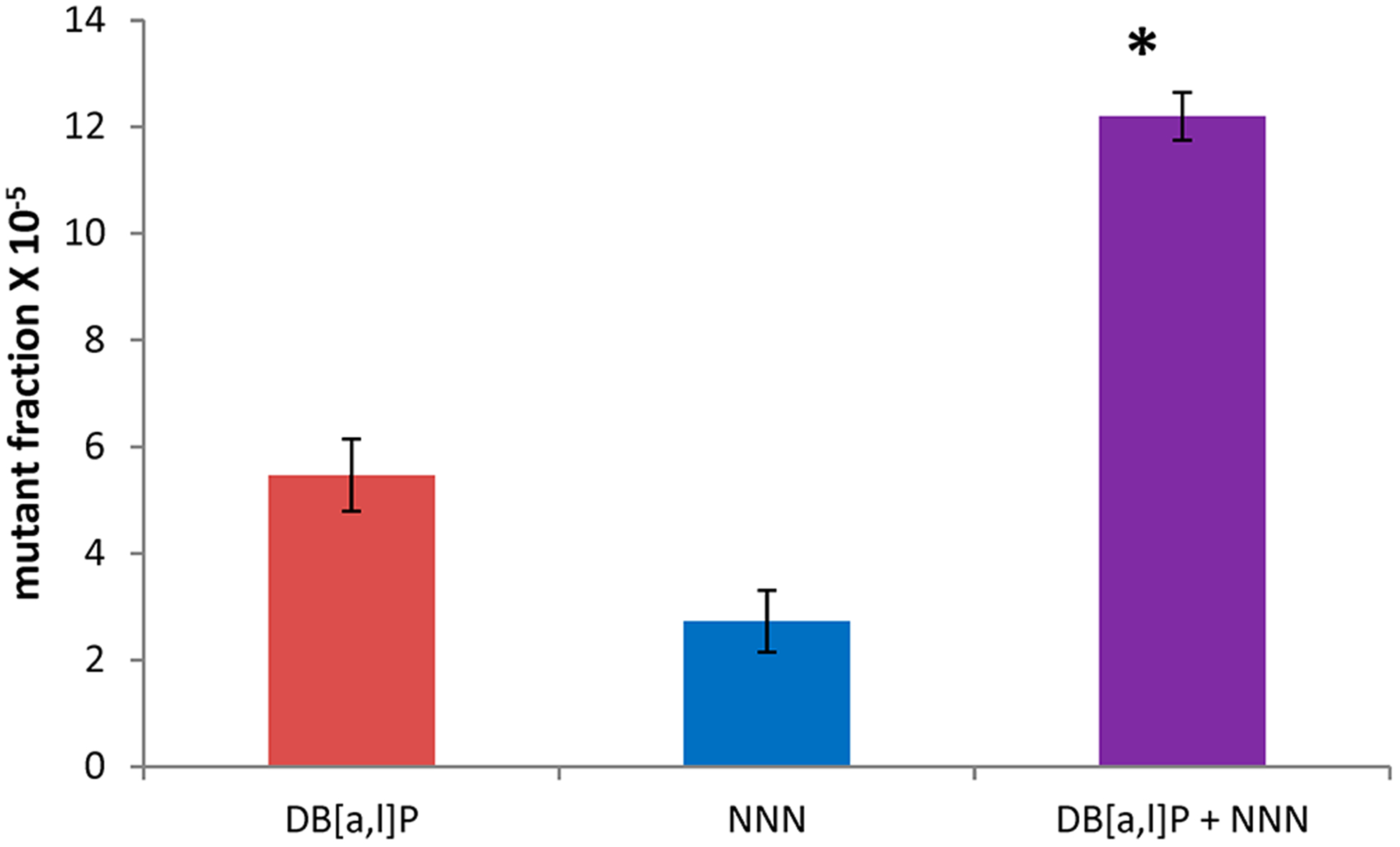
Mutant fractions (in pfu) induced by DB[a,l]P, NNN, and a combination of the two in the tongue of lacI mice. Background mutants (MF, 1.3 ± 0.6 pfu) were subtracted from the values in the figure. *, P < 0.05 versus the sum of individual mutant fractions of DB[a,l]P and NNN.
Mutation Fractions of Individual Mutation Classes.
Because the sum of the individual MFs of NNN and DB[a,l]P was different from that expected for the NNN + DB[a,l]P group, we were interested in determining the reason for this difference. We then sequenced about 50 mutants from each group and determined the MFs of seven mutation classes in groups treated with (1) NNN, (2) DB[a,l]P, and (3) NNN + DB[a,l]P. This was done by multiplying the percentage of each mutation class by the MF of each group. In addition, we calculated the expected MFs induced by NNN and DB[a,l]P by taking the sum of the values of the individual agents (Table 1). The most noticeable difference between the observed and expected was the large increase in observed versus expected mutations for the GC > AT transitions of 3.7 × 10−5 (5.12 vs 1.42, from Table 1). Other changes were much smaller, with the next greatest change (1.3 × 10−5 pfu) coming from GC > CG transversions (1.95 vs 0.65). The observed MF for the GC > AT mutation class was significantly different than that expected (P < 0.05), and those for AT > GC and AT > TA were near-significant (P < 0.1).
Table 1.
Mutant Fractions of Different Classes of Mutations Induced by NNN, DB[a,l]P, and Their Combination in the Tongue of lacI Mice
| NNN | DB[a,l]Pa | DB[a,l]P + NNN (obs.)b | DB[a,l]P + NNN (exp.)b | |||||
|---|---|---|---|---|---|---|---|---|
| class | mutants | MF × 10−5 | mutants | MF × 10−5 | mutants | MF × 10−5 | mutants | MF × 10−5 |
| G:A | 9 | 0.49 | 7 | 0.93 | 21 | 5.12d | 16 | 1.42 |
| G:T | 8 | 0.44 | 14 | 1.81 | 7 | 1.71 | 22 | 2.24 |
| G:C | 5 | 0.27 | 3 | 0.38 | 8 | 1.95 | 8 | 0.65 |
| A:G | 5 | 0.27 | 3 | 0.38 | 1 | 0.24 | 8 | 0.65 |
| A:C | 2 | 0.11 | 4 | 0.55 | 2 | 0.49 | 6 | 0.66 |
| A:T | 15 | 0.82 | 6 | 0.77 | 6 | 1.46 | 21 | 1.58 |
| indelc | 5 | 0.33 | 5 | 0.66 | 5 | 1.22 | 10 | 0.98 |
| 49 | 2.72 | 42 | 5.47 | 50 | 12.2 | 91 | 8.19 | |
Results from DB[a,l]P were taken from ref 7.
Obs. and exp. mutations in each class of mutation were compared using a Fisher’s exact test. There were three redundant mutations in the NNN group and two in the NNN + DB[a,l]P group. Three samples failed to yield a readable sequence.
indel, insertion or deletion.
p < 0.05 versus DB[a,l]P + NNN expected.
Mutational Profiles.
Using the values in Table 1, we calculated the percentages of each class of mutation and the expected percentages for the sum of the MFs of NNN individually and the observed values. The results are expressed in tabular form (Table 2) and graphically (Figure 4). There were indeed obvious differences between the observed and expected percentages of the individual NNN and DB[a,l]P treatments and the combination treatment. Driving the changes was the large and statistically significant increase in GC > AT transitions. This was largely at the expense of AT > TA and GC > TA substitutions, but the differences between observed and expected percentages did not reach significance. There was also a clear change in the percentage of AT > GC transitions, but this was a minor class of mutations, and the percentages were based on relatively small numbers of mutants. Similar considerations hold for GC > CG trans-versions, where the difference did not quite reach statistical significance. It may be relevant that the percentage of GC > AT transitions is ~40% in mutations in P53 in human head and neck cancers,25 which is similar to the observed value for the combination NNN + DB[a,l]P.
Table 2.
Mutational Profiles of NNN, DB[a,l]P, and Their Combination in lacI Mouse Tonguea
| percentage of mutations | ||||
|---|---|---|---|---|
| class | NNN | DB[a,l]P | NNN + DB[a,l]P obs. | NNN + DB[a,l]P exp. |
| G:A | 18 | 16.7 | 42.0b | 17.4 |
| G:T | 16 | 33.3 | 14.0 | 23.9 |
| G:C | 10 | 7.1 | 16.0 | 8.7 |
| A:G | 10 | 7.1 | 2.0 | 8.7 |
| A:C | 4 | 9.5 | 4.0 | 6.5 |
| A:T | 30 | 14.3 | 12.0 | 22.8 |
| indel | 12 | 11.9 | 10.0 | 12.0 |
| 100 | 100 | 100 | 100 | |
To test whether there were any statistically significant differences between the observed and expected percentages for each mutation class, an “N − 1” chi-squared test was employed.
p < 0.05 for the difference between the observed and expected percentages.
Figure 4.
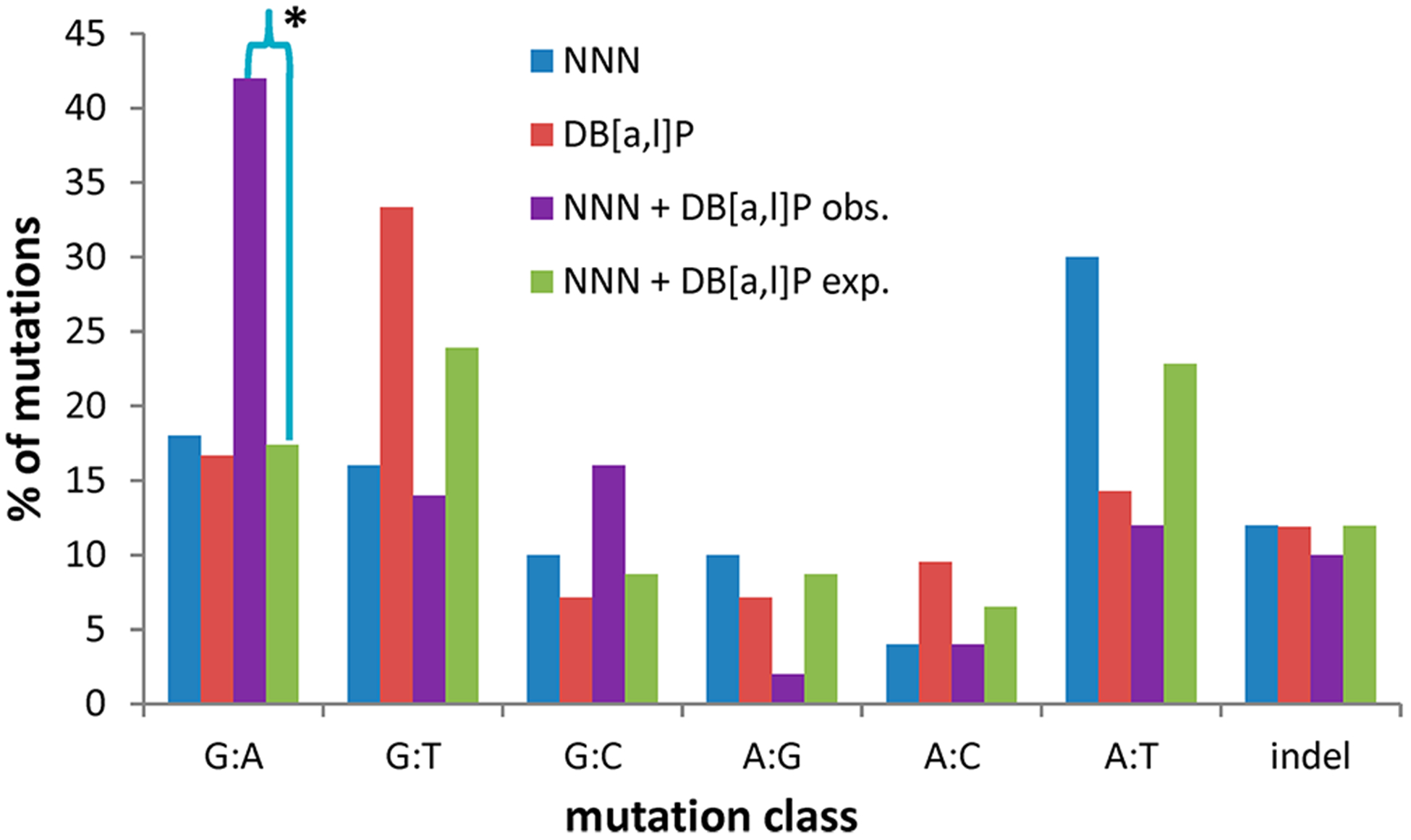
Mutational profiles of NNN and DB[a,l]P and the observed and expected profiles of NNN + DB[a,l]P in lacI mouse tongue. *, P < 0.05 for the difference between the observed and expected percentages of the NNN + DB[a,l]P mutation classes.
DNA Adducts.
Because mutagenesis induced by genotoxic agents largely results from DNA adducts, we attempted to understand the origin of the unexpected mutagenesis observations by measuring DNA adduct levels. We began by studying carcinogens using doses based on previous experiments where DB[a,l]P26 and NNN (unpublished results) each produced measurable levels of DNA adducts. NNN yields a pyridyloxobutylating intermediate that reacts with DNA to produce a variety of adducts with deoxyguanosine, deoxythymidine, and deoxycytidine.27 Acid hydrolysis of several pyridyloxobutyl adducts releases HPB.27 Thus HPB-releasing DNA adducts are a measure of the pyridyloxobutylation of DNA by NNN. This assay has been used to detect evidence of pyridyloxobutylation in the oral cavity of smokers.19,28 For DB[a,l]P adducts, we monitored levels of DB[a,l]PDE-N(6)-dA. We have previously reported that this is the major adduct detected in the oral tissue of mice treated with DB[a,l]P.18
We compared the levels of HPB in MSK cells treated with NNN and treated with NNN + DB[a,l]P. No significant difference in the adduct levels between the two groups was observed (Figure 5A). We also compared DB[a,l]PDE-N(6)-dA levels in cells treated with DB[a,l]P and in cells treated with DB[a,l]P + NNN. Again, no significant differences in adduct levels between the two groups was observed (Figure 5B).
Figure 5.
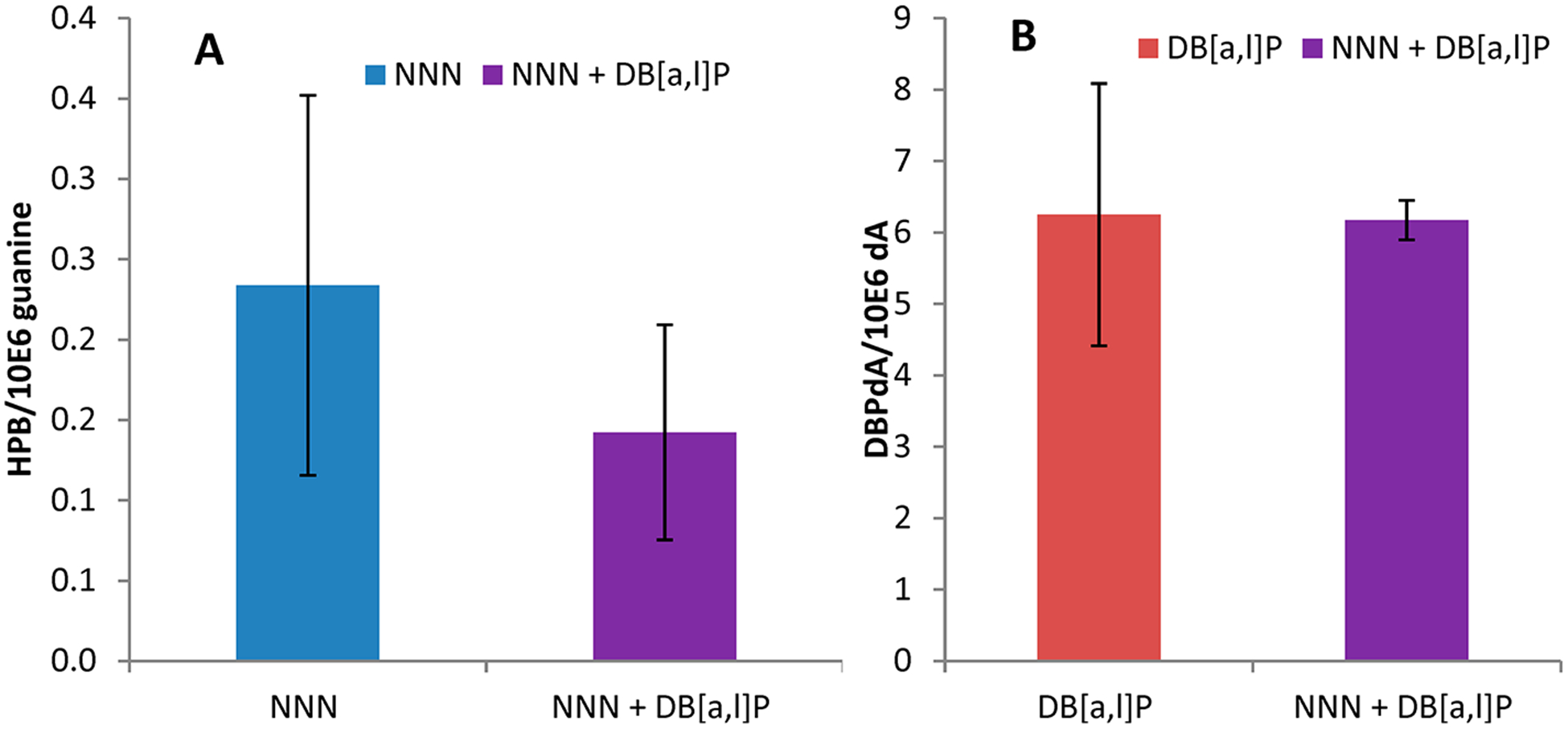
DNA adducts produced in MSK-Leuk1 cells by treatment with NNN, DB[a,l]P, and NNN + DB[a,l]P. (A) HPB levels. (B) DB[a,l]P-dA levels.
DISCUSSION
The origin of the difference in the MF and the mutational profile between the observed results for the combination treatment and the expected results from the individual treatments is not apparent, but it seems possible that DNA adducts that play smaller roles in leading to mutations induced by NNN or DB[a,l]P are individually more important when the treatment is by a combination of these two agents. The mutational efficiency of DNA adducts can vary greatly,29 and minor adducts may be important contributors to mutagenesis. Because the MF of GC > AT transitions is greatly enhanced in the combination treatment, the adduct is presumably at a guanine or cytosine residue. DB[a,l]P alone produces adducts at guanine residues and induces GC > AT transitions,18,30 and NNN also produces guanine adducts31,32 and induces GC > AT transitions (Table 2). We failed to see any changes in the levels of NNN or DB[a,l]P adducts when cells treated with these agents were combined compared with the treatment by each agent separately. It is important to recognize that cells were treated with a single dose, whereas mice were treated with multiple doses (three times per week for 5 weeks and sacrificed 4 weeks after the last carcinogen administration). In addition, we did not examine levels of DNA adducts in the lacI mice because based on our previous studies the levels of BPDE-dA could not be accurately quantified 4 weeks after the last administration.18 The levels of dA and dG adducts derived from DB[a,l]P may vary depending on the carcinogen dose, differences in metabolic capacities of various cell types in vitro and in vivo, treatment duration, as well as time points of these measurements.18 Clearly, our findings of this initial experiment support the need for additional in vivo and in vitro studies using other cell types to analyze adducts produced at lower levels and with varied structures. Future studies are planned to identify and quantify minor DNA adducts such as deoxyguanosine adducts derived from DB[a,l]P and O6-alkylguanine adducts derived from NNN in mice treated with these carcinogens individually and in combination.
If the initial damage deposition by the combination of DBP + NNN is unchanged compared with what is expected when cells are treated with NNN or DB[a,l]P alone, the processing of DNA adducts produced by either agent might be altered by additional DNA damage resulting from the second carcinogen. There are a number of proteins recruited to DNA damage sites,33 and the physical blockage of an adduct that is usually efficiently repaired may be precluded when additional DNA damage resulting from the second agent is present. A more specific type of interference in DNA repair may occur with O6-pyridyloxobutyl guanine adducts. These can be produced by NNN.31 O6-alkyl adducts generally give rise to mutations with high efficiency34,35 and almost exclusively GC > AT transitions.35 These adducts can be repaired by O6-alkylguanine-DNA alkyltransferase (AGT)36 and possibly other DNA repair systems. AGT is irreversibly deactivated at a reactive sulfhydryl by O6-alkylguanines,36,37 and also by other reactive molecules such as aldehydes.38 Polycyclic aromatic hydrocarbons such as DB[a,l]P are not likely to be a source of aldehydes because they are polynuclear with no alkyl side chains. However, they give rise to ortho-quinones that can react with sulfhydryls and may also result in the formation of DNA adducts, depurinating adducts, and oxidatively modified DNA lesions.39 It may also be relevant that carcinogenesis in the rat oral cavity and esophagus induced by (S)-NNN was greatly enhanced by the weakly carcinogenic (R)-NNN.31
Other explanations for the altered mutational profile seen in the combination of NNN + DB[a,l]P can be advanced. For instance, the levels of the cytochrome P450s that activate DB[a,l]P and NNN to ultimate mutagens may be altered relative to their levels when only one carcinogen is present, with a concomitant alteration in the distribution of DNA adducts.
In conclusion, we have extended our model for tobacco-induced oral cancer to now include two of the major known classes (polycyclic aromatic hydrocarbons and tobacco-specific nitrosamines) of oral carcinogens in tobacco. The observation that the percentage of the class of mutations induced by DB[a,l]P + NNN is similar to that seen in the P53 gene in human head and neck cancers supports the use of this animal model to investigate tobacco-smoke-induced cancer and cancer prevention in the oral cavity.
ACKNOWLEDGMENTS
The Penn State Cancer Institute Organic Synthesis Shared Resource provided the DB[a,l]P, DBPDE, and NNN used in this study. We thank Dongxiao Sun of the Penn State College of Medicine Mass Spectrometry Core Facility for conducting the analysis of HPB.
Funding
This project is supported by NIH grant no. CA173465.
ABBREVIATIONS
- DB[a,l]P
dibenzo[a,l]pyrene
- NNN
N′-nitrosonornicotine
- DBPDE
(±)-anti-11,12-dihydroxy-13,14-epoxy-11,12,13,14-tetrahydrodibenzo[a,l]pyrene
- OSCC
oral squamous cell carcinoma
- NNK
4-(methylnitrosamino)-1-(3-pyridyl)-1-butamone
- BaP
benzo[a]pyrene
- HPB
4-hydroxy-1-(3-pyridyl)-1-butanone
- MF
mutant fraction
- MSKLeuk1
human oral epithelial cell line
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Siegel RL, Miller KD, and Jemal A (2019) Cancer statistics, 2019. Ca-Cancer J. Clin 69, 7–34. [DOI] [PubMed] [Google Scholar]
- (2).Jethwa AR, and Khariwala SS (2017) Tobacco-related carcinogenesis in head and neck cancer. Cancer Metastasis Rev. 36, 411–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Blot WJ, McLaughlin JK, Winn DM, Austin DF, Greenberg RS, Preston-Martin S, Bernstein L, Schoenberg JB, Stemhagen A, and Fraumeni JF Jr. (1988) Smoking and drinking in relation to oral and pharyngeal cancer. Cancer Res. 48, 3282–3287. [PubMed] [Google Scholar]
- (4).Gillison ML (2007) Current topics in the epidemiology of oral cavity and oropharyngeal cancers. Head Neck 29, 779–792. [DOI] [PubMed] [Google Scholar]
- (5).Wyss A, Hashibe M, Chuang SC, Lee YC, Zhang ZF, Yu GP, Winn DM, Wei Q, Talamini R, Szeszenia-Dabrowska N, Sturgis EM, Smith E, Shangina O, Schwartz SM, Schantz S, Rudnai P, Purdue MP, Eluf-Neto J, Muscat J, Morgenstern H, Michaluart P Jr., Menezes A, Matos E, Mates IN, Lissowska J, Levi F, Lazarus P, La Vecchia C, Koifman S, Herrero R, Hayes RB, Franceschi S, Wunsch-Filho V, Fernandez L, Fabianova E, Daudt AW, Dal Maso L, Curado MP, Chen C, Castellsague X, de Carvalho MB, Cadoni G, Boccia S, Brennan P, Boffetta P, and Olshan AF (2013) Cigarette, cigar, and pipe smoking and the risk of head and neck cancers: pooled analysis in the International Head and Neck Cancer Epidemiology Consortium. Am. J. Epidemiol 178, 679–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).El-Bayoumy K, Chen KM, Zhang SM, Sun YW, Amin S, Stoner G, and Guttenplan JB (2017) Carcinogenesis of the Oral Cavity: Environmental Causes and Potential Prevention by Black Raspberry. Chem. Res. Toxicol 30, 126–144. [DOI] [PubMed] [Google Scholar]
- (7).Guttenplan JB, Kosinska W, Zhao ZL, Chen KM, Aliaga C, DelTondo J, Cooper T, Sun YW, Zhang SM, Jiang K, Bruggeman R, Sharma AK, Amin S, Ahn K, and El-Bayoumy K (2012) Mutagenesis and carcinogenesis induced by dibenzo[a,l]-pyrene in the mouse oral cavity: a potential new model for oral cancer. Int. J. Cancer 130, 2783–2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Chen KM, Guttenplan JB, Zhang SM, Aliaga C, Cooper TK, Sun YW, DelTondo J, Kosinska W, Sharma AK, Jiang K, Bruggeman R, Ahn K, Amin S, and El-Bayoumy K (2013) Mechanisms of oral carcinogenesis induced by dibenzo[a,l]pyrene: an environmental pollutant and a tobacco smoke constituent. Int. J. Cancer 133, 1300–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Balbo S, James-Yi S, Johnson CS, O’Sullivan MG, Stepanov I, Wang M, Bandyopadhyay D, Kassie F, Carmella S, Upadhyaya P, and Hecht SS (2013) (S)-N’-Nitrosonornicotine, a Constituent of Smokeless Tobacco, is a Powerful Oral Cavity Carcinogen in Rats. Carcinogenesis 34, 2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Khariwala SS, Hatsukami D, and Hecht SS (2012) Tobacco carcinogen metabolites and DNA adducts as biomarkers in head and neck cancer: potential screening tools and prognostic indicators. Head Neck 34, 441–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).von Pressentin M. d. M., Chen M, and Guttenplan JB (2001) Mutagenesis induced by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone-4-(methylnitrosamino)-1-(3- pyridyl)-1-butanone and N-nitrosonornicotine in lacZ upper aerodigestive tissue and liver and inhibition by green tea. Carcinogenesis 22, 203–206. [DOI] [PubMed] [Google Scholar]
- (12).Kassie F, Melkamu T, Endalew A, Upadhyaya P, Luo X, and Hecht SS (2010) Inhibition of lung carcinogenesis and critical cancer-related signaling pathways by N-acetyl-S-(N-2-phenethylthiocarbamoyl)-l-cysteine, indole-3-carbinol and myo-inositol, alone and in combination. Carcinogenesis 31, 1634–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Hecht SS, Isaacs S, and Trushin N (1994) Lung tumor induction in A/J mice by the tobacco smoke carcinogens 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and benzo[a]pyrene: a potentially useful model for evaluation of chemopreventive agents. Carcinogenesis 15, 2721–2725. [DOI] [PubMed] [Google Scholar]
- (14).Hu MW, Bondinell WE, and Hoffmann D (1974) Synthesis of carbon-14 labelled myosamine, norrnicotine, and N’-nitrosonornicotine. J. Labelled Compd 10, 79–88. [Google Scholar]
- (15).Sharma AK, Kumar S, and Amin S (2004) A Highly Abbreviated Synthesis of Dibenzo[def,p]chrysene and Its 12-Methoxy Derivative, a Key Precursor for the Synthesis of the Proximate and Ultimate Carcinogens of Dibenzo[def,p]chrysene. J. Org. Chem 69, 3979–3982. [DOI] [PubMed] [Google Scholar]
- (16).Kochhar A, Kopelovich L, Sue E, Guttenplan JB, Herbert BS, Dannenberg AJ, and Subbaramaiah K (2014) p53 modulates Hsp90 ATPase activity and regulates aryl hydrocarbon receptor signaling. Cancer Prev. Res 7, 596–606. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- (17).Sacks PG (1996) Cell, tissue and organ culture as in vitro models to study the biology of squamous cell carcinomas of the head and neck. Cancer Metastasis Rev. 15, 27–51. [DOI] [PubMed] [Google Scholar]
- (18).Zhang SM, Chen KM, Sun YW, Aliaga C, Lin JM, Sharma AK, Amin S, and El-Bayoumy K (2014) Simultaneous detection of deoxyadenosine and deoxyguanosine adducts in the tongue and other oral tissues of mice treated with Dibenzo[a,l]pyrene. Chem. Res. Toxicol 27, 1199–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Stepanov I, Muzic J, Le CT, Sebero E, Villalta P, Ma B, Jensen J, Hatsukami D, and Hecht SS (2013) Analysis of 4-Hydroxy-1-(3-pyridyl)-1-butanone (HPB)-Releasing DNA Adducts in Human Exfoliated Oral Mucosa Cells by Liquid Chromatography-Electrospray Ionization-Tandem Mass Spectrometry. Chem. Res. Toxicol 26, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Ma B, Ruszczak C, Jain V, Khariwala SS, Lindgren B, Hatsukami DK, and Stepanov I (2016) Optimized Liquid Chromatography Nanoelectrospray-High-Resolution Tandem Mass Spectrometry Method for the Analysis of 4-Hydroxy-1-(3-pyridyl)-1-butanone-Releasing DNA Adducts in Human Oral Cells. Chem. Res. Toxicol 29, 1849–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Jakubczak JL, Merlino G, French JE, Muller WJ, Paul B, Adhya S, and Garges S (1996) Analysis of genetic instability during mammary tumor progression using a novel selection-based assay for in vivo mutations in a bacteriophage lambda transgene target. Proc. Natl. Acad. Sci. U. S. A 93, 9073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Swiger RR (2001) Just how does the cII selection system work in Muta Mouse? Environ. Mol. Mutagen 37, 290–296. [DOI] [PubMed] [Google Scholar]
- (23).Guttenplan JB, Zhao ZL, Kosinska W, Norman RG, Krzeminski J, Sun YW, Amin S, and El-Bayoumy K (2007) Comparative mutational profiles of the environmental mammary carcinogen, 6-nitrochrysene and its metabolites in a lacI mammary epithelial cell line. Carcinogenesis 28, 2391–2397. [DOI] [PubMed] [Google Scholar]
- (24).Boyiri T, Guttenplan J, Khmelnitsky M, Kosinska W, Lin JM, Desai D, Amin S, Pittman B, and El-Bayoumy K (2003) Mammary carcinogenesis and molecular analysis of in vivo cII gene mutations in the mammary tissue of female transgenic rats treated with the environmental pollutant 6-nitrochrysene. Carcinogenesis 25, 637–643. [DOI] [PubMed] [Google Scholar]
- (25).p53 Mutations in Head and Neck Cancer, 2008. http://p53.free.fr/Database/p53_cancer/p53_oral.html (accessed June 6, 2019).
- (26).Guttenplan JB, Chen KM, Sun YW, Lajara B, Shalaby NAE, Kosinska W, Benitez G, Gowda K, Amin S, Stoner G, and El-Bayoumy K (2017) Effects of Black Raspberry Extract and Berry Compounds on Repair of DNA Damage and Mutagenesis Induced by Chemical and Physical Agents in Human Oral Leukoplakia and Rat Oral Fibroblasts. Chem. Res. Toxicol 30, 2159–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Hecht SS (2008) Progress and challenges in selected areas of tobacco carcinogenesis. Chem. Res. Toxicol 21, 160–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Khariwala SS, Ma B, Ruszczak C, Carmella SG, Lindgren B, Hatsukami DK, Hecht SS, and Stepanov I (2017) High Level of Tobacco Carcinogen-Derived DNA Damage in Oral Cells Is an Independent Predictor of Oral/Head and Neck Cancer Risk in Smokers. Cancer Prev. Res 10, 507–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Guttenplan JB (1990) Mutagenesis by N-nitroso compounds: relationships to DNA adducts, DNA repair, and mutational efficiencies. Mutat. Res., Fundam. Mol. Mech. Mutagen 233, 177–187. [DOI] [PubMed] [Google Scholar]
- (30).Spencer WA, Singh J, and Orren DK (2009) Formation and differential repair of covalent DNA adducts generated by treatment of human cells with (±)-anti-dibenzo[a,l]pyrene-11,12-diol-13,14-epoxide. Chem. Res. Toxicol 22, 81–89. [DOI] [PubMed] [Google Scholar]
- (31).Yang J, Villalta PW, Upadhyaya P, and Hecht SS (2016) Analysis of O(6)-[4-(3-Pyridyl)-4-oxobut-1-yl]-2’-deoxyguanosine and Other DNA Adducts in Rats Treated with Enantiomeric or Racemic N’-Nitrosonornicotine. Chem. Res. Toxicol 29, 87–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Zhao L, Balbo S, Wang M, Upadhyaya P, Khariwala SS, Villalta PW, and Hecht SS (2013) Quantitation of pyridyloxobutyl-DNA adducts in tissues of rats treated chronically with (R)- or (S)-N’-nitrosonornicotine (NNN) in a carcinogenicity study. Chem. Res. Toxicol 26, 1526–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Friedberg EC, Aguilera A, Gellert M, Hanawalt PC, Hays JB, Lehmann AR, Lindahl T, Lowndes N, Sarasin A, and Wood RD (2006) DNA repair: from molecular mechanism to human disease. DNA Repair 5, 986–996. [DOI] [PubMed] [Google Scholar]
- (34).Guttenplan JB (1984) Mutagenesis and O6-ethylguanine levels in DNA from N-nitroso-N-ethylurea-treated Salmonella typhimurium: evidence for a high mutational efficiency of O6-ethylguanine. Carcinogenesis 5, 155–159. [DOI] [PubMed] [Google Scholar]
- (35).Loechler EL, Green CL, and Essigmann JM (1984) In vivo Mutagenesis by O6-methylguanine Built Into a Unique Site in a Viral Genome. Proc. Natl. Acad. Sci. U. S. A 81, 6271–6275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Demple B, Jacobsson A, Olsson M, Robins P, and Lindahl T (1982) Repair of alkylated DNA in Escherichia coli. Physical properties of O6-methylguanine-DNA methyltransferase. J. Biol. Chem 257, 13776–13780. [PubMed] [Google Scholar]
- (37).Mijal RS, Kanugula S, Vu CC, Fang Q, Pegg AE, and Peterson LA (2006) DNA sequence context affects repair of the tobacco-specific adduct O(6)-[4-Oxo-4-(3-pyridyl)butyl]guanine by human O(6)-alkylguanine-DNA alkyltransferases. Cancer Res. 66, 4968–4974. [DOI] [PubMed] [Google Scholar]
- (38).Krokan H, Grafstrom RC, Sundqvist K, Esterbauer H, and Harris CC (1985) Cytotoxicity, thiol depletion and inhibition of O6-methylguanine-DNA methyltransferase by various aldehydes in cultured human bronchial fibroblasts. Carcinogenesis 6, 1755–1759. [DOI] [PubMed] [Google Scholar]
- (39).Penning TM (2010) Polycyclic Aromatic Hydrocarbons: Multiple Metabolic Pathways and the DNA Lesions Formed. The Chemical Biology of DNA Damage, 131–155. [Google Scholar]