

SUMMARY
The chromatin-associated protein WDR5 is a promising pharmacological target in cancer, with most drug discovery efforts directed against an arginine-binding cavity in WDR5 called the WIN site. Despite a clear expectation that WIN site inhibitors will alter the repertoire of WDR5 interaction partners, their impact on the WDR5 interactome remains unknown. Here, we use quantitative proteomics to delineate how the WDR5 interactome is changed by WIN site inhibition. We show that the WIN site inhibitor alters the interaction of WDR5 with dozens of proteins, including those linked to phosphatidylinositol 3-kinase (PI3K) signaling. As proof of concept, we demonstrate that the master kinase PDPK1 is a bona fide high-affinity WIN site binding protein that engages WDR5 to modulate transcription of genes expressed in the G2 phase of the cell cycle. This dataset expands our understanding of WDR5 and serves as a resource for deciphering the action of WIN site inhibitors.
Graphical Abstract
In Brief
Pharmacological inhibition of the WIN site of WDR5 is a promising anti-cancer strategy. Guarnaccia et al. use quantitative proteomics to characterize how inhibiting the WIN site alters the WDR5 interactome. This resource expands understanding of WDR5 and the action of WIN site inhibitors.
INTRODUCTION
WDR5 is a conserved WD40-repeat protein that rose to prominence through its role in epigenetic complexes, including the KMT2 (MLL/SET) enzymes that deposit histone H3 lysine 4 (H3K4) methylation and the NSL (non-specific lethal) complex that lays down H4 lysine 16 acetylation. But WDR5 has functions outside these complexes, including recruiting MYC to chromatin (Thomaset al., 2015), controlling expression of genes linkedto protein synthesis (Bryan et al., 2020), enabling rapid gene reactivation upon exit from mitosis (Oh et al., 2020), and promoting faithful assembly of the mitotic spindle (Ali et al., 2017). Why and how WDR5 participates in so many processes are unclear, as is the extent to which its moonlighting capabilities have been revealed.
Understanding WDR5 has become important in recent years, driven by observations linking it to cancer and by the idea that it could be targeted to develop anti-cancer therapies (Aho et al., 2019b). WDR5 is overexpressed in leukemia (Ge et al., 2016), bladder cancer (Chen et al., 2015), hepatocellular carcinoma (Cui et al., 2018), and breast cancer (Dai et al., 2015). It has been implicated in malignant processes, such as the epithelial-to-mesenchymal transition (Wu et al., 2011) and metastasis (Punzi et al., 2019), and serves as an important co-factor for oncogenic drivers, such as MYC and the retinoic acid receptor (Vilhais-Neto et al., 2017). Proof-of-concept experiments have demonstrated that pharmacologically inhibiting WDR5 can inhibit the growth of cancer cells in vitro, including those derived from KMT2A (MLL1)-rear-ranged (Aho et al., 2019a; Cao et al., 2014) and C/EBPα mutant (Grebien et al., 2015) leukemias, neuroblastoma (Bryan et al., 2020), and breast cancers (Punzi et al., 2019), as well as those expressing p53 gain-of-function variants (Zhu et al., 2015).
Most WDR5 inhibitors target the WIN (WDR5-interaction) site on WDR5, a pocket that binds to an arginine-containing WIN motif in partner proteins (Ali et al., 2017; Dias et al., 2014; Song and Kingston, 2008). These inhibitors were pursued with the intent of blocking the histone methyltransferase (HMT) function of KMT2A complexes, the catalytic activity of which is dependent on insertion of a WIN motif in KMT2A into the WIN site of WDR5 (Patel et al., 2008b). Accordingly, most actions of WIN site inhibitors in cells are assumed to be mediated by displacement of KMT2A from WDR5 and resulting changes in H3K4 methylation that reprogram the epigenome. But in KMT2A-rearranged cancer cells, which are the prototype for this response, we showed that WIN site inhibitors act independent of changes in H3K4 methylation, instead displacing WDR5 from chromatin at genes connected to protein synthesis and killing cells via induction of nucleolar stress (Aho et al., 2019a). Clearly, assumptions about how WIN site inhibitors act thwart understanding oftheir mechanism of action, particularlywhenthere is no systematic insight into the ways, beyond KMT2A inhibition and eviction from chromatin, in which they alter WDR5 behavior.
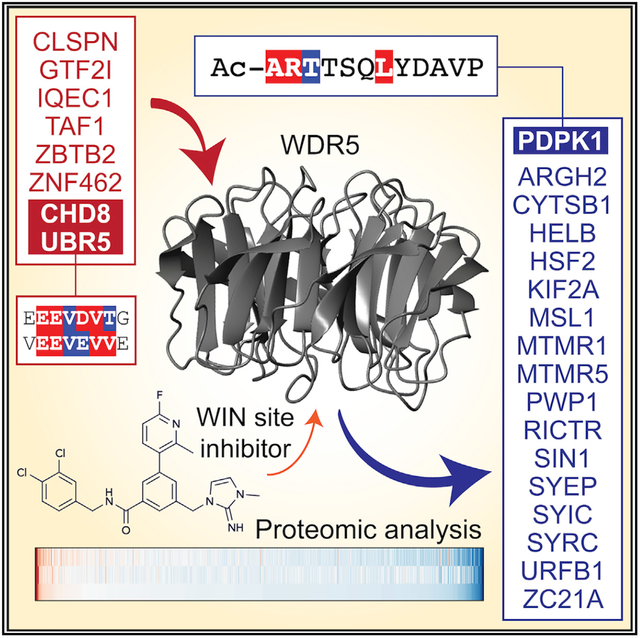
Defining the impact of the WIN site inhibitor on the WDR5 interactome will accelerate understanding of WDR5 and focus mechanism of action studies for clinical implementation of WIN site inhibitors. We took a quantitative proteomic approach to delineate how the WDR5 interactome changes when its WIN site is inhibited. We identify a collection of WIN site-sensitive WDR5 binders, many of which have links to growth factor signaling. As proof of concept, we validate the kinase PDPK1 (3-phosphoinositide-dependent protein kinase 1) as a WIN site binder and establish a role for the PDPK1-WDR5 interaction in controlling transcription of cell-cycle-regulated genes. In the process, we also demonstrate how modification of the amino terminus of PDPK1 creates an unusually high-affinity WIN motif. This resource expands our understanding of the roles of WDR5 and hones definition of what constitutes an avid WIN site binder.
RESULTS AND DISCUSSION
Effect of WIN site inhibitor on known WDR5 interaction partners
Proteins that interact with WDR5 do so through either a hydrophobic cleft called the “WBM” site or an arginine-binding pocket called the “WIN” site (Figure 1A; Figure S1A), both of which engage motifs in partner proteins (Guarnaccia and Tansey, 2018). The WBM motif, present in proteins such as MYC and RBBP5, is defined as [ED]-[ED]-[IVL]-D-V-[VT] (Odho et al., 2010). The WIN motif, present in KMT2 proteins, as well as histone H3, KANL1, Mbd3c, and KIF2A, is defined as [GV]-[SCA]-A-R-[AST]-[EKR] (Ali et al., 2017; Dias et al., 2014; Song and Kingston, 2008). Both sites are focal points for drug discovery, but because the WIN site is the subject of most efforts, we asked how the WIN site inhibitor C6 (Aho et al., 2019a) influences the interaction properties of WDR5.
Figure 1. WIN site inhibitor selectively displaces proteins from WDR5.
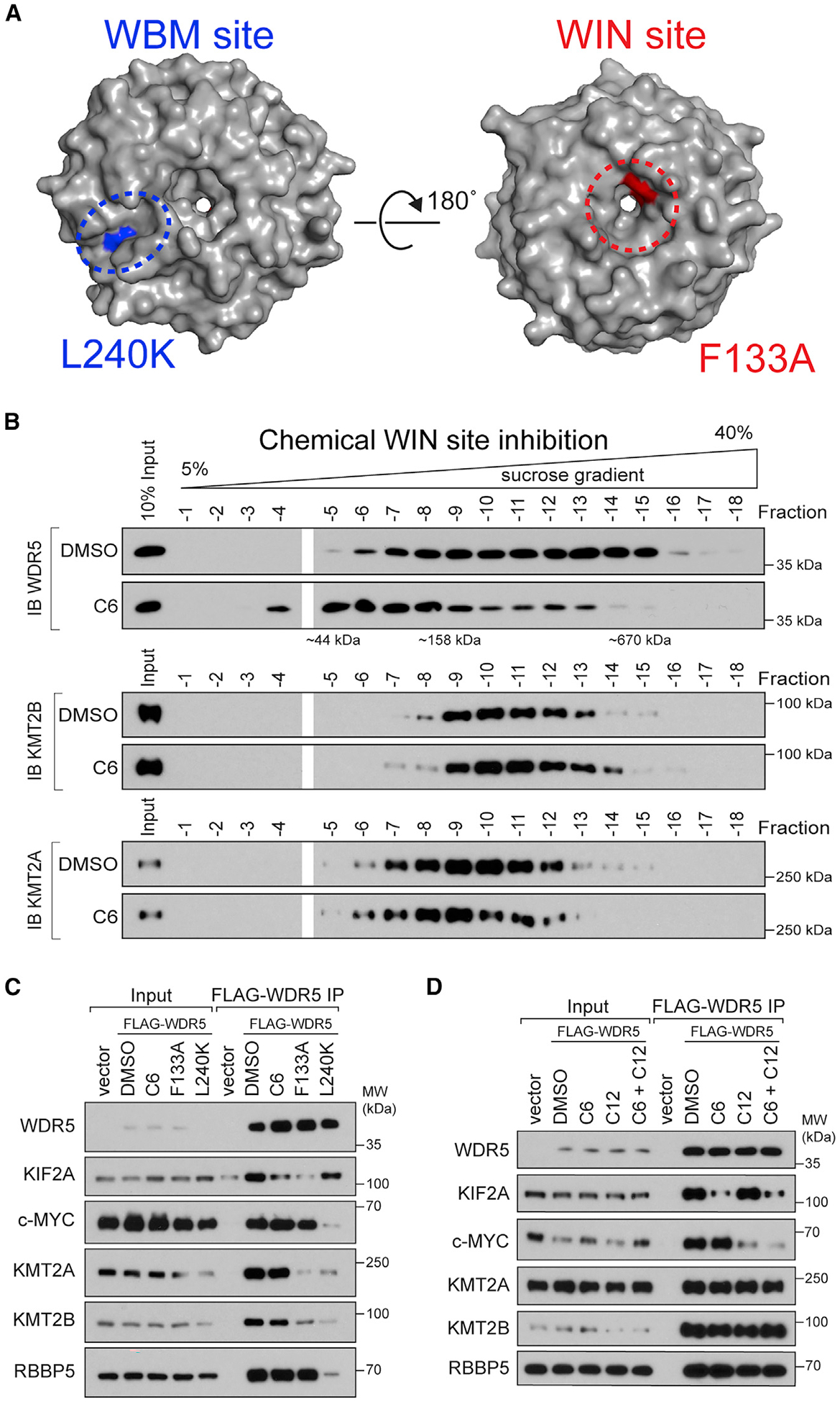
(A) Crystal structure of WDR5 (PDB: 2H14) outlining the location of the WBM site (blue) and the WIN site (red); locations of the L240K and F133A mutations are also shown.
(B) Density sedimentation analysis of HEK293 cells treated for 5 h with 30 μM C6 or DMSO. After treatment, cells were lysed and extracts analyzed by sucrose gradient density sedimentation followed by immunoblotting (IB) for WDR5 (top), KMT2B (middle), or KMT2A (bottom). Positions of molecular weight markers are indicated. n = 3 biological replicates.
(C) HEK293 cells stably expressing wild-type (WT) FLAG-tagged WDR5, or the indicated mutant, were treated with DMSO or 30 μM C6 for 5 h, WDR5 was recovered by FLAG IP, and the co-precipitating proteins detected by IB. Inputs are 5% for RBBP5 and WDR5, 3% for KMT2A and KMT2B, and 1% for KIF2A and c-MYC; n = 3 biological replicates.
(D) Lysates from cells stably expressing WT FLAG-tagged WDR5 were treated with DMSO (0.1%), 5 μM C6, 50 μM C12, or both 5 μM C6 and 50 μM C12, for 5 h; WDR5 was recovered by anti-FLAG IP and IB performed for the indicated proteins. Inputs are 5% for RBBP5 and WDR5, 3% for KMT2A and KMT2B, and 0.5% for KIF2A and c-MYC; n = 4 biological replicates.
See also Figure S1.
First, we tested the impact of C6 on the mobility of WDR5 in sucrose gradient sedimentation assays. We compared WDR5 with KMT2A and KMT2B, two proteins that bind the WIN site and one of which (KMT2A) is enzymatically inhibited by WIN site blockade (Aho et al., 2019a; Patel et al., 2008b). We used a high concentration of C6 in these experiments (30 μM) to ensure maximal inhibition, but treated for only 5 h to minimize secondary effects. In vehicle-treated HEK293 cells, WDR5 migrates beyond the 670-kDa marker (Figure 1B), consistent with assembly into multiprotein complexes. In C6-treated cells, however, WDR5 is absent from fractions above 670 kDa (Figures 1B and S1B), with most material migrating below the 158-kDa marker. The impact of C6 on complex formation is an authentic consequence of WIN site blockade, because mutation of phenylalanine 133 within the WIN site (F133A) has a comparable impact on the distribution of WDR5 (Figures S1C and S1D). Despite these effects, however, the migration of KMT2A/B is only subtly altered by C6, demonstrating that overall integrity of KMT2 complexes is not perturbed by WIN site inhibition, and that these complexes play little role in governing the bulk of high-molecular-weight WDR5 species. Supporting this point, an inactivating mutation in the WBM site (L240K), which disrupts interaction with both RBBP5 and KMT2A/B (Figure 1C), has little effect on WDR5 mobility in these gradients (Figures S1C and S1D).
To explore the impact of WIN site inhibitor on known WDR5 interaction partners, we used co-immunoprecipitation (coIP) assays to compare the effects of C6 with mutations in the WBM and WIN sites, both in cells treated with inhibitor (Figure 1C) and by treatment of lysates in vitro (Figure 1D). For the latter, we also tested the WBM site inhibitor C12 (Chacón Simon et al., 2020). These analyses revealed that not all WDR5 interaction partners comport with expectations. MYC and KIF2A, on one hand, behave as expected. Interaction of MYC with WDR5 is sensitive to genetic (L240K) and chemical (C12) disruption of the WBM site but insensitive to perturbations (F133A/C6) at the WIN site. And the opposite is true for the WIN site binder KIF2A. KMT2 complex members, on the other hand, do not behave as expected. Interaction of WDR5 with KMT2A, KMT2B, and RBBP5 is insensitive to both C6 and C12 (alone or in combination), and although RBBP5 is displaced by the WBM mutation, so too are KMT2A and KMT2B, both of which bind WDR5 through the WIN site. Based on the structure of the C6-WDR5 complex (Aho et al., 2019a), it is unlikely that its interaction with WDR5 could displace some WIN motifs but not others. Rather, it is possible that multivalent interactions among KMT2 complex members retain association of KMT2A/B with WDR5 even when the WIN site is blocked. These interactions could also explain why mutation of the WBM site disrupts interaction of WDR5 with the WIN site binding KMT2A/B proteins. Regardless of mechanism, these data show that C6 disrupts a majority of WDR5-containing protein complexes, and that these are distinct from complexes involving KMT2A/B and RBBP5. By extension, they also suggest that much of the impact of WIN site inhibition on the WDR5 interactome affects interaction partners that have yet to be characterized.
Impact of WIN site inhibitor on the WDR5 interactome
To learn how WIN site inhibition alters the ensemble of proteins with which WDR5 interacts, we used SILAC (stable isotope labeling of amino acids in cell culture) to compare WDR5 complexes treated with 5 μM C6 or its inactive analog C6nc (Aho et al., 2019a). We treated lysates from “heavy” and “light” HEK293 cells expressing FLAG-tagged WDR5, recovered proteins by FLAG immunoprecipitation (IP), and analyzed samples by MudPIT liquid chromatography-tandem mass spectrometry (LC-MS/MS) (Figure 2A; Figure S2A). The experiment was performed in duplicate, with label swap, and a total of 747 proteins were quantified in the IP samples (Figures S2B and S2C; Table S1).
Figure 2. Identification of WDR5 interaction partners that are sensitive to WIN site inhibitor.
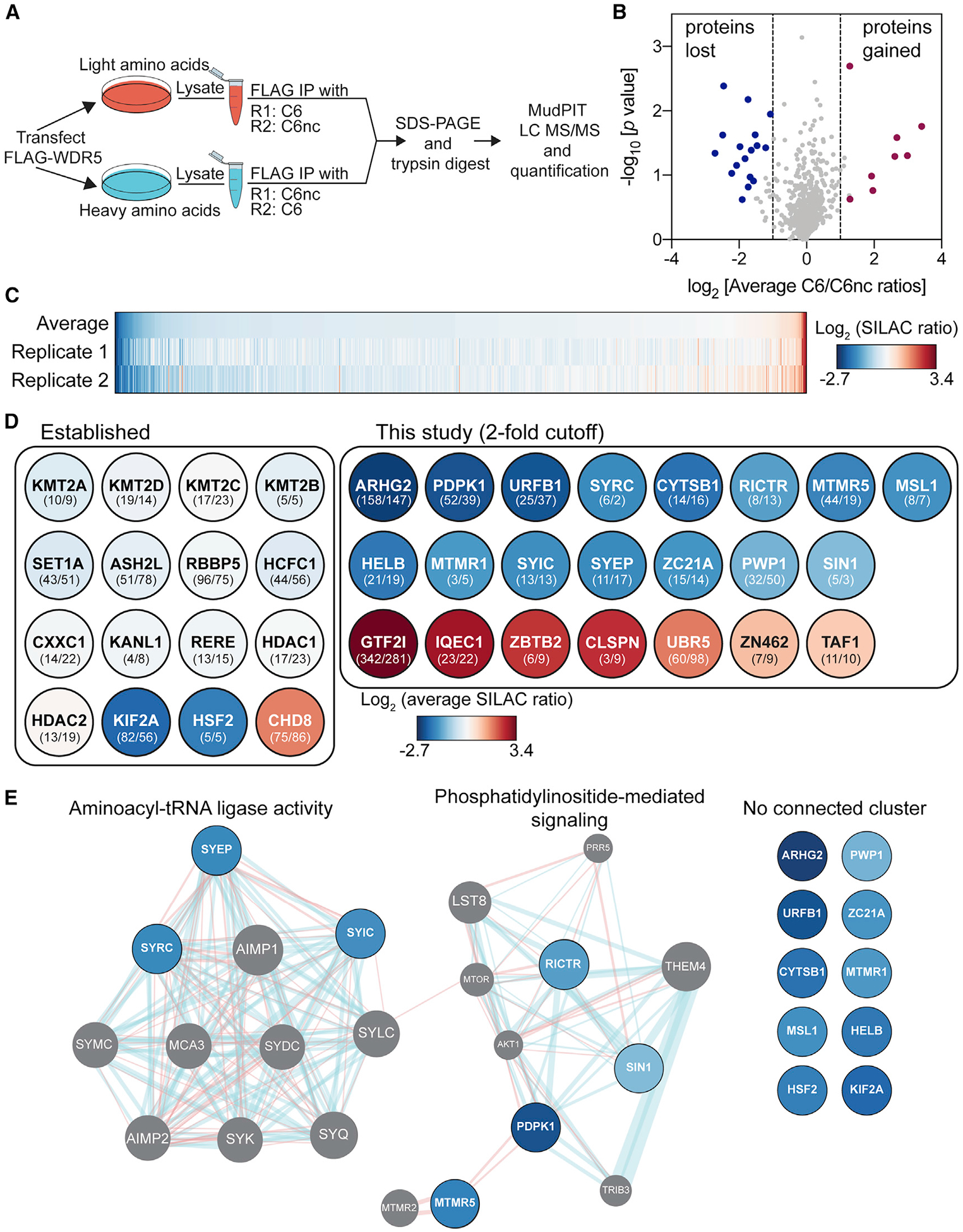
(A) Schematic of SILAC setup. The experiment was performed in duplicate (R1 and R2) with label swap.
(B) Volcano plot of the SILAC data plotting log2 (average ratio) against the p value from one-sample t test. Proteins meeting a 2-fold cutoff in both replicates are highlighted.
(C) Heatmap of the log2-transformed SILAC ratios for the 747 proteins quantified in both SILAC replicates and ranked by average ratio.
(D) Impact of C6 on the interaction of established (left) or novel (right) proteins with WDR5. The color of each circle corresponds to the average log2 (SILAC ratio) from the heatmap in (C). Numbers in parentheses are spectral counts from the two replicates (R1/R2). UniProt names are used throughout.
(E) GeneMANIA (Warde-Farley et al., 2010) was used to predict functional nodes among depleted proteins, identifying “aminoacyl tRNA ligase activity” (false discovery rate [FDR] = 2.04e–18) and “phosphatidylinositol-mediated signaling” (FDR = 4.00e–4). Blue lines represent pathway interactions; red lines indicate physical interactions. Gray circles represent proteins identified by GeneMANIA as connected functionally or physically to the 17 input proteins (blue). Proteins on the right failed to cluster.
Enforcing a 2-fold cutoff of SILAC ratios (C6nc/C6), 25 proteins are altered in their ability to interact with WDR5 by C6, 17 of which are reduced and 8 of which are increased (Figures 2B and 2C). As predicted from experiments in Figure 1, most canonical WDR5 interaction partners are recalcitrant to WIN site inhibition, including members of KMT2 and NSL complexes (Figure 2D; Figure S2D; Table S2). Also, as predicted, most of the WDR5-associated proteins affected by C6 have not been studied in detail. Some of these proteins were identified in previous large-scale screens (Hauri et al., 2016; Huttlin et al., 2017), but others (URFB1, MTMR5, MTMR1, ZC21A, PWP1) are exclusive to this dataset. Within the 17 decreased proteins, seven have relationships that cluster in two nodes: “aminoacyl tRNA ligase activity” and “phosphatidylinositol mediated signaling” (Figure 2E). The tRNA ligase node is represented by SYIC, SYEP, and SYRC, which are components of the multi-tRNA synthetase complex (Rajendran et al., 2018). The signaling node is represented by RICTR and SIN1, subunits of mTORC2 (mTOR complex 2) (Saxton and Sabatini, 2017), and by PDPK1, a kinase that, together with mTORC2, phosphorylates AKT (Gagliardi et al., 2018; Mora et al., 2004). These same themes are reinforced by results of Reactome pathway and ontology analyses (Figures S2E and S2F). The eight enriched proteins, in contrast, have few connections, and represent processes such as DNA replication (CLSPN), transcription (GTF2I, TAF1), ubiquitylation (UBR5), and chromatin remodeling (CHD8).
From these data, we conclude that WIN site inhibition bidirectionally alters the WDR5 interactome, resulting in decreased interactions with some proteins and increased interactions with others. We conclude that a majority of the impact of WIN site inhibitor is on proteins that have not previously been connected to WDR5 in a substantive way. And we conclude that some proteins displaced from WDR5 by WIN site inhibitor have links to tRNA synthetases or phosphatidylinositol 3-kinase (PI3K)/AKT signaling.
Validation
Because many of the proteins that are lost (Figure 3A) or gained (Figure 3B) from WDR5 in response to C6 have little to do with the known functions of WDR5, we validated some of the more interesting candidates. We found that IP of endogenous WDR5 from HEK293 cells recovers endogenous PDPK1, HELB, and MTMR1 (Figure 3C), as well as RICTR, SIN1, GTF2I, and UBR5 (Figure 3D). We confirmed that interaction of FLAG-tagged WDR5 with PDPK1, RICTR, SIN1, HELB, SYRC, SYIC, and MTMR1 is sensitive to C6 (Figure 3E) and to mutation of the WIN site of WDR5 (Figures S3A and S3B). And for these proteins, with the exception of MTMR1, we saw that interaction with WDR5 is insensitive to ethidium bromide (Figure 3E), indicating that they are not bridged by contaminating DNA in the IP samples (Lai and Herr, 1992). By extension, we infer that the MTMR1-WDR5 interaction may be caused by DNA contamination, and suggest investigators perform such experiments before pursuing MTMR1 or unvalidated proteins in our list of 25.
Figure 3. Validation of C6-sensitive WDR5 interaction partners.
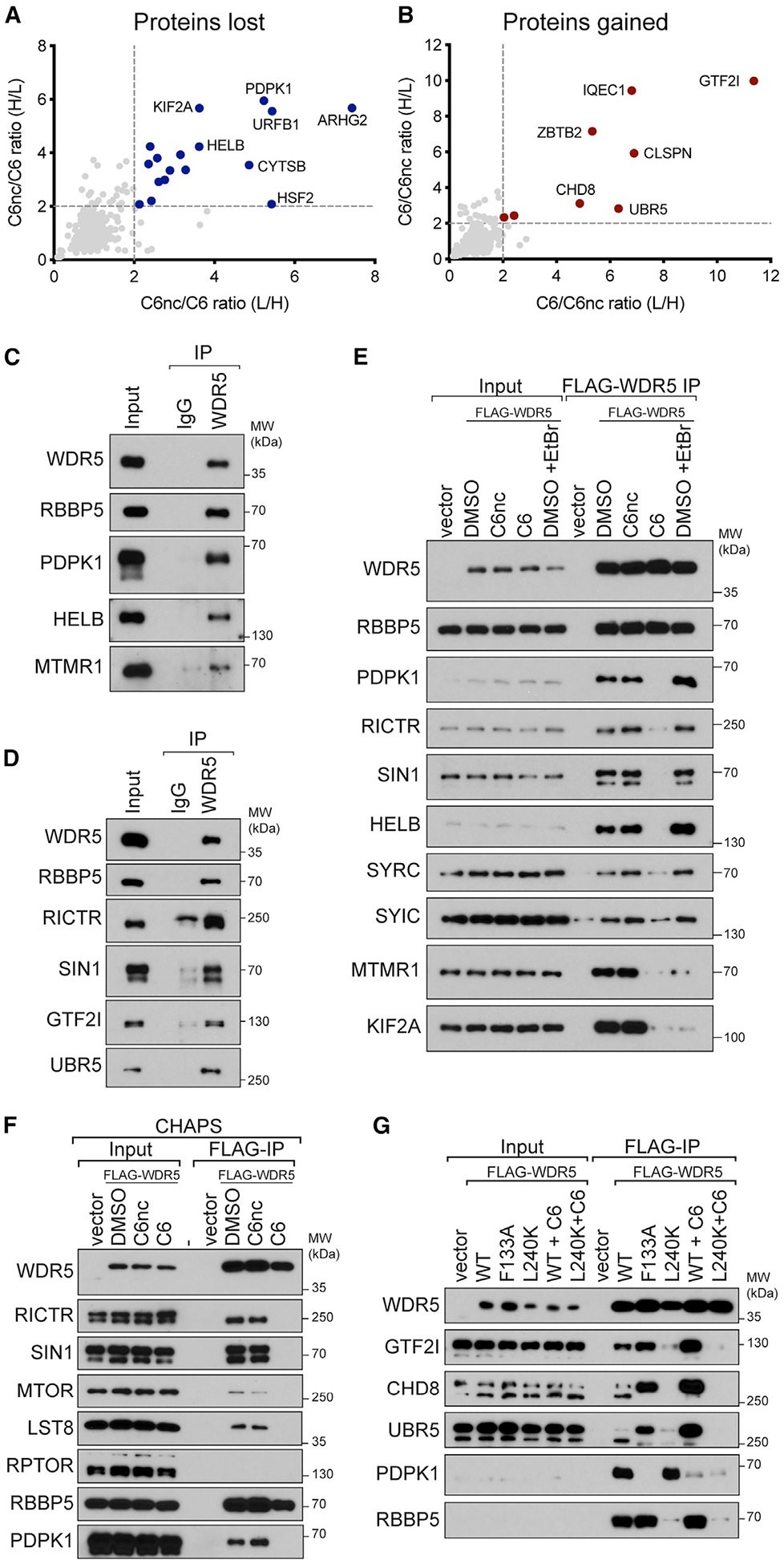
(A) Comparison of C6nc/C6 ratios for the two SILAC replicates. Depleted proteins that met a 2-fold cutoff in both replicates are highlighted in blue.
(B) As in (A) except for enriched proteins (red).
(C) Extracts from HEK293 cells were subject to IP with a polyclonal antibody against WDR5 or an immunoglobulin G (IgG) control. IP samples were probed with antibodies against the indicated endogenous proteins. Inputs are 2% for WDR5 and RBBP5 and 0.3% for others. n = 3 biological replicates.
(D) As in (C) but for different candidate proteins. Inputs are 5% for WDR5, RBBP5, and UBR5 and 0.3% for others. n = 3 biological replicates.
(E) HEK293 cells stably expressing FLAG-tagged WDR5 were treated for 4 h with 30 μM C6 or C6nc prior to lysis and subsequent FLAG IP. For ethidium bromide (EtBr) treatment, 200 μg/mL EtBr was added to the lysate for the duration of the experiment. Candidate WDR5 interaction partners were probed by IB. Inputs are 5% for WDR5 and RBBP5, 0.1% for SYRC and SYIC, and 1% for all others; n = 3 biological replicates.
(F) HEK293 cells stably expressing FLAG-tagged WDR5 were treated for 4 h with 30 mM C6 or C6nc prior to lysis and subsequent FLAG IP in buffer using CHAPS detergent. IP samples were probed with antibodies against the indicated proteins. Inputs are 10% for WDR5 and RBBP5 and 1% for others; n = 3 biological replicates.
(G) HEK293 cells stably expressing FLAG-tagged WDR5 proteins were treated for 4 h with 30 μM C6 (where indicated) prior to lysis and FLAG IP. IP samples were probed with antibodies against the indicated proteins. Inputs are 10% for WDR5 and 1% for others; n = 3 biological replicates.
See also Figure S3.
Because we recovered only two subunits of mTORC2 in our SILAC experiment, we asked whether WDR5 interacts with these subunits alone, or if it is capable of interacting with the remaining mTORC2 components LST8 and MTOR (Saxton and Sabatini, 2017). We suspected that it was our use of Triton X-100, rather than CHAPS, as a detergent that prevented recovery of LST8 and the MTOR kinase (Sarbassov et al., 2004), and repeated our FLAG-WDR5 coIPs in the presence of CHAPS. Now, WDR5 associates with all four mTORC2 components in a manner that is sensitive to C6 (Figure 3F) and the F133A mutation (Figure S3C). Importantly, WDR5 does not interact with the mTORC1-specific component RPTOR (Figure 3F: Hara et al., 2002; Kim et al., 2002), revealing that WDR5 interacts selectively with mTORC2.
Finally, to understand how C6 promotes association of WDR5 with partner proteins, we looked more closely at three gained interactors: CHD8, GTF2I, and UBR5. We confirmed that all three bind at higher levels to WDR5 upon inhibition or mutation of the WIN site (Figure 3G). Interestingly, we also observed that all three interact with WDR5 in a manner that is sensitive to mutation of the WBM site, and that two, CHD8 and UBR5, contain WBM motifs (Figure S3D). Thus, it appears as though disruption of protein binding to the WIN site of WDR5 can promote selective loading of proteins at the WBM site. WIN site perturbation may induce an unexpected conformational change in WDR5 that facilitates interaction with CHD8 and UBR5, or perhaps a steric clash with complexes tethered to the WIN site normally limits interaction of WDR5 with these proteins. Regardless, these findings establish that WIN site inhibitors influence more than just local protein-protein interactions at the WIN site, and raise the possibility that actions of WIN site inhibitors on cells are mediated, in part, by promoting association of WDR5 with partner proteins.
PDPK1 binds WDR5 in the nucleus and via an N-terminal WIN motif
We selected PDPK1 for further study because its role in growth factor signaling in the cytosol (Bayascas, 2010) is disparate from the known functions of WDR5 in the nucleus (Guarnaccia and Tansey, 2018). We confirmed by coIP that endogenous PDPK1 and WDR5 interact in multiple cell lines (Figure 4A; Figure S4A), and that the interaction in U2OS cells is sensitive to C6, as determined by proximity ligation assay (Figure 4B; Figure S4B). We demonstrated that the interaction is unaffected by treatment of cells with the PDPK1 kinase inhibitor GSK2334470 (Najafov et al., 2011: Figure 4C) and, conversely, that its disruption by C6 has no obvious effect on growth factor signaling by PDPK1, including AKT and S6 kinase phosphorylation (Figure S4C). Interestingly, although most PDPK1 is in the cytosolic fraction (S2) and most WDR5 is in the nuclear fraction (P3; Figure 4D), the interaction is detected only in IPs from nuclear lysates (Figure 4E). Based on these observations, we conclude that PDPK1 is a bona fide WDR5 interaction partner, that the interaction occurs in the nucleus, and that it likely has little to do with the role of PDPK1 in growth factor signaling.
Figure 4. PDPK1 interacts with WDR5 in nuclear lysates.
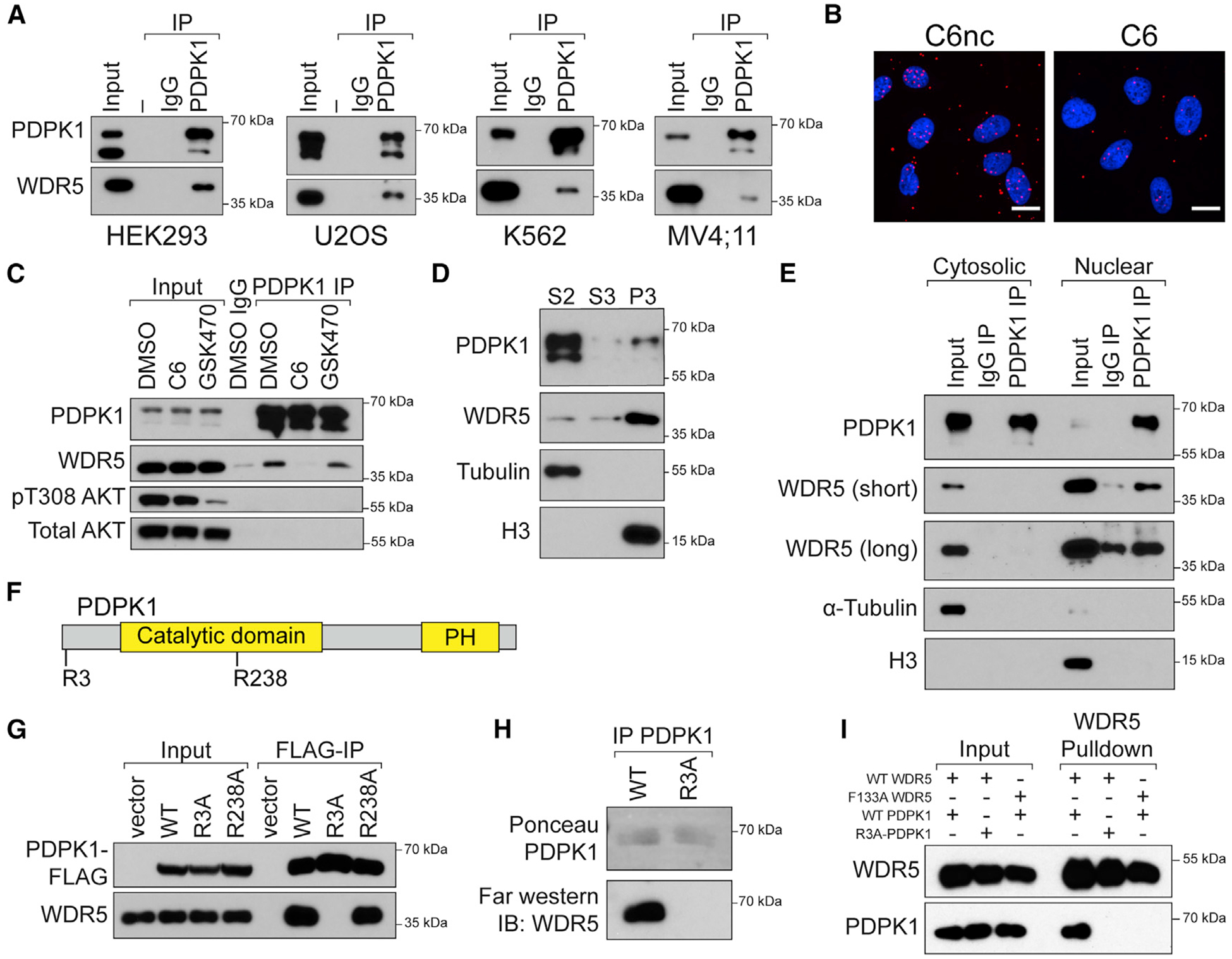
(A) Endogenous PDPK1 was recovered from lysates of the indicated cell lines and probed for co-precipitating WDR5 by IB. Inputs for PDPK1 are 10%–20%. Inputs for WDR5 are 1%–5%. n = 3 biological replicates.
(B) Proximity ligation assay with FLAG and WDR5 antibodies in U2OS cells stably expressing FLAG-tagged PDPK1. Cells were treated overnight (30 μM C6/C6nc) before analysis; scale bar, 20 μm. n = 3 biological replicates.
(C) HEK293 cells were treated overnight with 30 μM C6 or 5 μM GSK470, lysates prepared, and a PDPK1 IP performed. IB was then performed for the indicated proteins. Inputs are 5% for PDPK1 and 1% for all others. n = 3 biological replicates.
(D) HEK293 cells were fractionated into cytosolic (S2), soluble nuclear (S3), and chromatin-associated (P3) fractions. Equal amounts of each fraction were analyzed by IB with the antibodies against the indicated proteins. H3 (nuclear) and α-tubulin (cytosolic) are controls for fractionation. n = 3 biological replicates.
(E) Cytosolic and nuclear lysates from HEK293 cells were subject to IP with PDPK1 antibody or an IgG control and immunoblotted with antibodies against the indicated proteins. A short and long exposure of the WDR5 IB are shown. n = 3 biological replicates.
(F) PDPK1 possesses two WIN-like motifs centered on R3 and R238.
(G) FLAG-tagged PDPK1 (WT and the R3A and R238A mutants) were transiently expressed in HEK293 cells; lysates were prepared and subject to IP with anti-FLAG beads. Immune complexes were probed for PDPK1 or endogenous WDR5 by IB. n = 3 biological replicates.
(H) FLAG-tagged PDPK1 (WT and the R3A) was transiently expressed in HEK293 cells, recovered by FLAG-IP, resolved by SDS-PAGE, and transferred to polyvinylidene fluoride (PVDF) membrane. Membranes were then incubated with recombinant WDR5 followed by anti-WDR5 antibody. n = 3 biological replicates.
(I) In vitro-transcribed and -translated PDPK1-FLAG variants were incubated with recombinant 6xHis-SUMO-WDR5 proteins, recovered with Ni-NTA agarose, and analyzed by IB. n = 2 biological replicates.
PH, pleckstrin homology domain. See also Figure S4.
PDPK1 does not carry a motif matching the full WIN consensus. It does, however, have sequences that match the motif core, centered on arginine 3 (R3: ART) and 238 (R238: ARA) (Figure 4F). Mutation of R238 to alanine has no effect on the PDPK1-WDR5 interaction, as measured by coIP (Figure 4G; R238A). Mutation of R3 to alanine, in contrast, reduces interaction with WDR5, suggesting that this amino-terminal WIN-like sequence mediates interaction with WDR5. Consistent with this notion, the R3A mutation disrupts the ability of PDPK1 to interact with WDR5 in vitro, as measured by far-western (Figure 4H) and by interaction of recombinant WDR5 with in vitro-translated PDPK1 (Figure 4I). Together, these data suggest that PDPK1 interacts with WDR5 via its amino-terminal WIN-like motif.
Finally, because PDPK1 and WDR5 interact in the nucleus, we asked whether the nuclear shuttling of PDPK1 (Lim et al., 2003; Scheid et al., 2005) is dependent on interaction with WDR5. We inhibited CRM1-dependent nuclear export with leptomycin B (LMB) and quantified the distribution of wild-type (WT) and R3A EGFP-tagged PDPK1 variants in U2OS cells (Figures S4D–S4F). We confirmed that PDPK1 accumulates in the nucleus upon LMB treatment but observed no difference in nuclear accumulation of the R3A mutant compared with WT PDPK1. Thus, nuclear shuttling of PDPK1 occurs independent from interaction with WDR5.
Acetylation of the amino terminus of PDPK1 creates a high-affinity WIN motif
The only confirmed WIN site binding protein with an N-terminal WIN motif is histone H3 (Couture et al., 2006; Dou et al., 2006; Ruthenburg et al., 2006), which is bereft of an initiator methionine. We asked if the PDPK1 WIN-like motif resembles H3. We confirmed that a PDPK1 N-terminal peptide lacking the initiator methionine binds to recombinant WDR5 in vitro (Figure S5A). We also purified endogenous PDPK1 from HEK293 cells (Figure S5B), analyzed AspN digestion products by MS/MS (Figure S5C), and saw that all spectra assigned to the N terminus of PDPK1 carry two modifications; they lack the initiator methionine and carry an amino-terminal acetyl moiety (Figure 5A).
Figure 5. PDPK1 is a high-affinity WIN site binding protein.
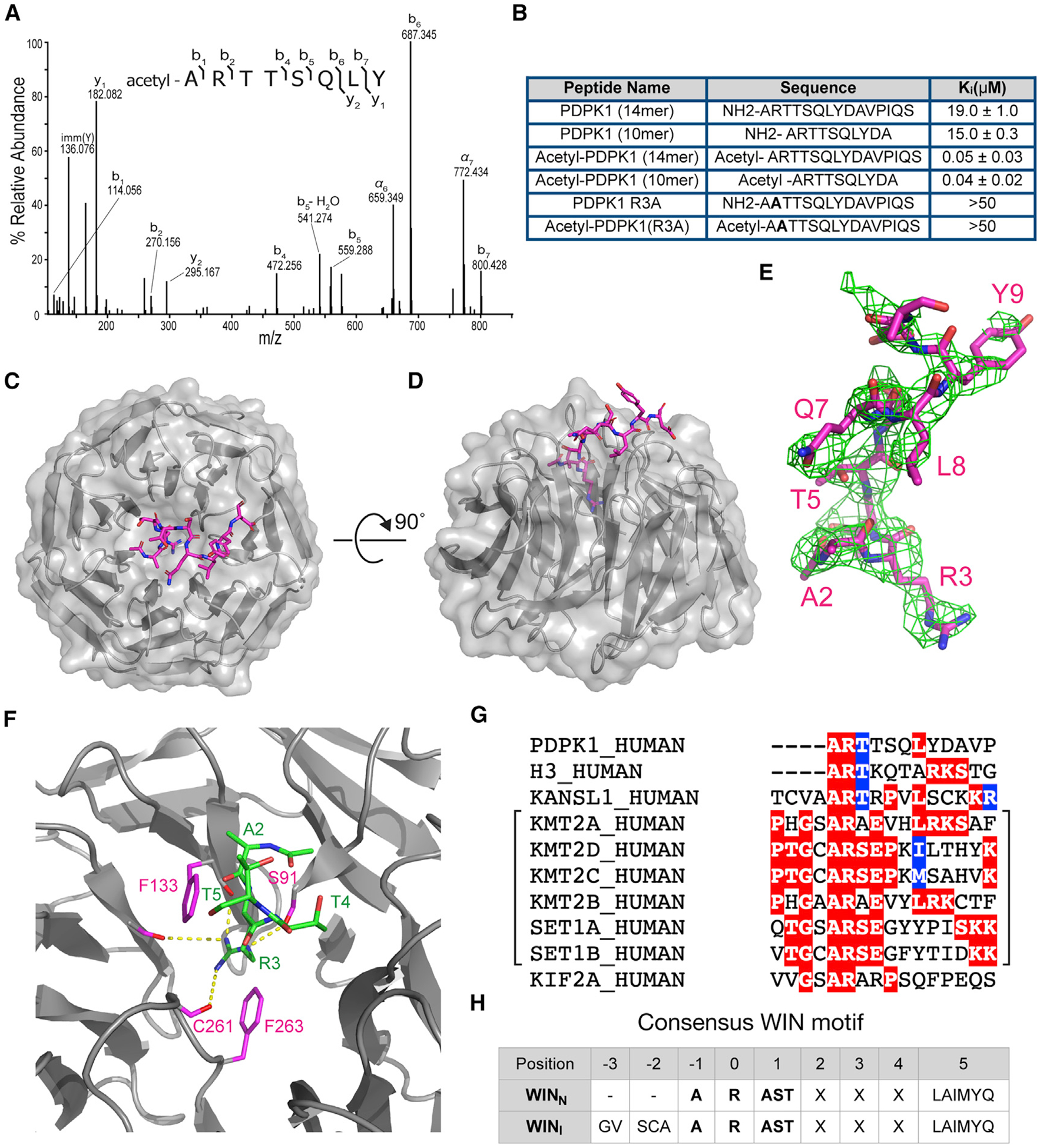
(A) Tandem mass spectrum of N-terminally acetylated PDPK1 peptide, residues 2–9. The doubly protonated precursor, [M+2H]+2, with m/z 491.2556 was fragmented with higher-energy collisional dissociation. The identified amino acid sequence is provided above the annotated spectrum; brackets indicate sites of dissociation at the peptide backbone. Observed product ions are assigned to their corresponding m/z peaks in the mass spectrum.
(B) Binding constants of PDPK1 peptides were determined using a TR-FRET-based KMT2A peptide competition assay. All peptides are amidated at the C terminus. Two or more repeats were obtained; average Ki values and standard deviations are reported.
(C) Structure of WDR5 in complex with the acetylated-PDPK1 WIN peptide. The PDPK1 WIN peptide is shown in stick representation (magenta, colored by atom type); WDR5 is shown as cartoon with semitransparent surface representation (gray). 2.7Å resolution.
(D) As in (C) but rotated along a 90°axis.
(E) The Fo-Fc omit map of PDPK1 peptide bound with WDR5 domain contoured at 2.0 σ level. PDPK1 peptide is shown in magenta sticks.
(F) Close-up of the first three residues of PDPK1 (ART) in the WIN site of WDR5. The PDPK1 peptide is green sticks; WDR5 is gray ribbons. Key WDR5 residues F133, S175, C261, and F263 are indicated in pink stick representation. Yellow dotted lines indicate intermolecular hydrogen bonds.
(G) WIN motif of PDPK1 aligned with established WIN motifs. The related histone methyltransferase enzymes are grouped with brackets.
(H) Table summarizing consensus sequences for internal (WINI) and N-terminal (WINN) WIN motifs.
Ectopic N-terminal acetylation has been shown to increase the affinity of H3 and KMT2A WIN motif peptides for WDR5 in vitro (Avdic et al., 2011b; Karatas et al., 2010), and we observe this phenomenon with H3 peptides in our TR-FRET (time-resolved fluorescence resonance energy transfer) assays (Figure S5D). But N-terminal acetylation is not a major modification of H3 (Huang et al., 2014; Yeom et al., 2017), and the KMT2A WIN motif is not N-terminal. To determine whether a naturally occurring WIN motif is impacted in this way, we measured how N-terminal acetylation influences the affinity of PDPK1 WIN peptides for WDR5 (Figure 5B). Compared with unmodified peptides, which bind WDR5 weakly (Ki = 15–19 μM), acetylated PDPK1 peptides bind tightly (Ki = 0.04–0.05 μM) and in a manner that is sensitive to the R3A mutation. Thus, acetylation of the N-terminal PDPK1 WIN motif increases its affinity for WDR5 by a factor of ~400.
Next, we determined the X-ray crystal structure of WDR5 in complex with an acetylated PDPK1 peptide (Figures 5C–5F; Table S3). In general, the WDR5-PDPK1 WIN peptide interaction resembles other WDR5-WIN peptide structures (Figure S5E), with residues A2 and R3 of PDPK1 making contacts with WDR5 deep in the WIN site (Figure S5F), and the guanidinium side chain of R3 sandwiched by two aromatic rings from F133 and F263 of WDR5 (Figure 5F). Like other WDR5-WIN peptide structures (Zhang et al., 2012), residues downstream of R3 lie along a crevice between blades three and four of the WDR5 b-propellor structure (Figure 5C), and like other structures, the exact conformation of these residues is unique (Figure S5E). Distinct from other structures, the side chain of L8 of PDPK1 makes contact with side chains of F149 and Y191 of WDR5 (Figure S5F). Although residues corresponding to L8 of PDPK1 are often hydrophobic in WIN motif proteins (Figure 5G) and make backbone contacts with WDR5, this is the only structure, so far, to show to side chain-side chain interactions at this position.
Looking at the PDPK1 N terminus, we see that the N-terminal acetyl group of PDPK1 fits into a WIN site adjacent pocket on WDR5, which is not large enough to accommodate a methionine at this position. Indeed, residues that occupy this position for other WIN site-binding peptides are small: alanine, serine, or cysteine (Figure 5G). Importantly, the acetyl group of the PDPK1 peptide forms an intramolecular interaction whereby the carbonyl of the acetyl group makes a hydrogen bond to the amide of T4 (Figure S5G). This intramolecular hydrogen bond stabilizes the conformation of the peptide and points the N-terminal amide of PDPK1 A2 toward the carboxylate of WDR5 D107 to form a salt bridge. These characteristics explain how removal of the initiator methionine and acetylation of the α-amino group of A2 enable the PDPK1 WIN motif to achieve its unusually high affinity.
Modification of the N terminus of PDPK1 likely occurs co-translationally and is predicted to be catalyzed by the action of methionine aminopeptidases and the ribosome-anchored NatA N-terminal acetyltransferase complex (Ree et al., 2018). Unlike other post-translational modifications, N-terminal acetylation is irreversible, meaning that, for any one molecule of PDPK1, whether or not it can interact tightly with WDR5 is fixed from its moment of synthesis. The proportion of modified PDPK1 molecules in a population, however, could be regulated; NatA complexes are subject to regulation and are often overexpressed in cancer (Koufaris and Kirmizis, 2020). Moreover, multiple PDPK1 splice variants have been described, several of which lack the amino-terminal WIN motif. These isoforms respond differently to upstream growth factor signals (Dong et al., 2002), suggesting that the interaction of PDPK1 with WDR5 could be controlled via alterations in PDPK1 isoform production. Further study is needed into whether and how the WDR5-PDPK1 interaction is regulated and potentially dysregulated in cancer.
Based on our findings, we suggest that definition of the WIN motif be revised to discriminate between those located internally (WINI) versus those located at the N terminus of a protein (WINN) (Figure 5H). Interestingly, the human proteome contains 67 proteins with potential WINN motifs (Figure S5H; Table S4), including PDPK1, 19 histone H3 variants, and HELB, one of the proteins that is displaced from WDR5 with the WIN site inhibitor. Whether any of these WINN motifs are capable of tight binding, like PDPK1, depends on removal of the initiator methionine and subsequent acetylation. Large-scale proteomic mapping of N-terminal acetylation (Lange et al., 2014; Yeom et al., 2017) has not detected this modification in any of the proteins in Figure S5H, with exception of a small percentage of H3, perhaps because trypsin (which cleaves after arginine residues) generates WINN fragments that are too small for robust detection. But as one-third of all human proteins are subject to this modification (Ree et al., 2018), chances are high that other high-affinity WIN site binding proteins await discovery.
The PDPK1-WDR5 interaction regulates G2/M-expressed genes
Interaction of PDPK1 and WDR5 in the nucleus, together with the transcriptional roles of WDR5, prompted us to ask whether PDPK1 and WDR5 influence expression of a common set of genes. We created U2OS cells in which endogenous PDPK1 or WDR5 are tagged with an FKBP12(F36V)-2xHA cassette (Figures S6A and S6B), permitting their degradation via the small molecule dTAG47 (Huang et al., 2017; Nabet et al., 2018: Figures S6C and S6D). For both proteins, depletion is stable over 6 days, and cell growth over this time is slowed (Figures S6E and S6F). Importantly, neither tag disrupts the PDPK1-WDR5 interaction by coIP (Figures S6G and S6H). We performed RNA sequencing (RNA-seq) after 24 h of dTAG47 treatment and found that PDPK1 depletion results in changes in the expression of ~1,100 genes (Figures 6A and 6B; Figure S6I). WDR5 depletion has more extensive effects, leading to changes in expression of ~7,400 genes (Figures 6C and 6D; Figure S6J). The overlap of significant gene expression changes between the two is ~660 (Figure S6K). For PDPK1 and WDR5 depletion, Gene Ontology (GO) enrichment analysis identified transcript changes consistent with the known functions of both proteins (Figures 6E and 6F; Figures S6L and S6M). Interestingly, however, we observed that several of the cell-cycle-related GO terms enriched in transcripts decreased by PDPK1 depletion are enriched in transcripts increased by WDR5 depletion (Figures 6E and 6F, red text). Gene set enrichment analysis (GSEA) (Subramanian et al., 2005) further strengthened these connections (Figures 6G–6I). To determine if these reciprocal enrichments are due to changes in a common set of genes, we overlaid gene lists and found that more than 40% (246) of genes whose expression is decreased by PDPK1 depletion are induced by depletion of WDR5 (Figure 6J). In general, changes in expression of these 246 genes are small (Figures 6K; Figure S6N) but significant (Figures S6O and S6P). Importantly, GO analysis on these common genes reinforced cell-cycle connections (Figure 6L), demonstrating that PDPK1 and WDR5 reciprocally regulate expression of a set of genes linked to the cell cycle.
Figure 6. PDPK1 and WDR5 oppositely influence the expression of cell-cycle genes.
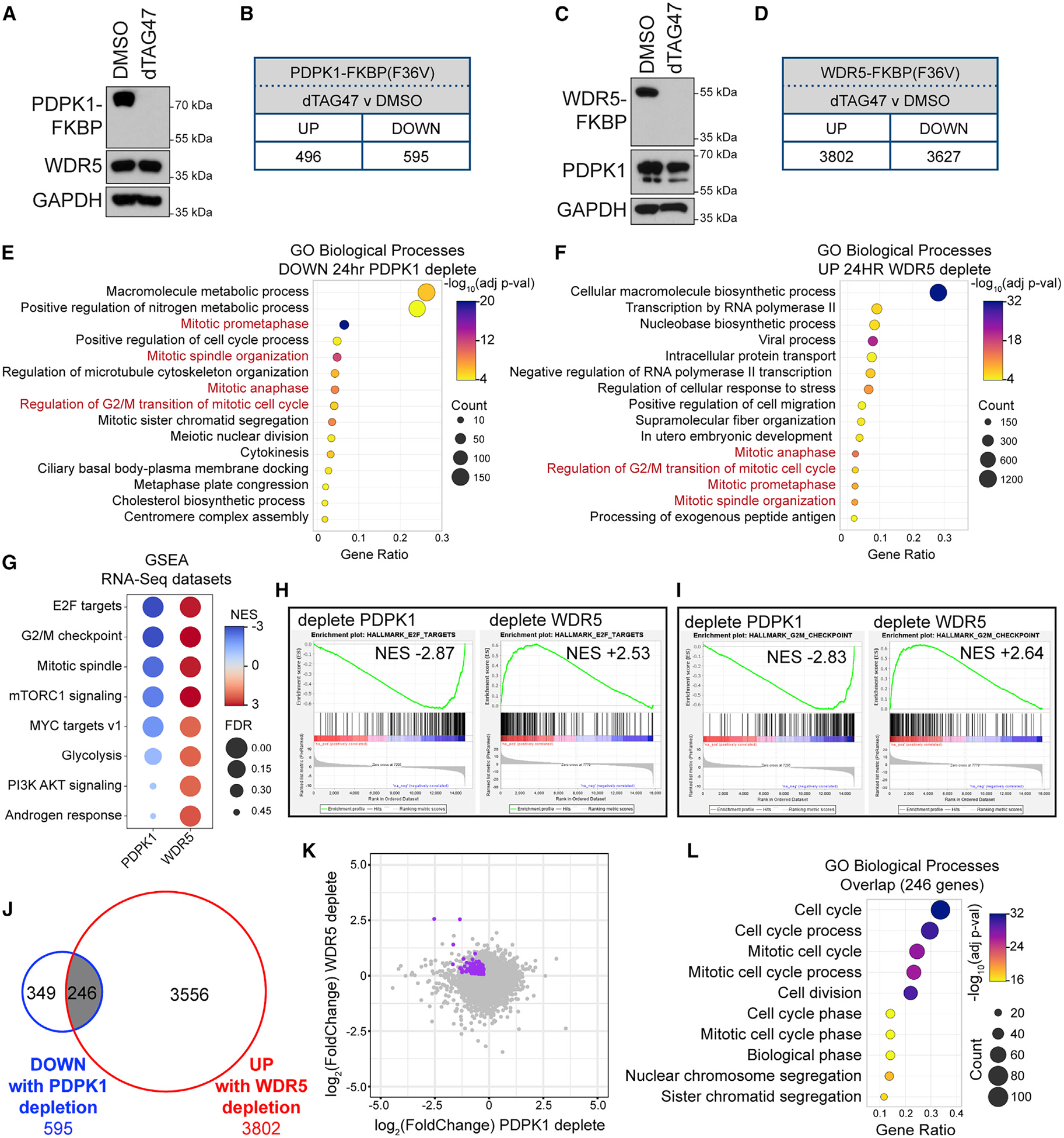
(A) U2OS cells expressing PDPK1-FKBP(F36V)-2xHA were treated for 24 h with 500 nM dTAG47 or DMSO, lysates prepared, and PDPK1, WDR5, and GAPDH levels determined by IB.
(B) Number of transcripts significantly (FDR < 0.05) altered by 24-h treatment of cells in (A) with 500 nM dTAG47, compared with DMSO control. n = 3 biological replicates.
(C) As in (A) but for the cells expressing WDR5-FKBP(F36V)-2xHA.
(D) Number of transcripts significantly (FDR < 0.05) altered by 24-h treatment of cells in (C) with 500 nM dTAG47, compared with DMSO control. n = 4 biological replicates.
(E) GO analysis of decreased transcripts identified by RNA-seq of U2OS cells depleted of PDPK1 for 24 h. Biological Process GO terms were ranked by adjusted p value, and the 15 most significant enriched terms are presented; the color indicates the Bonferroni-corrected Fisher exact p value; the size indicates the number of genes in that category; the x axis is the ratio of genes in the category over total analyzed genes.
(F) GO term analysis of increased transcripts identified by RNA-seq of U2OS cells depleted of WDR5 for 24 h. Ranking and presentation are as in (E).
(G) Enriched Hallmark gene sets (Liberzon et al., 2015), determined by GSEA of RNA-seq from 24-h PDPK1 or WDR5 depletion. Eight of the top Hallmarks are shown. Color indicates the normalized enrichment score (NES); size indicates the FDR value.
(H and I) Examples of GSEA enrichment plots summarized in (G). FDR = 0.000 for all plots shown.
(J) Overlap of transcripts that are decreased with PDPK1 depletion and increased with WDR5 depletion.
(K) Scatterplot of RNA-seq data from PDPK1 and WDR5 depletions. The 246 genes from (J) are highlighted in purple.
(L) GO term analysis of the 246 genes represented in (J). Biological Process GO terms were sorted hierarchically, and the most specific subclasses were ranked by adjusted p value. The 10 most significantly enriched subclasses are presented. Presented as in (E).
See also Figure S6.
To ask if any of these gene expression changes are due to disruption of the WDR5-PDPK1 interaction, we introduced the R3A mutation into endogenous PDPK1 loci in HEK293 cells (Figures S7A and S7B). Unfortunately, mutation of the N terminus of endogenous PDPK1 is accompanied by a decrease in PDPK1 expression (Figure S7A), preventing comparison of the R3A and WT parental cell lines. We therefore engineered R3A PDPK1 mutant cells with empty vector, or vectors overexpressing WT PDPK1 or the R3A PDPK1 mutant (Figure 7A), and performed RNA-seq. Compared with the WT PDPK1 reconstitution, we identified 429 significantly changed transcripts for vector cells and 136 significantly changed transcripts for R3A PDPK1 cells (Figures S7C and S7D), 110 of which overlap (Figure 7B). GO analysis on these 110 genes again shows enrichment for cell cycle and mitotic categories (Figure 7C), driven in large part by the genes reciprocally altered by WDR5 and PDPK1 depletion (see below).
Figure 7. Disrupting the PDPK1-WDR5 interaction induces transcription of cell-cycle genes.
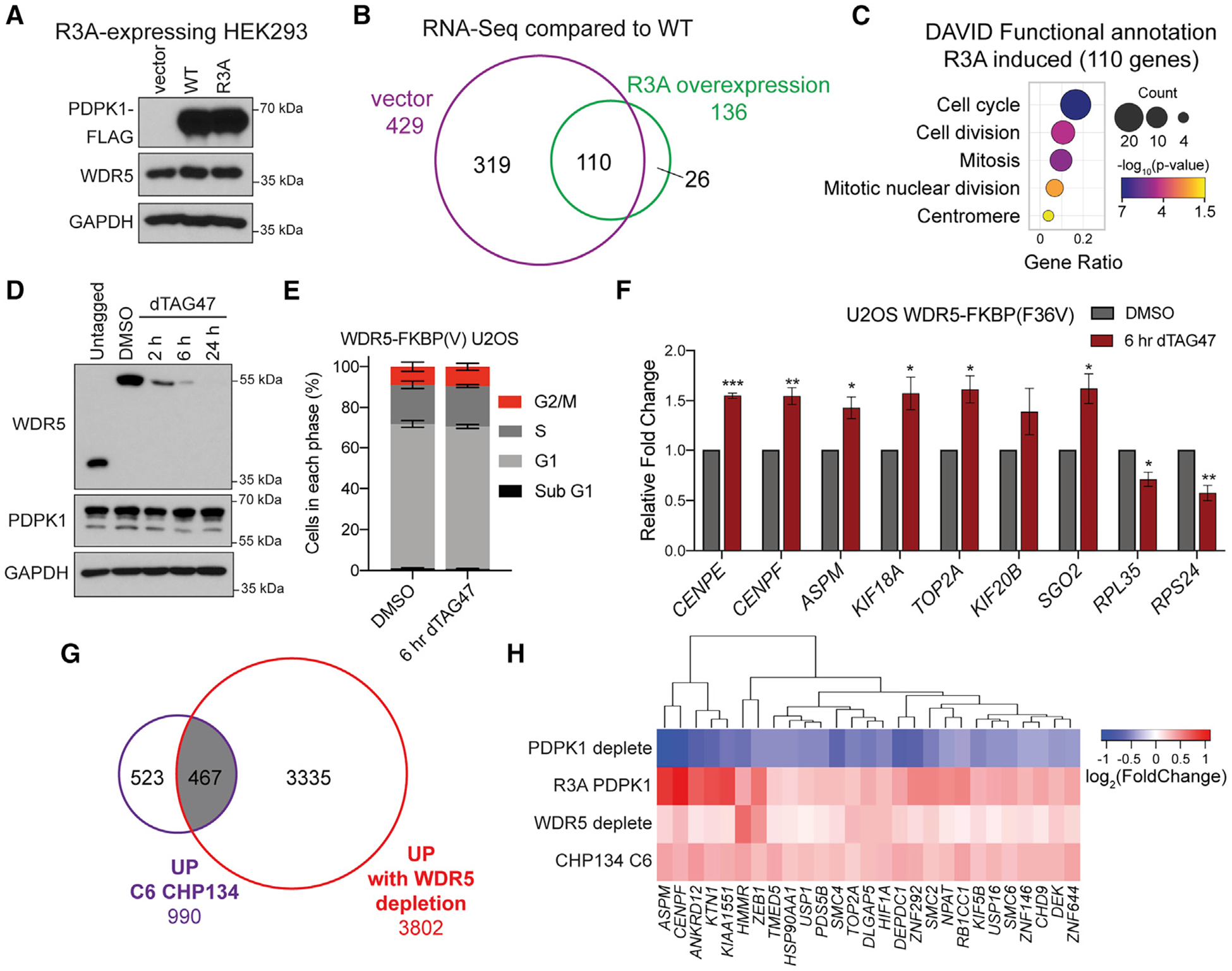
(A) IB of lysates from HEK293 cells expressing the PDPK1 R3A mutant and transduced with vector control, WT, and R3A PDPK1.
(B) Venn diagram of RNA-seq, comparing low-expressing (vector) and high-expressing R3A PDPK1, normalized to WT PDPK1-expressing cells. All 110 transcripts common to both samples are increased. n = 2 biological replicates for each condition.
(C) GO analysis on the 110 overlapping genes performed using DAVID Bioinformatic Resource (Huang et al., 2009a, 2009b); the color indicates the Fisher exact p value; the dot size indicates the number of genes in that category; the x axis represents the Gene Ratio, the ratio of genes in the category to total analyzed genes.
(D) IB of WDR5 depletion time course with 500 nM dTAG47 in U2OS cells expressing WDR5-FKBP(F36V)-2xHA and compared with untagged cells.
(E) Distribution of cell-cycle phases as determined by flow cytometry for WDR5-FKBP(F36V)-2xHA U2OS cells treated for 6 h with 500 nM dTAG47 or DMSO vehicle control. Data are represented as mean ± SEM; no significance between treatments by unpaired two-tailed t test. n = 3 biological replicates.
(F) Nuclear run-on analysis of nascent transcripts from cells treated with DMSO control or 500 nM dTAG47 for 6 h. Signal is normalized to nascent ACTB transcripts. Data are represented as mean ± SEM; n = 3 independent biological replicates. ***p < 0.001, **p < 0.01, *p < 0.05 by unpaired two-tailed t test.
(G) Venn diagram showing the overlap between gene expression changes that are increased with 24-h WDR5 depletion in U2OS cells and increased with 24-h C6 treatment in CHP134 cells (Bryan et al., 2020).
(H) Hierarchical clustering of log2 (fold change) in gene expression for genes significantly decreased for U2OS PDPK1-FKBP(F36V)-2xHA and increased for HEK293 R3A PDPK1, U2OS WDR5-FKBP(F36V)-2xHA, and CHP134 24-h 5 μM C6.
Degradation of WDR5 and PDPK1 leads to changes in cell-cycle distribution that could produce changes in cell-cycle gene expression (Figures S7E–S7G). To separate cause from effect, we arrested cells in G1 with palbociclib (Fry et al., 2004) or G2 with RO-3306 (Vassilev et al., 2006; Figure S7H) and examined the impact of WDR5 degradation on the expression of a set of G2/M-induced genes. Here, we observed that transcript levels from the G2/M genes are unaffected by WDR5 depletion in G1-arrested cells but are induced by WDR5 depletion in the G2 arrested state (Figure S7I). We also used nuclear run-on to ask if transcription of G2 genes is induced 6 h after dTAG47 addition, a time point at which most WDR5 is degraded (Figure 7D), but there is no impact on cell-cycle distribution (Figure 7E). Here, we see that transcription of seven representative G2 genes is induced by WDR5 degradation (Figure 7F), and that this induction is gene selective, because ribosome protein genes RPL35 and RPS24 show the expected decrease in transcription (Bryan et al., 2020). Together, these data argue that WDR5-dependent changes in G2/M gene expression drive changes in cell-cycle distribution and not vice versa.
Finally, we asked whether dysregulation of genes controlled by the WDR5-PDPK1 interaction could explain any of the aspects of the transcriptional response to WIN site inhibitor. Remarkably, we see that almost half (467) of the genes induced by C6 in CHP134 cells (Bryan et al., 2020) are induced by WDR5 degradation in U2OS cells, despite the different cell lines (Figure 7G). Ninety-two of these common genes (Table S5) show reduced expression in response to PDPK1 depletion (Figures S7J and S7K), and 27 are also induced by the R3A mutation in PDPK1 (Figure 7H). Many of these 27 genes are connected to the cell cycle and specifically to mitosis, including the mitotic spindle component ASPM, the centromere component CENPF, the segregation-critical topoisomerase TOP2A, and the condensin component SMC2. This analysis reinforces the concept that the WDR5 and PDPK1 together control the expression of G2/M connected genes and demonstrates that part of the response of cells to WIN site inhibitor C6 is due to disruption of the WDR5-PDPK1 interaction.
Although the nuclear functions of PDPK1 are not well understood, it is known to play an important role in the G2/M phase of the cell cycle (Nakamura et al., 2008) and can influence the transcription of G2/M-expressed genes via regulation of FoxM1 (Wang et al., 2010). Our data suggest that WDR5 impacts this aspect of PDPK1 function. What is interesting about this idea, however, is that WDR5 inhibits the ability of PDPK1 to activate these genes. Given the role of WDR5 in control of protein synthesis genes, one possibility is that WDR5 links nuclear functions of PDPK1 to protein synthesis capacity, restricting PDPK1 activity until a sufficient level of ribosome production is achieved, or new ribosome synthesis is completed, during G2. By extension, this notion predicts that WIN site inhibitors could act, at least in part, by allowing cells to enter mitosis without an adequate ribosome inventory. Further experimentation will be needed to determine when and how the WDR5-PDPK1 interaction controls events during G2/M and how this contributes to the response of cancer cells to WIN site inhibitors.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, William Tansey (william.p.tansey@vanderbilt.edu).
Materials availability
Plasmids and cell lines generated in this study are available upon request to the Lead Contact.
Data and code availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org/cgi/GetDataset) via the PRIDE (Perez-Riverol et al., 2019) partner repository. The accession number for the proteomics data reported in this paper is PRIDE: PXD019209. RNA-Seq data are deposited at the Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/). The accession number for the RNA-Seq data reported in this paper is GEO: GSE150400. X-ray crystal structure is deposited at the Protein Data Bank (https://www.rcsb.org). The accession number for the structural data reported in this paper is PDB: 6WJQ.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines
HEK293 (RRID:CVCL_0045), HEK293T (RRID:CVCL_1926), and U2OS (RRID:CVCL_0042) cells were cultured in DMEM with 10% FBS and 10 U/ml Penicillin-Streptomycin (GIBCO 15140122). CHP134 cells (RRID:CVCL_1124) were cultured in RPMI with 10% FBS and 10 U/ml Penicillin-Streptomycin (GIBCO 15140122). All cells were cultured at 37°C and 5% CO2 and split every 2–4 days. HEK293, HEK293T, and U2OS were purchased from ATCC. CHP134 cells were purchased from Sigma. All cell lines were tested for mycoplasma. HEK293, HEK293T, and U2OS are female. CHP134 cells are male.
Bacteria
The E. coli strain used for protein expression for biochemical assays is Rosetta 2-BL21; for protein purification for crystallization we used BL21-Gold (DE3) E. coli.
METHOD DETAILS
Generation of Stable Cell Lines
HEK293T (500,000) cells were plated in 60 mm dishes and the next day cells were transfected by the calcium phosphate method. For retroviral vectors, pBabe vector of interest and pCL10A vector were co-transfected. For lentiviral transfections, lentiviral vector of interest, psPAX2, and pMD2 were co-transfected. Transfected cells were grown overnight, media replaced with fresh media and grown for 24 hours. Virus-containing media was collected by filtering through 0.45 μm filter and either immediately applied to target cells or stored in aliquots at −80°C. To transduce target cells, 1 mL viral media and 2 mL fresh media were combined with 8 μg/ml polybrene, incubated for 5 minutes, then applied to 1–2 million target cells. This was repeated with new virus the next day. After two rounds of transduction, cells were selected with 1 μg/ml puromycin. Proper expression was confirmed by western blotting.
Genome Editing for Knock-in of Degradable Tag
Targeting vectors were constructed as described under “Plasmid constructions.” The gRNA targeting PDPK1 for C-terminal tagging binds the sense strand and cuts five bases upstream of the stop codon: CAGGCCACGTCACTGCACAG. The gRNA targeting WDR5 for C-terminal tagging binds the antisense strand and cuts eight bases downstream of the stop codon: CTCTCGCGGGCAGGAGCAAA. Chemically modified sgRNAs were synthesized (Synthego) and CRISPR reagents and targeting vectors were delivered to cells using the Neon Electroporation Transfection System (Invitrogen). Reactions of Cas9-sgRNA ribonucleoprotein (RNP) complexes were formed three at a time by combining recombinant Cas9 (Synthego) and sgRNA at a 1:3 ratio in Neon buffer R (Invitrogen) and incubating at room temperature for 10 min. RNP complexes were stored at 4°C until use. Electroporation reactions were performed in triplicate so that each triplicate of reactions included 10 pmol Cas9 with 30 pmol sgRNA, 900,000 U2OS cells, 12.5 μg targeting vectors (1:1 BFP:mCherry) brought to approximately 35 μl with Neon buffer R. Using this mixture, three electroporation rounds were performed using 10 μl Neon tips with the conditions of 1230 V, 10 ms pulse width, and 4 pulses. Cells were immediately placed into warm, antibiotic-free DMEM supplemented with FBS and allowed to recover for two days. After expansion and at least five days in culture, cells were analyzed by flow cytometry for expression of fluorescent markers as a proxy for proper integration. Cells were counter stained with Zombie NIR viability dye and resuspended in 0.5% BSA in PBS. Cells were analyzed using a BD LSRII Fortessa (BD Biosciences-US) instrument for expression of BFP and mCherry fluorescent markers. After confirmation of BFP/mCherry positive cells, a population of double positive cells was sorted using a BD FACSAria III (BD Biosciences-US) and analyzed by western blotting.
Genome Editing for R3A Point Mutation
Plasmid pX459 containing Cas9 linked to puromycin resistance marker was a gift from Feng Zhang (Addgene # 62988) (Ran et al., 2013) and was modified by digesting with BbsI and ligation of annealed gRNA sequences. Three gRNAs targeting the N terminus of PDPK1 were inserted into pX459 using BbsI sites, and proper insertion was confirmed by Sanger sequencing. The sequences of the gRNAs are listed in the Key Resources Table. A 200 bp single stranded oligonucleotide targeting the N-terminal region of PDPK1 was designed to mutate the third codon from AGG to GCG (R3A) and silence the PAM sequences. Proper repair with this ssODN also introduced a SpeI site in codons 5 and 6 for screening purposes. The 200 bp oligo was ordered from IDT and is listed in the Key Resources Table. For transfections, 500,000 HEK293 cells were plated one day prior, and then transfected using Lipofectamine 3000 to deliver 0.5 μg pMAX-GFP, 0.5 μg pX459 with gRNA, 1 μg pBluescript filler plasmid and 1 μl 10μM ssODN template. One day after transfection cells were selected for 48 hours with 1 μg/ml puromycin to enrich for cells expressing Cas9. Individual genetic variants were isolated by single cell dilutions and analyzed for introduction of the SpeI restriction marker. Genomic DNA from the individual clones was purified with Purelink genomic DNA mini kit (Invitrogen), and DNA was analyzed by PCR amplification with OneTaq (NEB) using GC Buffer and 10% GC enhancer. Primers for this amplification are listed in the Key Resources Table. PCR products were then analyzed by SpeI digest to screen for cells with homozygous integration. Clones carrying the R3A mutation were confirmed by Sanger sequencing and analyzed by western blotting.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-FLAG M2 (HRP conjugate) | Sigma-Aldrich | Cat# A8592; RRID:AB_439702 |
| Mouse monoclonal DYKDDDDKTag (9A3) | Cell Signaling Technology | Cat# 8146; RRID:AB_10950495 |
| Rabbit monoclonal anti-WDR5 (D9E1I) (used for western blotting) | Cell Signaling Technology | Cat# 13105; RRID:AB_2620133 |
| Rabbit polyclonal anti-WDR5 (used for IP and PLA) | Bethyl Laboratories | Cat# A302–429A; RRID:AB_1944302 |
| Rabbit polyclonal anti-KIF2A | Bethyl Laboratories | Cat# A300–914A; RRID:AB_2280872 |
| Rabbit monoclonal anti-c-Myc antibody [Y69] | Abcam | Cat# ab32072; RRID:AB_731658 |
| Rabbit monoclonal anti-MLL1 (D6G8N) (Carboxy-terminal Antigen) (anti-KMT2A) | Cell Signaling Technology | Cat# 14197; RRID:AB_2688010 |
| Rabbit monoclonal anti-MLL2/KMT2B (D6X2E) (Carboxy-terminal Antigen) | Cell Signaling Technology | Cat# 63735; RRID:AB_2737357 |
| Rabbit polyclonal anti-RBBP5 | Bethyl Laboratories | Cat# A300–109A; RRID:AB_210551 |
| Rabbit monoclonal anti-PDPK1 antibody [EP569Y] (used for western blotting) | Abcam | Cat# ab52893; RRID:AB_881962 |
| Rabbit polyclonal anti-PDK1 antibody (used for IP and western blotting) | Bethyl Laboratories | Cat# A302–130A; RRID:AB_1720395 |
| Rabbit monoclonal anti-PDK1 (D4Q4D) (used for western blotting) | Cell Signaling Technology | Cat# 13037; RRID:AB_2798095 |
| Rabbit monoclonal anti-RICTOR (53A2) (anti-RICTR) | Cell Signaling Technology | Cat# 2114; RRID:AB_2179963 |
| Rabbit polyclonal anti-Rictor Antibody (anti-RICTR) | Bethyl Laboratories | Cat# A300–459A RRID:AB_2179967 |
| Rabbit monoclonal anti-SIN1 (D7G1A) | Cell Signaling Technology | Cat# 12860; RRID:AB_2798048 |
| Rabbit polyclonal anti-DNA helicase B (anti-HELB) | Bethyl Laboratories | Cat# A304–686A; RRID:AB_2620881 |
| Rabbit polyclonal anti-RARS (anti-SYRC) | Bethyl Laboratories | Cat# A304–749A; RRID:AB_2620944 |
| Rabbit polyclonal anti-IARS (anti-SYIC) | Bethyl Laboratories | Cat# A304–747A; RRID:AB_2620942 |
| Rabbit polyclonal anti-MTMR1 | Bethyl Laboratories | A304–917A; RRID:AB_2621112 |
| Rabbit polyclonal anti-RAPTOR (anti-RPTOR) | Bethyl Laboratories | Cat# A300–553A, RRID:AB_2130793 |
| Rabbit polyclonal anti-mTOR | Cell Signaling Technology | Cat# 2972; RRID:AB_330978 |
| Rabbit monoclonal anti-GbetaL (anti-LST8) | Cell Signaling Technology | Cat# 3274; RRID:AB_823685 |
| Rabbit polyclonal anti-GTF2I/TFII-I | Bethyl Laboratories | Cat# A301–330A; RRID:AB_938033 |
| Rabbit polyclonal anti-CHD8 | Bethyl Laboratories | Cat# A301–224A; RRID:AB_890578 |
| Rabbit polyclonal anti-EDD1 (anti-UBR5) | Bethyl Laboratories | Cat# A300–573A; RRID:AB_2210189 |
| Rabbit monoclonal anti-phospho-Akt (Thr308) (D25E6) XP | Cell Signaling Technology | Cat# 13038; RRID:AB_2629447 |
| Rabbit monoclonal anti-Akt (pan) (C67E7) | Cell Signaling Technology | Cat# 4691; RRID:AB_915783 |
| Rabbit monoclonal anti-phospho-Akt (Ser473) (D9E) XP | Cell Signaling Technology | Cat# 4060; RRID:AB_2797780 |
| Rabbit monoclonal anti-p70 S6 Kinase (49D7) | Cell Signaling Technology | Cat# 5707; RRID:AB_10694087 |
| Rabbit polyclonal anti-phospho-p70 S6 Kinase (Thr389) | Cell Signaling Technology | Cat# 9205; RRID:AB_330944 |
| Rabbit monoclonal anti-GAPDH (D16H11) XP (HRP conjugate) | Cell Signaling Technology | Cat# 8884; RRID:AB_11129865 |
| Rabbit monoclonal anti-α-tubulin (11H10) (HRP conjugate) | Cell Signaling Technology | Cat# 9099; RRID:AB_10695471 |
| Rabbit monoclonal anti-Histone H3 (D1H2) XP (HRP conjugate) | Cell Signaling Technology | Cat# 12648; RRID:AB_2797978 |
| Rat monoclonal anti-HA (clone 3F10) | Roche | Cat# 12013819001; RRID:AB_390917 |
| Chicken polyclonal anti-GFP | Vanderbilt Antibody and protein resource | N/A |
| Mouse monoclonal anti-C23 (MS-3) (anti-nucleolin) | Santa Cruz Biotechnology | Cat# sc-8031 |
| Anti-FLAG(R) M2 Affinity Gel antibody | Sigma-Aldrich | Cat# A2220; RRID:AB_10063035 |
| Goat Anti-Mouse IgG, Light Chain Specific secondary antibody (HRP conjugate) | Jackson ImmunoResearch Labs | Cat# 115-035-174; RRID:AB_2338512 |
| Goat anti-Rabbit IgG Fc Secondary Antibody (HRP conjugate) | Invitrogen | Cat# 31463 |
| Normal Rabbit IgG | Cell Signaling Technology | Cat# 2729; RRID:AB_1031062 |
| Rabbit monoclonal anti-HA (C29F4) (used for ChIP) | Cell Signaling Technology | Cat# 3724; RRID:AB_1549585 |
| Monoclonal anti-6xHIS-Terbium cryptate Gold | Cisbio | Cat# 61HI2TLF |
| Bacterial and Virus Strains | ||
| NEB 5-alpha Competent E. coli (High Efficiency) | New England Biolabs | Cat# C2987 |
| XL1-Blue Competent Cells | Agilent | Cat# 200249 |
| Rosetta 2 Competent Cells | Millipore-Sigma | Cat# 71402 |
| BL21-Gold (DE3) | Agilent | Cat# 230132 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| C6, IUPAC name N-(3,4-Dichlorobenzyl)-3-(6-fluoro-2-methylpyridin-3-yl)-5-((2-imino-3-methyl-2,3-dihydro-1H-imidazol-1- yl) methyl)benzamide | (Aho et al., 2019a) | VU0808641 |
| C6nc, IUPAC name N-(3,4-dichlorobenzyl)-3-(6-fluoro-2-methylpyridin-3-yl)-4-((2-imino-3-methyl-2,3-dihydro-1H-imidazol-1- yl)methyl) benzamide | (Aho et al., 2019a) | VU0817566 |
| C12, 5-bromo-3-chloro-N-(1-cyclopentyl-2-(methylsulfonyl)-1H-imidazol-4-yl)-2-hydroxybenzenesulfonamide2 | (Chacón Simon et al., 2020) | VU0830838 |
| Ethidium Bromide Solution | BioRad | Cat# 1610433 |
| GSK2334470 (PDPK1 inhibitor) | Sigma-Aldrich | Cat# SML0217; CAS 1227911-45-6 |
| dTAG47 | Vanderbilt University Chemical Synthesis Core | (Huang et al., 2017) |
| PhosSTOP | Roche | Cat# 4906845001 |
| cOmplete, EDTA-free Protease Inhibitor Cocktail | Roche | Cat# 05056489001 |
| Dimethyl sulfoxide (DMSO) | Sigma-Aldrich | Cat# D2650; CAS 67-68-5 |
| SpeI-HF | New England Biolabs | Cat# R3133S |
| BbsI | New England Biolabs | Cat# R0539S |
| L-Arginine | Sigma-Aldrich | Cat# A6969; CAS 1119-34-2 |
| L-Lysine | Sigma-Aldrich | Cat# L8662; CAS 657-27-2 |
| L-Proline | Sigma-Aldrich | Cat# P0380; CAS 147-85-3 |
| 13C6 15N2 L-Arginine | Sigma-Aldrich | Cat# 608033; CAS 202468-25-5 |
| 13C615N4 L-Lysine | Sigma-Aldrich | Cat# 608041 |
| DMEM for SILAC | Thermo Scientific | Cat# 88364 |
| Dialyzed FBS | Gemini Bio-Products | Cat# 100–108 |
| Supersignal West Pico Plus Chemiluminescent Substrate | Thermo Fisher Scientific | Cat# 34580 |
| Pierce 16% Formaldehyde (w/v), Methanol-free | Thermo Fisher Scientific | Cat# 28908 |
| Q5 High-Fidelity DNA Polymerase | New England Biolabs | Cat# M0491L |
| Poly D-Lysine | Sigma-Aldrich | Cat# P7280; CAS 27964-99-4 |
| OneTaq DNA Polymerase | New England Biolabs | Cat# M0480 |
| ProLong Diamond Antifade Mountant with DAPI | Invitrogen | Cat# P36966 |
| Ni-NTA Agarose | QIAGEN | Cat# 30210 |
| Puromycin | Sigma-Aldrich | Cat# P7255; CAS 58-58-2 |
| Leptomycin B solution | Sigma-Aldrich | Cat# L2913; CAS 87081-35-4 |
| Isopropyl-beta-D-thiogalactoside (IPTG) | Research Products International | Cat# I56000; CAS 367-93-1 |
| Protein Assay Dye Reagent Concentrate | Bio-Rad | Cat# 5000006 |
| Protein A-agarose | Roche | Cat# 11134515001 |
| Pierce Streptavidin Agarose | Thermo Fisher Scientific | Cat# 20353 |
| Streptavidin-HRP | Cell Signaling Technology | Cat# 3999, RRID:AB_10830897 |
| FLAG peptide | Millipore | Cat# F3290 |
| Peptide PDPK1 WT: NH2-ARTTSQLYDAVPIQSS-GGY-K(biotin) (peptide pulldown) | Genscript | Custom order |
| Peptide PDPK1 R3A: NH2-AATTSQLYDAVPIQSS-GGY-K(biotin) (peptide pulldown) | Genscript | Custom order |
| Peptide PDPK1 WT 15-mer: NH2-ARTTSQLYDAVPIQS-amidated (TR-FRET) | Genscript | Custom order |
| Peptide acetyl PDPK1 WT 15-mer: Acetyl-ARTTSQLYDAVPIQS-amidated (TR-FRET and crystal structure) | Genscript | Custom order |
| Peptide PDPK1 WT 10-mer: NH2-ARTTSQLYDA-amidated (TR-FRET) | Genscript | Custom order |
| Peptide acetyl PDPK1 WT 10-mer: Acetyl - ARTTSQLYDA-amidated (TR-FRET) | Genscript | Custom order |
| Peptide PDPK1 R3A 15-mer: NH2-AATTSQLYDAVPIQS-amidated (TR-FRET) | Genscript | Custom order |
| Peptide acetyl PDPK1 R3A 15-mer: Acetyl-AATTSQLYDAVPIQS-amidated (TR-FRET) | Genscript | Custom order |
| Peptide acetyl H3 15-mer: Acetyl-ARTKQTARKSTGGK-amidated (TR-FRET) | Genscript | Custom order |
| MLL-5FAM = acetylation-ARTEVHLRKS-AHX-AHX-K(5-FAM)-amidation AHX = aminohexanoic acid (FPA assay) | Genscript | Custom order |
| Palbociclib (PD-0332991) HCl | Selleck Chemicals | Cat# S1116 |
| RO-3306 | Sigma-Aldrich | Cat# SML0569 |
| Dynabeads MyOne Streptavidin T1 | Thermo Fisher Scientific | Cat# 65601 |
| Biotin-11-CTP | Perkin Elmer | Cat# NEL54200 |
| SUPERase In RNase Inhibitor | Invitrogen | Cat# AM2694 |
| Critical Commercial Assays | ||
| SuperBlue Ultra Coomassie Stain | Protea Biosciences | Cat# SB-G250X-KIT |
| PureLink Genomic DNA Mini Kit | Invitrogen | Cat# K182001 |
| Gibson Assembly Cloning Kit | New England Biolabs | Cat# E5510S |
| Duolink PLA Starter Kit Mouse/Rabbit | Sigma Aldrich | Cat# DUO92102 |
| Human Cell-Free Protein Expression System | Takara Bio USA | Cat# 3281 |
| Direct-zol RNA Miniprep Kits | Zymo Research | Cat# R2050 |
| Zombie NIR Fixable Viability Kit | BioLegend | Cat# 423105 |
| Neon Transfection System 10 μL Kit | Thermo Fisher Scientific | Cat# MPK1025 |
| LunaScript RT SuperMix Kit | New England Biolabs | Cat# E3010S |
| KAPA SYBR FAST qPCR Master Mix (2X) Universal | KAPA Biosystems | Cat# KK4601 |
| Deposited Data | ||
| Mass spectrometry proteomics data | This paper, http://proteomecentral.proteomexchange.org/cgi/GetDataset | PRIDE: PXD019209 |
| RNA-seq data | This paper https://www.ncbi.nlm.nih.gov/geo/ | GEO: GSE150400 |
| RNaseq CHP134 C6 treatment | (Bryan et al., 2020) | GEO: GSE136451 |
| Crystal structure of WDR5 and PDPK1 N terminus | This paper, https://www.rcsb.org | PDB: 6WJQ |
| Crystal structure of WDR5 and unmodified H3 | (Schuetz et al., 2006) | PDB: 2H9M |
| Crystal structure of WDR5, MLL1 (KMT2A) and RBBP5 | (Avdic et al., 2011a) | PDB: 3P4F |
| Crystal structure of WDR5 and MLL1 (KMT2A) | (Patel et al., 2008a) | PDB: 3EG6 |
| Crystal structure of WDR5 and MLL2 (KMT2D) | (Zhang et al., 2012) | PDB: 3UVK |
| Crystal structure of WDR5 and MLL3 (KMT2C) | (Zhang et al., 2012) | PDB: 3UVL |
| Crystal structure of WDR5 and MLL4 (KMT2B) | (Zhang et al., 2012) | PDB: 3UVM |
| Crystal structure of WDR5 and SET1A | (Zhang et al., 2012) | PDB: 3UVN |
| Crystal structure of WDR5 and SET1B | (Zhang et al., 2012) | PDB: 3UVO |
| Crystal structure of WDR5 and KANL1 and KANL2 | (Dias et al., 2014) | PDB: 4CY2 |
| Crystal structure of WDR5 (apo-form) | (Couture et al., 2006) | PDB: 2H14 |
| Experimental Models: Cell Lines | ||
| HEK293T | ATCC | ATCC Cat# CRL-11268, RRID:CVCL_1926 |
| HEK293 | ATCC | Cat# CRL-1573, RRID:CVCL_0045 |
| U2OS | ATCC | Cat# HTB-96, RRID:CVCL_0042 |
| CHP134 | Sigma Aldrich | Cat# 06122002, RRID:CVCL_1124 |
| HEK293-pBabe-puro vector | This study | N/A |
| HEK293-pBabe-FLAG-WDR5 WT | This study | N/A |
| HEK293-pBabe-FLAG-WDR5 F133A | This study | N/A |
| HEK293-pBabe-FLAG-WDR5 L240K | This study | N/A |
| HEK293-pBabe-vector | This study | N/A |
| HEK293-pBabe-PDPK1-FLAG WT | This study | N/A |
| HEK293-pBabe-PDPK1-FLAG R3A | This study | N/A |
| U2OS-pBabe-puro vector | This study | N/A |
| U2OS-pBabe-puro PDPK1-EGFP-FLAG WT | This study | N/A |
| U2OS-pBabe-puro PDPK1-EGFP-FLAG R3A | This study | N/A |
| U2OS-pBabe-puro PDPK1-EGFP-FLAG D10N | This study | N/A |
| U2OS-pBabe-puro PDPK1-FLAG WT | This study | N/A |
| U2OS WDR5-FKBP(F36V)-2xHA | This study | N/A |
| U2OS PDPK1-FKBP(F36V)-2xHA | This study | N/A |
| HEK293 PDPK1 (R3A mutant) | This study | N/A |
| Oligonucleotides | ||
| See Table S6 for primer sequences | N/A | N/A |
| PDPK1 N-terminal gRNA 1: CCGACGCGGGGCCCATGGCCAGG | This study | N/A |
| PDPK1 N-terminal gRNA 2: TGGCTGGTGGTCCTGGCCATGGG | This study | N/A |
| PDPK1 N-terminal gRNA 3: CACCAGCTGGCTGGTGGTCCTGG | This study | N/A |
| R3A SSODN: GGCCATTGCTGGGGCTCCGCTTCGGGGAGGAGGACGCTGAGGAGGCGCCGAGCCGCGCAGCGCTGCGGGGGAGGCGCCCGCGCCGACGCGGGGGCCatgGCTGCGACCACTAGTCAGCTGGTGAGCGCGCGGCGGCGGACTGGACGCGCCGGTTTGTTACCCTGCCGGGTCCGGCGGCCGCCCGGGTCCGGCGAGGCGGG | Integrated DNA Technologies | Custom order |
| R3A CRISPR PCR amplification primer Forward: ACTAGCAAAGTTGCGCCTCTGAGT | Sigma Aldrich | Custom order |
| R3A CRISPR PCR amplification primer Reverse: CGCCAAGCCGAAAACAAACTTTC | Sigma Aldrich | Custom order |
| Chemically modified PDPK1 gRNA C-terminal (target: CAGGCCACGTCACTGCACAG) | Synthego | Custom order |
| Chemically modified WDR5 gRNA C-terminal (target: CTCTCGCGGGCAGGAGCAAA) | Synthego | Custom order |
| Recombinant DNA | ||
| pFLAG-WDR5 | (Thomas et al., 2015) | N/A |
| pSpCas9(BB)-2A-Puro (PX459) V2.0 | (Ran et al., 2013) | Addgene plasmid Cat# 62988 |
| pBluescript II SK(+) | Agilent | Cat# 212205 |
| pMAX GFP | (Thomas et al., 2015) | N/A |
| pCL10A | Novus Biologicals | Cat# NBP2–29542 |
| pCMV-PAX2 (GAG and POL) | Gift from A. Reynolds | N/A |
| pMD2 (VSV-G Env) | Gift from A. Reynolds | N/A |
| pBabe-puro | (Morgenstern and Land, 1990) | N/A |
| pBabe-puro-FLAG-WDR5 | (Thomas et al., 2015) | N/A |
| pBabe-puro-FLAG-WDR5 F133A | (Aho et al., 2019a) | N/A |
| pBabe-puro-FLAG-WDR5 L240K | (Aho et al., 2019a) | N/A |
| PDPK1 in pcDNA3.1+/C-(K)DYK | Genscript | Cat# OHu13008 |
| pcDNA3.1+/C-(K)DYK PDPK1-FLAG R3A | This study | N/A |
| pcDNA3.1+/C-(K)DYK PDPK1-FLAG R238A | This study | N/A |
| pBabe-puro-PDPK1-FLAG | This study | N/A |
| pBabe-puro-PDPK1-FLAG R3A | This study | N/A |
| pBabe-puro PDPK1-EGFP-FLAG WT | This study | N/A |
| pBabe-puro PDPK1-EGFP-FLAG R3A | This study | N/A |
| pBabe-puro PDPK1-EGFP-FLAG Δ10N | This study | N/A |
| pAW62.YY1.FKBP.knock-in.mCherry | (Weintraub et al., 2017) | Addgene plasmid Cat# 104370 |
| pAW63.YY1.FKBP.knock-in.BFP | (Weintraub et al., 2017) | Addgene plasmid Cat# 104371 |
| pAG.PDPK1.FKBP.knock-in.mCherry | This study | N/A |
| pAG.PDPK1.FKBP.knock-in.BFP | This study | N/A |
| pAG.WDR5.FKBP.knock-in.mCherry | This study | N/A |
| pAG.WDR5.FKBP.knock-in.BFP | This study | N/A |
| pSUMO 6xHis-SUMO-WDR5 | (Cao et al., 2014) | N/A |
| pSUMO 6xHis-SUMO-WDR5 F133A | (Aho et al., 2019a) | N/A |
| pET6xHis-SUMO-WDR5 | (Aho et al., 2019a | N/A |
| pT7-IRES | Takara Bio USA | Included with Cat# 3281 |
| pT7-IRES-PDPK1-FLAG WT | This study | N/A |
| pT7-IRES-PDPK1-FLAG R3A | This study | N/A |
| Software and Algorithms | ||
| MaxQuant v 1.3.0.5 | (Cox and Mann, 2008) | http://www.coxdocs.org/doku.php?id=:maxquant:start; RRID:SCR_014485 |
| Perseus v 1.5.8.5 | (Tyanova et al., 2016) | https://maxquant.net/perseus/; RRID:SCR_015753 |
| Scaffold 4.3.2 | N/A | http://www.proteomesoftware.com/products/scaffold/ RRID:SCR_014345 |
| SEQUEST Cluster | Thermo Scientific | N/A |
| Universal Protein Resource (UniProt) | EMBL-EBI | https://www.uniprot.org/ RRID: SCR_002380 |
| Prism v 8 | GraphPad | https://www.graphpad.com:443/; RRID:SCR_002798 |
| PANTHER 15.0 | (Mi et al., 2019a, 2019b; Mi and Thomas, 2009) | http://pantherdb.org |
| FIJI (ImageJ) version 2.0.0 | (Schindelin et al., 2012) | https://imagej.net/Fiji; RRID SCR_002285 |
| Intensity Ratio Nuclei Cytoplasm Tool | ImageJ Macros | http://dev.mri.cnrs.fr/projects/imagej-macros/wiki/Intensity_Ratio_Nuclei_Cytoplasm_Tool |
| Seaborn | https://seaborn.pydata.org/index.html | N/A |
| DAVID Bioinformatic Resources v 6.8 | (Huang et al., 2009a, 2009b) | https://david.ncifcrf.gov/ |
| PyMOL v2.3 | Schrodinger, Inc. | https://pymol.org/2/; RRID: SCR_000305 |
| Phaser | (McCoy et al., 2007) | https://www.phenix-online.org/documentation/reference/phaser.html RRID: SCR_014219 |
| Cutadapt | (Martin, 2011) | https://cutadapt.readthedocs.io/en/stable/ |
| STAR | (Dobin et al., 2013) | http://code.google.com/p/rna-star/ |
| FeatureCounts | (Liao et al., 2014) | https://bioconductor.org/packages/release/bioc/html/Rsubread.html |
| DESeq2 | (Love et al., 2014) | http://www.bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Other | ||
| PolyScreen PVDF Hybridization Transfer Membrane | PerkinElmer | Cat# NEF1002 |
| 5 mL, Open-Top Thinwall Polypropylene Tube, 13 × 51mm | Beckman Coulter | Cat# 326819 |
| Fisherbrand Cover Glasses - Circles No. 1.5; Thickness: 0.16 to 0.19mm | Fisher Scientific | Cat# 12-545-81 |
| Q Exactive mass spectrometer | Thermo Scientific | Cat# IQLAAEGAAPFALGMAZR |
| Q Exactive Plus mass spectrometer | Thermo Scientific | Cat# IQLAAEGAAPFALGMBDK |
Density Sedimentation Analyses
For stably expressing samples, FLAG-WDR5 HEK293 cells were plated one day prior to analysis. For treated samples, HEK293 cells were plated one day prior and then treated with DMSO or 30 μM C6 for five hours prior to analysis. Cells were washed twice in cold PBS and then lysed in Kischkel buffer (50 mM Tris pH 8.0, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100) supplemented with Roche cOmplete Protease Inhibitor Cocktail and 1 mM PMSF. Protein concentrations of the lysates were measured by Bio-Rad Protein Assay Dye Reagent and normalized. Equal amounts of lysates were carefully loaded onto 5%–40% sucrose gradients prepared in 5mL, 13 × 51 mm polypropylene centrifuge tubes (Cat# 326819, Beckman Coulter). Samples were centrifuged in a Beckman L-90K ultracentrifuge with a SW 55 Ti rotor at 4°C for 14 hr at 50,000 rpm (accelerate max; decelerate no brake). 0.5 mL fractions were collected and resuspended in SDS sample buffer supplemented with β-mercaptoethanol. Samples were heated at 95°C for five minutes and then analyzed by western blotting.
Generating Lysates for Western Blotting
Cells were collected by scraping into PBS. Cell pellets were lysed in RIPA buffer (10mM Tris pH 8.0, 0.5 mM EDTA, 1% NP-40, 0.1% deoxycholate, 0.1% SDS, 140 mM NaCl) supplemented fresh with Roche cOmplete Protease Inhibitor Cocktail, 1 mM PMSF, and Roche PhosSTOP inhibitor tablet. Lysates were incubated on ice for at least 20 minutes and insoluble material was cleared by 10 minutes of centrifugation at 4°C. Protein concentrations were measured by Bio-Rad Protein Assay Dye Reagent, normalized, and taken forward for western blotting analysis.
Western Blotting Analysis
Samples were boiled in SDS sample buffer supplemented with β-mercaptoethanol and run on homemade acrylamide gels. After transferring proteins to PVDF membrane (PerkinElmer), the membranes were blocked in 5% milk for at least one hour, and then incubated with primary antibodies overnight. The antibodies used are detailed in the Figures and the Key Resources Table. Membranes were washed three times with TBST and incubated with HRP-conjugated secondary antibodies. Blots were developed by ECL with Supersignal West Pico Plus Chemiluminescent Substrate (Thermo Fisher Scientific).
FLAG Immunoprecipitations
For transient transfections, cells were transfected by the calcium phosphate method three days prior to experiments. Stable pBabe HEK293 cells were plated two days prior to experiments. To treat cells prior to IPs, media was removed and replaced with media containing DMSO, 30 μM C6, or 30 μM C6nc for 4 hours unless otherwise indicated in the figure legend. Cells were washed twice in cold PBS, then scraped into cold Kischkel buffer (50 mM Tris pH 8.0, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100) or CHAPS buffer (40 mM HEPES pH 7.5, 120 mM NaCl, 1 mM EDTA, 0.3% CHAPS) supplemented with Roche cOmplete Protease Inhibitor Cocktail and 1 mM PMSF. Whole cell extracts were sonicated for 15 s and then clarified by 10 minutes of centrifugation at 4°C. For ethidium bromide treatment, ethidium bromide was added to lysates at 200 μg/ml prior to sonication and maintained at 200 μg/ml for the duration of the IP. Anti-FLAG M2 affinity gel was equilibrated in lysis buffer and blocked at room temperature for at least 20 minutes with 1 mg/ml BSA in lysis buffer. Protein concentrations of lysates were measured with Bio-Rad Protein Assay Dye Reagent and normalized, and then a 20 μl bed volume of BSA-blocked anti-FLAG M2 affinity gel was added to each sample. For FLAG IPs from treated lysates, treatments were added to the lysates and incubated overnight simultaneously with the M2 affinity gel. IPs were incubated on a rotator overnight at 4°C. The next day, IPs were centrifuged at 2500 rpm, and washed four times for five minutes in cold lysis buffer. After last wash, the remaining liquid was aspirated with a 27-gauge needle and bead samples were boiled in SDS sample buffer supplemented with β-mercaptoethanol. Samples were taken forward for western blotting.
SILAC Media and Cell Culture Conditions
Heavy and light media for SILAC was prepared using DMEM for SILAC (Thermo Scientific) and adding 0.79 mM heavy or light (13C6; 15N2) lysine, 0.39 mM heavy or light (13C6;15N4) arginine, and 3.5 mM light proline. Media was then sterile filtered and supplemented with 10% dialyzed FBS and 10 U/ml Penicillin-Streptomycin.
SILAC Sample Preparation
For each sample three plates of 3×106 heavy or light HEK293 cells were plated. The next day cells were transfected with 5 μg pFLAG-WDR5 and 1 μg pMAX-GFP using the calcium phosphate method. When cells were confluent (2–3 days), cells were lysed in Kischkel buffer supplemented with Roche cOmplete Protease Inhibitor Cocktail and 1 mM PMSF. Whole cell extracts were sonicated for 15 s and then clarified by 10 minutes of centrifugation. Anti-FLAG M2 affinity gel (Sigma) was equilibrated in Kischkel buffer and blocked at room temperature for at least 20 minutes with 1 mg/ml BSA in Kischkel buffer. Protein concentrations were measured by Bio-Rad Protein Assay Dye Reagent. Lysates were rotated overnight at 4°C with 5 μM C6 or C6nc and 20 μl bed volume of BSA-blocked anti-FLAG M2 affinity gel. The next day IPs were washed four times for five minutes with cold Kischkel buffer. Samples were transferred to new tubes and eluted twice with 30 μl 100 ng/μl FLAG peptide in TBST by agitation on low speed mixer for 15 minutes at room temperature. Samples were analyzed by western blotting for even levels of heavy and light samples before being taken forward for mass spectrometry.
SILAC-Based Quantitative Mass Spectrometry
SILAC samples were mixed 1:1 and partially separated by SDS-PAGE. Gel regions were excised and cut into 1mm3 cubes and treated with 45 mM DTT for 30 minutes. Available Cys residues were carbamidomethylated with 100mM iodoacetamide for 45 minutes. After destaining with 50% MeCN in 25mM ammonium bicarbonate, proteins were digested with trypsin (10ng/uL) in 25mM ammonium bicarbonate overnight at 37°C. Peptides were then extracted by gel dehydration with 60% MeCN, 0.1% TFA, vacuum dried, and reconstituted in 0.1% formic acid.
Peptides were analyzed by LC-coupled tandem mass spectrometry (LC-MS/MS) using MudPIT analysis with an 8 step salt pulse gradient. Peptides were loaded onto a self-packed biphasic C18/SCX MudPIT column using a Helium-pressurized cell. The MudPIT column consisted of 360 × 150 μm i.d. fused silica, which was fritted with a filter-end fitting (IDEX Health & Science) and packed with 5 cm of Luna SCX material (5 μm, 100Å) followed by 4 cm of Jupiter C18 material (5 μm, 300Å, Phenomenex). Once the sample was loaded, the MudPIT column was connected using an M-520 microfilter union (IDEX Health & Science) to an analytical column (360 μm × 100 μm i.d.), equipped with a laser-pulled emitter tip and packed with 20 cm of C18 reverse phase material (Jupiter, 3 μm beads, 300Å, Phenomenex). Using a Dionex Ultimate 3000 nanoLC and autosampler, MudPIT analysis was performed with an 8-step salt pulse gradient (25, 50, 100, 200, 300, 500, 750, and 1M ammonium acetate). Following each salt pulse, peptides were gradient-eluted from the reverse analytical column at a flow rate of 350 nL/min, and the mobile phase solvents consisted of 0.1% formic acid, 99.9% water (solvent A) and 0.1% formic acid, 99.9% acetonitrile (solvent B). For the peptides from the first 7 SCX fractions, the reverse phase gradient consisted of 2% to 50% B in 83 min, 50% B from 83–84 min, 50% down to 2% B from 84–85 min, and column equilibration at 2% B from 85–95 min. For the last SCX-eluted peptide fraction, the peptides were eluted from the reverse phase analytical column using a gradient of 2% to 98%B in 83 min, 98% B from 83–84 min, 98 to 2% B from 84–85 min, and 2% B from 85–95 min. Peptides were introduced via nanoelectrospray into a Q Exactive mass spectrometer (Thermo Scientific) operating in a data-dependent mode. The instrument method consisted of MS1 using an MS AGC target value of 3×106, followed by up to 20 MS/MS scans of the most abundant ions detected in the preceding MS scan. The MS2 intensity threshold was set to 5×104, dynamic exclusion was set to 20 s, and peptide match and isotope exclusion were enabled.
SILAC MS Data Analysis
For peptide and protein identification, data were analyzed using the Maxquant software package (Cox and Mann, 2008). MS/MS spectra were searched against a human subset of the UniprotKB protein database. Precursor mass tolerance was set to 6 ppm, and variable modifications included oxidation of methionine and carbamidomethylation of cysteine. Enzyme specificity was set to trypsin/P, and a maximum of 2 missed cleavages were allowed. The target-decoy false discovery rate (FDR) for peptide and protein identification was set to 1% for both peptides and proteins. A multiplicity of 2 was used, and Arg10 and Lys8 heavy labels were selected. For SILAC protein ratios, a minimum of 2 unique peptides and a minimum H/L ratio count of 2 were required, and normalized ratios were considered for all presented analysis. In total, 754 proteins are quantified by these criteria in both replicates. The label swap revealed seven contaminating keratinous proteins (included in Table S1) which were removed (resulting in 747 proteins) before further analysis. SILAC data were also assembled in Scaffold to view protein sequence coverage and assigned spectra for identified peptides. Heatmaps of these data were generated using Seaborn. Pearson correlation analysis one sample t test of the SILAC data was performed using Perseus software package (Tyanova et al., 2016).
Immunoprecipitations of Endogenous Proteins
Cells were plated to be confluent two days later. If cells were treated, media was changed to media containing the appropriate treatment for the indicated time. Each plate was rinsed twice with PBS, and then scraped into Kischkel buffer supplemented with Roche cOmplete Protease Inhibitor Cocktail, 1 mM PMSF, and Roche PhosSTOP inhibitor tablet. For suspension cells, cells were pelleted, washed twice in PBS, and then resuspended in lysis buffer. Lysates were sonicated for 15 s and cleared by centrifugation for 10 minutes at 4°C. Protein concentrations were measured by Bio-Rad Protein Assay Dye Reagent. For each IP 1–5 mg of lysate was used as total input. Antibodies used for IPs were 6–10 μg anti-WDR5 (Bethyl A302–429A), 4–6 μg anti-PDPK1 (Bethyl A302–130A), and an equivalent amount of Normal Rabbit IgG (Cell Signaling Technologies #2729S). Antibodies and lysates were rotated at 4°C overnight, and the next day a 20 μl bed volume of Roche Protein A agarose, blocked for at least 20 minutes with 1 mg/ml BSA in Kischkel buffer, was added to each sample. IPs were incubated with protein A agarose for 2–6 hours and then washed four times for five minutes with 1 mL cold Kischkel buffer, transferring to new tubes before last wash. Samples were eluted with SDS sample buffer supplemented with β-mercaptoethanol and taken forward for western blotting analysis.
Subcellular Fractionation
Subcellular fractionation was performed similar to as described (Méndez and Stillman, 2000). A confluent plate of U2OS cells was washed twice in PBS, scraped into PBS and pelleted. Cells were resuspended in 200 mL Buffer A (10 mM HEPES, pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol, 1 mM DTT, Roche cOmplete Protease Inhibitor Cocktail and 1 mM PMSF) and incubated on ice for 8 minutes. Samples were centrifuged at 1,300 × g at 4°C for five minutes. The supernatant (S1 fraction) and pellet (P1 fraction) were separated and S1 was clarified by high-speed centrifugation at 4°C for 10 minutes. The resulting supernatant (S2 fraction) was collected and the pellet (P2 fraction) was discarded. The P1 fraction was washed once with 500 mL Buffer A and centrifuged 1 minute at 1,300 × g. The P1 fraction was lysed by resuspending in 100 mL Buffer B (3 mM EDTA, 0.2 mM EGTA, Roche cOmplete Protease Inhibitor Cocktail and 1 mM PMSF) and incubated for 30 minutes on ice, followed by centrifugation at 1,700 × g at 4°C for five minutes. The resulting supernatant (S3 fraction) was separated from the chromatin-enriched pellet (P3 fraction). P3 was washed once with 500 mL Buffer B and resuspended in 400μl SDS sample buffer. All samples were brought to 400 μl in SDS sample buffer and boiled for five minutes. Equal volumes of each fraction were taken forward for western blotting.
EGFP Imaging Experiments
U2OS cells stably expressing PDPK1-EGFP fusions were plated onto coverslips coated with poly-D lysine and cultured overnight in DMEM. Cells were then treated with 20 nM leptomycin B (LMB) or an equivalent volume of 70% methanol vehicle control in DMEM for four hours. Cells were fixed in 4% paraformaldehyde for 10 min at room temperature then washed three times for five minutes with PBS. Cells were permeabilized in PBS containing 0.1% (v/v) Triton X-100 for five min then washed three times for five minutes with PBS. Coverslips were then mounted in ProLong Diamond Antifade Mountant with DAPI. Confocal images were acquired using an Andor DU-897 EMCCD camera mounted on a Nikon Spinning Disk Microscope.
Proximity Ligation Assay
Retroviral pBabe-puro U2OS cells stably expressing PDPK1-FLAG were plated onto coverslips pretreated with poly D-lysine. After plating, cells were treated overnight with 30μM C6 or C6nc. Cells were fixed in 4% methanol-free formaldehyde and permeabilized with 0.5% Triton. Proximity ligation assays were performed with the Duolink PLA mouse/rabbit kit (Sigma) according to the manufacturer’s instructions. Primary antibodies used were mouse anti-FLAG and rabbit anti-WDR5 (Bethyl 429A). Confocal images were acquired using an Andor DU-897 EMCCD camera mounted on a Nikon Spinning Disk Microscope.
Treatment for Targeted Protein Degradation
To deplete cells of FKBP(F36V)-tagged proteins, cells were first plated in normal media, and the next day media was changed to media containing 500 nM dTAG47 bifunctional small molecule, synthesized through the Vanderbilt Chemical Synthesis Core. DMSO vehicle control was 0.01% DMSO. After the time point indicated, cells were collected for the relevant analysis.
Preparation of RNA for RNA-Seq
Cells were plated at sub-confluence and collected after 1–2 days. Where appropriate, media on cells was changed to contain the indicated treatments for the indicated time frames. Cells were collected in Trizol and RNA was purified with Direct-Zol RNA Miniprep kit (Zymo) with on-column DNaseI treatment. RNA was submitted to Genewiz or the Vanderbilt Technologies for Advanced Genomics Core Laboratory for library preparation and deep sequencing.
RNA Extraction and RT-qPCR Analysis
Cells were plated at sub-confluence and the next day media was changed to contain the appropriate treatment (ex. 500 nM dTAG47 or 0.01% DMSO vehicle control). Cells were collected in Trizol and RNA was purified with Direct-Zol RNA Miniprep kit (Zymo) with on-column DNaseI treatment. RNA was reverse transcribed with LunaScript RT SuperMix Kit (New England Biolabs) and analyzed by qPCR using gene-specific primers (Table S6) and KAPA SYBR FAST qPCR 2x Master Mix.
Purifying Recombinant WDR5 for Binding Assays
pSUMO plasmids containing N-terminal 6xHis-SUMO tagged WDR5 (amino acids 22–334), WT or F133A mutant, were transformed into Rosetta DE3 competent cells. Bacterial cultures were grown in LB medium supplemented with 50 μg/ml kanamycin. A 50 mL starter culture was grown overnight and used to inoculate a 500 mL culture. When the culture reached OD of approximately 0.8, it was induced with 1 mM isopropyl-beta-D-thiogalactoside (IPTG) for 3 hours. Bacteria were aliquoted, pelleted, and stored at −80°C. To purify 6xHis-SUMO-WDR5 protein, bacterial pellets were thawed on ice and resuspended in 5 mL SUMO lysis buffer (50 mM NaH2PO4, 300 mM NaCl, 3 mM imidazole, adjusted to pH 8.0) supplemented with Roche cOmplete Protease Inhibitor Cocktail and 1 mM PMSF. Bacteria were lysed by sonication and cleared by centrifugation. Proteins were purified by incubating bacterial lysates with Ni-NTA agarose (QIAGEN). After incubation, agarose was washed 3x with 5 mL SUMO lysis buffer. Proteins were either left on beads, or eluted with SUMO elution buffer (1x PBS, 250 mM imidazole, 10% glycerol, 2mM DTT). Protein concentration and purity were assessed by SDS–PAGE and Coomassie Blue staining alongside BSA standards.
Far Western
PDPK1-FLAG (WT and R3A) were FLAG immunoprecipitated from HEK293 pBabe retroviral stable cell lines. IP samples were run on an 8% gel and transferred to PVDF. This membrane was stained with Ponceau, destained in water, and imaged before blocking in 5% milk for one hour. Membrane was then incubated overnight with recombinant 6xHis-SUMO-WDR5 in 2% milk, 10% glycerol, 1mM DTT, and 0.5 mM EDTA. Membrane was washed three times in TBST, then probed with anti-WDR5 antibody.
In Vitro PDPK1 Pulldown
In vitro transcription and translation of PDPK1 variants was performed using the Takara Human Cell-Free Protein Expression System (Takara #3281) following the manufacturer’s protocol. Yield of the in vitro reaction was quantified by western blotting alongside a FLAG-tagged standard. For binding reactions each in vitro reaction was incubated with 20 mg of 6xHis-SUMO-WDR5 (22–334) bound to NTA-Ni beads. After combining PDPK1 and WDR5, inputs were removed and binding reactions were performed in a 500μl volume in Kischkel buffer for 2 hours at 4°C. Beads were washed with 1 mL cold Kischkel buffer four times for two minutes and transferred to new tubes before the last wash. Samples were eluted by boiling in SDS sample buffer supplemented with β-mercaptoethanol and analyzed by western blotting.
Peptide Pulldown Experiments
Biotinylated peptides were pre-bound to streptavidin agarose by adding an excess of the indicated peptide (60 μg, approximately 3x excess to binding capacity of streptavidin beads) to a 20 μl bed volume of Pierce Streptavidin Agarose and rotating at 4°C for one hour. Beads were washed three times for five minutes with cold Kischkel buffer and transferred to new tubes. 20 μg of 6xHis-SUMO-WDR5 purified protein was added and samples were rotated for two hours at 4°C. Beads were washed four times for two minutes with cold Kischkel buffer, eluted in SDS sample buffer supplemented with β-mercaptoethanol, and analyzed by western blotting.
Purification of PDPK1 for MS Analysis
HEK293 cells were lysed in Kischkel buffer supplemented with Roche cOmplete Protease Inhibitor Cocktail, 1 mM PMSF, and 5 mM sodium orthovanadate. IP reactions were performed as for an endogenous IP with 5 mg lysate and 10 μg anti-PDPK1 (Bethyl A302–130A) or Normal Rabbit IgG (Cell Signaling Technologies #2729S). Antibodies were incubated with lysates overnight at 4°C on a rotator, and the next day a 20 μl bed volume of BSA-blocked Roche Protein A agarose was added to each IP. IPs were incubated with protein A agarose for 3 hours and then washed four times for five minutes with 1 mL cold Kischkel buffer, transferring to new tubes before last wash. Samples were eluted with SDS sample buffer supplemented with β-mercaptoethanol. Samples were run on a gel and stained with SuperBlue Ultra Coomassie Stain. The band corresponding to PDPK1 was cut out and taken forward for analysis by mass spectrometry.
Mass Spectrometry Analysis of PDPK1
Gel band was cut out and diced into 1mm3 cubes. Proteins were treated for 30 minutes with 45 mM DTT, and available Cys residues were carbamidomethylated with 100mM iodoacetamide for 45 minutes. After destaining with 50% MeCN in 25mM ammonium bicarbonate, proteins were digested with trypsin or AspN (10ng/uL) in 25mM ammonium bicarbonate overnight at 37°C. Peptides were extracted by gel dehydration with 60% MeCN, 0.1% TFA, the extracts were dried by speed vac centrifugation, and reconstituted in 0.1% formic acid for LC-MS/MS. An analytical column was packed with 20cm of C18 reverse phase material (Jupiter, 3 μm beads, 300Å, Phenomenox) directly into a laser-pulled emitter tip. Peptides were loaded on the capillary reverse phase analytical column using a Dionex Ultimate 3000 nanoLC and autosampler. The mobile phase solvents consisted of 0.1% formic acid, 99.9% water and 0.1% formic acid, 99.9% acetonitrile. Peptides were gradient-eluted at a flow rate of 350 nL/min, using a 90-minute gradient. The gradient consisted of the following: 1–72 min, 2%–40% B; 72–78 min, 40%–90% B; 78–79 min, 90% B; 79–80 min, 90%–2% B; 80–90min (column re-equilibration), 2% B. A Q Exactive Plus mass spectrometer (Thermo Scientific) was used to mass analyze the eluting peptides using a data-dependent method. The instrument method consisted of MS1 using an MS AGC target value of 3×106, followed by up to 16 MS/MS scans of the most abundant ions detected in the preceding MS scan. For identification of peptides, tandem mass spectra were searched with Sequest (Thermo Fisher Scientific) against a Homo sapiens subset database created from the UniprotKB protein database. Variable modification of +15.9949 on Met (oxidation), +57.0214 on Cys (carbamidomethylation), and +42.01056 on the N terminus (acetylation) were included for database searching. Search results were assembled using Scaffold proteome software.
TR-FRET Based Peptide Competition Assays
TR-FRET (time-resolved fluorescence resonance energy transfer) emissions were recorded on a BioTek Cytation 3 instrument and assays were performed with the indicated peptides. Two or more repeats were obtained, and average Ki values are reported. The PDPK1 and H3 peptides were unlabeled and the KMT2A (MLL1) peptide was a labeled 10-mer-Thr-FAM (ARTEVHLRKS-(Ahx-Ahx)(Lys-(5-FAM))) (Karatas et al., 2010). 100 nM KMT2A-5FAM peptide, 4 nM 6xHis-tagged WDR5 (24–334) protein and 1 nM anti HisTag-terbium antibody (Cisbio) were combined in FRET Buffer (1X Phosphate Buffered Saline, 300mM NaCl, 0.5mM TCEP, 0.1% CHAPS, pH 7.3). The indicated PDPK1 and H3 peptides were diluted in FRET Buffer and dispensed into 384-well, white, flat-bottom plates in a 10-point, 5x serial dilution scheme. Diluted peptides were incubated with the KMT2A-5FAM/WDR5/anti-His for one hour. The change in TR-FRET signal (Delta F) was measured on the Biotek Cytation 3 equipped with a filter cube containing an Ex 340/30 nM Em 620/10 filter and an Ex 340/30 Em 520 filter. Measurement plates were excited at a wavelength of 340 nm, and emission wavelengths of 495 and 520 nm were used. The 520 / 495 emission ratios (TR-FRET) were used to calculate an IC50 (peptide concentration at which 50% of the KMT2A-5FAM bound peptide is displaced) by fitting the inhibition data using XLFit software (Guilford, UK) to a four parameter dose-response (variable slope) equation. This was converted into a binding inhibition/displacement constant (Ki) using the formula (Nikolovska-Coleska et al., 2004):
where [I]50 is the concentration of the free peptide at 50% inhibition, [L]50 is the concentration of the free labeled ligand at 50% inhibition, [P]0 is the concentration of the free protein at 0% inhibition, and represents the dissociation constant of the 10-mer-Thr-FAM probe.
Purification of WDR5 for Structural Studies
Human WDR5 (aa: 22–334) was cloned into a modified pET vector (pBG104) with a 6xHis-SUMO tag at the N terminus. The plasmid was then transformed into E. coli BL21-Gold (DE3) cells. One hundred milliliters of LB starter was used to inoculate a 10 L fermentation culture (BioFlo 415, New Brunswick Scientific) and grown at 37°C. Fermentation growth media contained KH2PO4 (4 g/L), K2HPO4 (6 g/L), Na2SO4 (2 g/L), K2SO4 (1 g/L), NaCl (0.5 g/L), Yeast Extract (5 g/L), glycerol (2 ml/L), Antifoam (0.2 ml/L), 5% LB medium, glucose (25 g/L), MgCl2 (2 mM), CaCl2 (0.1 mM), NH4Cl (2.5 g/L), and Kanamycin (50 mg/ml). When the cell density reached OD600 = 2.0, the temperature was lowered to 30°C, and WDR5 expression induced by treatment with 1 mM isopropyl-beta-D-thi-ogalactoside (IPTG) overnight. Cell pellets were collected, dissolved in lysis buffer containing 1XPBS plus 300 mM NaCl, 20 mM imidazole, 5 mM BME, and 10% glycerol, and lysed by homogenization (APV-2000, APV). The lysate was cleared by centrifugation, filtered, and then applied to the Ni-column (140 mL, ProBond, Invitrogen). Bound protein was eluted using an imidazole gradient (0–300 mM). The His-SUMO-tag was cleaved by SUMO protease during dialysis and subsequently eliminated through a second Ni-column. WDR5 protein was then purified by size-exclusion chromatography (HiLoad 26/60, Superdex 75, GE Healthcare) using crystallization buffer (20 mM HEPES, pH 7.0, 250 mM NaCl, and 5 mM DTT). The purity of protein was assessed using SDS–PAGE. Purified WDR5 was then concentrated to 10 mg/mL, and stored at −80°C.
Protein Crystallization and Data Processing
Purified WDR5 was crystallized in the presence of a five-fold molar excess of the acetylated PDPK1 15-mer peptide under conditions containing 25% PEG 8K, 100 mM Tris, and 1 mM TCEP. The complex crystallized at 18°C in the P21 space group (cell dimensions a = 54.52Å, b = 47.23Å, c = 118.97Å, β = 90.92° with 2 molecule in the asymmetric unit). A single flash-cooled crystal diffracted to 2.5Å, and data were collected on the Life Sciences Collaborative Access Team (LS-CAT) 21-ID-F beamline at the Advanced Photon Source (APS), Argonne National Laboratory. The WDR5–PDPK1 peptide structure was determined by molecular replacement method using the WDR5-RBBP5 peptide complex (PDB: 2XL2) as the search molecule in Phaser (McCoy et al., 2007). The model was refined to a final R and R-free values of 21% and 25%, respectively. The refined models and corresponding structure factor amplitudes were deposited in the Research Collaboratory for Structural Biology under accession number PDB: 6WJQ. Figures were prepared with PyMOL. 6WJQ was aligned with other WDR5 cocrystal structures using the PyMOL ‘super’ command.
Cell Cycle Analysis by Flow Cytometry
U2OS cell lines were plated at approximately 50% confluence and the next day media was exchanged to contain the indicated treatment. At the indicated time point, cells were collected by trypsinization and pelleted by centrifugation. Cell pellet was resuspended in PBS and fixed by mixing drop-wise into 70% ethanol. Cells were stored in ethanol at 20°C for at least 2 hours before washing once in PBS and resuspending in propidium iodide staining buffer (1x PBS, 10 μg/mL propidium iodide (PI), 100 μg/mL RNaseA, 2 mM MgCl2). Samples were stained overnight in the dark at 4°C before straining through 35 μm nylon Falcon 5 mL Round Bottom Polystyrene Test Tubes. Cell cycle phases were analyzed by DNA content at the Vanderbilt University Flow Cytometry Shared Resource using a Becton Dickinson LSR Fortessa instrument. For each sample, at least 10,000 single cells were analyzed using forward and side scatter to select single cells.
Treatment for Cell Synchronization
U2OS cells expressing WDR5-FKBP(F36V) were plated at 50% confluence and the next day media was exchanged to contain the appropriate treatment. 1 μM Palbociclib CDK4/6 inhibitor (Fry et al., 2004) was used to synchronize cells in G1 and 10 μM RO-3306 CDK1 inhibitor (Vassilev et al., 2006) was used to synchronize cells in G2/M. Cells were treated for 20 hours with or without simultaneous treatment with 500 nM dTAG47. After treatment cells were collected and taken forward for the appropriate analysis.
Nuclear Run-On RT-qPCR
Protocol was adapted from (Roberts et al., 2015). 4×106 U2OS cells expressing WDR5-FKBP(F36V) were plated one day prior to the experiment. Cells were treated for six hours with 500 nM dTAG47 or DMSO vehicle control. Cells were trypsinized, counted, and resuspended in cold PBS. 4×106 cells were pelleted, and then resuspended in 1 mL NP-40 lysis buffer (10 mM Tris, pH 7.4, 300 mM sucrose, 10 mM KCl, 5 mM MgCl2, 0.5% NP-40, Roche cOmplete Protease Inhibitor Cocktail, and 0.8 U/μl SUPERase·In RNase Inhibitor) and incubated on ice for 5 minutes. Nuclei were pelleted at 300 × g for 4 minutes at 4°C. Pelleted nuclei were washed once with 1 mL of NP-40 lysis buffer and immediately pelleted again at 300 × g for 4 minutes at 4°C. Nuclei were resuspended in 40 μl Nuclei Storage Buffer (50 mM Tris, pH 8.0, 25% glycerol, 5 mM MgCl2, 0.1 mM EDTA, Roche cOmplete Protease Inhibitor Cocktail) resulting in a volume of approximately 60 μl. 60 μl of 2x Transcription Buffer (20 mM Tris, pH 8.0, 5 mM MgCl2, 5 mM DTT, 300 mM KCl, 0.5 mM Bio-11-CTP, 1 mM ATP, 1mM GTP, 0.5 mM CTP, 1 mM UTP, 1% sarkosyl, and 0.8 U/μl SUPERase·In RNase Inhibitor) was added to each sample, mixed by pipetting, and incubated at 30°C for 30 minutes. Samples were resuspended in 300 μl Trizol LS (Thermo Fisher Scientific) and rotated at room temperature for 5 minutes. RNA was extracted using a Direct-Zol RNA Miniprep kit (Zymo) with on-column DNaseI treatment. RNA was eluted in 25 μl of water and stored overnight at −80°C.
For biotin pulldown, Dynabeads MyOne Streptavidin T1 were first prepared by washing for two minutes at room temperature once with Bead Wash Buffer 1 (0.1 M NaOH, 50 mM NaCl) and then twice with Bead Wash Buffer 2 (100 mM NaCl). Beads were resuspended in Binding Buffer (10 mM Tris, pH 7.4, 300 mM NaCl, 0.1% Triton) and added to RNA samples. RNA samples were rotated at room temperature for 20 minutes. Beads were recovered on a magnetic rack, and the liquid was removed. Beads were washed briefly by resuspending in High Salt Buffer (50 mM Tris, pH 7.4, 2 M NaCl, 0.5% Triton), then Binding Buffer, and finally Low Salt Buffer (5 mM Tris, pH 7.4, 0.1% Triton). In between washes, beads were recovered on a magnetic rack and liquid was removed by aspiration. RNA was extracted from beads by resuspending beads in 300 μl Trizol, rotating five minutes at room temperature, and extracting with 60 μl of chloroform. After recovering the aqueous fraction, bead pellet was extracted a second time with Trizol and chloroform. Aqueous phases were combined and precipitated with 3x volume of ice cold ethanol and 1 μl Invitrogen Ambion GlycoBlue Coprecipitant. Samples were incubated at −20°C for at least ten minutes, and then centrifuged at 4°C for at least 20 minutes at 13,000 rpm. Nucleic acid pellet was washed with 500 μl ice cold 75% ethanol, dried, and then resuspended in 25 μl water. RNA was reverse transcribed with LunaScript RT SuperMix Kit (New England Biolabs), diluted 1:4 in water, and analyzed by qPCR. qPCR was performed using KAPA SYBR FAST qPCR 2x Master Mix and gene-specific primers designed for detection of early transcripts, i.e., primers spanning an intron-exon boundary to avoid contaminating total mRNA (see Table S6 for primer sequences).
Plasmid Constructions
Molecular cloning was performed using XL1Blue (Agilent) or NEB 5-alpha Competent E. coli. PCR amplifications were performed using Q5 High-Fidelity DNA Polymerase (NEB).
pcDNA3.1 containing the PDPK1 ORF and a C-terminal FLAG epitope tag was purchased from Genscript. Point mutations were generated by reverse mutagenesis or Gibson assembly modification. The PDPK1-FLAG sequences were then amplified and cloned into pBabe-puro (Morgenstern and Land, 1990) by Gibson assembly. PDPK1-EGFP fusions were similarly constructed by Gibson assembly.
Targeting vectors for endogenous FKBP(F36V) tagging were modified from pUC19-based targeting vectors that were a gift from Richard Young (Addgene #104370 and #104371) (Weintraub et al., 2017). Vectors contain FKBP12(F36V)-P2A-BFP or FKBP12(F36V)-P2A-mCherry, and were modified by Gibson Assembly to include asymmetrical homology arms. Homology arms were amplified from U2OS genomic DNA purified with Purelink genomic DNA mini prep kit (Invitrogen). Homology arms used for PDPK1 are 200 bp 5′ and 900 bp 3′ surrounding and not including the stop codon. Homology arms used for WDR5 are 200 bp 5′ (up to the stop codon) and 800 bp 3′ (starting 17bp after the stop codon to ensure deletion of PAM sequence). Plasmids were verified by Sanger sequencing and prepped with the QIAGEN Midi-prep kit.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical comparisons between replicates for image quantification, RT-qPCR, and cell cycle profile analysis were performed with GraphPad Prism software 8.0. The n indicates number of biological replicates. The n, error bar representations, and details of statistical tests can be found in the figure legends or under the specific Methods heading.
Analysis of Density Sedimentation Data
Scans of western blots were analyzed in FIJI (ImageJ) (Schindelin et al., 2012) by drawing a box around the area of interest, inverting the image, and analyzing the plot profile of the area within the box. Pixel intensities were plotted against pixel distance. Images were unadjusted and boxes of equal pixel size were used for the comparisons of blots.
Image Analysis
Statistical comparisons between two groups for proximity ligation assay analyses were performed with GraphPad Prism software using a two-tailed unpaired t test. PDPK1-EGFP localization images were analyzed in batch mode using FIJI (ImageJ) software with the Intensity Ratio Nuclei Cytoplasm Tool. The experiment was performed in triplicate with at least four fields of view analyzed per replicate. Statistical analysis was performed with GraphPad Prism software 8.0.
Ontology and Categorization
Gene ontology (GO) enrichment analysis was performed with PANTHER 15.0 with Gene Ontology version 1.2, 2020-02-21 release (Mi et al., 2019a, 2019b; Mi and Thomas, 2009). Unless otherwise stated in the figure legend, analysis type is PANTHER Overrepresentation Test with “GO biological function complete” Annotation Dataset. Analysis was also performed using DAVID Bioinformatic Resource v 6.8 (Huang et al., 2009a, 2009b). Protein categorization was performed using PANTHER 15.0 Protein Class ontology tool, 2019_04 reference proteome. Dot plots of the GSEA and GO analyses were generated using Seaborn.
Identifying WIN and WBM Motifs
Motifs were identified using the MOTIF2 Search online tool available through GenomeNet at https://www.genome.jp/tools/motif/MOTIF2.html. Patterns used for searches are: WIN motif A-R-[AST]; WBM motif [ED]-[ED]-[IVL]-D-V-[VT].
RNA-Seq Data Analysis
After adaptor trimming by Cutadapt (Martin, 2011), RNA-Seq reads were aligned to the hg19 genome using STAR (Dobin et al., 2013) and quantified by featureCounts (Liao et al., 2014). Differential analysis were performed by DESeq2 (Love et al., 2014), which estimated the log2 fold changes, Wald test p-values, and adjusted p-values (FDR) by the Benjamini-Hochberg procedure. The significantly changed genes were chosen with the criteria FDR < 0.05.
Proteomic Data Analysis
This information can be found under the appropriate methods heading.
Supplementary Material
Highlights.
WIN site inhibitor C6 bidirectionally modulates the WDR5 interactome
N-terminal acetylation enhances affinity for the WIN site
The signaling kinase PDPK1 directly binds to the WIN site
PDPK1-WDR5 interaction contributes to the regulation of mitotic gene expression
ACKNOWLEDGMENTS
For reagents we thank David Cortez, Richard Young, and Feng Zhang. For assistance and advice we thank Monica Bomber, David Flaherty, Vivian Gama, Scott Hiebert, Emily Hodges, Brittany Matlock, Francis Prael III, Kristy Stengel, and Andrea Wojciechowski. dTAG47 was synthesized by the Vanderbilt Chemical Synthesis Core. The VUMC Flow Cytometry Shared Resource is supported by the Vanderbilt-Ingram Cancer Center (P30CA68485) and the Vanderbilt Digestive Disease Research Center (DK058404). The VANTAGE Shared Resource is supported by the CTSA Grant (5UL1 RR024975-03), the Vanderbilt-Ingram Cancer Center (P30 CA68485), the Vanderbilt Vision Center (P30 EY08126), and NIH/NCRR (G20 RR030956). The Nikon Center of Excellence within the Vanderbilt Cell Imaging Shared Resource is supported by NIH grants CA68485, DK20593, DK58404, DK59637, and EY08126. This work was supported by awards from the NIH/NCI, under Chemical Biology Consortium Contract No. HHSN261200800001E (to S.W.F. and W.P.T.), F31CA225065 (to A.D.G), T32CA009582 (to A.D.G.), CA200709 (to W.P.T.), and T32CA119925 (to A.M.W., and W.P.T.), as well as grants from the Robert J. Kleberg, Jr., and Helen C. Kleberg Foundation (to W.P.T. and S.W.F.), Edward P. Evans Foundation (to W.P.T.), Rally Foundation for Childhood Cancer Research Fellowship (to A.M.W.), Open Hands Overflowing Hearts co-funded research fellowship (to A.M.W.), and American Association for Cancer Research Basic Cancer Research Fellowship (to A.M.W.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108636.
DECLARATION OF INTERESTS
Two patents related to work presented in this paper are as follows: S.W.F., S.R. Stauffer, J.M. Salovich, W.P.T., F. Wang, J. Phan, E.T.O., inventors. WDR5 inhibitors and modulators. United States Patent US 10,501,466. 10 December 2019; S.W.F., S.R. Stauffer, W.P.T., E.T.O., J. Phan, F. Wang, K. Jeon, R.D. Gogliotti, inventors. WDR5 inhibitors and modulators. United States Patent US 10,160,763. 25 December 2018.
SUPPORTING CITATIONS
The following references appear in the supplemental information: Lorton et al. (2020); Migliori et al. (2012).
REFERENCES
- Aho ER, Wang J, Gogliotti RD, Howard GC, Phan J, Acharya P, Macdonald JD, Cheng K, Lorey SL, Lu B, et al. (2019a). Displacement of WDR5 from Chromatin by a WIN Site Inhibitor with Picomolar Affinity. Cell Rep. 26, 2916–2928.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aho ER, Weissmiller AM, Fesik SW, and Tansey WP (2019b). Targeting WDR5: A WINning Anti-Cancer Strategy? Epigenet. Insights 12, 2516865719865282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali A, Veeranki SN, Chinchole A, and Tyagi S (2017). MLL/WDR5 Complex Regulates Kif2A Localization to Ensure Chromosome Congression and Proper Spindle Assembly during Mitosis. Dev. Cell 41, 605–622.e7. [DOI] [PubMed] [Google Scholar]
- Avdic V, Zhang P, Lanouette S, Groulx A, Tremblay V, Brunzelle J, and Couture JF (2011a). Structural and biochemical insights into MLL1 core complex assembly. Structure 19, 101–108. [DOI] [PubMed] [Google Scholar]
- Avdic V, Zhang P, Lanouette S, Voronova A, Skerjanc I, and Couture JF (2011b). Fine-tuning the stimulation of MLL1 methyltransferase activity by a histone H3-based peptide mimetic. FASEB J. 25, 960–967. [DOI] [PubMed] [Google Scholar]
- Bayascas JR (2010). PDK1: the major transducer of PI 3-kinase actions. Curr. Top. Microbiol. Immunol 346, 9–29. [DOI] [PubMed] [Google Scholar]
- Bryan AF, Wang J, Howard GC, Guarnaccia AD, Woodley CM, Aho ER, Rellinger EJ, Matlock BK, Flaherty DK, Lorey SL, et al. (2020). WDR5 is a conserved regulator of protein synthesis gene expression. Nucleic Acids Res. 48, 2924–2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao F, Townsend EC, Karatas H, Xu J, Li L, Lee S, Liu L, Chen Y, Ouillette P, Zhu J, et al. (2014). Targeting MLL1 H3K4 methyltransferase activity in mixed-lineage leukemia. Mol. Cell 53, 247–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chacón Simon S, Wang F, Thomas LR, Phan J, Zhao B, Olejniczak ET, Macdonald JD, Shaw JG, Schlund C, Payne W, et al. (2020). Discovery of WD Repeat-Containing Protein 5 (WDR5)-MYC Inhibitors Using Fragment-Based Methods and Structure-Based Design. J. Med. Chem 63, 4315–4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Xie W, Gu P, Cai Q, Wang B, Xie Y, Dong W, He W, Zhong G, Lin T, and Huang J (2015). Upregulated WDR5 promotes proliferation, self-renewal and chemoresistance in bladder cancer via mediating H3K4 trimethylation. Sci. Rep 5, 8293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couture JF, Collazo E, and Trievel RC (2006). Molecular recognition of histone H3 by the WD40 protein WDR5. Nat. Struct. Mol. Biol 13, 698–703. [DOI] [PubMed] [Google Scholar]
- Cox J, and Mann M (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- Cui Z, Li H, Liang F, Mu C, Mu Y, Zhang X, and Liu J (2018). Effect of high WDR5 expression on the hepatocellular carcinoma prognosis. Oncol. Lett 15, 7864–7870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai X, Guo W, Zhan C, Liu X, Bai Z, and Yang Y (2015). WDR5 Expression Is Prognostic of Breast Cancer Outcome. PLoS ONE 10, e0124964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias J, Van Nguyen N, Georgiev P, Gaub A, Brettschneider J, Cusack S, Kadlec J, and Akhtar A (2014). Structural analysis of the KANSL1/WDR5/KANSL2 complex reveals that WDR5 is required for efficient assembly and chromatin targeting of the NSL complex. Genes Dev 28, 929–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong LQ, Ramos FJ, Wick MJ, Lim MA, Guo Z, Strong R, Richardson A, and Liu F (2002). Cloning and characterization of a testis and brain-specific isoform of mouse 3′-phosphoinositide-dependent protein kinase-1, mPDK-1 beta. Biochem. Biophys. Res. Commun 294, 136–144. [DOI] [PubMed] [Google Scholar]
- Dou Y, Milne TA, Ruthenburg AJ, Lee S, Lee JW, Verdine GL, Allis CD, and Roeder RG (2006). Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat. Struct. Mol. Biol 13, 713–719. [DOI] [PubMed] [Google Scholar]
- Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade M, Trachet E, Al-bassam M, Zheng X, Leopold WR, Pryer NK, and Toogood PL (2004). Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol. Cancer Ther 3, 1427–1438. [PubMed] [Google Scholar]
- Gagliardi PA, Puliafito A, and Primo L (2018). PDK1: At the crossroad of cancer signaling pathways. Semin. Cancer Biol 48, 27–35. [DOI] [PubMed] [Google Scholar]
- Ge Z, Song EJ, Kawasawa YI, Li J, Dovat S, and Song C (2016). WDR5 high expression and its effect on tumorigenesis in leukemia. Oncotarget 7, 37740–37754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grebien F, Vedadi M, Getlik M, Giambruno R, Grover A, Avellino R, Skucha A, Vittori S, Kuznetsova E, Smil D, et al. (2015). Pharmacological targeting of the Wdr5-MLL interaction in C/EBPa N-terminal leukemia. Nat. Chem. Biol 11, 571–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarnaccia AD, and Tansey WP (2018). Moonlighting with WDR5: A Cellular Multitasker. J. Clin. Med 7, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara K, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S, Tokunaga C, Avruch J, and Yonezawa K (2002). Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 110, 177–189. [DOI] [PubMed] [Google Scholar]
- Hauri S, Comoglio F, Seimiya M, Gerstung M, Glatter T, Hansen K, Aebersold R, Paro R, Gstaiger M, and Beisel C (2016). A High-Density Map for Navigating the Human Polycomb Complexome. Cell Rep. 17, 583–595. [DOI] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, and Lempicki RA (2009a). Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, and Lempicki RA (2009b). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Huang H, Sabari BR, Garcia BA, Allis CD, and Zhao Y (2014). SnapShot: histone modifications. Cell 159, 458–458.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang HT, Seo HS, Zhang T, Wang Y, Jiang B, Li Q, Buckley DL, Nabet B, Roberts JM, Paulk J, et al. (2017). MELK is not necessary for the proliferation of basal-like breast cancer cells. eLife 6, e26693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttlin EL, Bruckner RJ, Paulo JA, Cannon JR, Ting L, Baltier K, Colby G, Gebreab F, Gygi MP, Parzen H, et al. (2017). Architecture of the human interactome defines protein communities and disease networks. Nature 545, 505–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karatas H, Townsend EC, Bernard D, Dou Y, and Wang S (2010). Analysis of the binding of mixed lineage leukemia 1 (MLL1) and histone 3 peptides to WD repeat domain 5 (WDR5) for the design of inhibitors of the MLL1-WDR5 interaction. J. Med. Chem 53, 5179–5185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, and Sabatini DM (2002). mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 110, 163–175. [DOI] [PubMed] [Google Scholar]
- Koufaris C, and Kirmizis A (2020). N-Terminal Acetyltransferases Are Cancer-Essential Genes Prevalently Upregulated in Tumours. Cancers (Basel) 12, 2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai JS, and Herr W (1992). Ethidium bromide provides a simple tool for identifying genuine DNA-independent protein associations. Proc. Natl. Acad. Sci. USA 89, 6958–6962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange PF, Huesgen PF, Nguyen K, and Overall CM (2014). Annotating N termini for the human proteome project: N termini and Na-acetylation status differentiate stable cleaved protein species from degradation remnants in the human erythrocyte proteome. J. Proteome Res 13, 2028–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, and Tamayo P (2015). The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim MA, Kikani CK, Wick MJ, and Dong LQ (2003). Nuclear translocation of 3′-phosphoinositide-dependent protein kinase 1 (PDK-1): a potential regulatory mechanism for PDK-1 function. Proc. Natl. Acad. Sci. USA 100, 14006–14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorton BM, Harijan RK, Burgos ES, Bonanno JB, Almo SC, and Shechter D (2020). A binary arginine methylation switch on histone H3 arginine 2 regulates its interaction with WDR5. Biochemistry 59, 3696–3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J 17, 10–12. [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007). Phaser crystallographic software. J. Appl. Cryst 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Méndez J, and Stillman B (2000). Chromatin association of human origin recognition complex, cdc6, and minichromosome maintenance proteins during the cell cycle: assembly of prereplication complexes in late mitosis. Mol. Cell. Biol 20, 8602–8612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, and Thomas P (2009). PANTHER pathway: an ontology-based pathway database coupled with data analysis tools. Methods Mol. Biol 563, 123–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Muruganujan A, Ebert D, Huang X, and Thomas PD (2019a). PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res 47 (D1), D419–D426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Muruganujan A, Huang X, Ebert D, Mills C, Guo X, and Thomas PD (2019b). Protocol Update for large-scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nat. Protoc 14, 703–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliori V, Müller J, Phalke S, Low D, Bezzi M, Mok WC, Sahu SK, Gunaratne J, Capasso P, Bassi C, et al. (2012). Symmetric dimethylation of H3R2 is a newly identified histone mark that supports euchromatin maintenance. Nat. Struct. Mol. Biol 19, 136–144. [DOI] [PubMed] [Google Scholar]
- Mora A, Komander D, van Aalten DM, and Alessi DR (2004). PDK1, the master regulator of AGC kinase signal transduction. Semin. Cell Dev. Biol 15, 161–170. [DOI] [PubMed] [Google Scholar]
- Morgenstern JP, and Land H (1990). Advanced mammalian gene transfer: high titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res. 18, 3587–3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabet B, Roberts JM, Buckley DL, Paulk J, Dastjerdi S, Yang A, Leggett AL, Erb MA, Lawlor MA, Souza A, et al. (2018). The dTAG system for immediate and target-specific protein degradation. Nat. Chem. Biol 14, 431–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najafov A, Sommer EM, Axten JM, Deyoung MP, and Alessi DR (2011). Characterization of GSK2334470, a novel and highly specific inhibitor of PDK1. Biochem. J 433, 357–369. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Sakaue H, Nishizawa A, Matsuki Y, Gomi H, Watanabe E, Hiramatsua R, Tamamori-Adachi M, Kitajima S, Noda T, et al. (2008). PDK1 regulates cell proliferation and cell cycle progression through control of cyclin D1 and p27Kip1 expression. J. Biol. Chem 283, 17702–17711. [DOI] [PubMed] [Google Scholar]
- Nikolovska-Coleska Z, Wang R, Fang X, Pan H, Tomita Y, Li P, Roller PP, Krajewski K, Saito NG, Stuckey JA, and Wang S (2004). Development and optimization of a binding assay for the XIAP BIR3 domain using fluorescence polarization. Anal. Biochem 332, 261–273. [DOI] [PubMed] [Google Scholar]
- Odho Z, Southall SM, and Wilson JR (2010). Characterization of a novel WDR5-binding site that recruits RbBP5 through a conserved motif to enhance methylation of histone H3 lysine 4 by mixed lineage leukemia protein-1. J. Biol. Chem 285, 32967–32976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh E, Mark KG, Mocciaro A, Watson ER, Prabu JR, Cha DD, Kampmann M, Gamarra N, Zhou CY, and Rape M (2020). Gene expression and cell identity controlled by anaphase-promoting complex. Nature 579, 136–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel A, Dharmarajan V, and Cosgrove MS (2008a). Structure of WDR5 bound to mixed lineage leukemia protein-1 peptide. J. Biol. Chem 283, 32158–32161. [DOI] [PubMed] [Google Scholar]
- Patel A, Vought VE, Dharmarajan V, and Cosgrove MS (2008b). A conserved arginine-containing motif crucial for the assembly and enzymatic activity of the mixed lineage leukemia protein-1 core complex. J. Biol. Chem 283, 32162–32175. [DOI] [PubMed] [Google Scholar]
- Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M, et al. (2019). The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 47 (D1), D442–D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punzi S, Balestrieri C, D’Alesio C, Bossi D, Dellino GI, Gatti E, Pruneri G, Criscitiello C, Lovati G, Meliksetyan M, et al. (2019). WDR5 inhibition halts metastasis dissemination by repressing the mesenchymal phenotype of breast cancer cells. Breast Cancer Res. 21, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendran V, Kalita P, Shukla H, Kumar A, and Tripathi T (2018). Aminoacyl-tRNA synthetases: structure, function, and drug discovery. Int. J. Biol. Macromol 111, 400–414. [DOI] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ree R, Varland S, and Arnesen T (2018). Spotlight on protein N-terminal acetylation. Exp. Mol. Med 50, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts TC, Hart JR, Kaikkonen MU, Weinberg MS, Vogt PK, and Morris KV (2015). Quantification of nascent transcription by bromouridine immunocapture nuclear run-on RT-qPCR. Nat. Protoc 10, 1198–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruthenburg AJ, Wang W, Graybosch DM, Li H, Allis CD, Patel DJ, and Verdine GL (2006). Histone H3 recognition and presentation by the WDR5 module of the MLL1 complex. Nat. Struct. Mol. Biol 13, 704–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, Tempst P, and Sabatini DM (2004). Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol 14, 1296–1302. [DOI] [PubMed] [Google Scholar]
- Saxton RA, and Sabatini DM (2017). mTOR Signaling in Growth, Metabolism, and Disease. Cell 169, 361–371. [DOI] [PubMed] [Google Scholar]
- Scheid MP, Parsons M, and Woodgett JR (2005). Phosphoinositide-dependent phosphorylation of PDK1 regulates nuclear translocation. Mol. Cell. Biol 25, 2347–2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuetz A, Allali-Hassani A, Martín F, Loppnau P, Vedadi M, Bochkarev A, Plotnikov AN, Arrowsmith CH, and Min J (2006). Structural basis for molecular recognition and presentation of histone H3 by WDR5. EMBO J. 25, 4245–4252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song JJ, and Kingston RE (2008). WDR5 interacts with mixed lineage leukemia (MLL) protein via the histone H3-binding pocket. J. Biol. Chem 283, 35258–35264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas LR, Wang Q, Grieb BC, Phan J, Foshage AM, Sun Q, Olejniczak ET, Clark T, Dey S, Lorey S, et al. (2015). Interaction with WDR5 promotes target gene recognition and tumorigenesis by MYC. Mol. Cell 58, 440–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, and Cox J (2016). The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740. [DOI] [PubMed] [Google Scholar]
- Vassilev LT, Tovar C, Chen S, Knezevic D, Zhao X, Sun H, Heimbrook DC, and Chen L (2006). Selective small-molecule inhibitor reveals critical mitotic functions of human CDK1. Proc. Natl. Acad. Sci. USA 103, 10660–10665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilhais-Neto GC, Fournier M, Plassat JL, Sardiu ME, Saraf A, Garnier JM, Maruhashi M, Florens L, Washburn MP, and Pourquié O (2017). The WHHERE coactivator complex is required for retinoic acid-dependent regulation of embryonic symmetry. Nat. Commun 8, 728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Ahmad A, Li Y, Banerjee S, Kong D, and Sarkar FH (2010). Forkhead box M1 transcription factor: a novel target for cancer therapy. Cancer Treat. Rev 36, 151–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warde-Farley D, Donaldson SL, Comes O, Zuberi K, Badrawi R, Chao P, Franz M, Grouios C, Kazi F, Lopes CT, et al. (2010). The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res 38, W214–W220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weintraub AS, Li CH, Zamudio AV, Sigova AA, Hannett NM, Day DS, Abraham BJ, Cohen MA, Nabet B, Buckley DL, et al. (2017). YY1 Is a Structural Regulator of Enhancer-Promoter Loops. Cell 171, 1573–1588.e28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu MZ, Tsai YP, Yang MH, Huang CH, Chang SY, Chang CC, Teng SC, and Wu KJ (2011). Interplay between HDAC3 and WDR5 is essential for hypoxia-induced epithelial-mesenchymal transition. Mol. Cell 43, 811–822. [DOI] [PubMed] [Google Scholar]
- Yeom J, Ju S, Choi Y, Paek E, and Lee C (2017). Comprehensive analysis of human protein N-termini enables assessment of various protein forms. Sci. Rep 7, 6599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Lee H, Brunzelle JS, and Couture JF (2012). The plasticity of WDR5 peptide-binding cleft enables the binding of the SET1 family of histone methyltransferases. Nucleic Acids Res. 40, 4237–4246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Sammons MA, Donahue G, Dou Z, Vedadi M, Getlik M, Barsyte-Lovejoy D, Al-awar R, Katona BW, Shilatifard A, et al. (2015). Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth. Nature 525, 206–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org/cgi/GetDataset) via the PRIDE (Perez-Riverol et al., 2019) partner repository. The accession number for the proteomics data reported in this paper is PRIDE: PXD019209. RNA-Seq data are deposited at the Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/). The accession number for the RNA-Seq data reported in this paper is GEO: GSE150400. X-ray crystal structure is deposited at the Protein Data Bank (https://www.rcsb.org). The accession number for the structural data reported in this paper is PDB: 6WJQ.