

Abstract
The mechanism by which transmembrane reductases use a single pair of cysteine residues to relay electrons between protein substrates across biological membranes is a long-standing mystery in thiol-redox biochemistry. Here we show the NMR structure of a reduced-state mimic of archaeal CcdA, a protein that transfers electrons across the inner membrane, by using a redox-active NMR sample. The two cysteine positions in CcdA are separated by 20 Å. Whereas one is accessible to the cytoplasm, the other resides in the protein core, thus implying that conformational exchange is required for periplasmic accessibility. In vivo mixed disulfide–trapping experiments validated the functional positioning of the cysteines, and in vitro accessibility results confirmed conformational exchange. Our NMR and functional data together show the existence of multiple conformational states and suggest a four-state model for relaying electrons from cytosolic to periplasmic redox substrates.
The bacterial periplasm is oxidizing, whereas the cytoplasm is reducing. Transport of reducing power from the cytoplasm to the periplasm by a transmembrane (TM) electron transporter is essential to periplasmic thiol-redox activity. The best-known TM reductase is Escherichia coli (Ec) DsbD, which has a TM domain (DsbDβ) and two periplasmic domains (DsbDα and DsbDγ), each containing a redox-active cysteine pair1,2 (Fig. 1a). Electrons are transferred from cytoplasmic thioredoxin A (TrxA) to DsbDβ, relayed across the membrane and through its periplasmic domains, and transferred to various periplasmic targets to maintain their reductive activity3–8. Redox activities (and their associated proteins) include protein disulfide isomerization (DsbC), cytochrome c maturation (CcmG) and defense against oxidative stress (DsbG)9–11.
Figure 1.
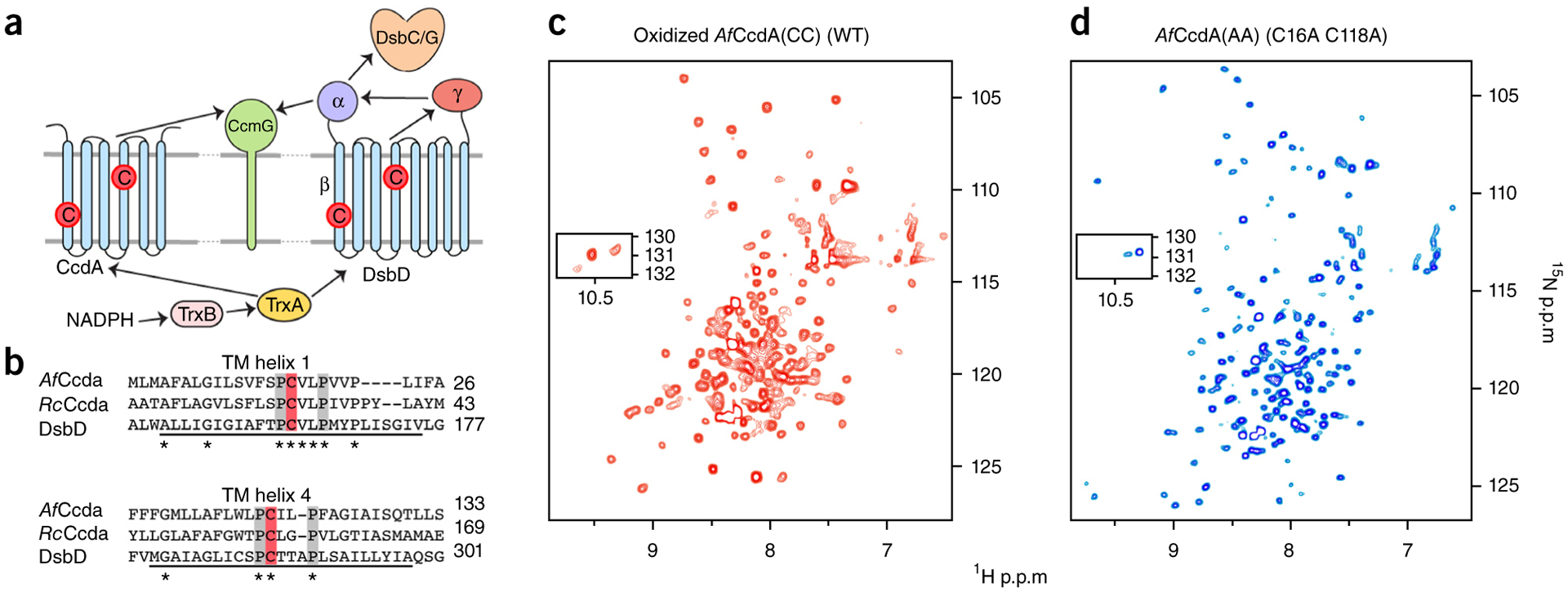
Transmembrane reductase topology and redox-active NMR system. (a) Schematic of CcdA and DsbD topology and reductive pathway from cytoplasmic NADPH to periplasmic targets via CcdA or DsbD. Arrows indicate the flow of two electrons, TrxB indicates Trx reductase, and Cs in red circles denote the redox-active cysteines. Slash denotes ‘or’. (b) Multiple sequence alignments (ClustalW2) of predicted TM helices 1 and 4 from A. fulgidus and R. capsulatus (Rc) CcdA and DsbD. Conserved residues are marked with an asterisk, functional cysteines are boxed in red, and the conserved flanking prolines are in gray. The DsbD TM helix prediction is underlined14. (c,d) 1H-15N TROSY-HSQC spectra of AfCcdA(CC) (oxidized) (c) and AfCcdA(AA) (reduced mimic) (d), recorded at 600 MHz.
CcdA is a minimal homolog of DsbDβ that has six TM helices instead of eight (DsbDβ) and can also reduce CcmG12,13 (Fig. 1a). Another DsbD homolog, ScsB, found in Salmonella typhimurium, also lacks the last two TM helices but maintains similar periplasmic domains and functionality, thus suggesting that the last two TM helices are dispensable in the function of these DsbD homologs14,15. Separate DsbDα homologs exist in many bacteria with CcdA only15. Furthermore, DsbD α, β and γ domains that are physically split and combined individually can reconstitute the function of DsbD3. Therefore, it has been proposed that CcdA has evolved into DsbD through fusion of the two periplasmic proteins to the TM domain, thus allowing greater electron-transfer efficiency9,12.
The transporter-like architecture of DsbDβ has been suggested from functional mutagenesis and biochemical studies probing solvent accessibility16,17. The two functional cysteines of DsbDβ are on predicted TM helices 1 and 4 (Fig. 1a and Supplementary Fig. 1). The sequence surrounding the cysteines is highly conserved, particularly the proline residues flanking each cysteine (Fig. 1b). Each proline in the PCX(2–3)P active sites is important for cysteine accessibility and redox activity16,18,19. DsbDβ has an inverted pseudo-symmetry, thus allowing access to the functional cysteines from both sides of the membrane16,20, which in turn allows the two cysteines to cycle through oxidized and reduced states and thus pass reducing power into the periplasm.
There are no known structures of any TM reductases. The TM reductases use no cofactors, bind soluble protein substrates on both sides of the membrane and must transfer reducing power via a single cysteine pair without disrupting membrane integrity21. To elucidate the molecular architecture allowing this mechanism, we chose the minimalist CcdA (190 amino acids) from the archaeon Archaeoglobus fulgidus (AfCcdA) for structure determination by NMR. AfCcdA with EcDsbDα fused at its N terminus (Dα-AfCcdA (Supplementary Fig. 1); DsbDα is removed during purification) can be produced in sufficient, stable quantities when expressed in a ΔtrxB strain22. In this strain, oxidized TrxA accumulates, owing to the thioredoxin reductase (TrxB) knockout; therefore, the AfCcdA disulfide bond is maintained. AfCcdA is highly homologous to the functionally characterized Rhodobacter capsulatus CcdA and DsbD around the functional cysteines12 (Fig. 1b). Its structure thus should be representative of the TM reductase family. We set out to reveal the molecular architecture of the TM reductase by characterizing the structure of the AfCcdA with NMR methods.
RESULTS
A functionally relevant NMR sample of AfCcdA
AfCcdA was solubilized and purified in dodecylphosphocholine (DPC) micelles. Several studies have used fos-choline detergents to reconstitute membrane-embedded enzymes and solute carriers that are both functional and able to generate high-resolution NMR spectra23–25. The 2D 1H-15N correlation spectrum of oxidized AfCcdA is typical of a helical membrane protein of its size (Fig. 1c). Reduction of the protein with Tris(2-carboxyethyl)phosphine (TCEP; Supplementary Fig. 2) dramatically changed the NMR spectrum, thus indicating that a global conformational change occurs just by the breaking of one disulfide bond. Removal of the disulfide bond by double mutation (C16A C118A; denoted AfCcdA(AA)) produced an equivalent spectrum (Fig. 1d). After removal of TCEP by dialysis, the reduced spectrum returned to the oxidized form (Supplementary Fig. 2). That AfCcdA could interchange between redox states in our detergent micelle conditions indicated that the protein purified for structural study was redox active.
NMR structure of the reduced-state mimic of AfCcdA
The reduced state represented by AfCcdA(AA) generated sufficiently high-quality NMR spectra to be chosen as the structural target. We determined the structure of AfCcdA(AA) by using 521 local and 101 long-range distance restraints derived from NOE measurements and independently validated them by paramagnetic relaxation enhancements (PREs) from four single spin labels (Supplementary Fig. 3a). We used the PRE restraints only for qualitative validation of the NOE-derived structure because AfCcdA displayed intrinsic conformational exchange (described below). Exchange with an alternate conformation, even as a small percentage, could lead to substantial PRE if the time scale of exchange were sufficiently fast. Indeed, for some spin-label positions, we observed weaker PREs at some remote positions in the structure (Supplementary Fig. 3a). These effects could be attributed to either equilibrium conformational exchange or transient protein-protein aggregation. The 15 lowest-energy structures out of 75 calculated converged to an r.m.s. deviation of 0.971 Å and 1.553 Å for backbone and all heavy atoms, respectively, for the structured regions of the protein (Fig. 2a and Table 1). As expected from sequence-based topology prediction, AfCcdA has six TM helices. The distribution of charged and hydrophobic residues on the protein periphery indicates that the structure would partition favorably in a 30-Å hydrophobic lipid bilayer (Fig. 2b). The architecture of AfCcdA is not represented in the Protein Data Bank.
Figure 2.
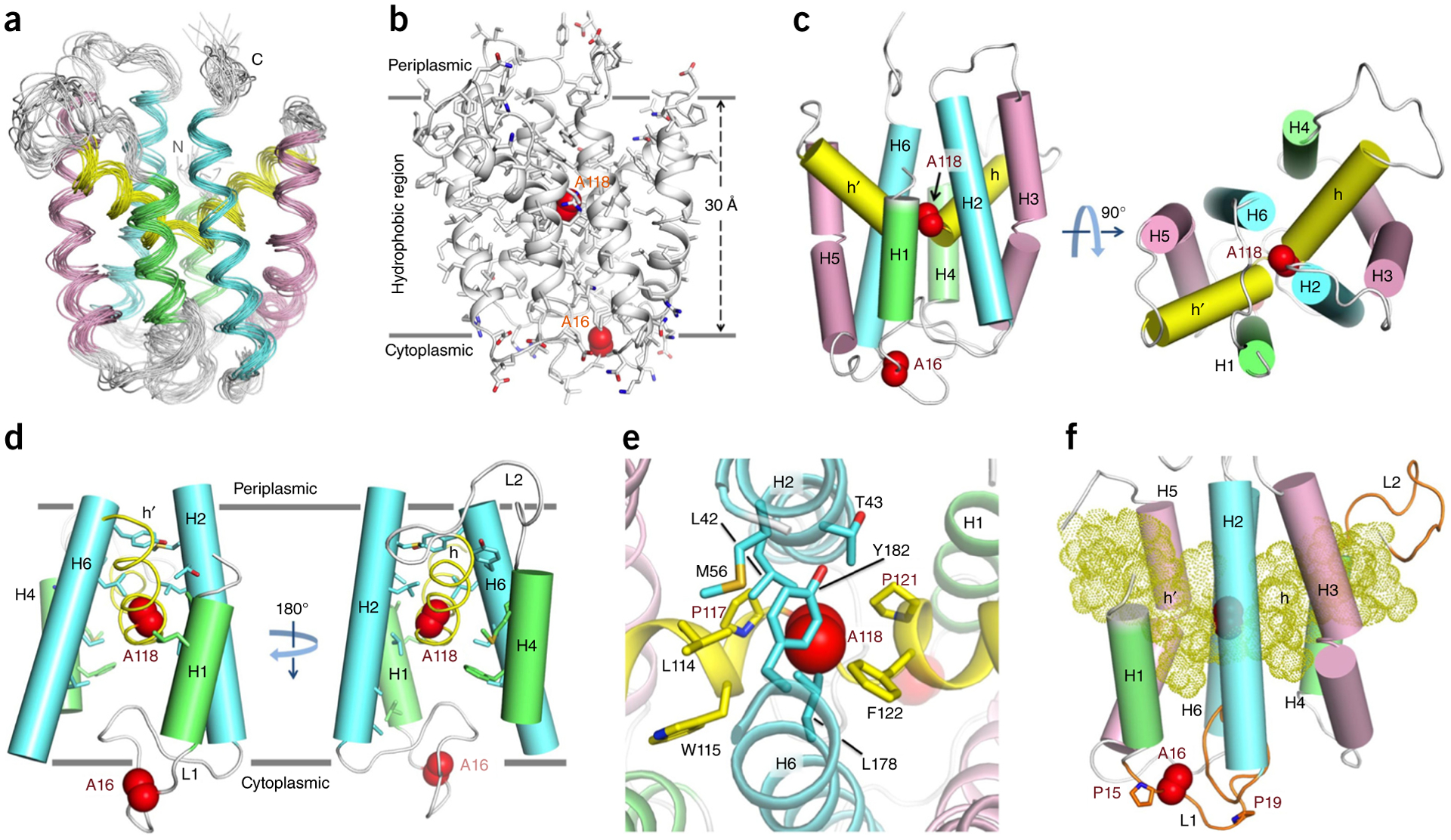
Structure of the transmembrane reductase AfCcdA. (a) Ensemble of 15 low-energy structures calculated with NMR-derived restraints (Table 1). (b) Representative structure showing the charged and polar amino acid distribution. Side chains are shown in gray except arginine, lysine, aspartate, glutamate, asparagine and glutamine. Two solid lines indicate the hydrophobic region, suggesting the position of AfCcdA in the lipid bilayer. C16A and C118A are shown as red spheres. (c) Cylinder representation illustrating AfCcdA structural symmetry. Helical segments related by rotational symmetry have the same color. (d) Cut-away side view showing the cytoplasmic splaying of the H2 and H6 central helices. (e) Protection of C118A by hydrophobic residues from the periplasmic side. (f) Side view showing the two long loops (orange). L1 is 16 residues long and contains C16A. L2 is 12 residues long. The internal helical segments are shown as dots.
Table 1.
NMR and refinement statistics
| AfCcdA(AA) | |
|---|---|
| NMR distance and dihedral constraints | |
| Distance constraints | |
| Total NOE | 622 |
| Intraresidue | 125 |
| Inter-residue | 497 |
| Sequential (|i - j| = 1) | 232 |
| Medium range (2 ≤ |i - j| ≤ 4) | 164 |
| Long range (|i - j| ≥ 5) | 101 |
| Total dihedral-angle restraints | 238 |
| ϕ | 119 |
| ψ | 119 |
| Structure statistics | |
| Violations (mean ± s.d.) | |
| Distance constraints (Å) | 0.147 ± 0.006 |
| Dihedral-angle constraints (°) | 0.352 ± 0.026 |
| Max. dihedral-angle violation (°) | 1.80 |
| Max. distance-constraint violation (A) | 1.07 |
| Deviations from idealized geometry | |
| Bond lengths (Å) | 0.006 ± 0.000 |
| Bond angles (°) | 0.863 ± 0.023 |
| Impropers (°) | 0.704 ± 0.027 |
| Average pairwise r.m.s. deviation (Å)a | |
| Heavy | 1.553 |
| Backbone | 0.971 |
Statistics are calculated and averaged over an ensemble of the 15 lowest-energy structures out of 75 calculated structures. The precision of the atomic coordinates is defined as the average r.m.s. difference between the 15 final structures and their mean coordinates. The calculation includes only the structured regions of the protein: residues 4–12, 31–49, 58–77, 84–94, 108–117, 120–130, 137–156 and 165–183.
AfCcdA(AA) has six TM helices, labeled from the N-terminal H1 to the C-terminal H6 (Fig. 2c), arranged in a helical-bundle architecture. The two central TM helices H2 and H6 are ~19 residues long. H3 and H5 are longer, with ~22 residues, but are severely kinked. H1 and H4 are short TM helices with only ~11 residues. Contrary to the TM topology prediction (Supplementary Fig. 1b), the structure has an unusual additional feature. The predicted TM H4 is instead two helical segments, broken by the prolines flanking C118A and forming a V-shaped internal helix. These helical segments, h and h′, are sandwiched by H1 and H3 on one side and H4 and H6 on the other. Conformation and orientation comparisons of the helical segments identified a quasi-two-fold rotational symmetry relating H1 to H4, H2 to H6, H3 to H5 and h to h′ (Fig. 2c).
Positions of the two cysteines in the reduced state
The C16A and C118A positions in the reduced-state structure are ~20 Å apart. This distance was unexpected because the two cysteines must be close (2.5 Å) to form the disulfide bond in the oxidized state. C16A is three residues C terminal to H1 on the protein periphery. C118A, however, is at the center of the protein core, sandwiched between the central TM helices H2 and H6. C118A also resides on the two-fold-symmetry axis, around which the helical segments fold (Fig. 2c). The reduced-state structure shows overall greater accessibility of the protein core from the cytoplasmic side because the central TM helices H2 and H6 associate near the periplasmic side and splay apart toward the cytoplasmic side (Fig. 2d). C118 is clearly the less accessible cysteine because its position is covered by an array of hydrophobic residues, which would block its access from the periplasmic side (Fig. 2e). C118 needs to be accessible at certain steps of the transport cycle requiring major rearrangements of the V-shaped internal helix and the surrounding TM helices. There are two relatively long loops that might function to allow these movements: loop L1, between H1 and H2, which contains the first cysteine, and loop L2, which connects H4 and h, the internal segment preceding the second cysteine (Fig. 2d,f). Whereas 90% of L2 residues were assigned, only ~70% of L1 residues were assigned. The relatively undefined structure of L1 could be attributed to the lack of NOE restraints. Rearrangement of the TM and internal helices and the loops may all be involved in accommodating alternating-access conformations with substrates for reductase function. The completely different oxidized-state spectrum supports such global conformational change (Fig. 1c,d).
The structure is consistent with in vivo cysteine reactivity
The positions of the cysteine-to-alanine mutations in AfCcdA(AA) suggested that C16 would interact with cytoplasmic Trx, and C118 would interact with the periplasmic substrate. We therefore tested the specific cysteine reactivity in vivo. First, we showed that AfCcdA could receive electrons from TrxA (like DsbD) and pass them to Af1675, a new Trx-like envelope protein identified in A. fulgidus (renamed AfTrxE), when the proteins were coexpressed in E. coli (Supplementary Fig. 4). We similarly tested the specific cysteine reactivity with single-cysteine mutants, AfCcdA C16A (AC) and AfCcdA C118A (CA), to trap the mixed disulfide intermediates with TrxA and AfTrxE (Fig. 3). As expected from the structure, AfCcdA(CA) trapped TrxA, and AfCcdA(AC) trapped AfTrxE. This trapping experiment provided direct evidence for the in vivo functional relevance of our AfCcdA(AA) structure and suggested that C118 becomes periplasm exposed. Although a previous in vitro study has proposed that only the C16 equivalent in DsbDβ reacts with both TrxA and DsbDγ (ref. 7), the specific reactivities of the AfCcdA cysteines shown in vivo in this study are consistent with those of comparable complexes detected between DsbDβ and its substrates shown in our previous in vivo studies3,16.
Figure 3.
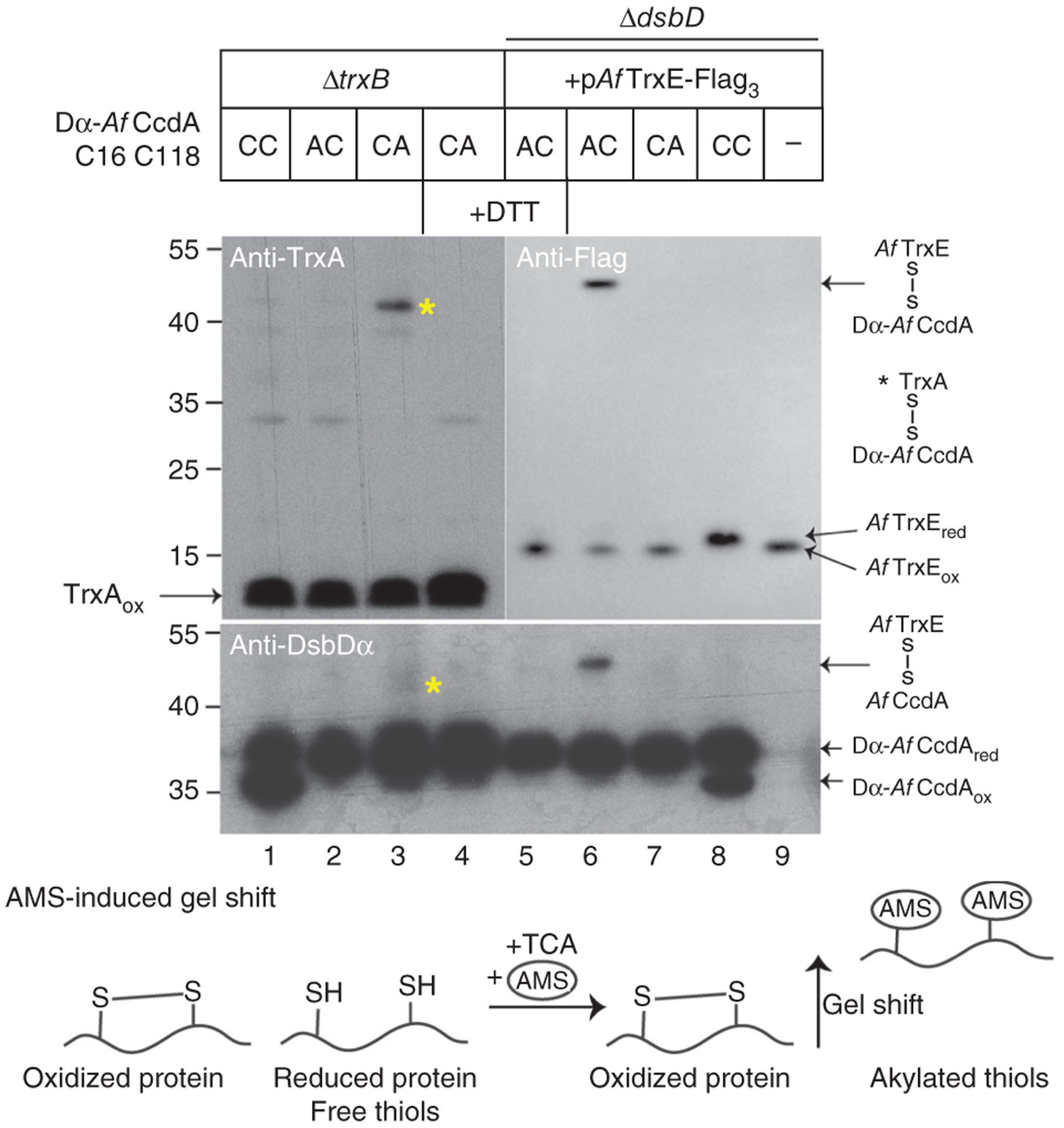
AfCcdA cysteine reactivity and specificity. In vivo reactivity of AfCcdA with TrxA and AfTrxE. Expression of Dα-AfCcdA variants in the E. coli ΔtrxB strain (lanes 1–4). Coexpression of Dα-AfCcdA and AfTrxE in the ΔdsbD strain (lanes 5–9). Each protein was detected with the indicated antibodies. Redox states were determined by 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid (AMS)-induced gel shift, as shown in the schematic. Red, reduced; ox, oxidized; TCA, trichloroacetic acid.
In vitro cysteine reactivity indicates an excited reduced state
Both cysteines of DsbDβ are accessible to small-molecule modification in the reduced state16,17. If this were also true for AfCcdA, it would imply that the reduced state, in which the C118 site is inaccessible, needs to sample another state that exposes it. To test this property in AfCcdA, we purified the AfCcdA(AC) and (CA) mutants and labeled them with 5-kDa methoxypolyethylene glycol maleimide (malPEG) in our NMR conditions22 (Fig. 4a). We monitored labeling over time and detected it by gel shift (Fig. 4b). We found that within 60 min, C16 of AfCcdA(CA) reacted completely with malPEG. Despite its position in the TM core, C118 of AfCcdA(AC) also reacted completely with malPEG, consistently with the accessibility previously observed in DsbDβ (refs. 16,17). Whereas the labeling for both mutants was complete after 60 min, it was evident from the early time points (for example, at 1 min) that the labeling of CcdA(AC) was slower than that of CcdA(CA) (Supplementary Fig. 5), results consistent with the decreased accessibility to the periplasmic cysteine site (C118) in the reduced-state structure.
Figure 4.
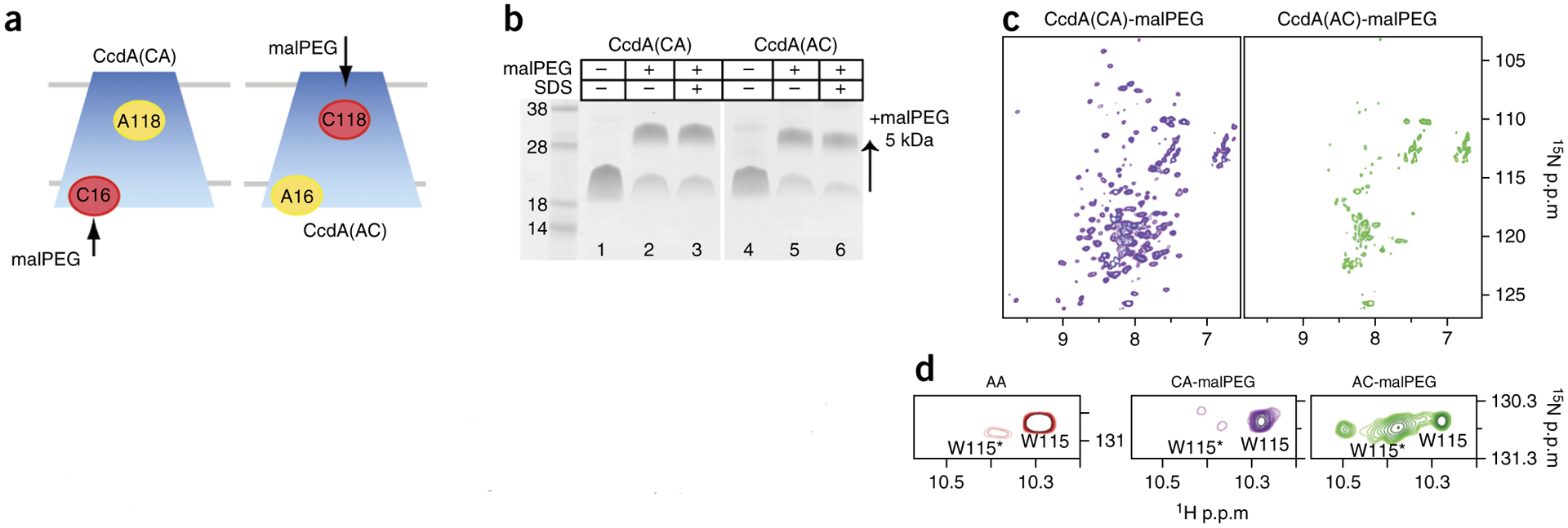
Cysteine accessibility by malPEG labeling. (a) In vitro labeling schematic of AfCcdA mutants with 5-kDa malPEG. (b) SDS-PAGE of AfCcdA(CA) and (AC) malPEG labeling after 60 min (lanes 2 and 5) versus negative (lanes 1 and 4) and positive (lanes 3 and 6) controls. (c) 1H-15N TROSY-HSQC spectra of malPEG-labeled AfCcdA(AC) (purple) and AfCcdA(CA) (green). (d) Major W115 and minor W115* side chain indole peaks.
The malPEG labeling results suggested that reduced AfCcdA must undergo transient conformational exchange to a C118-accessible state. To address this hypothesis, we recorded NMR spectra of malPEG-labeled AfCcdA to determine the effect of labeling on the AfCcdA structure. The AfCcdA(CA)-malPEG spectrum was nearly identical to that of AfCcdA(AA) (Figs. 4c and 1d). Labeling C16 with the large PEG molecule did not disturb the reduced-state structure, consistently with C16 being positioned in the open cytoplasmic face of the protein. Conversely, the spectrum of AfCcdA(AC)-malPEG was dramatically different, and the majority of resonances were either missing or diminished (Fig. 4c). This suggested that the malPEG reaction chemically trapped an excited state of reduced AfCcdA. This state appeared to be structurally unstable because its NMR spectrum showed severe exchange broadening.
W115 is adjacent to C118 in the internal helix and experiences conformational changes around this cysteine site. The W115 side chain indole amine resonance therefore should report multiple functional conformations of AfCcdA. In the AfCcdA(AA) spectrum, the W115 indole amine had a dominant peak at 10.29 p.p.m. and a minor peak at 10.38 p.p.m. (denoted W115*) (Fig. 4d). The W115 peak distribution in AfCcdA(CA)-malPEG was similar to that of AfCcdA(AA), thus indicating little change at C118. In AfCcdA(AC)-malPEG, however, the minor-peak relative intensity increased, and an additional peak appeared (unassigned). The W115 peak distributions indicated that a small reduced-state population naturally samples a conformation that is trapped by the malPEG linkage at C118. AfCcdA must therefore be a dynamic protein, exchanging major and minor conformations through its functional cycle.
DISCUSSION
The NMR structure of AfCcdA(AA) is consistent with results of our functional tests, both in vivo and in vitro, thus indicating that this structure represents a relevant conformation in the reductase mechanism. On the basis of the structural and functional data, we propose a four-state mechanism for transporting reducing power across the membrane (Fig. 5).
Figure 5.
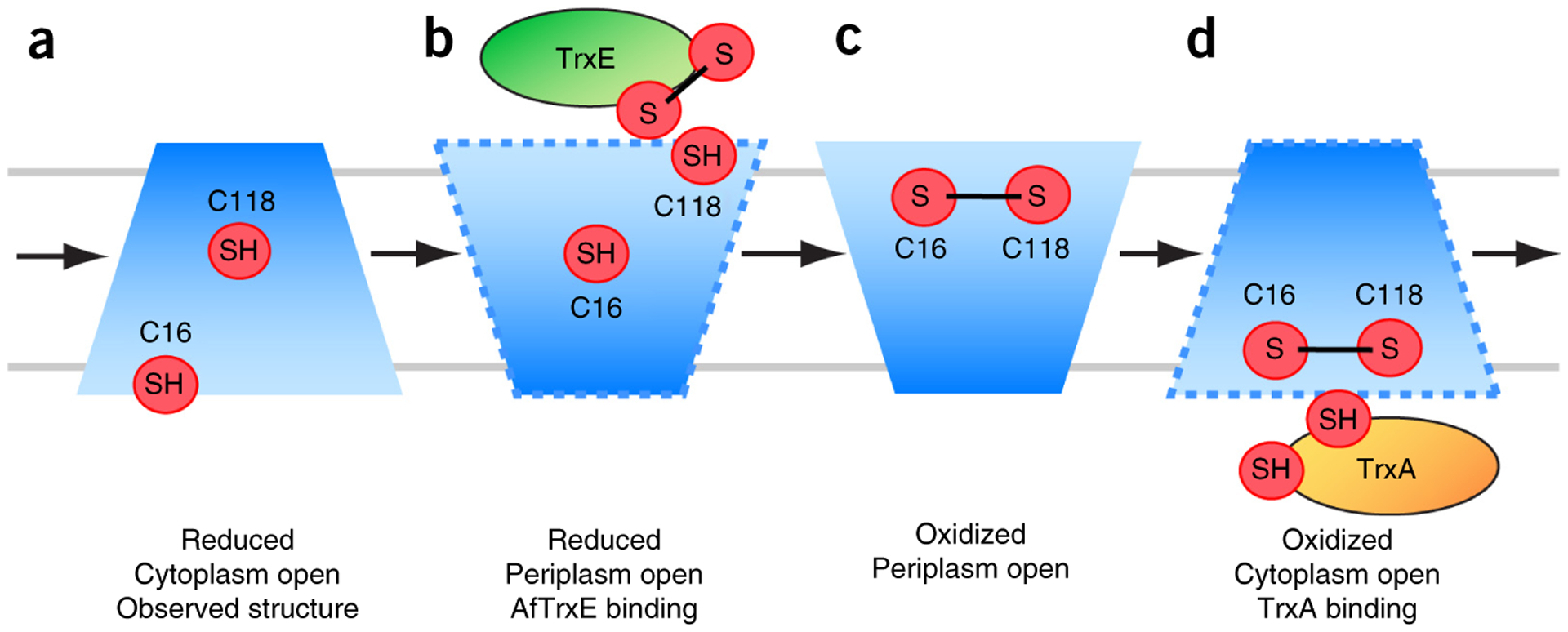
Four-state transmembrane reductase mechanism. (a) CcdA is in a ground reduced state after reduction by cytoplasmic TrxA. The core is in the cytoplasm-open state, with C16 cytoplasm exposed, and C118 in the middle of the TM core. (b) The excited reduced state, in which C118 becomes accessible to the periplasmic substrate (TrxE) and C16 moves into the protein core. (c) After oxidation by TrxE, CcdA is in a ground oxidized state, in which the core is periplasmic open. (d) Reduced Trx may stabilize an excited oxidized state that opens CcdA toward the cytoplasm to access the disulfide-bonded C16 and C118. TrxA reduces CcdA, and the cycle repeats.
The AfCcdA(AA) structure represents a reduced, cytoplasm-open state (Fig. 5a). This state was sufficiently stable for structure determination and thus should be a low-energy or ground reduced state. After reduction by cytoplasmic TrxA, C16 remains near the cytoplasm, and C118 is buried in the TM core. The next step in the reduction pathway requires AfCcdA to reduce AfTrxE via interaction with C118. Because the C118 is inaccessible from the periplasm, an excited reduced state is required to expose C118 to the periplasm, as evidenced by the malPEG-labeling experiments (Fig. 4b–d). The severe exchange broadening of the AfCcdA(AC)-malPEG NMR resonances suggested that this chemically trapped state is structurally unstable. We believe that AfTrxE binding at the periplasmic face may stabilize this excited reduced state (Fig. 5b). Next, C118 forms a mixed disulfide bond with AfTrxE (Fig. 3), which must be resolved by C16, thus creating the C16-C118 disulfide bond (Fig. 5c). Opening toward the periplasm would therefore also require H1 to slide up, bringing C16 close to C118. By the inverted-access argument, we propose that the oxidized state represented by the spectrum in Figure 1c is the ground oxidized state, in which the disulfide-bonded cysteines are mostly inaccessible from the cytoplasm. Moreover, an excited oxidized state should exist to open the protein core to the cytoplasm, thus allowing the disulfide bond to move to the cytoplasmic face (Fig. 5d). In this case, TrxA binding should stabilize the excited oxidized state to carry out the reducing reaction.
Although we do not have direct structural evidence showing inverted substrate accessibility of the ground reduced and oxidized states, there is indirect evidence in the limited NMR data recorded for the AfCcdA(CC) sample. One piece of evidence comes from the backbone resonance assignment and chemical shift–derived secondary structures (with TALOS+26). An obvious structural feature of the ground reduced state is that the two central helices H2 and H6 associate near their C-terminal ends and splay apart toward their N-terminal ends (Fig. 2d). This feature is partially responsible for stabilizing the structure around C118 as well as the C-terminal ends of H2 and H6. If the inverted-access model were true, this interaction would be the opposite in the ground oxidized state. For both H2 and H6, assigned 13Cα and 13Cβ chemical shifts indicated that the two helices are more structured at the C-terminal end (periplasmic side) in the reduced state than the oxidized state, and for H2, the N-terminal end (cytoplasmic side) is more structured in the oxidized state than the reduced state (Supplementary Fig. 1b). Moreover, the well-structured helical segment (h) preceding C118 could not be assigned, owing to disappearance of these peaks, which was probably due to unstable structure and exchange broadening. These results are consistent with a model of the ground oxidized state in which H2 and H6 dissociate at the periplasmic end, destabilizing the structures in the region including the h segment and the C-terminal ends of H2 and H6. Destabilization of the region would also be consistent with an overall periplasmic open conformation in the ground oxidized state.
Solvent-accessibility data obtained indirectly with Gd-DOTA (a water-soluble paramagnetic probe) provided additional insight into the ground oxidized conformation. For both reduced and oxidized AfCcdA, we measured PRE upon Gd-DOTA titration, using the 2D TROSY-HSQC spectra as readout. Because of the severe resonance overlap in the 2D spectrum, we could assign PREs of both reduced and oxidized states for only a small number of residues, and among them a few residues showed interesting properties. Leu73 of H3 and Thr149 of H5, both in the cytoplasmic and the more open part of the reduced structure, showed strong PRE (~0.0) in the reduced state but weak PRE (>0.7) in the oxidized state (Supplementary Fig. 3b), thus suggesting that a structural rearrangement makes them less accessible in the oxidized state. In contrast, Arg61 of H3 near the periplasmic side showed strong PRE (~0.0) in the oxidized state but much weaker PRE (~0.5) in the reduced state.
The most striking feature of our reduced structure is the V-shaped internal helix (helical segments h and h′) in place of the predicted TM helix 4 (Fig. 1b). H4 is instead in the predicted H3-H4 loop (Supplementary Fig. 1b). H4 does not show sequence conservation but shows quite strong amphipathicity20. These helices have properties unlike those of traditional TM helices, which may allow them to move through the TM or switch between order and disorder. These features would be very important in the dynamic conformational change toward a periplasmic open state, which may have architecture antisymmetrical from that of the current AfCcdA(AA) structure. Indeed, DsbDβ shows inverse pseudosymmetrical accessibility in the TM1 and TM4 helices16,17.
As described above, the V-shaped helix is broken into two helical segments by the conserved and functionally important prolines flanking C118. We speculate that the role of the prolines is to provide the flexibility around C118, for example, the PCX(2–3)P sequence could function as a pivot allowing the h and h′ segments to switch between the ground and excited reduced states. Similarly, the conserved proline sequence surrounding C16 should be important for the structural rearrangement of H1 and L1 when cycling between the reduced and oxidized states. It has been shown for DsbDβ that when the prolines around the cysteine equivalent to C118 are mutated, the cysteine becomes less accessible, whereas the opposite effect has been observed when the prolines around the cysteine of C16 equivalent are mutated16.
The proposed four-state mechanism accommodates the essential properties of a transmembrane reductase: relaying reducing electrons across the membrane in a substrate-specific pathway while maintaining the integrity of the membrane. The mechanism is similar to the alternating-access mechanism that governs the activity of most solute transporters27–30. CcdA is unusual in that it does not transport small molecules but instead ‘transports’ two reactive cysteines back and forth in a manner coordinated with protein-substrate access on alternating sides of the membrane.
METHODS
Methods and any associated references are available in the online version of the paper.
ONLINE METHODS
Bacterial strains and media.
The bacterial strains used in this study are listed in Supplementary Table 1a. The E. coli BL21(DE3) derivative C43(DE3) was used as a background strain31. The trxB-deletion mutant (SEN212) and dsbD–transposon insertion mutant (SEN256) were constructed with the alleles from previous studies5,32 by P1 phage transduction. The cells were grown in LB medium (for in vivo tests) and M9 minimal medium (for NMR samples) at 37 °C. When needed, 200 μg/mL of ampicillin, 30 μg/mL of chloramphenicol, 100 μg/mL of spectinomycin, or 50 μg/mL kanamycin was added.
Plasmids.
Plasmids used in this study are shown in Supplementary Table 1a. Primers used to create the plasmids are listed in Supplementary Table 1b. E. coli codon-optimized A. fulgidus ccdA was synthesized (Epoch Biolabs) and cloned in pET22b plasmid with the primers AfCcdA(NdeI)_F and AfCcdA(XhoI)_R, thus yielding pSC180. However, the expression level of AfCcdA in SEN212 was not sufficient for structural determination by NMR methods. Because AfCcdA is a heterologous protein expressed in E. coli and notably has short loops between transmembrane helices, we reasoned that an EcDsbDα (the N-terminal soluble domain of EcDsbDβ, a homologus domain of CcdA) fusion including the signal sequence at the N terminus would aid in the expression and membrane insertion of AfCcdA. Therefore, we generated an EcDsbDα-AfCcdA protein fusion (Supplementary Fig. 1). First, the gene encoding EcDsbDα with a thrombin-cleavage site at its C terminus was cloned into pET28a with the primers DsbDα(NcoI)_F and DsbDα(BamHI)_R, thus yielding pET28a:: DsbDαthrombin (pSC124). Then A. fulgidus ccdA was cloned into pSC124 with the primers AfCcdA(BamHI)_F and AfCcdA(XhoI)_R, thus generating pSC148. In AfCcdA(BamHI)_F, the nucleotides encoding the peptide (QEQPTAQL) from the linker region between EcDsbDα and EcDsbDβ (ref. 18) were included before AfCcdA. During protein purification in detergent micelles, the added length from the linker region was necessary to increase thrombin digestion of the fusion protein.
pSC148AC and pSC148CA were generated by site-directed mutagenesis with the primer pairs (AF9Ccda C1F and AF9Ccda C1R) and (AF9Ccda C2F and AF9Ccda C2R), respectively. pSC148AA was made in two steps with both primer sets. Single-cysteine mutants of pSC148AA for PRE measurements were generated with the primers listed in Supplementary Table 1b.
The Af1675 gene (described below) was PCR-amplified with the primers Af1675(NheI)_F and Af1675(XbaI)_R from the genomic DNA of A. fulgidus DSM4304 (ATCC) and cloned into pSC209 (ref. 15), which is a pBAD43 derivative containing the triple Flag tag (3× Flag), thus generating pSC175.
NMR sample preparation.
pSC148 and its variants were transformed into SEN212 cells and grown in isotopically labeled M9 minimal medium at 37 °C until the optical density at 600 nm (OD600) reached 0.6–0.7. After induction with 0.2 mM isopropyl-β-d-thiogalactopyranoside (IPTG), protein was expressed for 3 h at 37 °C. Protein purification was performed at 4C, unless noted. All buffer pH values were measured at room temperature (RT). Cells were harvested by centrifugation and resuspended in buffer A (20 mM Tris base, 300 mM NaCl, pH 8). Cells were frozen, thawed, and lysed in a microfluidizer with the addition of 1 μL benzonase (Novagen). The membrane component of the cell lysate was collected by centrifugation at 150,000g for 1 h and homogenized in buffer A. 1% n-dodecylphosphocholine (DPC) (Anatrace) was added, and membrane extraction proceeded for 2 h at 4 °C with moderate stirring. Insoluble material was removed by centrifugation at 150,000g for 30 min. The clarified, solubilized membrane component was incubated with Ni-NTA agarose beads (Qiagen) for 2 h at 4 °C with gentle agitation and then added to a gravity-flow column. The nickel beads were washed with ten column volumes (CV) of buffer A containing 15 mM imidazole and 4.5 mM DPC and eluted with 3 CV buffer B (20 mM sodium phosphate, 40 mM NaCl, pH 7.5) plus 200 mM imidazole and 4.5 mM DPC and concentrated five-fold in a 10,000 Da–MWCO Amicon Ultra centrifugal filter. To remove the EcDsbDα domain, 500 U thrombin (Sigma) was added to the eluted protein, and the entire reaction was dialyzed overnight at 37 °C against buffer B. To remove the cleaved EcDsbDα domain, the dialyzed, digested protein solution was applied to nickel beads in a gravity-flow column, washed with 5 CV buffer B plus 15 mM imidazole and 4.5 mM DPC and eluted with 3 CV buffer B plus 200 mM imidazole and 4.5 mM DPC. The eluted protein was concentrated to 1 mL in a 3,000 Da–MWCO Amicon Ultra centrifugal filter and purified by size-exclusion chromatography (SEC) over a HiLoad 16/60 Superdex 200 column (GE Healthcare) equilibrated with buffer C (20 mM MES, 100 mM NaCl, pH 6) plus 3 mM DPC. Elution fractions containing AfCcdA were pooled and concentrated. NMR samples were diluted during concentration to reduce the NaCl concentration. D2O was added to the sample before loading into a Shigemi tube, thus yielding a final NMR condition of 20 mM MES, 50 mM NaCl, pH 6, 100–150 mM DPC, and 5–10% D2O.
Assignment of NMR resonances.
All NMR experiments were conducted at 30 °C on Bruker or Agilent spectrometers equipped with cryogenic probes. NMR spectra were processed with NMRPipe33 and analyzed with ccpNMR34 and Xeasy35. Sequence-specific assignment of backbone 1HN, 15N, 13Ca, 13Cb and 13C′ chemical shifts was achieved with the TROSY versions of HNCA, HN(CO)CA, HNCACB, HN(CA)CO and HNCO36,37. The assignments were validated with two 3D (HN, HN) HSQC-NOESY-TROSY experiments: one with 15N, 15N and 1HN evolution in the t1, t2 and t3 dimensions, respectively; and the other with 1H, 15N and 1HN evolution in the t1, t2 and t3 dimensions, respectively, both recorded with an NOE mixing time (tNOE) of 200 ms. These experiments were performed with (15N, 13C, 2H)-labeled AfCcdA(AA) on a 600-MHz spectrometer. In addition to using NMR connectivity experiments, we used samples with selectively 15N-labeled leucine, isoleucine, valine, phenylalanine or cysteine to aid in assignment. Owing to fast relaxation, possibly due to exchange, the NMR signals in triple-resonance experiments are generally weak. By combining the triple-resonance data with the use of NOESY and selective amino acid labeling, we were able to confidently assign 82.7% of nonproline residues.
Protein side chain aliphatic and aromatic resonances were assigned with a combination of 2D 13C HSQC, 3D 15N-edited NOESY-TROSY (tNOE = 60 ms and 120 ms) and 3D 13C-edited NOESY-HSQC (tNOE = 150 ms) optimized for methyl groups, recorded on an 800-MHz spectrometer. These experiments were performed with (15N, 13C)-labeled AfCcdA(AA) in deuterated detergent. We note that for helical regions, matching NOEs in the 15N-edited and 13C-edited NOESYs with multiple mixing times is an effective way to associate the backbone amide assignments with the methyl resonances because intraresidue NOEs are usually much stronger than inter-residue NOEs. For fragile samples that generate very marginal NMR spectra such as the AfCcdA, through-bond experiments for correlating methyl resonances with backbone amide resonances generated insufficient NMR signals.
With the assignments of the backbone and side chain resonances, we were able to quickly identify a set of long-range NOEs from the hydrophobic core of the protein centered at the Ala118 and use them to generate an initial model. More long-range NOEs were assigned iteratively, as described below. The same 15N-edited NOESY-TROSY and 13C-edited NOESY-HSQC as described above were used to assign local- and long-range NOEs.
PRE measurements.
For PRE measurements, four single-cysteine AfCcdA mutants (Supplementary Fig. 3a) were labeled with 1-oxyl-2,2,5,5-tetramethyl-Δ3-pyrroline-3-methyl methanethiosulfonate (MTSL)38. Protein was purified as described above with the addition of 5 mM β-mercaptoethanol (BME) in all purification buffers. Before SEC purification, 10 mM dithiothreitol (DTT) was added to remove any unwanted disulfide interactions at the introduced cysteine site. SEC was run in 20 mM sodium phosphate, 100 mM NaCl, 3 mM DPC, and 5 mM BME, pH 7.5 (the subsequent MTSL reaction requires a higher pH). Fractions containing AfCcdA were pooled and concentrated to 2.5 mL. BME was removed with a PD-10 column (GE Healthcare) with buffer B. All subsequent MTSL procedures were performed in the dark at RT. Immediately after desalting, MTSL was added to the protein at 10× the protein concentration (~0.8–1 mM MTSL). After 30 min, a second dose of MTSL was added, and the reaction was incubated overnight. To remove free MTSL, the reaction was applied to 2 mL Ni-NTA agarose in a gravity-flow column and washed with 10 CV buffer B plus 4.5 mM DPC. The labeled protein was eluted with 10 mL buffer B with 200 mM imidazole and 4.5 mM DPC. The protein was concentrated and buffer-exchanged into the standard NMR conditions. PRE measurements were performed in 3D HNCO mode. Labeling efficiency was close to 100%, as evidenced by the complete broadening of resonances of neighboring residues. The PRE effect was calculated as the ratio of peak intensities before and after reduction of MTSL with 20 mM sodium ascorbate (prepared as a 1 M stock in 100 mM MES, pH 6).
Solvent accessibility of AfCcdA was probed by titration with the water-soluble paramagnetic gadolinium (III) (1,4,7,10-tetraazacyclododecane)-1,4,7,10-tetraacetate (Gd-DOTA, Macrocyclics)39. 15N-labeled AfCcdA(CC) and AfCcdA(AA) were purified as described above, and paramagnetic free reference 1H-15N TROSY-HSQC spectra were recorded. Gd-DOTA was added from a 613 mM stock in water to 50 μM, 100 μM, 200 μM, 500 μM, 1 mM, 2 mM, 5 mM, 10 mM and until a spectrum could no longer be recorded at 20 mM Gd-DOTA. The PRE effect was calculated as the ratio of the assigned resonance intensities to the paramagnetic free reference intensities and plotted as a function of Gd-DOTA concentration (data not shown). The greatest dispersion of paramagnetic effects was seen at 5–10 mM Gd-DOTA. All Gd-DOTA experiments were recorded at 600 MHz.
Structure calculation.
Structures were calculated with XPLOR-NIH40. The combined analysis of local NOE restraints and backbone chemical shifts (with TALOS+26) precisely defined the helical and loop regions of the protein. Therefore, the helical regions of the protein were first defined in XPLOR-NIH with strong α-helical dihedral restraints and local NOE restraints. In the second step, the preliminary fold was generated after incorporating ~30 unambiguous long-range NOE restraints in the 15N-edited NOESY-TROSY and 13C-edited NOESY-HSQC spectra. These long-range NOEs mostly involve the core residues of the protein. The initial tertiary model was then used as a test model to guide the assignment of more long-range NOEs in the same 3D NOE spectra, and an improved model was calculated with the newly assigned NOEs. This process was repeated until the calculated models converged to a backbone r.m.s. deviation of <1 Å. The NOE-derived structure was then independently validated with PRE restraints (Supplementary Fig. 3a). A total of 75 structures were calculated with a simulated annealing protocol in which the bath temperature was cooled from 1,000 to 100 K, and 15 low-energy structures were selected as the structural ensemble (refinement statistics in Table 1). Ramachandran-plot statistics for the structure ensemble (excluding the unstructured loops), calculated with PROCHECK41, are as follows: most favored (97.6%), additionally allowed (1.0%), generously allowed (0.9%) and disallowed (0.6%).
Analyses of thiol-redox state and mixed disulfide complexes.
For in vivo redox states and trapping mixed disulfide complex, protein expression was induced for 1 h and 20 min by addition of 0.25 mM IPTG (AfCcdA) or 0.2% l-arabinose (Af1675) when cells reached an OD600 of 0.2. To analyze the in vivo redox states of the proteins, free thiols were acid-trapped by trichloroacetic acid (TCA) and alkylated with 3 mM 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid (AMS) (Invitrogen) as previously described1. When indicated, 50 mM DTT was added to reduce samples, or AMS and DTT were not added to samples to obtain oxidized control proteins. Anti-EcDsbDα (ref. 1), anti-EcTrxA (T0803, Sigma), anti-Flag M2 monoclonal antibody (F1804, Sigma), and horseradish peroxidase-conjugated anti-His antibody (34460, Qiagen) were used for western blot analysis. Validation of the commercial antibodies is provided on the manufacturers’ web-sites. Uncropped images of gels are shown in Supplementary Data Set 1.
Bioinformatics analysis.
To predict the TM helices, we used TMpred analysis (http://www.ch.embnet.org/software/TMPRED_form.html). To find homologs of EcTrx-fold proteins (TrxA, TrxC, GrxA, DsbA, DsbC, CcmG, and DsbD) in A. fulgidus, we performed position-specific iterated blast (PSI-BLAST) analysis42. Genes encoding interacting proteins often exist as neighbors. Thus, to find the envelope partner protein of CcdA, we analyzed gene organization around ccdA in bacteria in the MicrobesOnline portal (http://www.microbesonline.org/). HRM2_42220 (locus tag in NCBI database) was selected as a query in PSI-BLAST to find its homologs in A. fulgidus. We found Af1675 (AfTrxE) as one of the top-scoring candidates ((expected value, 2 × 10−6; identities, 16/63 (25%); positives: 33/63(52%)). This protein is predicted to have a Trx-like fold and has four cysteines (of which two are in CXXC and the others form a structural disulfide bond (Supplementary Fig. 4b)). To predict the signal sequence and the transmembrane segment of AfTrxE, SignalP4.1 (http://www.cbs.dtu.dk/services/SignalP/) and TMpred analyses were used. The prediction showed that AfTrxE has an N-terminal TM helix or signal sequence, thus indicating its role in the periplasm. When the predicted N-terminal hydrophobic sequence was replaced by the signal sequence of DsbA and expressed in a ΔdsbD strain together with AfCcdA, DsbAsignal sequence-AfTrxE could not be reduced (data not shown) as it could in the wild-type AfTrxE, thus suggesting the presence of an N-terminal TM helix and its importance in the interaction with AfCcdA.
MalPEG labeling.
Single-cysteine mutants of AfCcdA were labeled with 5-kDa malPEG (Sigma) to determine cysteine accessibility. AfCcdA(CA) (C16A) and AfCcdA(AC) (C118A) were expressed and purified as described above for the PRE mutants. SEC was run in 20 mM MES, pH 6.5, 100 mM NaCl, 3 mM DPC, and 5 mM BME. This pH was chosen as the lowest pH permitting the malPEG reaction while staying as close to the standard NMR conditions as possible. SEC fractions containing AfCcdA were pooled and concentrated to 2.5 mL, and the BME was removed with a PD-10 column with 50 mM MES, 50 mM NaCl, and 3 mM DPC, pH 6.5. Before labeling, the mutants were concentrated, and a 1H-15N HSQC reference spectrum was recorded. MalPEG labeling was optimized in a series of 50-μL reactions containing ~50 μM AfCcdA and 5 mM malPEG. Reactions were incubated at 37 °C and quenched with 5 mM BME after 0–16 h. The negative control contained protein only (no malPEG), and the positive control was denatured first with 1% SDS. An additional control reaction used AfCcdA(AA) to rule out nonspecific labeling (data not shown). The extent of labeling (PEGylation) was detected by the 5-kDa shift by SDS-PAGE. To prepare the NMR samples, the PEGylated AfCcdA mutants were incubated with 5 mM malPEG overnight at 37 °C, quenched with 5 mM BME, diluted to 15 mL with buffer B plus 4.5 mM DPC, and then applied to 2 mL Ni-NTA agarose in a gravity-flow column and washed with 10 CV buffer B plus 4.5 mM DPC. The labeled protein was eluted with 10 mL buffer B plus 200 mM imidazole and 4.5 mM DPC. The protein was concentrated and buffer-exchanged into the standard NMR conditions. 1H-15N TROSY-HSQC spectra of the PEGylated mutants were recorded at 700 MHz.
Supplementary Material
ACKNOWLEDGMENTS
We thank T. Rapoport for initiating the structural investigation of CcdA along with members of the Chou and Beckwith laboratories for scientific discussion throughout the entire project. This work was supported by a grant from the European Research Council (ERC) (FP7/2007–2013) Independent Researcher Starting Grant 282335 – Sulfenic to J.-F.C. and US National Institutes of Health (NIH) grants GM094608 to J.J.C. and GMO41883 to J.R.B. The NMR facility used for this study was supported by NIH grant P41 EB-002026.
Footnotes
Accession codes. Coordinates and structure factors have been deposited in the Protein Data Bank under accession code PDB 2N4X. The chemical-shift values have been deposited in the Biological Magnetic Resonance Data Bank under accession code 25685.
Any Supplementary Information and Source Data files are available in the online version of the paper.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Stewart EJ, Katzen F & Beckwith J Six conserved cysteines of the membrane protein DsbD are required for the transfer of electrons from the cytoplasm to the periplasm of Escherichia coli. EMBO J. 18, 5963–5971 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chung J, Chen T & Missiakas D Transfer of electrons across the cytoplasmic membrane by DsbD, a membrane protein involved in thiol-disulphide exchange and protein folding in the bacterial periplasm. Mol. Microbiol 35, 1099–1109 (2000). [DOI] [PubMed] [Google Scholar]
- 3.Katzen F & Beckwith J Transmembrane electron transfer by the membrane protein DsbD occurs via a disulfide bond cascade. Cell 103, 769–779 (2000). [DOI] [PubMed] [Google Scholar]
- 4.Bessette PH, Cotto JJ, Gilbert HF & Georgiou G In vivo and in vitro function of the Escherichia coli periplasmic cysteine oxidoreductase DsbG. J. Biol. Chem 274, 7784–7792 (1999). [DOI] [PubMed] [Google Scholar]
- 5.Rietsch A, Bessette P, Georgiou G & Beckwith J Reduction of the periplasmic disulfide bond isomerase, DsbC, occurs by passage of electrons from cytoplasmic thioredoxin. J. Bacteriol 179, 6602–6608 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Collet JF, Riemer J, Bader MW & Bardwell JC Reconstitution of a disulfide isomerization system. J. Biol. Chem 277, 26886–26892 (2002). [DOI] [PubMed] [Google Scholar]
- 7.Malojčić G, Geertsma ER, Brozzo MS & Glockshuber R Mechanism of the prokaryotic transmembrane disulfide reduction pathway and its in vitro reconstitution from purified components. Angew. Chem. Int. Edn Engl 51, 6900–6903 (2012). [DOI] [PubMed] [Google Scholar]
- 8.Rozhkova A et al. Structural basis and kinetics of inter- and intramolecular disulfide exchange in the redox catalyst DsbD. EMBO J. 23, 1709–1719 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cho SH & Collet JF Many roles of the bacterial envelope reducing pathways. Antioxid. Redox Signal 18, 1690–1698 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hatahet F, Boyd D & Beckwith J Disulfide bond formation in prokaryotes: history, diversity and design. Biochim. Biophys. Acta 1844, 1402–1414 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Verissimo AF & Daldal F Cytochrome c biogenesis System I: an intricate process catalyzed by a maturase supercomplex? Biochim. Biophys. Acta 1837, 989–998 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Katzen F, Deshmukh M, Daldal F & Beckwith J Evolutionary domain fusion expanded the substrate specificity of the transmembrane electron transporter DsbD. EMBO J. 21, 3960–3969 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deshmukh M, Brasseur G & Daldal F Novel Rhodobacter capsulatus genes required for the biogenesis of various c-type cytochromes. Mol. Microbiol 35, 123–138 (2000). [DOI] [PubMed] [Google Scholar]
- 14.Gupta SD, Wu HC & Rick PD A Salmonella typhimurium genetic locus which confers copper tolerance on copper-sensitive mutants of Escherichia coli. J. Bacteriol 179, 4977–4984 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cho SH et al. A new family of membrane electron transporters and its substrates, including a new cell envelope peroxiredoxin, reveal a broadened reductive capacity of the oxidative bacterial cell envelope. MBio 3, e00291–11 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cho SH, Porat A, Ye J & Beckwith J Redox-active cysteines of a membrane electron transporter DsbD show dual compartment accessibility. EMBO J. 26, 3509–3520 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cho SH & Beckwith J Two snapshots of electron transport across the membrane: insights into the structure and function of DsbD. J. Biol. Chem 284, 11416–11424 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cho SH & Beckwith J Mutations of the membrane-bound disulfide reductase DsbD that block electron transfer steps from cytoplasm to periplasm in Escherichia coli. J. Bacteriol 188, 5066–5076 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hiniker A, Vertommen D, Bardwell JC & Collet JF Evidence for conformational changes within DsbD: possible role for membrane-embedded proline residues. J. Bacteriol 188, 7317–7320 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kimball RA, Martin L & Saier MH Jr. Reversing transmembrane electron flow: the DsbD and DsbB protein families. J. Mol. Microbiol. Biotechnol 5, 133–149 (2003). [DOI] [PubMed] [Google Scholar]
- 21.Rozhkova A & Glockshuber R Thermodynamic aspects of DsbD-mediated electron transport. J. Mol. Biol 380, 783–788 (2008). [DOI] [PubMed] [Google Scholar]
- 22.Katzen F & Beckwith J Role and location of the unusual redox-active cysteines in the hydrophobic domain of the transmembrane electron transporter DsbD. Proc. Natl. Acad. Sci. USA 100, 10471–10476 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Van Horn WD et al. Solution nuclear magnetic resonance structure of membrane-integral diacylglycerol kinase. Science 324, 1726–1729 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jaremko L, Jaremko M, Giller K, Becker S & Zweckstetter M Structure of the mitochondrial translocator protein in complex with a diagnostic ligand. Science 343, 1363–1366 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Berardi MJ & Chou JJ Fatty acid flippase activity of UCP2 is essential for its proton transport in mitochondria. Cell Metab. 20, 541–552 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shen Y, Delaglio F, Cornilescu G & Bax A TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 44, 213–223 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abramson J et al. Structure and mechanism of the lactose permease of Escherichia coli. Science 301, 610–615 (2003). [DOI] [PubMed] [Google Scholar]
- 28.Kumar H et al. Structure of sugar-bound LacY. Proc. Natl. Acad. Sci. USA 111, 1784–1788 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krishnamurthy H & Gouaux E X-ray structures of LeuT in substrate-free outward-open and apo inward-open states. Nature 481, 469–474 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guan L, Mirza O, Verner G, Iwata S & Kaback HR Structural determination of wild-type lactose permease. Proc. Natl. Acad. Sci. USA 104, 15294–15298 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miroux B & Walker JE Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J. Mol. Biol 260, 289–298 (1996). [DOI] [PubMed] [Google Scholar]
- 32.Gon S et al. A novel regulatory mechanism couples deoxyribonucleotide synthesis and DNA replication in Escherichia coli. EMBO J. 25, 1137–1147 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Delaglio F et al. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 (1995). [DOI] [PubMed] [Google Scholar]
- 34.Vranken WF et al. The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins 59, 687–696 (2005). [DOI] [PubMed] [Google Scholar]
- 35.Bartels C, Xia TH, Billeter M, Guntert P & Wuthrich K The program XEASY for computer-supported NMR spectral analysis of biological macromolecules. J. Biomol. NMR 6, 1–10 (1995). [DOI] [PubMed] [Google Scholar]
- 36.Kay LE, Ikura M, Tschudin R & Bax A Three-dimensional triple resonance NMR spectroscopy of isotopically enriched proteins. J. Magn. Reson 89, 496–514 (1990). [DOI] [PubMed] [Google Scholar]
- 37.Pervushin K, Riek R, Wider G & Wuthrich K Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc. Natl. Acad. Sci. USA 94, 12366–12371 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Battiste JL & Wagner G Utilization of site-directed spin labeling and high-resolution heteronuclear nuclear magnetic resonance for global fold determination of large proteins with limited nuclear overhauser effect data. Biochemistry 39, 5355–5365 (2000). [DOI] [PubMed] [Google Scholar]
- 39.Hilty C, Wider G, Fernandez C & Wuthrich K Membrane protein-lipid interactions in mixed micelles studied by NMR spectroscopy with the use of paramagnetic reagents. ChemBioChem 5, 467–473 (2004). [DOI] [PubMed] [Google Scholar]
- 40.Schwieters CD, Kuszewski J, Tjandra N & Clore GM The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson 160, 65–73 (2003). [DOI] [PubMed] [Google Scholar]
- 41.Laskowski RA, MacArthur MW, Moss DS & Thornton JW PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr 26, 283–291 (1993). [Google Scholar]
- 42.Altschul SF et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.