

Abstract
We present a case of 50-year-old man with history of ulcerative right axillary mass for 6 months. Axillary lymphadenopathy and organomegaly were absent. Microscopic examination showed sheets of pleomorphic cells which were mitotically active. Distinctive myxoid change was seen throughout the tumor. These cells were strongly positive for CD30 and vimentin but were negative for CD3, CD5, CD20, CD15, anaplastic lymphoma kinase protein (ALK), CD56, cytokeratin, melan A, desmin, myogenin, CD68, S100, epithelial membrane antigen and CD34. The final diagnosis of primary cutaneous ALK-negative T-cell anaplastic large cell lymphoma (PCALCL), myxoid variant was made. Work-up revealed no systemic involvement. The patient received eight cycles of cyclophosphamide, doxorubicin, vincristine, prednisone and etoposide chemotherapy with complete resolution of disease. This case report highlights that a high index of suspicion is necessary in patients of PCALCL due to varied clinical presentation, and to discuss in brief the histopathologic and immunophenotypic features of this entity along with its differential diagnosis.
Keywords: pathology, malignant and benign haematology, dermatology
Background
Anaplastic lymphoma kinase protein (ALK)-negative anaplastic large cell lymphoma (ALCL) is defined by the WHO as a CD30-positive T-cell neoplasm that are not reproducibly distinguishable from ALK-positive ALCL except on the basis of absence of ALK protein.1 The most common presentations of primary cutaneous ALCL are as an ulcerating mass or a nodule and less commonly as multiple nodules limited to a single area on the skin with the usual size smaller than 2.5 cm.2 3 The peak incidence of ALK-negative ALCL is in fifth and sixth decades with the median age of 58 years, unlike ALK-positive forms which usually occurs in children and young adults. These ALK-negative ALCLs have male preponderance and worse prognosis. They can involve lymph nodes as well as extranodal sites including bone, soft tissue and skin.4 The systemic form of ALCL commonly presents with lymphadenopathy as well as extranodal involvement. ALK-negative forms are less likely to involve extranodal sites as opposed to ALK-positive forms.5 Most patients present with advanced stage disease with peripheral and/or abdominal lymphadenopathy and/or bone marrow involvement and ‘B’ symptoms including high-grade fever and weight loss.6 7 It has also been noticed that there is an associated cutaneous involvement in nearly 20% of ALK-positive cases of systemic ALCL.8 The myxoid variant of ALK-negative ALCL has been described seldom, either in the primary cutaneous form or systemic form.9–11 With the best of our knowledge Chan et al in 1990 reported the very first case of a primary cutaneous ALCL in a 45-year-old man with prominent sarcomatoid and myxoid features occurring as a solitary tumour nodule on leg.9
Case presentation
A 50-year-old man presented to the physician with an ulcerative right axillary mass which was present for the past 6 months. The mass was progressively increasing in size and was associated with pain and tenderness. There were no complaints of cough, dyspnoea or evening rise of temperature. The patient had no history of diabetes. There was no relevant family history present. On physical examination, an ulcerated, irregular and firm mass of approximately 5 cm was noted in the right axilla (figure 1). Axillary lymphadenopathy and organomegaly were absent. An excisional biopsy of the mass was performed and sent to us for histopathological examination.
Figure 1.
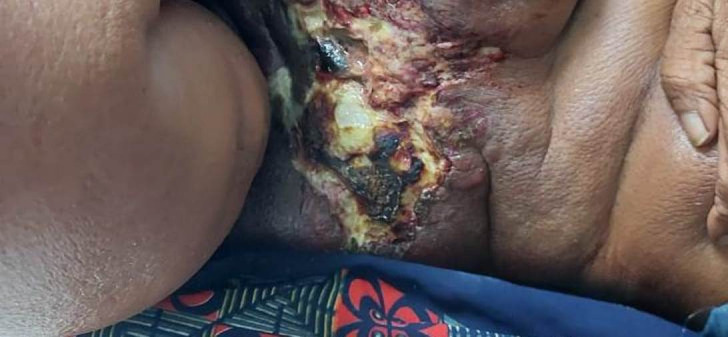
Clinical photograph of an ulcerated lesion over right axilla.
Investigations
Non-contrast computerised tomography and contrast-enhanced CT of the thorax, neck and abdomen showed normal scans. For histopathological examination, we received a single greyish brown soft tissue piece measuring 3×2.5×1.5 cm without the overlying skin. On light microscopy, sections showed sheets of pleomorphic population of large polygonal to round cells having hyperchromatic nuclei and abundant eosinophilic cytoplasm. These neoplastic cells were mitotically active and at places were arranged in perivascular pattern. Distinctive myxoid change was seen throughout the tumour. Eosinophils, neutrophils, small lymphocytes and histiocytes were intimately admixed with the cells throughout the lesion (figures 2 and 3).
Figure 2.
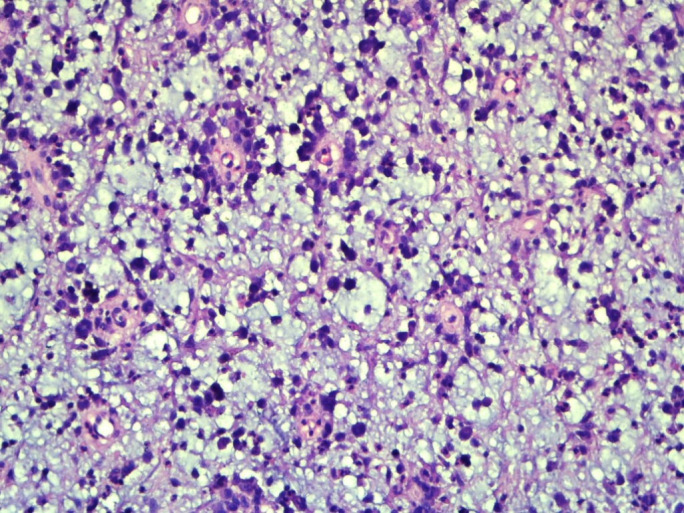
×100 magnification image showing large atypical cells with perivascular arrangement and diffuse myxoid areas in the background.
Figure 3.
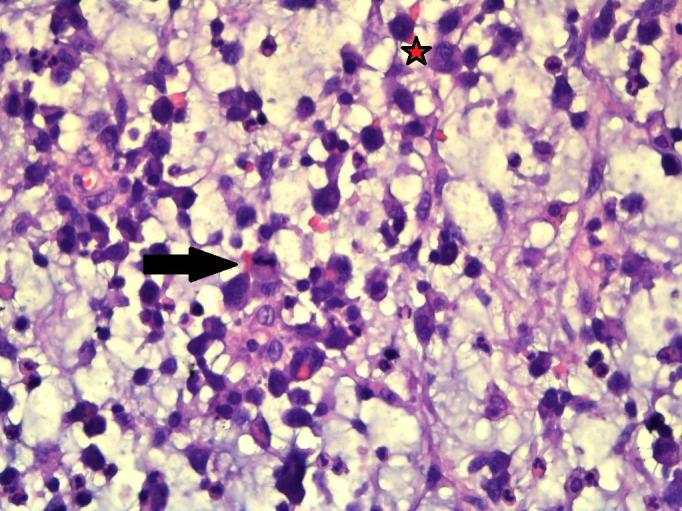
×400 magnification shows abundant mucin, which is easily seen intermingled with the inflammatory cells comprising of eosinophils, neutrophils and small lymphocytes along with binucleated cells (star) with mitotic figures (arrow head).
Differential diagnosis
The differentials that were considered based on light microscopy findings were myxoinflammatory fibroblastic sarcoma, inflammatory myofibroblastic tumour, myxoid liposarcoma, myxofibrosarcoma and malignant melanoma.
On immunohistochemistry (IHC), the neoplastic cells were negative for cytokeratin, melan A, desmin, myogenin, CD68, S100, epithelial membrane antigen (EMA) and CD34 with positivity for vimentin only (figure 4). Retrospective examination of the tumour showed cells having horseshoe-shaped (hallmark cells) and wreath-like nuclei along with few binucleated cells (figures 3 and 5). The second panel of IHC showed that the neoplastic cells were strongly positive for CD30. The cells were negative for CD3, CD5, CD20, CD15, CD56 and ALK-1 (figure 4). The final diagnosis of primary cutaneous ALK-negative T-cell ALCL, myxoid variant was made.
Figure 4.

Immunohistochemistry Images: vimentin and CD30 show positive immunostaining, while anaplastic lymphoma kinase (ALK), epithelial membrane antigen (EMA), S100, CD68, cytokeratin, myogenin and desmin do not show immunoreactivity.
Figure 5.
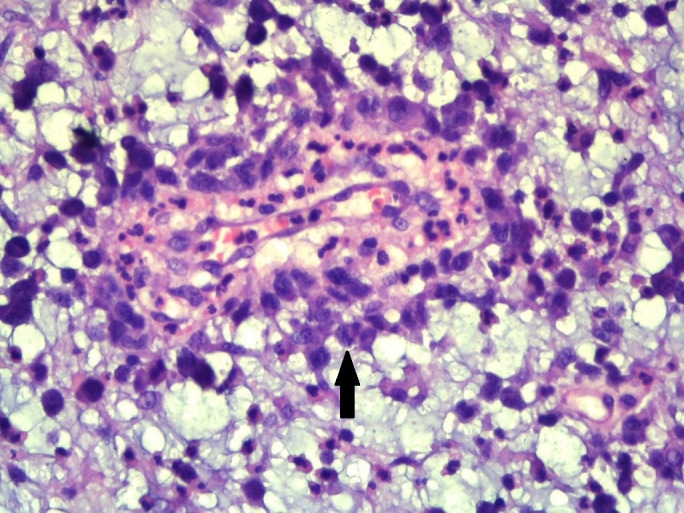
×400 magnification shows large atypical cells with hallmark cells (arrow head).
The involvement of lymph node was never confirmed because the specimen was diffusely involved by the myxoid process. Moreover, the apparent absence of axillary lymphadenopathy suggested that the process was primarily cutaneous rather than systemic.
Treatment
The patient received eight cycles of cyclophosphamide, doxorubicin, vincristine, prednisone and etoposide (CHOP-E) chemotherapy.
Outcome and follow-up
During 6-month follow-up period, there was complete resolution of symptoms with no residual cutaneous disease. Bone marrow examination showed no evidence of infiltration by tumour cells.
Discussion
Primary cutaneous T-cell lymphomas (CTCLs) include clinically and biologically heterogenous group of non-Hodgkin lymphomas defined by clonal proliferation of skin-homing malignant T lymphocytes.12 Primary cutaneous CD30+ lymphoproliferative disorders account for 30% of the CTCL.13 Under the group of primary cutaneous CD30+ lymphoproliferative disorders, the following diseases are included: primary cutaneous anaplastic large cell lymphoma (PCALCL), lymphomatoid papulosis (LyP) and borderline cases. The criteria for the diagnosis of PCALCL include: (a)>75% infiltration of CD30+ large anaplastic cells in skin biopsy, (b) no clinical history of LyP, mycosis fungoides or other cutaneous lymphomas and (c) no extracutaneous localisation after extensive investigations at presentation.14
This case represents a unique example of a myxoid variant of ALK-negative primary cutaneous T-cell ALCL. Until today myxoid variant has been described rarely.9–11 This tumour can be easily misdiagnosed as high-grade sarcoma. In our case also, ALCL was not the leading diagnostic consideration initially because of the prominent myxoid change, the mixed cellular infiltrate and pleomorphic population of large polygonal to round cells. A myxoid sarcoma was initially considered because of the deep dermal and subcutaneous involvement, the nodular growth pattern and the observation that myxoid change is commonly seen in many soft tissue sarcomas. The differentials that were initially considered were myxoinflammatory fibroblastic sarcoma, inflammatory myofibroblastic tumour, myxoid liposarcoma, myxofibrosarcoma and malignant melanoma. It was only after the negative findings of the first IHC panel (cytokeratin, melan A, desmin, myogenin, CD68, S100, EMA, and CD34) that the morphology was reviewed and hallmark cells were identified, after which the possibility of myxoid ALCL was considered
PCALCL is a chemosensitive and radiosensitive lymphoma. Treatment strategies include excision, local radiotherapy, CHOP chemotherapy or combination of these modalities.15 16 The overall complete response of PCALCL to CHOP therapy is 83%.16
Learning points.
Primary cutaneous myxoid anaplastic large cell lymphoma (ALCL) is a diagnostic challenge for both clinicians and histopathologists. Establishing a proper diagnosis is possible based on thorough medical history, clinical and imaging examinations, and histopathological evaluation of the biopsy sample from the lesion site.
We suggest including myxoid ALCL in the histopathologic differential diagnosis of cutaneous lesions with a prominent myxoid background.
Careful examination for the presence of a hallmark cells and the use of immunohistochemistry, particularly CD30, can help avoid a misdiagnosis which can lead to inappropriate therapy and result in disease progression or unnecessary harm to the patient.
Acknowledgments
Dr. Preeti Agarwal: supervision, conceptualisation, analysis of data, language editing and proofreading. Dr. Sumaira Qayoom: supervision, conceptualisation and analysis of data. Dr. Geeta Yadav: supervision, conceptualisation and analysis of data. Dr. Mohit Mishra: technical editing, language editing and proofreading.
Footnotes
Contributors: Conceptualisation, acquisition, analysis of data and final approval of version published: all authors. Drafting the manuscript: MM. Revising it critically for important intellectual content: MM, SR and MMG.
Funding: The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests: None declared.
Patient consent for publication: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Swerdlow SH Who classification of tumours of haematopoietic and lymphoid tissues. WHO classification of tumours 2008;22008:439. [PubMed] [Google Scholar]
- 2.Bekkenk MW, Geelen FA, Vader PC, et al. Primary and secondary cutaneous CD30+ lymphoproliferative disorders: a report from the Dutch cutaneous lymphoma group on the long-term follow-up data of 219 patients and guidelines for diagnosis and treatment. Blood, The Journal of the American Society of Hematology 2000;95:3653–61. [PubMed] [Google Scholar]
- 3.Zackheim HS, Amin S, Kashani-Sabet M, et al. Prognosis in cutaneous T-cell lymphoma by skin stage: long-term survival in 489 patients. J Am Acad Dermatol 1999;40:418–25. 10.1016/S0190-9622(99)70491-3 [DOI] [PubMed] [Google Scholar]
- 4.Savage KJ, Harris NL, Vose JM, et al. ALK- anaplastic large-cell lymphoma is clinically and immunophenotypically different from both ALK+ ALCL and peripheral T-cell lymphoma, not otherwise specified: report from the International peripheral T-cell lymphoma project. Blood 2008;111:5496–504. 10.1182/blood-2008-01-134270 [DOI] [PubMed] [Google Scholar]
- 5.ten Berge RL, Oudejans JJ, Ossenkoppele GJ, et al. Alk expression in extranodal anaplastic large cell lymphoma favours systemic disease with (primary) nodal involvement and a good prognosis and occurs before dissemination. J Clin Pathol 2000;53:445–50. 10.1136/jcp.53.6.445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Falini B, Pileri S, Zinzani PL, et al. ALK+ lymphoma: clinico-pathological findings and outcome. Blood 1999;93:2697–706. [PubMed] [Google Scholar]
- 7.Brugières L, Deley MC, Pacquement H, et al. CD30(+) anaplastic large-cell lymphoma in children: analysis of 82 patients enrolled in two consecutive studies of the French Society of Pediatric Oncology. Blood 1998;92:3591–8. [PubMed] [Google Scholar]
- 8.Stein H, Foss HD, Dürkop H, et al. Cd30+ anaplastic large cell lymphoma: a review of its histopathologic, genetic, and clinical features. Blood, The Journal of the American Society of Hematology 2000;96:3681–95. [PubMed] [Google Scholar]
- 9.Chan JK, Buchanan R, Fletcher CD. Sarcomatoid variant of anaplastic large-cell Ki-1 lymphoma. Am J Surg Pathol 1990;14:983–8. 10.1097/00000478-199010000-00013 [DOI] [PubMed] [Google Scholar]
- 10.Chan JK Anaplastic large cell lymphoma: redefining its morphologic spectrum and importance of recognition of the ALK-positive subset. Adv Anat Pathol 1998;5:281–313. [PubMed] [Google Scholar]
- 11.Gable AD, Clark SH, Magro CM. Myxoid variant of anaplastic large cell lymphoma involving the skin: a case report. J Cutan Pathol 2012;39:787–90. 10.1111/j.1600-0560.2012.01911.x [DOI] [PubMed] [Google Scholar]
- 12.Rosen ST, Querfeld C. Primary cutaneous T-cell lymphomas. Hematology 2006;2006:323–30. 10.1182/asheducation-2006.1.323 [DOI] [PubMed] [Google Scholar]
- 13.Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood 2005;105:3768–85. 10.1182/blood-2004-09-3502 [DOI] [PubMed] [Google Scholar]
- 14.Gould JW, Eppes RB, Gilliam AC, et al. Solitary primary cutaneous CD30+ large cell lymphoma of natural killer cell phenotype bearing the t(2;5)(p23;q35) translocation and presenting in a child. Am J Dermatopathol 2000;22:422–8. 10.1097/00000372-200010000-00007 [DOI] [PubMed] [Google Scholar]
- 15.Shehan JM, Kalaaji AN, Markovic SN, et al. Management of multifocal primary cutaneous CD30 anaplastic large cell lymphoma. J Am Acad Dermatol 2004;51:103–10. 10.1016/j.jaad.2003.12.028 [DOI] [PubMed] [Google Scholar]
- 16.Liu HL, Hoppe RT, Kohler S, et al. Cd30+ cutaneous lymphoproliferative disorders: the Stanford experience in lymphomatoid papulosis and primary cutaneous anaplastic large cell lymphoma. J Am Acad Dermatol 2003;49:1049–58. 10.1016/S0190-9622(03)02484-8 [DOI] [PubMed] [Google Scholar]