

Abstract
Metabolomics is emerging as an important field in life sciences. However, a weakness of current mass spectrometry (MS) based metabolomics platforms is the time-consuming analysis and the occurrence of severe matrix effects in complex mixtures. To overcome this problem, we have developed an automated and fast fractionation module coupled online to MS. The fractionation is realized by the implementation of three consecutive high performance solid-phase extraction columns consisting of a reversed phase, mixed-mode anion exchange, and mixed-mode cation exchange sorbent chemistry. The different chemistries resulted in an efficient interaction with a wide range of metabolites based on polarity, charge, and allocation of important matrix interferences like salts and phospholipids. The use of short columns and direct solvent switches allowed for fast screening (3 min per polarity). In total, 50 commonly reported diagnostic or explorative biomarkers were validated with a limit of quantification that was comparable with conventional LC–MS(/MS). In comparison with a flow injection analysis without fractionation, ion suppression decreased from 89% to 25%, and the sensitivity was 21 times higher. The validated method was used to investigate the effects of circadian rhythm and food intake on several metabolite classes. The significant diurnal changes that were observed stress the importance of standardized sampling times and fasting states when metabolite biomarkers are used. Our method demonstrates a fast approach for global profiling of the metabolome. This brings metabolomics one step closer to implementation into the clinic.
Metabolomics is increasingly important in the field of life sciences. It is used for the screening of inborn errors of metabolism,1 precision medicine,2 and discovery of new biomarkers for health, disease, and intervention.3 To accommodate this increased interest, there is a need for fast and comprehensive screening of the metabolome.4 Mass spectrometry (MS) is a highly sensitive technique, and MS-based methods can screen a large range of metabolites in a single run.5 This makes MS highly suitable for comprehensive metabolomics. The downside of MS is that it often requires extensive sample preparation and separation to reduce interferences of complex biological samples at the ionization source.6
Flow injection analysis coupled to mass spectrometry (FIA–MS) is an appealing approach in fast and comprehensive screening since there is no chromatography that discriminates against compound classes or decreases the throughput.7 The sample preparation of these methodologies is often a “dilute-and-shoot” approach, whereby dilution is applied to decrease the interference of the sample matrix at the ionization source. However, these methods often suffer in terms of sensitivity because the analytes are also diluted or high abundant matrix interferences still cause severe ion suppression.8 Therefore, sample preparation remains an important aspect in fast MS analysis to decrease the sample complexity while maintaining a sufficient analyte concentration. Liquid–liquid extraction (LLE) has been performed in parallel and coupled to FIA–MS to improve throughput and coverage.9 However, solid-phase extraction (SPE) has been coupled online to mass spectrometry in the RapidFire system resulting in analyses times of around 8.5 s.10 By using LLE or different SPE sorbents in parallel, however, the cleanup efficiency remains limited. Generally, these approaches only result in two fractions (water/organic fraction in LLE and flow-through/elution fraction in SPE) and fractions are ionized at once without within-fraction separation.
In this work, we demonstrate a comprehensive and fast sample preparation method coupled online to MS. The method utilizes two important chemical properties of the metabolome: polarity and charge. Three consecutive high performance (particle size ≤5 μm) SPE columns, consisting of a reversed phase, mixed-mode cation exchange, and mixed-mode anion exchange sorbent chemistry, are coupled online to a mass spectrometer. This ensured the allocation of metabolites into different fractions (flow-through; polar/neutral, reversed phase; apolar, cation exchange; polar and positive, anion exchange; polar and negative). Moreover, it also removed known ion suppressors from different fractions minimizing their adverse effects during electrospray ionization. Phospholipids and salts are held responsible for a majority of signal suppression during electrospray ionization of plasma samples.11 By using a fractionation approach based on polarity and charge, phospholipids are retained on the reversed phase column, whereas positive and negative salt ions are trapped on and eluted from the cation and anion exchange, respectively. Another benefit of serially coupled columns is the flow-through fraction, which is cleaned by three sorbent chemistries instead of one in conventional single-column methods. The advantage of online fractionation over offline fractionation is that it allows for some separation between compounds within a fraction prior to electrospray ionization. Hereby, retained ion suppressors could elute at another time than retained analytes. To our knowledge, this is the first publication that reports the use of serially coupled high performance SPE columns to realize an online fractionation including some separation prior to MS analysis. The strength of this platform is emphasized by the use of short analytical columns which allow for fast solvent switches while still benefiting from chromatographic separation.
We have developed a targeted platform for the analysis of 50 commonly reported diagnostic or explorative biomarkers.12−14 These compounds belong to the following compound classes: amino acids, amines, purines, sugars, acylcarnitines, organic acids, and fatty acids. We present a fast online sample preparation method that fractionates these compound classes in plasma. Several online SPE columns have been evaluated for their ability to fractionate plasma prior to MS analysis. The optimized methods for both positive and negative electrospray ionization mode have been validated and applied in a study investigating the effect of circadian rhythm and food intake on several metabolite classes. This study should give insight into the diurnal variations of the studied biomarkers. These variations are important to assess because they could potentially be misinterpreted as disease or intervention related variations. This misinterpretation compromises the diagnostic and explorative power of a potential biomarker.
Materials and Methods
Chemicals
An overview of the used (internal) standards and concentrations is provided in the Supporting Information (SI Tables S1 and S2). Water was obtained from an arium pro UF/VF water purification system with a Sartopore 2 0.2 μm filter. Methanol (Ultra-LC–MS grade) was purchased from Actu-All (Oss, The Netherlands). Ammonium hydroxide (28–30 wt % solution of ammonia in water) and formic acid (98%) were purchased from Acros Organics (Bleiswijk, The Netherlands). Ammonium acetate (≥99.0%) and ammonium formate (≥99.995%) were purchased from Sigma-Aldrich (Zwijndrecht, The Netherlands).
Method Development
We have used polymeric mixed-mode ion exchange columns because they provide a superior pH stability over other ion exchange sorbent types. Several ion exchange columns have been evaluated according to the retention, trapping, and elution performances of representative standards. We tested four low performance (particle size >5 μm), four high performance Sepax (particle size 1.7–5 μm), and four high performance Zirchrom (particle size 3 μm) SPE columns. The low performance, Sepax, and Zirchrom SPE columns were composed of four mixed-mode ion exchange types (strong cation exchange (SCX), strong anion exchange (SAX), weak cation exchange (WCX), and weak anion exchange (WAX)). Similar loading and elution buffers were used for each type of ion exchange. The evaluated ion exchange columns, loading, and elution buffers explored during development can be found in the SI (Table S3). The selected ion exchange columns were coupled to a reversed phase column and ordered in a way that was most beneficial in terms of matrix effect reduction and peak shape. The reversed phase column was a ZORBAX Extend-C18, 2.1 × 5 mm, 1.8 μm guard column from Agilent Technologies Netherlands (Waldbronn, The Netherlands).
Five cationic compounds were used to represent different types of cations (leucine, glutamic acid, arginine, hypoxanthine, and choline) and four anionic compounds were used to represent different types of anions (lactic acid, malic acid, citric acid and indoxyl sulfate). The amino acids consisted of cationic and anionic functional groups. Glucose functioned as a neutral marker and indicated whether ions were efficiently removed from the column flow-through.
Validation
Individual stock solutions and calibration mixtures were stored at −80 °C. In each specific fraction, there was at least one internal standard present. In total seven calibration points were used (C1–7). The highest calibration concentration is referred to as C7 (SI Table S1) and the subsequent concentrations were 1:1 dilutions of the previous concentration. All calibration standards were included in the same stock solution and all calibration solutions were composed of 69% methanol in water. C0 was prepared by adding 69% MeOH without standards. Within the calibration range, C4 and the internal standard concentration were set to mimic the physiological concentration of the analyte found on the Human Metabolome Database (HMDB).15 Calibration curves were constructed by standard addition of the calibration standards to plasma samples. The repeatability of the method was determined by the relative standard deviation of three replicates of three different concentrations (C0, C2, and C4). The intermediate precision was determined by the relative standard deviation of three different concentrations (C0, C2, and C4) on three different days (N = 9). The matrix effect was determined by the ratio of the peak area of the internal standard in a plasma and water sample.16 Ion suppression was determined by subtracting 100% by the matrix effect. Ion suppression of ion enhanced compounds was set at 0% when calculating the mean ion suppression.
| 1 |
| 2 |
The carryover was evaluated as the ratio of the peak area in a blank sample and the peak area in a pooled plasma sample that was analyzed just before the blank (N = 3). Ten concentration levels of internal standards were used to determine the limit of detection (LOD) and lower limit of quantification (LLOQ). The highest concentration was C6 which was four times the physiological value of the unlabeled counterpart (SI Table S2) and the subsequent concentrations were 1:1 dilutions of the previous concentration. The LOD (formula 3) and LLOQ (formula 4) were determined by the following formula which used the peak area of a blank, the standard deviation (SD) of the lowest concentration with a S/N greater than 3 (Clow) and the response factor (RF), which was calculated by the ratio of the peak area and concentration of Clow.
| 3 |
| 4 |
Sample Preparation
During the method validation, 30 μL EDTA plasma aliquots, 30 μL of calibration standard and 30 μL of the internal standard solution, H2O and MeOH were mixed reaching a total volume of 195 μL and 71% MeOH. The mixture was vigorously vortexed and centrifuged (10 min, 16 100g, 4 °C). After centrifugation, 100 μL of the supernatant was transferred into an autosampler vial containing a 150 μL insert. Study samples were prepared by mixing 15 μL EDTA plasma, 15 μL of internal standard solution, H2O, and MeOH reaching a total volume of 97.5 μL and 71% MeOH (same ratios as during method validation). The vortex and centrifuge step remained the same, and 50 μL of the supernatant was transferred into an autosampler vial containing a 150 μL insert.
The flow injection analysis (FIA) sample preparation was adapted from Carducci et al.17 Ten microliters of EDTA plasma and internal standard solution were mixed with methanol, water, and acetic acid to reach a final solution of 80% methanol, 0.1% acetic acid and a plasma dilution ratio of 100. This dilution ratio was found to give the highest sensitivity after testing plasma dilution ratios of 10 to 500. An adjusted Bligh and Dyer LLE was also performed prior to the FIA.18 Ten microliters of EDTA plasma and internal standard solution were extracted with methanol, dichloromethane, and water (v/v/v, 2/2/1.8) reaching a total volume of 1000 μL. 200 μL of the apolar and 200 μL of the polar fraction were evaporated and separately reconstituted in 200 μL 0.1% acetic acid in 80% MeOH.
Fractionation and Mass Spectrometry
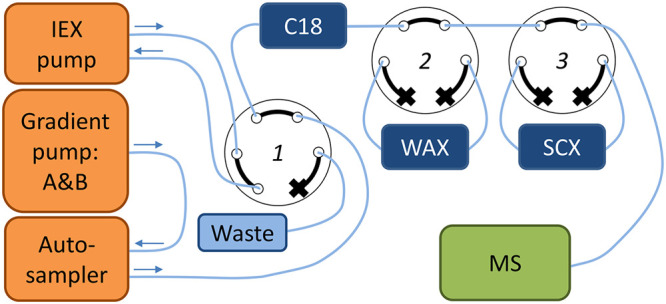
A Shimadzu Nexera UHPLC (Darmstadt, Germany) was connected to a Sciex X500R QToF (Darmstadt, Germany). The setup was extended by a stand-alone Agilent 1260 Infinity Isocratic Pump (Waldbronn, Germany) and two VICI six-port valves (Rotterdam, The Netherlands). Figure 1 shows a schematic overview of the setup.
Figure 1.
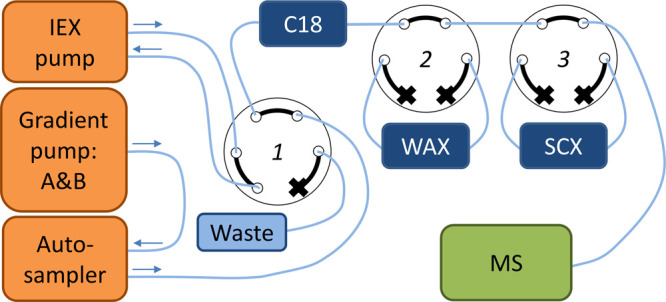
Online fractionation setup. Valve 1, which was located on the mass spectrometer, was used to change between the IEX pump and the gradient pump. Valves 2 and 3, VICI valves, were used to switch the mixed-mode ion exchange columns in or out of line.
The injection volume of the fractionation method was set at 1 μL and the flow rate at 800 μL/min. In positive mode, the C18, WAX, and SCX columns were loaded consecutively. The mobile phases consisted of 0.2% formic acid in water for loading (gradient pump: A), 2 mM ammonium acetate in methanol for the C18 elution (gradient pump: B) and 100 mM ammonium acetate pH 10 for ion exchange elution (IEX pump). In negative mode, the C18 and WAX columns were loaded consecutively. The mobile phases consisted of 2 mM ammonium acetate in water for loading (gradient pump: A), 2 mM ammonium acetate in methanol (gradient pump: B) for the C18 elution and 100 mM ammonium formate pH 10.5 for ion exchange elution (IEX pump). When the gradient pump was selected, the IEX pump pumped the solvent back to the solvent bottle. When the IEX pump was selected, the gradient pump flow was directed to waste. By using two other six-port valves, the IEX columns could be switched in and out of the line of the LC flow. The total runtime was 3 min and the detailed timetable of the fractionation in positive and negative mode can be found in the SI (Table S4 and S5).
The flow injection analysis (FIA) method was adapted from Carducci et al.17 The injection volume was set at 20 μL and the flow rate at 80 μL/min. The mobile phase consisted of 80% methanol in water. Although the mobile phase contained no additives, the sample diluent contained 0.1% acetic acid which was sufficient to promote ionization. At 0.8 min, the flow rate was increased to 800 μL/min for 0.5 min to flush the system and at 1.3 min the flow rate returned to 80 μL/min. The total analysis time was 1.4 min. The MS parameters can be found in the SI (Table S6).
The data were processed in Analytics of Sciex OS 1.6. For the targeted processing, the analytes were integrated by integrating the signal of the M+H (in positive mode) and M–H (in negative mode) ion with an XIC width of 0.01 Da. Glucose was measured as an M+Na ion and choline was measured as an M+ ion. The untargeted data processing was performed using the “Nontargeted Peaks” function in Analytics (see detailed information in the SI Table S11C).
Effect of Circadian Rhythm and Food Intake on Metabolite Classes
The effect of circadian rhythm and food intake on the metabolite classes was evaluated for 10 healthy male volunteers (aged 18–45 years). The clinical study was approved by the Ethical Committee of the Centre for Human Drug Research Leiden and all volunteers signed an informed consent form. The study design has previously been published.19 In short, blood samples were collected over 24 h under uniform conditions for food intake, physical activity, and night rest. At each time point, 20 mL of blood was drawn into two 10 mL BD Vacutainer K2EDTA tubes and kept on ice. The tubes were gently inverted multiple times and centrifuged (1000g, 15 min, 4 °C). Plasma samples were aliquoted and stored at −80 °C prior to analysis. A quality control (QC) was prepared by pooling 15 μL of every individual study sample. A QC sample was analyzed every 10 samples. Metabolites with an RSD below 15% throughout the QC samples were included in the data analysis.
Each metabolite was normalized on the first time point and subsequently log-transformed using the natural logarithm. Then, the metabolites were allocated to six different compound classes (amino acids, amines, hexose, acylcarnitines, organic acids, or fatty acids). An overview of the compound classes is provided in the SI (Table S7). Within each compound class, all metabolite concentrations were averaged per time point and volunteer. A Wilcoxon Signed Rank test was used to assess the change in this mean per time point relative to the baseline.20 A multiple comparisons correction (Benjamini–Yekutieli, < 0.1) was used to adjust the p-values for multiple testing.21 All statistical analyses were performed in R (version 3.4.3).22
Results and Discussion
Method Development
This study aims to develop an efficient and fast methodology to minimize matrix effects, focusing on salt and (phospho)lipid removal. Lipid removal was accomplished by a reversed phase column and salt removal by mixed-mode ion exchange columns. An Agilent ZORBAX Extend-C18 UPLC guard column was selected as the reversed phase column because it demonstrated superior separation and peak shape over low performance SPE columns.
Table 1 provides an overview of the performance of the evaluated ion exchangers. The grading scheme is depicted by numbers and colors indicating good (positive and green) or bad (negative and red) performances. Table 1 indicates that the WCX columns had a relatively low trapping efficiency as most of the analytes eluted at the dead time (grade 0). Most of the analytes were efficiently retained or trapped (grades 1 and 2, respectively) by the SCX columns. However, choline could not be eluted in the Hysphere column and arginine caused breakthrough (grade −1) in the Zirchrom column indicating a superior performance of the Sepax column. The right part of Table 1 shows that all SAX columns did not allow the desorption of indoxyl sulfate (grade −3) indicating that this type of anion exchanger could be exhausted over time due to the irreversible binding of analytes. The Sepax WAX was suitable for all representative analytes, whereas the Oasis column was too strong (grade −3 for indoxyl sulfate) and the Zirchrom column repeatedly resulted in extreme tailing (grade −2). The Sepax SCX and WAX columns were unsurpassed in terms of retention and trapping and allowed for the analysis of all representative compounds. Therefore, we selected these columns for the trapping of the ionic species. The combination of a WAX and SCX also provided the possibility to use a similar elution buffer for both columns. The elution from a WAX column requires a high pH to remove the positive charge on the sorbent, whereas the high pH removes the positive charge of the analytes during the elution of an SCX column. Besides, the high pH is accomplished by the use of ammonia, which is a suitable counterion for an SCX column.
Table 1. Evaluation of Different Mixed-Mode Cation and Anion Exchange Columnsa.
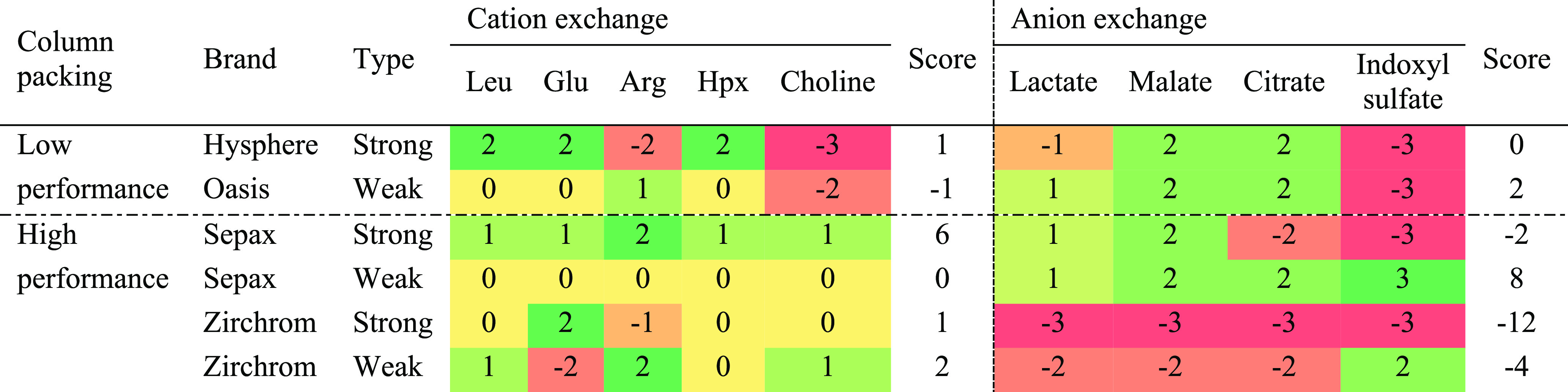
The grading scheme is as follows: elution at dead time: 0; retention: 1; trapped and eluted: 2; trapped and separated during elution: 3; no peak visible: −3; extreme tailing: −2; breakthrough: −1).
The silica material of the ZORBAX Extend-C18 guard column was end-capped with methyl groups which made the sorbent resistant to high pH. Therefore, this particular column could be permanently in line with the flow. In contrast, the IEX columns were switched out of the line during C18 elution because this improved retention. In negative mode, the WAX elution profile was better in the absence of the SCX column. Since the SCX column did not contribute to the reduction of ion suppression in negative mode, this column was permanently switched out of the line during the analysis in negative mode. The IEX methods were further optimized to improve retention and peak shapes and to minimize carry-over.
Fractionation Characteristics
Figure 2 shows the chromatograms of a pooled plasma sample measured with the final fractionation methods in positive and negative mode. The chromatogram contains three different fractions in positive mode (flow-through: polar neutral/positive; IEX: polar positive, and C18: apolar) and three fractions in negative mode (flow-through: polar neutral/negative; IEX: polar negative and C18: apolar). An overview of the fractions and charge of the analytes during loading is supplied in the Supporting Information (Table S7). The elution profile of the phospholipids in the negative fractionation method is measured in positive MS polarity (because of ionization efficiency) and shown in the SI (Figure S1). The phospholipids are separated from both the acylcarnitines and the fatty acids and therefore could not suppress their ionization. This stresses the importance of the combined online fractionation and separation. If these fractions were collected offline and subsequently injected into the MS, then the phospholipids would have been ionized simultaneously with the fatty acids and acylcarnitines. The salts were most likely divided over the mixed-mode ion exchangers (SCX and WAX in positive mode and WAX in negative mode) and eluted during the ion exchange elution. By allocating these known ion suppressors over different fractions, we minimized the ion suppression in a limited amount of time.
Figure 2.
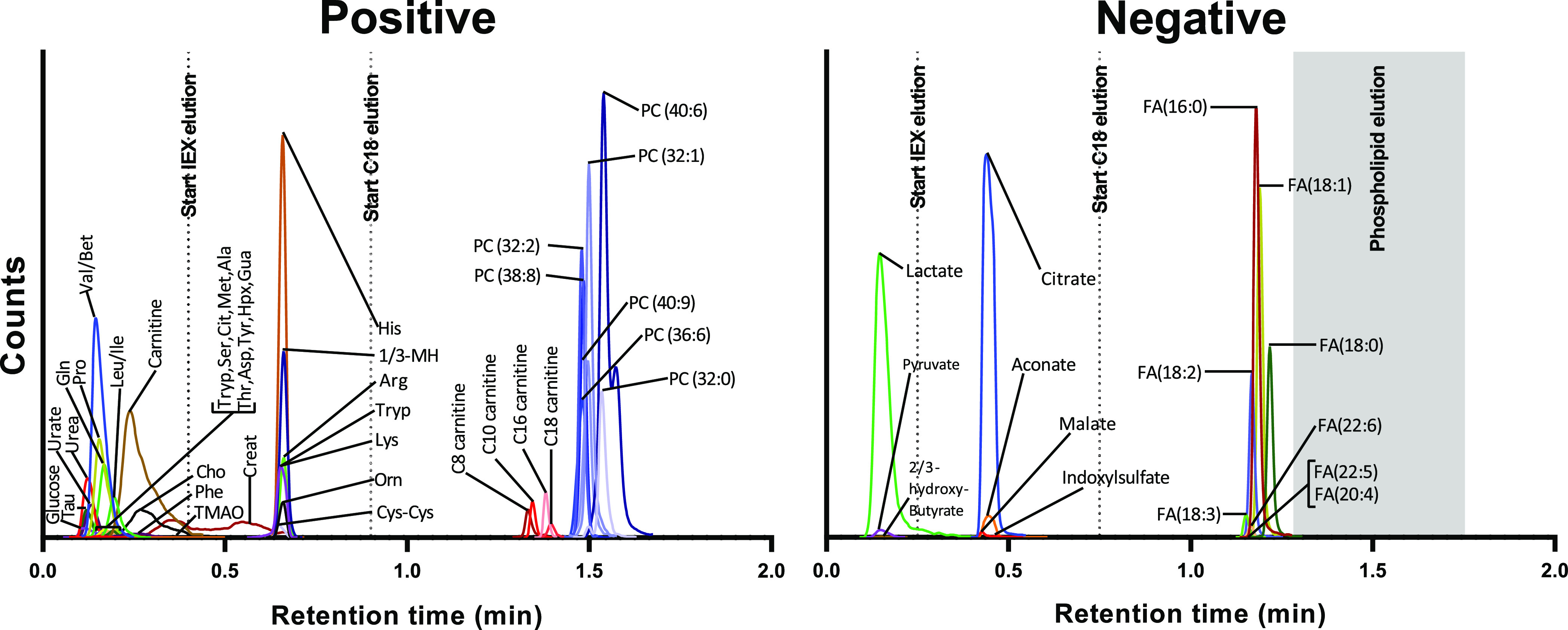
Extracted ion chromatogram of a pooled plasma sample measured by the fractionation method in positive and negative mode. The phospholipid elution window (phospholipid elution profile shown in SI Figure S1) in negative mode is indicated by the gray area. All the ions are measured by M+H in positive mode and M–H in negative mode, apart from hexose which was measured as a sodium adduct. For visualization purposes, the phospholipids and fatty acids were extracted using the one 13C m/z value.
In general, the flow-through fraction contained analytes that were polar and consisted of a zero and/or one net charge during loading. Singly charged compounds experienced some retention in positive mode, but no retention in negative mode. The lack of retention might be explained by the counterion effect of the high concentrations of salts in plasma. In positive mode, a remaining negative charge on the acids might have impaired the retention of amino acids. The second fraction comprised all the components that were trapped on the ion exchange columns. A compound was efficiently trapped on the IEX column if it consisted of multiple net charges or was in equilibrium between one net charge and multiple charges at the pH during loading. The third fraction consisted of all the apolar compounds, which were efficiently trapped on and eluted from the C18 column.
Creatinine was strongly retained but not trapped on the SCX column. Creatinine had one positive net charge and two additional neutral nitrogen atoms, which could have potentially increased the interaction with the stationary phase. We did not find any other compounds that resulted in multiple peaks due to breakthrough or multiple trappings. Nongaussian shaped peak areas were obtained by integrating the area under the curve between the two intersections with the baseline. These compounds were corrected by their corresponding internal standard because their peak shape and retention time were similar (see SI Figure S2 for the example of creatinine(-D3)). Other analytes were corrected either by their corresponding internal standard or by an internal standard that coeluted.
Method Validation
The validation was performed by assessing the repeatability, intermediate precision, carryover, LOD, LLOQ, and the matrix effect of the method. The results of the validation can be found in the SI (Table S8).
The mean repeatability and intermediate precision were 6.0 and 7.1%, respectively. The relative standard deviation of 48 compounds was below 15% and two components varied more than 15%: TMAO and guanine. This was most likely caused by the low signal of these analytes due to the low physiological concentration and the low molecular weight. In total, 1071 injections were performed on the same set of columns with a sufficient repeatability as is shown in the validation (first injections) and biological application (last injections). The coefficient of determination (R2) was on average 0.995, which indicated a good linearity of the fractionation method. The linearity of 47 compounds was higher than 0.99 and three compounds revealed a linearity lower than 0.99. The linearity of C16- and C18-carnitine was compromised by matrix interferences since a calibration curve constructed in water demonstrated a sufficient linearity (>0.99). All the acylcarnitines were corrected by the same internal standard, i.e., octanoylcarnitine-d3. This internal standard corrected well for coeluting analytes C8- and C10-carnitine. C16- and C18-carnitine were more strongly retained and eluted further away from the internal standard and closer to the (phospho)lipids. Therefore, the linearity of these analytes would be improved by the correction of a more apolar internal standard. The lower linearity of docosapentanoic acid was found for both plasma and water samples. The reason for this was unknown.
The LOD and LLOQ were determined by spiking several internal standards in plasma. This was done because the analytes of interest were endogenous and differences in chromatography were observed between water and plasma samples. Figure 3 demonstrates that physiological blood levels as reported in literature were higher than the calculated LLOQ indicating a sufficient sensitivity of the method. The average carryover was 0.5% when a blank sample was measured after a QC sample. In total 48 compounds demonstrated a lower carryover than 2%. There were two compounds with a higher carryover: methionine (5.3%) and decanoylcarnitine (2.4%). The carryover of methionine can be explained by the fact that sulfur sticks to stainless steel.23 The reason for the carryover of decanoylcarnitine was unclear. Although a slight carryover has been observed, we expect no problems with respect to the quantification of study samples. The analytes of interest are endogenous compounds, which are present in every studied person. This will ensure that a small carryover will have a limited effect on the quantification values of the analytes.
Figure 3.
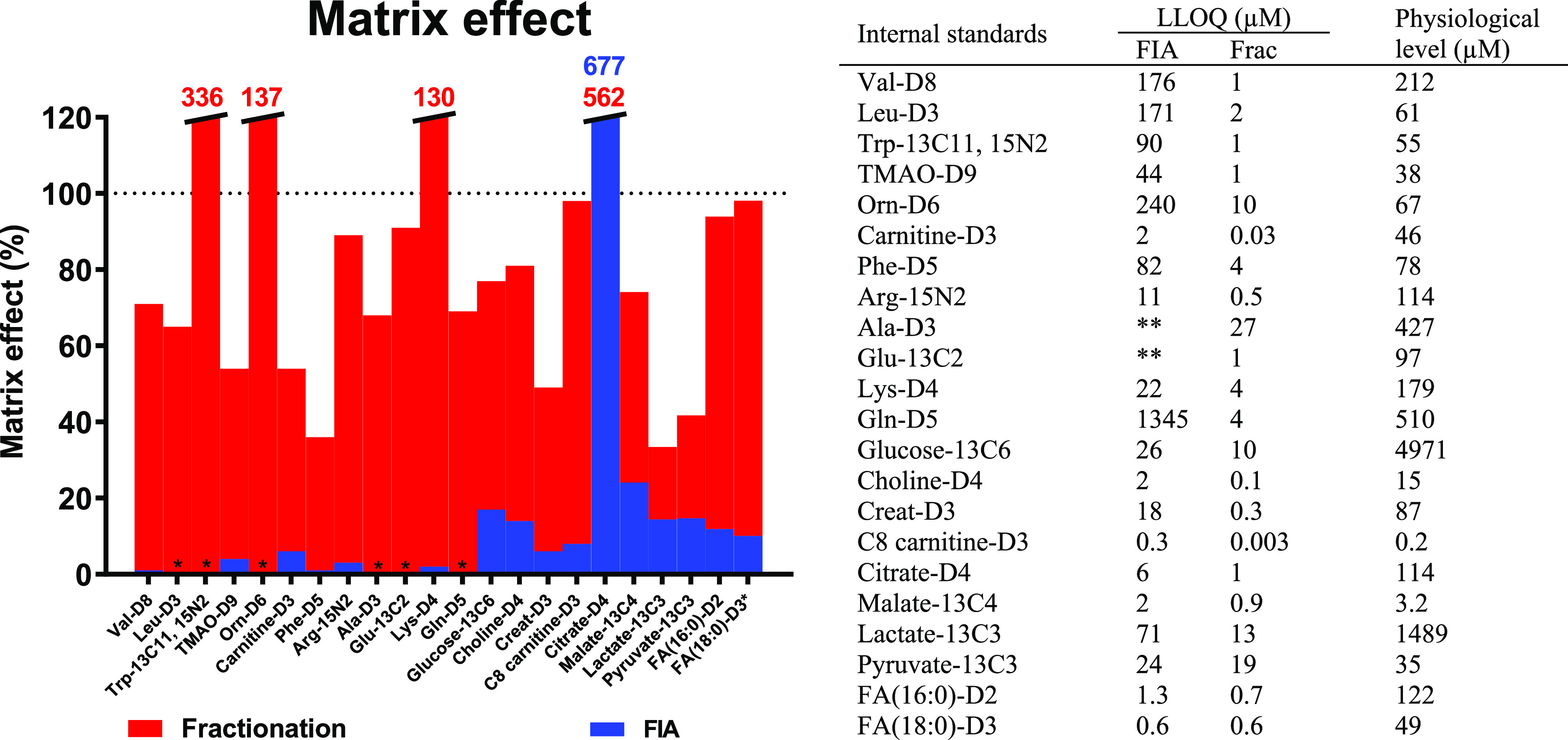
Performance comparison of the fractionation (Frac) method and flow injection analysis (FIA). The graph shows the matrix effect for each internal standard measured by either the fractionation method (red) or FIA (blue). Compounds with 0% matrix effect (indicated by *) were not detected at C4 levels. Compounds that experienced ion enhancement (matrix effect >100%) were cut off at a matrix effect of 120% (values are indicated in corresponding colors). The table on the right shows the lower limit of quantification (LLOQ) of FIA and fractionation as well as the physiological plasma levels (HMDB values).29 (** = not detected at C7 levels).
Fractionation versus Flow Injection Analysis and Conventional Liquid Chromatography
In order to demonstrate the cleanup efficiency of the fractionation method, we measured spiked internal standards in plasma and water. Hereby, the matrix effect, ion suppression, and LLOQ were determined for the fractionation and an FIA method. Figure 3 shows that the mean ion suppression of the fractionation method was 25%, whereas the mean ion suppression in the FIA method was 89%. We have previously reported the effects of salts and phospholipids on the ESI.11 The fractionation method provides a fast solution to minimize ion suppression caused by these matrix interferences. The use of three orthogonal columns allocated phospholipids, negative and positive salts into three different fractions. The online elution into the MS and the use of high performance SPE columns allowed for the separation between analytes and matrix interferences within a fraction. An additional LLE step prior to the FIA decreased the ion suppression to 80% (see SI Table S9). This decrease in ion suppression was predominantly observed for compounds in the apolar fraction, i.e., fatty acids and acylcarnitines. However, the ion suppression of these compounds was still considerably less in our fractionation method. For metabolites in the polar fraction, the ion suppression was comparable with FIA without LLE. LLE demonstrates little cleanup efficiency because samples are only fractionated based on polarity, and the obtained fractions are analyzed at once without further separation.
The fractionation method demonstrated a superior sensitivity in comparison with FIA. The mean LLOQ of the fractionation method was 21 times lower which ensured a sufficient sensitivity to measure physiological levels in plasma. In contrast, 9 out of 22 analytes could not be quantified using the FIA method due to insufficient sensitivity (LLOQ higher than physiological levels). The substantial difference in ion suppression was most likely responsible for the differences in sensitivity. The performance improvement was mainly reflected in positive mode. In negative mode, the improvement in ion suppression and sensitivity was smaller. This is in accordance to other studies, in which was shown that ion suppression is less occurring in negative ionization mode.24,25 Although the FIA method is faster (1.4 versus 3 min), the findings in Figure 3 emphasize the necessity of online fractionation prior to electrospray ionization.
We have also compared the LLOQ of the ISTDs with the LLOQ of conventional LC–MS analyses reported in literature (see SI Table S10). These findings demonstrated that the sensitivity of fractionation and LC–MS is in a similar range. This was also expected because of the limited ion suppression in the fractionation method and a comparable peak width, injection volume, and flow rate with regard to general LC–MS. However, differences in, for example, LLOQ determinations, used mass spectrometer (tandem and high-resolution) and derivatization might complicate this comparison. It does indicate that we are at least in a comparable sensitivity range relative to LC–MS. This is also emphasized by the coverage of the fractionation method in comparison with conventional reversed phase (RP) and hydrophilic interaction chromatography (HILIC) separations. The number of unique retention time and m/z features was 2289, 3475, and 3529 for fractionation, RP and HILIC, respectively (the methodologies are presented in the SI Table S11). The difference in coverage is mostly explained by the additional isomeric separation that is experienced in conventional chromatography as the number of unique m/z features was practically similar (2089, 2465, and 2325 for fractionation, RP and HILIC, respectively).
Our fractionation approach enables the analysis of multiple compound classes in 3 min per polarity, whereas conventional LC–MS usually requires a gradient time of around 3–30 min per compound class (see Table S10). The analysis time of LC–MS can be reduced by the use of faster gradients. However, in order to realize a comprehensive targeting of the metabolome, multiple LC separations would be needed (e.g., HILIC and RP for polar and apolar, respectively). The inclusion of multiple chromatographic gradients drastically decreases the overall throughput of the analysis. Moreover, the equilibration and flushing time of conventional LC columns (3–15 cm) is substantially higher in comparison with short chromatographic columns (0.5–1 cm). The benefit of an integrated fractionation approach is due to the use of multiple short chromatographic columns, which allow for an efficient separation, while little time is spent on gradients and column equilibration/flushing. The challenge of using a fractionation approach instead of conventional chromatography is the lack of isomeric separation. This could be overcome by the use of ion-mobility and MS/MS experiments.
Effect of Circadian Rhythm and Food Intake on Metabolite Classes
It is known that there are trends in metabolite levels due to the circadian rhythm and food intake.26 These fluctuations are important to take into account when metabolites are studied or used as biomarkers. Different sampling times throughout the day could cause variations in metabolite levels that are not attributable to a studied disease or intervention. For this, we profiled 10 healthy volunteers on 10 different time points on a time scale of 24 h. This study should clarify the significance of these diurnal changes.
After the data acquisition, 47 compounds were included in the data analysis and three compounds were excluded. Fatty acid 16:0 and 18:0 had an RSD of more than 15% due to fluctuating background levels. C18 carnitine also had an RSD of more than 15%. The reason for this was unclear. Figure 4 shows that our validated platform allowed us to demonstrate significant changes of metabolite classes throughout the day (false discovery rate (FDR) adjusted p-values are listed in the SI Table S12). All compound classes changed significantly from the baseline, apart from the amines. The amines (quaternary amines, creatinine, urea, and uric acid) did not reveal a significant difference over a period of 24 h. This is in accordance with our prior work, in which we demonstrated that gut metabolites (quaternary amines) were not affected by the fasting state of an individual.11 The amino acid levels started to rise after wake time. The levels remained high throughout the morning/afternoon and decreased again toward baseline levels just before dinner. After dinner, the amino acids increased again and subsequently returned to baseline levels during night rest. The increase in amino acids after wake time and in the afternoon/evening has also been observed in prior studies.20
Figure 4.
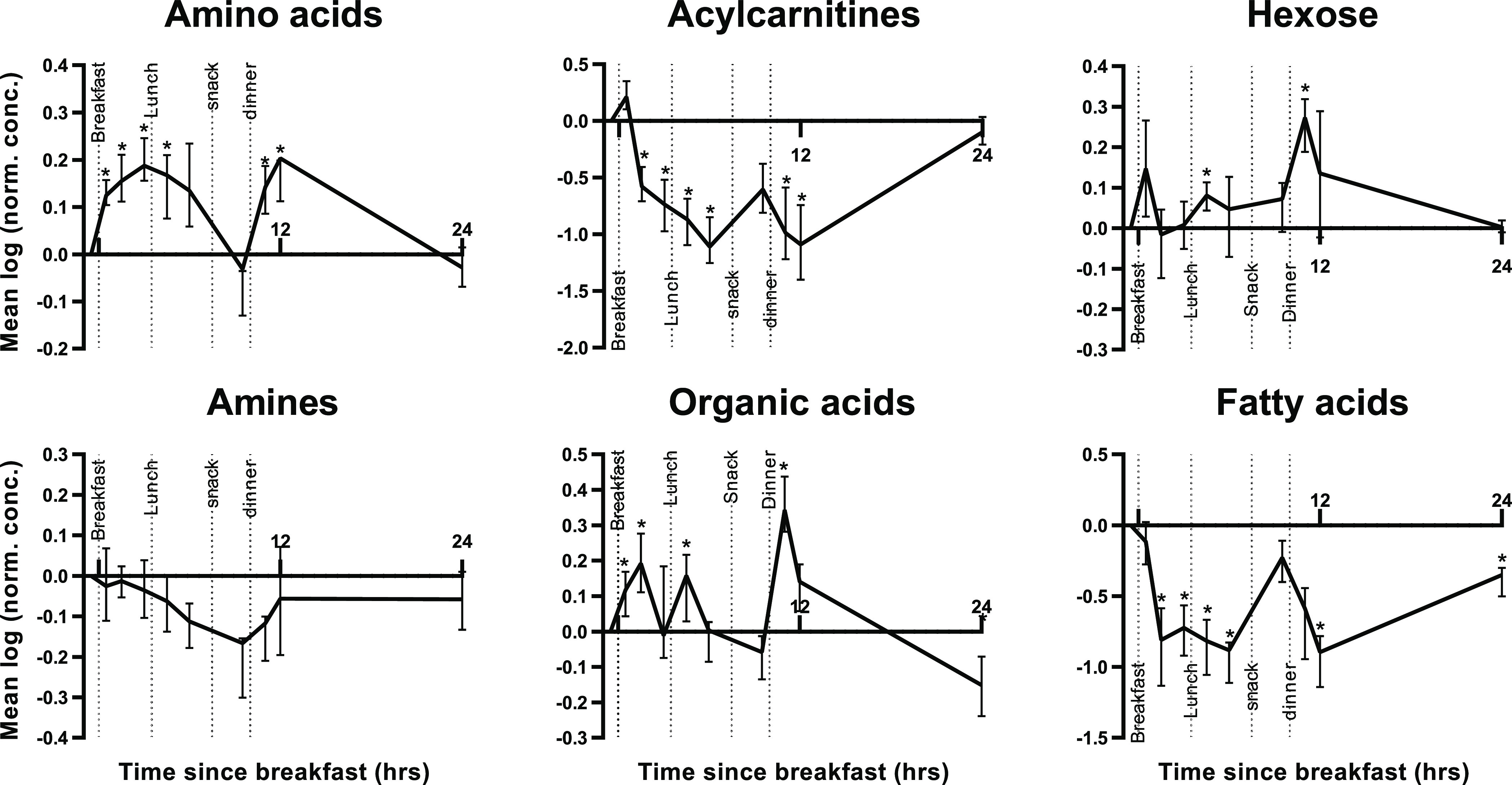
Mean natural logarithm of metabolite concentrations over time. Normalization was performed on the first time point. Within each compound class, metabolites were averaged per time point and volunteer. The mean of these curves over the 10 volunteers are depicted and the pointwise interquartile range (IQR) of the volunteers is presented in the error bars. Time points that are significantly different from the baseline are indicated (* FDR adjusted p-values <0.1). The time frame comprises four standardized feeding times and meals and one night rest. The time is presented with respect to the breakfast time. Individual trends are shown in the SI Figure S3.
The hexose and organic acid levels significantly increased after the feeding times (except for hexose after breakfast which did not reach FDR corrected significance). When sugar is available, glucose is the main source of the citric acid cycle. This explains the similarities of the hexose and organic acid trends since organic acids are the main constituents in the citric acid cycle. The fatty acid concentrations decreased throughout the day and increased just before dinner and after 24 h, which has been observed before.26 During (overnight) fasting, glucose is mainly depleted, switching the main energy source to fatty acids. In this state, fatty acids are released from triglycerides by lipolysis, which explains the high fatty acid levels prior to dinner and after a night rest.27 In order to accommodate the increased demand for fatty acids, acylcarnitines are put in place to transport the fatty acids into the mitochondria for β-oxidation.28 This explains the similarities between the fatty acid and acylcarnitine profile. Sampling time is an indispensable parameter to take into account when metabolites are used or studied as biomarkers. Food intake and circadian rhythm significantly change compound classes from baseline levels. Therefore, sampling times and fasting states should be standardized when metabolites are used for diagnosis, clinical studies or biomarker discovery. This should further strengthen the use of discovered metabolite biomarkers in personalized health care.
Conclusions
Although much progress has been made in the analysis of metabolites, fast and global profiling of the metabolome in complex matrices remains a challenging aspect. For this purpose, we demonstrated a fast and comprehensive fractionation method coupled online to mass spectrometry. The three serially coupled high performance SPE columns resulted in a fractionation based on polarity, charge, and removed important ion suppressors from different fractions. The online and orthogonal setup realized a flow-through which was cleaned by three different sorbent chemistries and a within-fraction separation of analytes and ion suppressors. The comparison with FIA emphasized the performance improvement achieved with the fractionation method. In a limited amount of time, the fractionation method drastically lowered the ion suppression as well as the detection limits. The online fractionation demonstrated similar quantification limits in comparison to the conventional LC–MS analyses. This proves that online fractionation enables the analysis of a large range of metabolites without suffering in terms of sensitivity. The developed fractionation method was able to demonstrate fluctuations of metabolite classes in blood samples from healthy volunteers on different time points throughout the day, which could be explained by underlying metabolic processes. These significant diurnal variations are important for clinicians when metabolites are used as biomarkers. Standardized sampling times and fasting states should minimize variations caused by food intake and circadian rhythm on the disease or intervention related variations. This work provides a methodology to target multiple metabolite classes within a single analytical platform without suffering in terms of analysis time. This development in comprehensive and fast metabolite screening should encourage researchers and clinicians to make full use of the field of metabolomics and to further investigate the value of potential prognostic and diagnostic biomarker metabolites.
Acknowledgments
The authors are grateful to receive funding for this research from The Netherlands Organization for Scientific Research (NWO) in the framework of the Technology Area TA-COAST (Fund New Chemical Innovations, project no. 053.21.118). This research was also part of The Netherlands X-omics Initiative and partially funded by NWO, project 184.034.019.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.0c01375.
Figure S1, extracted ion chromatogram of phospholipids in negative mode; Figure S2, chromatogram of creatinine(-D3); Figure S3, individual mean natural logarithm of metabolite classes over time; Table S1, overview of calibration standards; Table S2, overview of internal standards; Table S3, solid-phase extraction column information; Table S4, LC and valve parameters for positive mode; Table S5, LC and valve parameters for negative mode; Table S6, mass spectrometry parameters; Table S7, overview of the different fractions; Table S8, method validation parameters; Table S9, matrix effect in Frac, LLE-FIA, and FIA; Table S10, LLOQ of literature LC–MS methods; Table S11, coverage of Frac, RP–LC and HILIC–LC; and Table S12, p-values of circadian rhythm and food intake application (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Mussap M.; Zaffanello M.; Fanos V. Metabolomics: A challenge for detecting and monitoring inborn errors of metabolism. Ann. Transl. Med. 2018, 6, 338. 10.21037/atm.2018.09.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balashova E.; Maslov D.; Lokhov P. A Metabolomics Approach to Pharmacotherapy Personalization. J. Pers. Med. 2018, 8, 28. 10.3390/jpm8030028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushik A. K.; DeBerardinis R. J. Applications of metabolomics to study cancer metabolism. Biochim. Biophys. Acta, Rev. Cancer 2018, 1870, 2–14. 10.1016/j.bbcan.2018.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miggiels P.; Wouters B.; Van Westen G. J.; Dubbelman A.; Hankemeier T. Novel technologies for metabolomics: more for less. TrAC, Trends Anal. Chem. 2019, 120, 115323. 10.1016/j.trac.2018.11.021. [DOI] [Google Scholar]
- Gowda G. A. N.; Djukovic D. Overview of Mass Spectrometry-Based Metabolomics: Opportunities and Challenges. Methods Mol. Biol. 2014, 1198, 3–12. 10.1007/978-1-4939-1258-2_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ismaiel O. A.; Halquist M. S.; Elmamly M. Y.; Shalaby A.; Karnes H. T. Monitoring phospholipids for assessment of matrix effects in a liquid chromatography-tandem mass spectrometry method for hydrocodone and pseudoephedrine in human plasma. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2007, 859, 84–93. 10.1016/j.jchromb.2007.09.007. [DOI] [PubMed] [Google Scholar]
- Enot D. P.; et al. Preprocessing, classification modeling and feature selection using flow injection electrospray mass spectrometry metabolite fingerprint data. Nat. Protoc. 2008, 3, 446–470. 10.1038/nprot.2007.511. [DOI] [PubMed] [Google Scholar]
- Nanita S. C.; Kaldon L. G. Emerging flow injection mass spectrometry methods for high-throughput quantitative analysis. Anal. Bioanal. Chem. 2016, 408, 23–33. 10.1007/s00216-015-9193-1. [DOI] [PubMed] [Google Scholar]
- Zhang J.; et al. High-throughput salting-out assisted liquid/liquid extraction with acetonitrile for the simultaneous determination of simvastatin and simvastatin acid in human plasma with liquid chromatography. Anal. Chim. Acta 2010, 661, 167–172. 10.1016/j.aca.2009.12.023. [DOI] [PubMed] [Google Scholar]
- Zhang X.; et al. SPE-IMS-MS: An automated platform for sub-sixty second surveillance of endogenous metabolites and xenobiotics in biofluids. Clin. Mass Spectrom. 2016, 2, 1–10. 10.1016/j.clinms.2016.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Laan T.; Kloots T.; Beekman M.; Kindt A.; Dubbelman A.-C.; Harms A.; van Duijn C. M.; Slagboom P. E.; Hankemeier T.; et al. Fast LC-ESI-MS/MS analysis and influence of sampling conditions for gut metabolites in plasma and serum. Sci. Rep. 2019, 9, 12370. 10.1038/s41598-019-48876-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trivedi D. K.; Hollywood K. A.; Goodacre R. Metabolomics for the masses: The future of metabolomics in a personalized world. New Horizons Transl. Med. 2017, 3, 294–305. 10.1016/j.nhtm.2017.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472, 57–63. 10.1038/nature09922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayo Clinic. Rochester 2018 Interpretive Handbook; 2018 10.1016/S1002-0721(13)60014-9. [DOI] [Google Scholar]
- Wishart D. S.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. 10.1093/nar/gkx1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matuszewski B. K.; Constanzer M. L.; Chavez-Eng C. M. Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC-MS/MS. Anal. Chem. 2003, 75, 3019–30. 10.1021/ac020361s. [DOI] [PubMed] [Google Scholar]
- Carducci C.; et al. Quantitative determination of guanidinoacetate and creatine in dried blood spot by flow injection analysis-electrospray tandem mass spectrometry. Clin. Chim. Acta 2006, 364, 180–187. 10.1016/j.cca.2005.06.016. [DOI] [PubMed] [Google Scholar]
- Bligh E. G.; Dyer W. J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911. 10.1139/y59-099. [DOI] [PubMed] [Google Scholar]
- Duisters K.; Ogino S.; Andou T.; Ito K.; Akabane T.; Harms A.; Moerland M.; Hashimoto Y.; Ando A.; Ohtsu Y.; et al. Intersubject and Intrasubject Variability of Potential Plasma and Urine Metabolite and Protein Biomarkers in Healthy Human Volunteers. Clin. Pharmacol. Ther. 2020, 107, 397. 10.1002/cpt.1606. [DOI] [PubMed] [Google Scholar]
- Thompson D. K.; et al. Daily Variation of Serum Acylcarnitines and Amino Acids. Metabolomics 2012, 8, 556–565. 10.1007/s11306-011-0345-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y.; Yekutieli D. The control of the false discovery rate in multiple testing under dependency. Ann. Stat. 2001, 29, 1165–1188. 10.1214/aos/1013699998. [DOI] [Google Scholar]
- R Core Team.. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2017; available at https://www.r-project.org/. [Google Scholar]
- Nagu M.; Abdulhadi A.; Huwaiji A.; Alanazi N. M. Effect of Elemental Sulfur on Pitting Corrosion of Steels Muthukumar. Insights Anal. Electrochem. 2018, 4 (1), 6. 10.21767/2470-9867.100030. [DOI] [Google Scholar]
- Ghosh C.; Shinde C. P.; Chakraborty B. S. Ionization Polarity as a Cause of Matrix Effects, its Removal and Estimation in ESI-LC-MS/MS Bio-analysis. J. Anal. Bioanal. Tech. 2010, 01 (02), 1–7. 10.4172/2155-9872.1000106. [DOI] [Google Scholar]
- Oldekop M. L.; Rebane R.; Herodes K. Dependence of matrix effect on ionization polarity during LC–ESI–MS analysis of derivatized amino acids in some natural samples. Eur. J. Mass Spectrom. 2017, 23, 245–253. 10.1177/1469066717711026. [DOI] [PubMed] [Google Scholar]
- Dallmann R.; Viola A. U.; Tarokh L.; Cajochen C.; Brown S. A. The human circadian metabolome. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 2625–2629. 10.1073/pnas.1114410109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali A. H.; et al. Free fatty acid storage in human visceral and subcutaneous adipose tissue: Role of adipocyte proteins. Diabetes 2011, 60, 2300–2307. 10.2337/db11-0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kompare M.; Rizzo W. B. Mitochondrial Fatty-Acid Oxidation Disorders. Semin. Pediatr. Neurol. 2008, 15, 140–149. 10.1016/j.spen.2008.05.008. [DOI] [PubMed] [Google Scholar]
- Human Metabolome Database . Available at http://www.hmdb.ca/.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.