

Abstract
Thirteen compounds were isolated from the methanolic extract of the leaves of Androcymbium palaestinum Baker (Colchicaceae). Of these, three were new, two were new natural products, and eight were known. The new isolated compounds were (+)-1-demethylandrocine (5), (−)-andropalaestine (8), and (+)-2-demethyl-β-lumicolchicone (10), while the new natural products were (+)-O-methylkreysigine-N-oxide (3) and (+)-O,O-dimethylautumnaline (9). Moreover, two known compounds are reported for the first time from this species, specifically (−)-colchicine (11) and (−)-3-demethyldemecolcine (13). The structures of the isolated compounds were elucidated using a series of spectroscopic and spectrometric techniques, principally HRESIMS, 1D-NMR (1H and 13C-NMR) and 2D-NMR (COSY, edited-HSQC, and HMBC). ECD spectroscopy was used for assigning the absolute configurations of compounds 3, 5, and 10. The cytotoxic activities of the isolated compounds were evaluated using the MDA-MB-435 (melanoma), MDA-MB-231 (breast), and OVCAR3 (ovary) cancer cell lines. Compound 11 was the most potent against all tested cell lines, with IC50 values of 12, 95 and 23 nM, respectively.
Keywords: Alkaloids, Androcymbium, Leaves, Absolute configuration, Cytotoxicity, Human cancer cell lines
1. Introduction
Throughout history natural products have contributed immensely to the drug discovery process [1–4]. In the area of cancer, for example, and over the time frame from 1940s to 2014, of the 175 small molecules approved, 131, or 75%, are other than synthetic, with 85, or 49%, actually being either natural products or directly derived therefrom [2]. Moreover, of the 13 natural product-derived drugs that were approved in the U.S. between 2005 and 2007, five were the first members of new classes [1].
Jordan, with its unique position in the heart of the Middle East, acts as a floral bridge between the continents of Asia, Africa, and Europe [5, 6]. This geographical position bestows the country with ecologically diverse habitats and a rich variety of wild plants [7, 8]. For example, more than 2000 plant species were reported to grow in the wild [6], with less than 5% explored previously for bioactive secondary metabolites.
The Colchicaceae, a family of 16 genera and more than 250 species [9], is represented in Jordan by two genera: Androcymbium and Colchicum [6]. While Colchicum sp. in Jordan have been investigated to some degree [10–15], members of the Androcymbium are less well studied. The genus Androcymbium is native to Africa, the Mediterranean, and the Middle East, and includes about 56 species [9, 16], many of which have been reported in folk medicine for the treatment of a variety of illnesses [17]. Several subclasses of alkaloids, including colchicinoids, dibenzocycloheptylamines, homoaporphines, and l-phenethyltetrahydroisoquinolines, have been reported previously from this genus [18–23].
Androcymbium palaestinum Baker (Colchicaceae), which is found flowering from December to February, is the only species reported to grow in Jordan [6]. It is a perennial plant with small underground corms covered with brown scales. It has white showy flowers with brown midribs. The leaves are basal folded in the midrib, wider at base and narrow towards the tip. It is known by local people as “Zanbaq Alghor”, which translates to lily of the Jordan Valley. It flourishes in warm sandy soils, such as those found in the Jordan Valley, near the Dead Sea, and in Wadi Araba and Wadi Rum [6]. Over 30 years ago, the corms of A. palaestinum were studied, yielding 14 compounds from three different alkaloid classes, specifically, the homoaporphine alkaloids: (+)-O-methylkreysigine, (+)-kreysigine, (+)-androcine, (+)-androcimine, (+)-androbine, (+)-nor-O-methylkreysigine, (+)-norandrobine, and (+)-szovistamine; the dibenzocycloheptylamine: (−)-androbiphenyline, K-3, and K-4; the colchicinoids: (−)-demecolcine and (−)-3-demethylcolchiceine; and the homomorphinandienone (−)-collutine (Table S1, Supplementary Data) [18, 19].
While the flower of A. palestinum is beautiful, there are no reports of the use of this plant by local people for traditional medicine; interestingly, it is avoided by grazing animals, such as goats (Personal Communication). Natural products chemistry studies on a methanolic extract of the leaves of this plant resulted in the isolation and identification of thirteen compounds; of which eight were known (1, 2, 4, 6, 7, and 11–13 ), two were new natural products: (+)-O-methylkreysigine-N-oxide (3) and (+)-O,O-dimethylautumnaline (9), and three were new compounds: (+)-1-demethylandrocine (5), (−)-andropalaestine (8), and (+)-2-demethyl-β-lumicolchicone (10). ECD spectroscopy was used to confirm the absolute configurations of the known compounds and to assign the absolute configurations of the new compounds 3, 5, and 10. A scheme has been added to the supplementary data file to summarize the biogenetic relationships between the various compounds reported in the current manuscript (Scheme S1, Supplementary Data). The isolated compounds (1-13) were tested for their cytotoxicity using three human cancer cell lines, specifically MDA-MB-435 (melanoma), MDA-MB-231 (breast), and OVCAR3 (ovarian).
2. Experimental
2.1. General experimental procedures
Optical rotations, UV data, and ECD spectra were obtained using a Rudolph Research Autopol III polarimeter (Rudolph Research Analytical), a Varian Cary 100 Bio UV‒vis spectrophotometer (Varian Inc.), and an Olis DSM 17 ECD spectrophotometer (Olis, Inc.). NMR data were collected using either a JEOL ECA-500 NMR spectrometer operating at 500 MHz for 1H and 125 MHz for 13C or a JEOL ECS-400 NMR spectrometer operating at 400 MHz for 1H and 100 MHz for 13C and equipped with a high sensitivity JEOL Royal probe and a 24-slot autosampler (both from JEOL Ltd.) or an Agilent 700 MHz NMR spectrometer (Agilent Technologies), equipped with a cryoprobe, operating at 700 MHz for 1H and 175 MHz for 13C. Residual solvent signals were utilized for referencing. HRMS data were acquired using a Thermo QExactive Plus mass spectrometer equipped with an electrospray ionization source (Thermo Fisher Scientific). Gemini‒NX C18 analytical (5 μm; 250 × 4.6 mm) and preparative (5 μm; 250 × 21.2 mm), Luna PFP C18 analytical (5 μm; 250 × 4.6 mm), semipreparative (5 μm; 250 × 10.0 mm), and preparative (5 μm; 250 × 19.0 mm) columns (all from Phenomenex) along with Waters Atlantis T3 C18 analytical (5 μm; 250 × 4.6 mm), semipreparative (5 μm; 250 × 10.0 mm), and preparative (5 μm; 250 ×19.0 mm) columns (all from Waters Corp.) were used on a Varian Prostar HPLC system equipped with ProStar 210 pumps and a Prostar 335 photodiode array detector (PDA), with data collected and analyzed using Galaxie Chromatography Workstation software (version 1.9.3.2, Varian Inc.). Flash chromatography was performed on a Teledyne ISCO CombiFlash Rf 200 using Silica Gold columns (both from Teledyne ISCO) and monitored by UV and evaporative light-scattering detectors. All other reagents and solvents were obtained from Fisher Scientific and were used without further purification.
2.2. Plant material
Leaves of A. palaestinum were collected during flowering stage in February/March 2016 from the Jordan Valley, Waqqas city (32°32’34.7712” N; 35°36’18.6624” E, 152.4 m below sea level). The plant material was identified by Dr. Mohammed Gharaibeh, Plant Taxonomist, Faculty of Agriculture, JUST. A voucher specimen (PHS-121) was deposited in the herbarium of the Faculty of Pharmacy, JUST, Irbid, Jordan. The collected plant material was air-dried in the shade away from direct sunlight. This dried plant material was ground into powder using a laboratory mill, stored at room temperature, and protected from light until required for extraction and analysis.
2.3. Extraction and isolation
Extraction and fractionation was carried out as reported by Alali et al (2005) [24]. Briefly, air-dried leaves (600 g) of A. palaestinum were extracted via infusion by soaking in MeOH (3 × ~5 L) at rt for three days followed by filtration to separate the marc. The filtrates were combined and dried under reduced pressure to yield a MeOH extract (111 g).
The MeOH extract was reconstituted in 1000 mL of 5% acetic acid. The acidic solutions were then extracted three times using petroleum ether (3 × 1000 mL) to yield fraction A (3.8 g), and then three times with diethyl ether (3 × 1000 mL) to yield fraction B (5 g). The acidic aqueous residues were then made alkaline (pH = 9) using 10% ammonium hydroxide (NH4OH), and extracted three times with dichloromethane (3 × 1000 mL) to yield fraction C (979 mg). The pH of the basic aqueous residues were adjusted to pH=12 using sodium hydroxide and extracted three times with dichloromethane (3 × 1000 mL) to yield fraction D (100 mg). Finally, the fractions were brought to dryness under vacuum.
The dried alkaloid rich fraction (Fraction C) of the leaves (~979 mg) was dissolved in CHCl3 and mixed with Celite 545. Normal-phase flash chromatography was performed using a gradient solvent system of hexanes-CHCl3-MeOH, at a flow rate of 30 mL/min, and 64.4 column volumes over a total run time of 36 min using a 12 g silica RediSep column to yield eight fractions, which were subjected to purifications using HPLC methods, both preparative and semipreprative, leading to the isolation of 1-13.
Fraction 3 (251.6 mg) of the normal-phase flash chromatography was subjected to preparative HPLC over a Gemini column using a gradient system of 40:60–60:40 of MeOH–H2O (0.1% formic acid) over 15 min at a flow rate of 21.24 mL/min to yield 16 subfractions. Subfraction 5 (32.4 mg) was subjected preparative HPLC over an Atlantis T3 column using a gradient system of 30:70–40:60 of MeOH–H2O (0.1% formic acid) over 30 min, no hold, at a flow rate of 17 mL/min to yield 6 subfractions. Subfraction 3 (9.3 mg) was subjected to preparative HPLC over a PFP column using a gradient system of 0:100–50:50 of CH3CN-H2O (10% formic acid) over 15 min at a flow rate of 21.24 mL/min to yield compound 12 (5.4 mg). Subfraction 11 that had resulted from fraction 3 of the normal-phase flash chromatography was subjected to semipreparative HPLC over an Atlantis T3 column using a gradient system of 40:60–70:30 of MeOH–H2O (0.1% formic acid) over 15 min, no hold, at a flow rate of 4.6 mL/min to yield 4 subfractions. Subfraction 3 (8.6 mg) was subjected to preparative HPLC over a PFP column using a gradient solvent system of 0:100–20:80 of CH3CN-H2O (10% formic acid) over 5 min, to 40:60 over 15 min at a flow rate of 21.24 mL/min to yield compound 11 (6.4 mg). Moreover, semipreparative HPLC purification of subfraction 1 (1.3 mg) over a PFP column using a gradient solvent system of 0:100–20:80 of CH3CN-H2O (10% formic acid) over 5 min to 40:60 over 15 min at a flow rate of 4.62 mL/min yielded compound 10 (0.7 mg). Subfraction 7 (9.6 mg) that had resulted from fraction 3 of the normal-phase flash chromatography was subjected to semipreparative HPLC over an Atlantis T3 column using a gradient solvent system of 40:60–70:30 of MeOH:H2O (0.1% formic acid) over 15 min, no hold, at a flow rate of 4.6 mL/min to yield 3 subfractions. Subfraction 1 (3.4 mg) was subjected to semipreparative HPLC over a PFP column using a gradient system of 0:100–10:90 of CH3CN:H2O (10% formic acid) over 5 min to 30:70 over 15 min at a flow rate of 4.62 to yield compound 9 (2.2 mg).
Fraction 4 (138 mg) of the normal-phase flash chromatography was subjected to preparative HPLC over a PFP column using a gradient system of 40:60–60:40 of MeOH–H2O (0.1% formic acid) over 15 min at a flow rate of 21.24 mL/min to yield 12 subfractions. Subfraction 4 (48.8 mg) was subjected to preparative HPLC over a PFP column using a gradient of 7:93 to 13:87 of CH3CN-H2O (0.1% formic acid) over 15 min at a flow rate of 21.24 mL/min to yield 3 subfractions. Subfraction 3 (12.9 mg) was subjected to preparative HPLC over a PFP column using a gradient system of 0:100–10:90 over 5 min to 20:80 over 15 min of CH3CN-H2O (10% formic acid) at a flow rate of 21.24 mL/min to yield compound 1 (5 mg). Subfraction 7 (31.6 mg) that had resulted from fraction 4 of the normal-phase flash chromatography was subjected to preparative HPLC over an Atlantis T3 column using a gradient system of 30:70–50:50 over 15 min, no hold, of MeOH-H2O (0.1% formic acid) at a flow rate of 17 mL/min to yield 5 subfractions. Subfraction 1 (10 mg) was subjected to preparative HPLC over a PFP column using a gradient system of 0:100–10:90 over 5 min to 30:70 over 15 min of CH3CN-H2O (10% formic acid) at a flow rate of 21.24 mL/min to yield compound 2 (5.4 mg). Subfraction 5 (16.6 mg) that had resulted from fraction 4 was subjected to preparative HPLC over a PFP column using a gradient system of 7:93–13:87 over 13 min of CH3CN:H2O (0.1% formic acid) at a flow rate of 21.24 mL/min to yield 5 subfractions. Subfraction 2 (2.1 mg) was subjected to semiprepartive HPLC over a PFP column using a gradient system of 0:100–10:90 of CH3CN:H2O (10% formic acid) over 5 min to 20:80 over 15 min at a flow rate of 4.62 mL/min to yield compound 13 (1.8 mg).
Fraction 5 (124.3 mg) of the normal-phase flash chromatography was subjected to preparative HPLC over a PFP column using a gradient mobile phase of 0:100–20:80 of CH3CN:H2O (0.1% formic acid) over 15 min, hold for 5 min, then to 50:50 over 15 min at a flow rate of 21.24 mL/min to yield 7 subfractions. Subfraction 4 (14.6 mg) was subjected to semipreparative HPLC over an Atlantis T3 column using a gradient system of 20:80–40:60 of MeOH:H2O (0.1% formic acid) over 15 min, no hold, at a flow rate of 4.6 mL/min to yield 4 subfractions. Subfraction 2 (4.5 mg) was subjected to preparative HPLC over a PFP column using a gradient system of 0:100–10:90 of CH3CN:H2O (10% formic acid) over 5 min to 30:70 over 15 min at a flow rate of 21.24 mL/min to yield compound 5 (4.1 mg). Subfraction 6 (25.9 mg) that had resulted from fraction 5 of the normal-phase flash chromatography was subjected to preparative HPLC over an Atlantis T3 column using a gradient mobile phase of 30:70–40:60 of MeOH:H2O (0.1% formic acid) over 15 min at a flow rate of 17 mL/min to yield 2 subfractions. Subfraction 2 (10.6 mg) was subjected to preparative HPLC over a PFP column using a gradient mobile phase of 0:100–10:90 of CH3CN:H2O (10% formic acid) over 5 min to 20:80 over 15 min at a flow rate of 21.24 mL/min to yield compound 4 (5.9 mg).
Fraction 6 (151.4 mg) of the normal-phase flash chromatography was subjected to preparative HPLC over a PFP column using a gradient system of 0:100–20:80 of CH3CN:H2O (0.1% formic acid) over 15 min, hold for 5 min, then to 50:50 over 15 min at a flow rate of 21.24 mL/min to yield 8 subfractions. Subfraction 4 (8.6 mg) was subjected preparative HPLC over a PFP column using a gradient mobile phase system of 10:90–15:85 of CH3CN:H2O (0.1% formic acid) over 15 min at a flow rate of 21.24 mL/min to yield 2 subfractions. Subfraction 2 (6 mg) was subjected to preparative HPLC over a PFP column using a gradient system of 0:100–15:85 CH3CN:H2O (10% formic acid) over 15 min at a flow rate of 21.24 mL/min to yield compound 7 (1.9 mg). Subfraction 8 (39.6 mg) that had resulted from fraction 6 of the normal-phase flash chromatography was subjected to preparative HPLC over an Atlantis T3 column using a gradient system of 20:80–40:60 of MeOH:H2O (0.1% formic acid) over 15 min, no hold, at a flow rate of 17 mL/min to yield 3 subfractions. Subfractions 2 (4.4 mg) and 3 (7 mg) were subjected to HPLC over a PFP column using a gradient system of 0:100–10:90 of CH3CN:H2O (10% formic acid) over 5 min to 20:80 over 15 min at a flow rate of 21.24 mL/min to yield compounds 6 (1.4 mg). and 8 (3.1 mg), from fractions 2 and 3, respectively.
Fraction 7 (28.8 mg) of the normal-phase flash chromatography was subjected to preparative HPLC over a PFP column using a gradient system of 0:100–20:80 of CH3CN:H2O (0.1% formic acid) over 15 min, hold for 5 min, then to 50:50 over 15 min at a flow rate of 21.24 mL/min to yield 9 subfractions. Subfraction 9 (12.8 mg) was subjected to preparative HPLC over an Atlantis T3 column using a system of 40:60–70:30 of MeOH:H2O (0.1% formic acid) over 15 min, no hold, at a flow rate of 17 mL/min to yield 4 subfractions. Subfraction 4 (2.1 mg) was subjected to semipreparative HPLC over a PFP column using a gradient system of 0:100–10:90 of CH3CN:H2O (10% formic acid) over 5 min to 30:70 over 15 min at a flow rate of 4.62 mL/min to yield compound 3 (0.8 mg).
2.3.1. (+)-O-Methylkreysigine-N-oxide (3)
White powder; ; UV (MeOH) λmax (log ε) 260 (3.18), 227 (3.46) nm; ECD (c 0.001 M, MeOH) λ (Δε) 223 (+4.10) nm, 259 (−6.50) nm (Fig. 3A); HRESIMS m/z 416.2065 [M+H]+ (calcd for C23H30NO6, 416.2068).
Fig. 3.
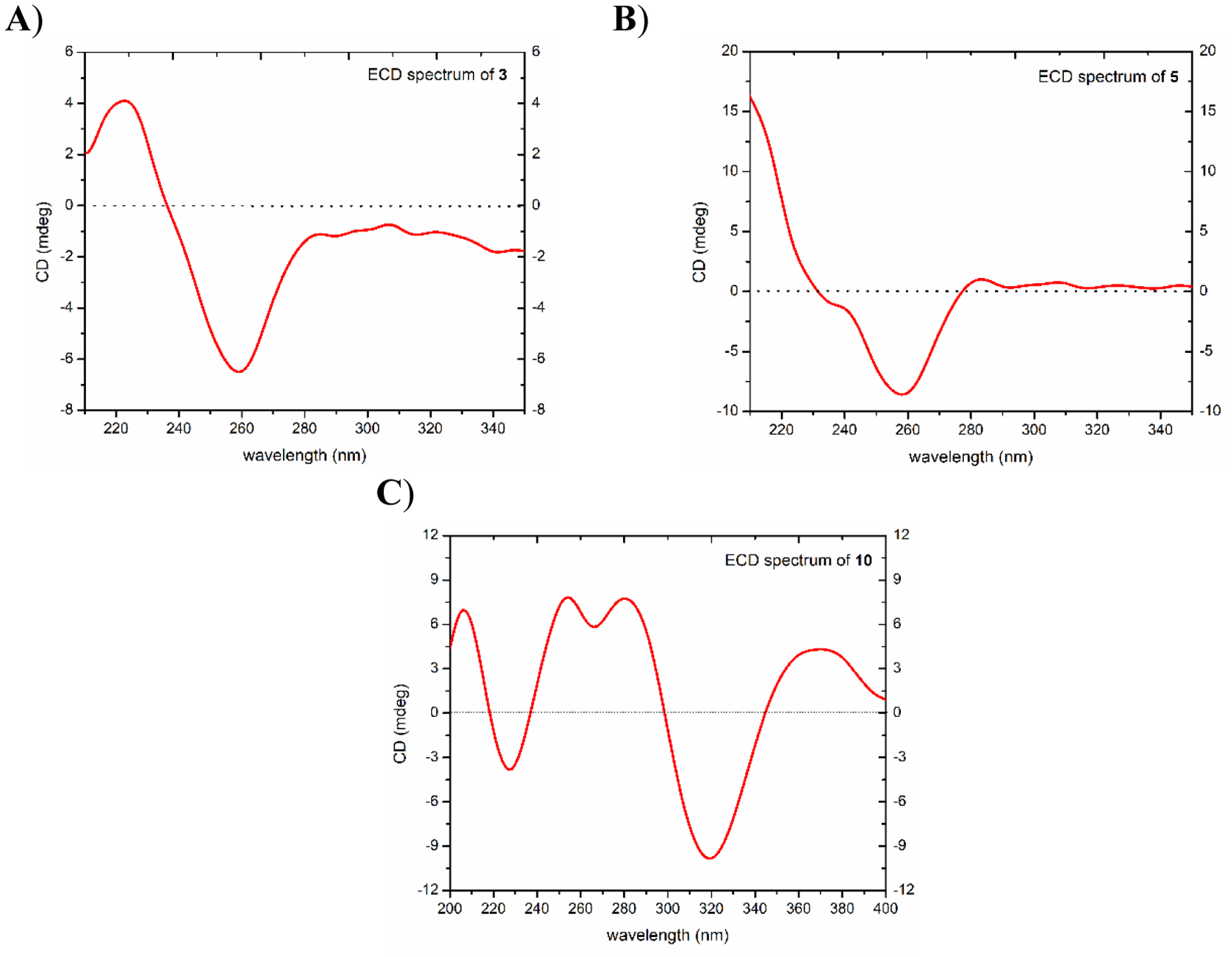
ECD spectra for compounds A) 3 (1 mM), B) 5 (0.3 mM), and C) 10 (0.3 mM) [MeOH, cell length 2 cm].
2.3.2. (+)-1-Demethylandrocine (5)
Light brown amorphous solid; ; UV (MeOH) λmax (log ε) 298 (2.15), 259 (2.44), 224 (2.63) nm; ECD (c 0.3 × 10−3 M, MeOH) λ (Δε) 258 (−8.58) nm, 283 (+1.02) nm (Fig. 3B); HRESIMS m/z 372.1802 [M+H]+ (calcd for C21H26NO5, 372.1805).
2.3.3. (−)-Andropalaestine (8)
Light brown amorphous solid; ; UV (MeOH) λmax (log ε) 281 (2.88), 236 (3.08) nm; HRESIMS m/z 388.2116 [M+H]+ (calcd for C22H30NO5, 388.2118).
2.3.4. (+)-O,O-Dimethylautumnaline (9)
White powder; ; UV (MeOH) λmax (log ε) 279 (2.88), 256 (3.28), 228 (3.35), 216 (3.26) nm; HRESIMS m/z 402.2276 [M+H]+ (calcd for C23H32NO5, 402.2275).
2.3.5. (+)-2-Demethyl-β-lumicolchicone (10)
Light yellow powder; ; UV (MeOH) λmax (log ε) 376 (2.99), 353 (3.01), 327 (2.94), 259 (2.93) nm; ECD (c 0.3 × 10−3 M, MeOH) λ (Δε) 205 (+6.99) nm, 226 (−3.82) nm, 253 (+7.83) nm, 266 (+5.82), 278 (+7.75) nm, 317 (−9.83) nm, 368 (+4.32) nm (Fig. 3C); HRESIMS m/z 343.1173 [M+H]+ (calcd for C19H19O6, 343.1176).
2.4. Cytotoxicity assay
Compounds 1‒13 were tested for cytotoxicity against human melanoma cancer cells MDA-MB-435 [25], human breast cancer cells MDA-MB-231, and human ovarian cancer cells OVCAR3 as described previously [26, 27]. Briefly, the cell lines were propagated at 37 °C in 5% CO2 in RPMI 1640 medium, supplemented with fetal bovine serum (FBS) (10%), penicillin (100 units/mL), and streptomycin (100 μg/mL). Cells in log phase growth were harvested by trypsinization followed by two washings to remove all traces of trypsin. A total of 5,000 cells were seeded per well of a 96-well clear, flat-bottom plate (Microtest 96, Falcon) and incubated overnight (37 °C in 5% CO2). Samples dissolved in DMSO were then diluted and added to the appropriate wells. The cells were incubated in the presence of test substance for 72 h at 37 °C and evaluated for viability with a commercial absorbance assay (CellTiter 96 AQueous One Solution Cell Proliferation Assay, Promega Corp, Madison, WI) that measured viable cells. IC50 values are expressed in μM relative to the solvent (DMSO) control. Taxol (paclitaxel) was used as a positive control.
3. Results and discussion
The dried leaves of A. palaestinum were extracted with MeOH, and the resulting crude extract was reconstituted in 5% acetic acid and defatted using, in order, petroleum ether and diethyl ether. The defatted acidic solution was then made alkaline and partitioned with dichloromethane. The resulting dried alkaloid-rich dichloromethane extract (i.e. fraction C) was fractionated using normal-phase flash chromatography. Extensive purifications of the resulting fractions using reversed-phase HPLC methods, both preparative and semipreparative, resulted in the isolation of thirteen compounds (1−13), most of which were alkaloids (Fig. 1).
Fig. 1.
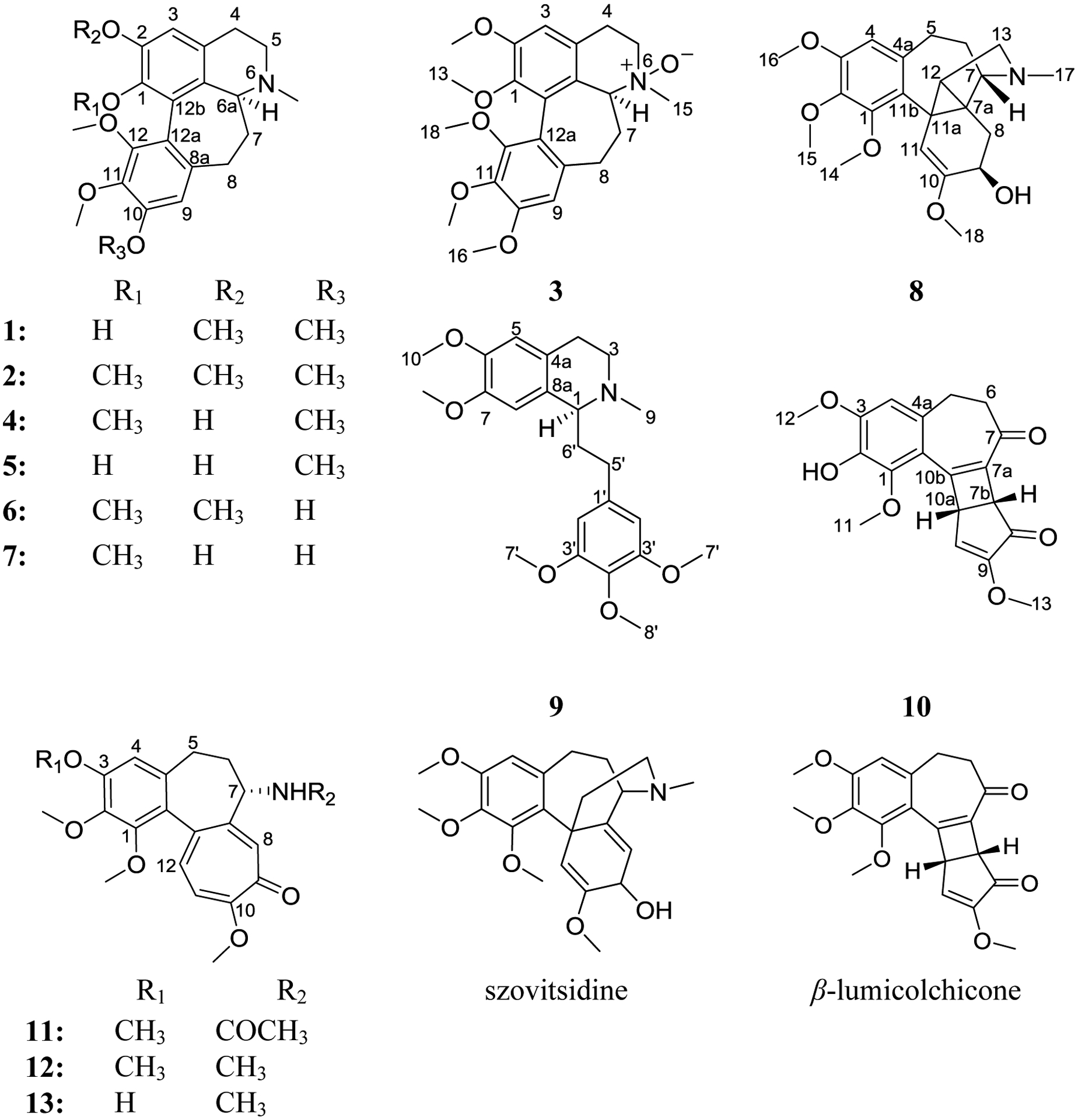
Structures of compounds 1–13, szovitsidine, and β-lumicolchicone.
Compounds (1, 2, 4, 6, 7, and 11-13) were identified as the known homoaporhine/colchicinoid alkaloids. Their structures were established by comparison of NMR (1D/2D), HRMS, specific rotation, and ECD data with literature values and were identified as: (+)-kreysigine (1) [22], (+)-O-methylkreysigine (2) [22], (+)-androcine (4) [22], (+)-androcimine (6) [22], (+)-androbine (7) [22], (−)-colchicine (11) [28, 29], (−)-demecolcine (12) [28, 30], and (−)-3-demethyldemecolcine (13) [28, 30] (Figs. S1–S4, S6, S8, S9, S13–S15, Table S2, Supplementary Data).
Compound 3 (0.8 mg) was obtained as a white powder with a molecular formula of C23H29NO6 as determined by HRESIMS (m/z 416.2065 [M+H]+, calcd 416.2068) and NMR data (Table 1, Figs. S1 and S5, Supplementary Data), establishing an index of hydrogen deficiency of 10. Analysis of the NMR data indicated a new homoaporphine alkaloid with structural similarity to 2 (Fig. 1). A key difference was an N-oxide moiety, consistent with a 16 amu difference in the HRMS data of compound 3 relative to 2, indicating oxidation of the tertiary nitrogen into N-oxide to give (+)-O-methylkreysigine-N-oxide, which was reported previously as a semisynthetic product [31]. Inspection of the NMR data of 3 showed signals characteristic of two singlet aromatic protons (δH/δC 6.74/111.4, H-3 and 6.59/107.3, H-9), nine aliphatic protons (δH 3.17, H2-4; 3.84 and 3.55, H2-5; 3.86, H-6a; 2.06 and 2.97, H2-7 and 2.22 and 2.58, H2-8), one N-methyl group (δH/δC 3.24/56.1), and five methoxy functionalities (δH/δC 3.56/60.6; 3.75/61.1; 3.88/61.1; 3.90/56.2 and 3.91/56.2) (Table 1, Fig. S5, Supplementary Data). HMBC correlations from the 1-OCH3, 2-OCH3, 10-OCH3, 11-OCH3 and 12-OCH3 protons to C-1 (δC 146.1), C-2 (δC 152.9), C-10 (δC 153.9), C-11 (δC 141.0), and C-12 (δC 151.5), respectively, confirmed their connectives (Fig. 2). HMBC correlations from H-3 to C-1 (δc 146.1), C-4 (δc 27.3), and C-3b (δc 123.6); H-9 to C-8 (δc 30.1), C-11 (δc 141.0), and C-12a (δc 119.0); H-6a to C-5 (δc 58.7), N-methyl, C-7 (δc 34.2) and C-12b (δC 128.9) were also observed confirming the structure of 3. COSY data identified two-spin systems as H-4/H-5 and H-6a/H-7/H-8 (Fig. 2).
Table 1:
NMR data for compound 3 (700 MHz for 1H and 175 MHz for 13C, CDCl3) and compound 5 (400 MHz for 1H and 125 MHz for 13C, CDCl3)
| position | 3 | 5 | ||
|---|---|---|---|---|
| δc, type | δH, mult (J in Hz) | δc, type | δH, mult (J in Hz) | |
| 1 | 146.1, C | - | 146.9, C | - |
| 2 | 152.9, C | - | 139.2, C | - |
| 3 | 111.4, CH | 6.74, s | 109.5, CH | 6.68, s |
| 3a | 123.0, C | - | 123.4 | - |
| 3b | 123.6, C | - | 122.2 | - |
| 4 | 27.3, CH2 | 3.17, m | 23.3, CH2 | 2.82, dd (15.9, 5.2) |
| 3.12, m | ||||
| 5 | 58.7, CH2 | 3.84, m | 43.7, CH2 | 3.11, m |
| 3.55, m | 3.40, m | |||
| 6a | 72.8, CH | 3.86, m | 58.7, CH | 3.63, dd (11.2, 6.2) |
| 7 | 34.2, CH2 | 2.06, m | 34.6, CH2 | 2.16, m |
| 2.97, m | 2.52, m | |||
| 8 | 30.1, CH2 | 2.22, m | 30.4, CH2 | 2.33, m |
| 2.58, m | 2.53, m | |||
| 8a | 134.1, C | - | 135.3, C | - |
| 9 | 107.3, CH | 6.59, s | 114.1, CH | 6.79, s |
| 10 | 153.9, C | - | 153.7, C | - |
| 11 | 141.0, C | - | 141.2, C | - |
| 12 | 151.5, C | - | 149.5, C | - |
| 12a | 119.0, C | - | 119.7, C | - |
| 12b | 128.9, C | - | 128.9, C | - |
| 13 | 60.6, CH3 | 3.56, s | 39.9, CH3 | 2.58, s |
| 14 | 56.2, CH3 | 3.90, s | 56.3, CH3 | 3.93, s |
| 15 | 56.1, CH3 | 3.24, s | 61.6, CH3 | 3.94, s |
| 16 | 56.2, CH3 | 3.91, s | 62.6, CH3 | 3.66, s |
| 17 | 61.1, CH3 | 3.88, s | ||
| 18 | 61.1, CH3 | 3.75, s | ||
Fig. 2.
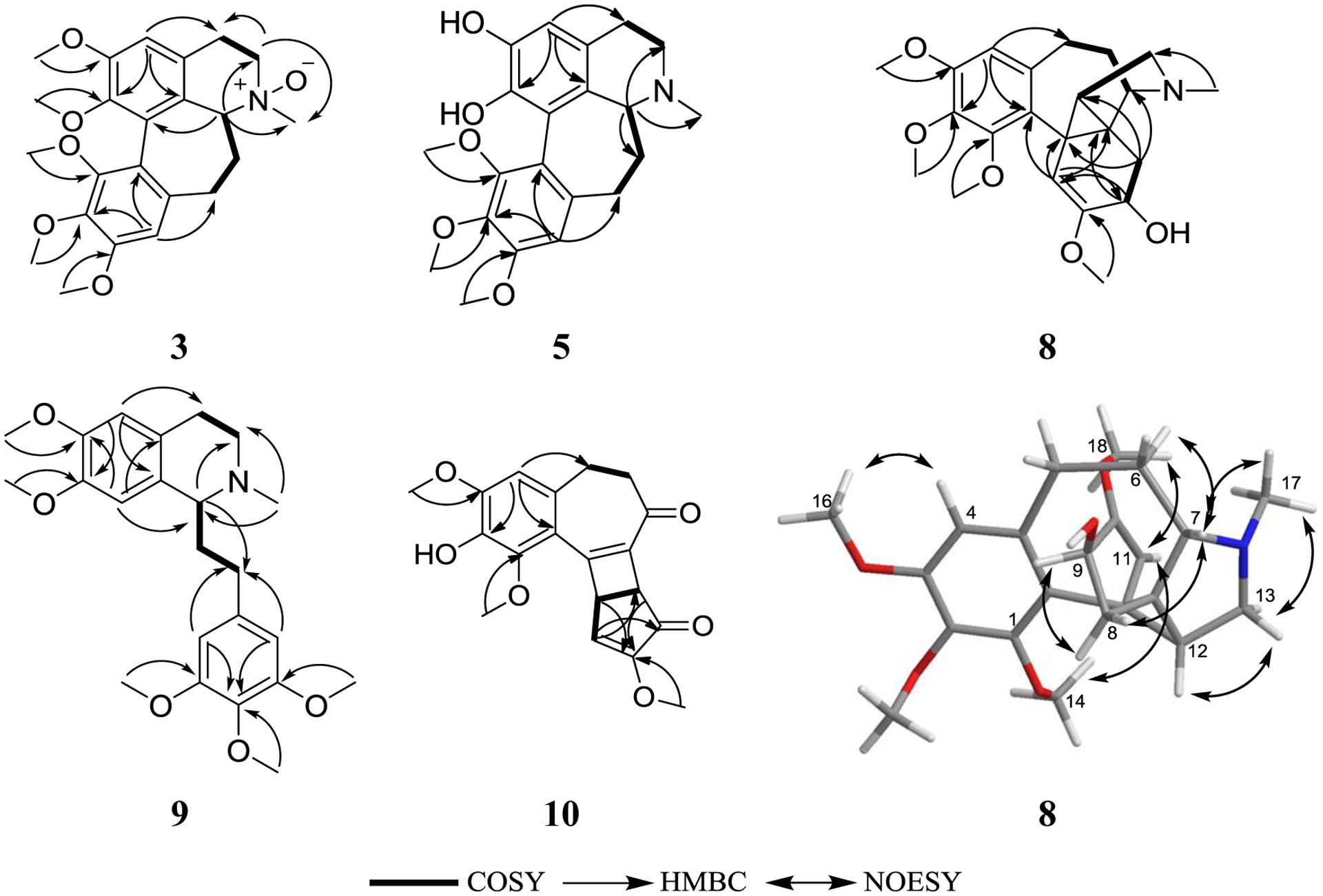
Key COSY and HMBC correlations of 3, 5, and 8–10, and NOESY correlations of 8.
The absolute configurations of homoaporphine alkaloids are assigned using specific rotation and electronic circular dichroism (ECD) spectroscopy [18, 22, 32], in which a clockwise rotation of plane polrized light (dextrorotatory) along with a negative Cotton effect in the 254–258 nm region of the ECD spectra are indicative of a C-6aS configuration [18, 22]. A specific rotation of [α]D24 = +20 (c 0.001, MeOH) for 3, along with a negative Cotton effect at 259 nm (Δε = −6.5) in the ECD spectrum, supported an S-configuration at C-6a (Fig. 3A).
Compound 5 (4.1 mg) was obtained as a light brown amorphous solid with a molecular formula of C21H25NO5 as determined by HRESIMS (m/z 372.1802 [M+H]+, calcd 372.1805) and NMR data (Table 1, Figs. S1 and S7, Supplementary Data), establishing an index of hydrogen deficiency of 10. Analysis of the NMR data indicated a new homoaporphine alkaloid with structural similarity to 4 (Fig. 1). Compound 5 lacked a methoxy group, which was replaced by an exchangeable proton, consistent with a 14 amu difference in the HRMS data of 5 relative to 4. Inspection of the NMR data showed signals characteristic of two singlet aromatic protons (δH/δC 6.68/109.5; H-3 and 6.79/114.1; H-9), nine aliphatic protons (δH 2.82 and 3.12, H2-4; 3.11 and 3.40, H2-5; 3.63, H-6a; 2.16 and 2.52, H2-7 and 2.33 and 2.53, H2-8), one N-methyl group (δH/δC 2.58/39.9) and three methoxy functionalities (δH/δC 3.66/62.6; 3.93/56.3, and 3.94/61.6) (Table 1, Fig. S7, Supplementary Data). HMBC correlations from the 10-OCH3, 11-OCH3, and 12-OCH3 protons to C-10 (δC 153.7), C-11 (δC 141.2), and C-12 (δC 149.5), respectively, confirmed their connectives (Fig. 2). HMBC correlations from H-3 to C-1 (δc 146.9), C-2 (δc 139.2), C-3b (δc 122.2), and C-4 (δc 23.3); H-9 to C-8 (δc 30.4), C-10 (δc 153.7), C-11 (δc 141.2), and C-12a (δc 119.7) and H-6a to C-3a (δc 123.4), C-3b and C-5 (δc 43.7) were also observed (Fig. 2). COSY data showed two-spin systems as H-4/H-5 and H-6a/H-7/H-8 (Fig. 2). The trivial name 1-demethylandrocine was assigned to compound 5. A negative Cotton effect at 258 nm (Δε = −8.58) in the ECD spectrum along with a specific rotation value of [α]D24 = +126 (c 0.001, MeOH), supported an S-configuration at C-6a (Fig. 3B).
Compound 8 (3.1 mg) was obtained as a light brown amorphous solid with a molecular formula of C22H29NO5 as determined by HRESIMS (m/z 388.2116 [M+H]+, calcd 388.2118) and analysis of NMR data (Table 2, Figs. S1 and S10, Supplementary Data), establishing an index of hydrogen deficiency of 9. Inspection of the NMR data showed signals characteristic of one singlet aromatic proton (δH/δC 6.34/107.2), one singlet olefinic proton (δH/δC 4.98/103.9), eight aliphatic protons displayed between 1.76 and 2.83 ppm, one N-methyl group (δH/δC 1.97/40.4), and four methoxy functionalities (δH 3.49; 3.83; 3.85, and 4.02) (δC 54.5; 55.9; 60.6, and 61.2) (Table 2, Fig. S10, Supplementary Data). HMBC correlations from the 1-OCH3, 2-OCH3, 3-OCH3, and 10-OCH3 protons to C-1 (δC 152.0), C-2 (δC 139.9), C-3 (δC 152.5), and C-10 (δC 153.1), respectively, confirmed their connectives. HMBC correlations from H-4 to C-2 (δC 139.9), C-3 (δC 152.5), C-5 (δC 30.7), and C-11b (δC 126.8); H-11 to C-9 (δc 65.9), C-11b, C-11a (δc 26.9), and C-7a (δc 33.6); H-9 to C-11 (δc 103.9), C-10 and C-7a; H-8 to C-10, C-7 (δC 68.4), C-11a and C-12 (δC 38.4) and H-12 to C-11, C-8 (δC 31.3) and C-13 (δC 56.9) were also observed (Fig. 2). COSY data showed three spin systems as H2-8/H-9, H2-5/H2-6/H-7, and H-12/H2-13 (Fig. 2). These data suggested that 8 was related to the androcymbines (homomorphinans/homomorphinandienone) class of alkaloids, showing structural similarity to szovitsidine, which was isolated from Colchicum szovitsii (Fig. 1) [33]. Compound 8 and szovitsidine showed identical molecular weight, molecular formula, and unsaturation number. However, the NMR data of 8 indicated the lack of the olefinic C7a-C8 bond in 8 relative to szovitsidine, which was replaced by a quaternary aliphatic carbon (δC 33.6 for C-7a) and a methylene group (δH/δC 2.20/31.3 for H2-8/C-8). Moreover, the methylene group at C-12 in szovitsidine was replaced by a methine (δH/δC 1.76/38.4 for H-12/C-12) in 8. These data, along with HMBC correlations from H2-8 to C-7 (δc 68.4), C-12, and C-11a (δc 26.9), indicated the formation of a cyclopropane ring (C-7a, C-11a, and C-12), which account for the unsaturation unit that was lost due to the saturation of the C7a-C-8 double bond in 8 relative to szovitsidine. An attempt to assign the absolute configuration of 8 using Mosher’s esters methodology [34] was unsuccessful. The relative configuration of 8 was assigned by NOESY NMR data (Fig. 2).
Table 2:
NMR data for compound 8 (500 MHz for 1H and 125 MHz for 13C, CDCl3).
| position | δc, type | δH, mult (J in Hz) | position | δc, type | δH, mult (J in Hz) |
|---|---|---|---|---|---|
| 1 | 152.0, CH | - | 10 | 153.1, C | - |
| 2 | 139.9, C | - | 11 | 103.9, CH | 4.98, s |
| 3 | 152.5, C | - | 11a | 26.9, C | - |
| 4 | 107.2, C | 6.34, s | 11b | 126.8, C | - |
| 4a | 136.5, C | - | 12 | 38.4, CH | 1.76, m |
| 5 | 30.7, CH2 | 2.30, ddd (13.3, 4.8, 2.1) | 13 | 56.9, CH2 | 2.49, dd (11.1, 5.7) |
| 2.83, m | 2.77, m | ||||
| 6 | 25.4, CH2 | 1.76, m | 14 | 61.2, CH3 | 4.02, s |
| 7 | 68.4, CH | 2.75, m | 15 | 60.6, CH3 | 3.85, s |
| 7a | 33.6, C | - | 16 | 55.9, CH3 | 3.83, s |
| 8 | 31.3, CH2 | 2.20, m | 17 | 40.4, CH3 | 1.97, s |
| 9 | 65.9, CH | 4.29, dd (4.0, 3.0) | 18 | 54.5, CH3 | 3.49, s |
Compound 9 (2.2 mg) was obtained as a white powder with a molecular formula of C23H31NO5 as determined by HRESIMS (m/z 402.2276 [M+H]+, calcd 402.2275) and NMR data (Table 3, Figs. S1 and S11, Supplementary Data), establishing an index of hydrogen deficiency of 9. Inspection of NMR data indicated 9 as a phenethylisoquinoline alkaloid with structural similarity to the known alkaloids (+)-autumnaline [35] and (+)-homolaudanosine [36]. However, compound 9 has an additional two methyl groups consistent with a 28 amu difference in the HRMS data of 9 relative to (+)-autumnaline and an extra methoxy group consistent with a 30 amu difference in the HRMS data of 9 relative to (+)-homolaudanosine (Table 3, Fig. S11, Supplementary Data). HMBC correlations from the 6-OCH3, 7-OCH3, 2 × 3´-OCH3, and 4´-OCH3 protons to C-6 (δC 147.51), C-7 (δC 147.47), 2 × C-3´ (δC 153.2), and C-4´ (δC 136.1), respectively, confirmed their connectives and established the structure of 9 (Fig. 1). Other HMBC correlations observed were from H-1 to C-3 (δC 48.1), C-4a (δC 126.8), C-8 (δC 110.3), and C-5´ (δC 32.2); H-5 to C-4 (δc 25.4), C-7 (δc 147.47) and C-8a (δc 129.7); H-8 to C-1 (δc 62.8), C-4a (δc 126.8), and C-6 (δc 147.51). H-6´ has HMBC correlations with C-5´, C-4´ (δC 136.1), C-3´ (δC 153.2), and C-2´ (δC 105.5), and H-2´ to C-5´, C-2´ (δC 105.5), C-3´ (δC 153.2) and C-4´ (Fig. 2). COSY data showed two spin systems as H-1/H-6´/H-5´ and H-3/H-4 (Fig. 2). Interestingly, compound 9 was reported previously by synthesis [32, 37, 38]. However, this is the first report of 9 from nature, to which the trivial name O,O-dimethylautumnaline was assigned. The specific rotation of 9 was determined as [α]D24 = +3 (c 0.001, MeOH) supporting an S-configuration at C-1, consistent with reported values in literature [32, 37, 38].
Table 3:
NMR data for compound 9 (400 MHz for 1H and 100 MHz for 13C, CDCl3).
| position | δc, type | δH, mult (J in Hz) | position | δc, type | δH, mult (J in Hz) |
|---|---|---|---|---|---|
| 1 | 62.8, CH | 3.46, dd (5.44, 4.24) | 10 | 56.2, CH3 | 3.83, s |
| 3 | 48.1, CH2 | 2.70, m | 11 | 56.0, CH3 | 3.86, s |
| 3.14, m | |||||
| 4 | 25.4, CH2 | 2.70, m | 1´ | 138.8, C | - |
| 4a | 126.8, C | - | 2´, 2´ | 105.5, CH | 6.40, s |
| 5 | 111.5, CH | 6.58, s | 3´, 3´ | 153.2, C | - |
| 6 | 147.51, C | - | 4´ | 136.1, C | - |
| 7 | 147.47, C | - | 5´ | 32.2, CH2 | 2.05, m |
| 8 | 110.3, CH | 6.54, s | 6´ | 37.1, CH2 | 2.50, m |
| 2.70, m | |||||
| 8a | 129.7, C | - | 7´, 7´ | 56.2, CH3 | 3.83, s |
| 9 | 42.8, CH3 | 2.50, s | 8´ | 61.0, CH3 | 3.81, s |
Compound 10 (0.70 mg) was obtained as a light yellow powder with a molecular formula of C19H18O6 as determined by HRESIMS (m/z 343.1173 [M+H]+, calcd 343.1176) and NMR data (Table 4, Figs. S1 and S12, Supplementary Data), establishing an index of hydrogen deficiency of 11. Analysis of the NMR data suggested 10 as a new colchicone derivative with a structural similarity to the known alkaloid β-lumicolchicone (Fig. 1), which was isolated from the tubers of Gloriosa superba [39]. Compound 10 lacked a methoxy group at C-2, which was replaced by an exchangeable proton (Table 4, Fig. S12, Supplementary Data), consistent with a 14 amu difference in the HRMS data of 10 relative to β-lumicolchicone. Inspection of the NMR data showed signals characteristic of one singlet aromatic proton (δH/δC 6.60/108.2), one singlet olefinic proton (δH/δC 6.56/125.0), six aliphatic protons that are displayed between 2.64 and 4.24, and three methoxy functionalities (δH 3.68; 3.95 and 4.04) (δC 57.0, 56.5, and 61.2) (Table 4, Fig. S12, Supplementary Data). HMBC correlations from the 1-OCH3, 3-OCH3 and 9-OCH3 protons to C-1 (δC 146.6), C-3 (δC 149.3), and C-9 (δC 158.3), respectively, confirmed their connectives (Fig. 2). HMBC correlations from H-4 to C-2 (δC 137.2), C-10c (δC 118.7), and C-5 (δC 30.8); H-7b to C-7a (δc 133.6), C-8 (δc 197.3), C-9 (δc 158.3) and C-10a (δc 43.8); H-10 to C-7b (δc 48.6), C-8 and C-10a; H-10a to C-9 and C-10 (δC 125.0) were also observed (Fig. 2). COSY data showed two spin systems as H-5/H-6 and H-10/H-10a/H-7b (Fig. 2). The trivial name 2-demethyl-β-lumicolchicone was assigned to 10, in deference to the known compound β-lumicolchicone. A NOESY correlation from H-7b to H-10a along with coupling constant value of 2.8 ppm confirmed the syn-fusion at C-7b/C-10a. The absolute configuration was established using CD spectra of structurally related compounds [40]. The CD spectrum of 9 was similar to that reported for β-lumicolchicine but opposite to that of γ-lumicolchicine establishing the configuration as 7bR,10aS (Fig. 3C) [40].
Table 4:
NMR data for compound 10 (700 MHz for 1H and 175 MHz for 13C, CDCl3).
| Position | δc, type | δH, mult (J in Hz) | position | δc, type | δH, mult (J in Hz) |
|---|---|---|---|---|---|
| 1 | 146.6, C | - | 8 | 197.3, C | - |
| 2 | 137.2, C | - | 9 | 158.3, C | - |
| 3 | 149.3, C | - | 10 | 125.0, CH | 6.56, d (3.4) |
| 4 | 108.2, CH | 6.60, s | 10a | 43.8, CH | 4.24, dd (3.4, 2.8) |
| 4a | 135.9, C | - | 10b | 158.3, C | - |
| 5 | 30.8, CH2 | 2.85, m | 10c | 118.7, C | - |
| 6 | 40.8, CH2 | 2.64, m | 11 | 61.2, CH3 | 4.04, s |
| 7 | 194.8, C | - | 12 | 56.5, CH3 | 3.95, s |
| 7a | 133.6, C | - | 13 | 57.0, CH3 | 3.68, s |
| 7b | 48.6, CH | 3.90, d (2.8) | 2-OH | 5.52, s |
The cytotoxicities of 1-13 were tested against MDA-MB-435 (melanoma), MDA-MB-231 (breast), and OVCAR3 (ovary) cancer cell lines. Colchicine (11) was the most potent, with IC50 values in the range of 10 to 100 nM, depending on the cell line (Table 5); these data were consistent with previous studies on this well-known alkaloid [41–43]. The cytotoxicity data facilitated conclusions on the structure-activity relationships (i.e. SARs). For instance, compounds 1-10, all of which lacked the tropolone ring, were inactive, demonstrating the importance of the tropolone moiety for cytotoxic activity. Replacing the N-acetyl group in 11 by an N-methyl group in 12 reduced the activity against MDA-MB-435 and OVCAR3 by factors of 2, and 3, respectively, although the activity vs MDA-MB-231 cells remained intact. Moreover, demethylation of the 3-OCH3 group, as noted in compounds 13 vs 12, diminished the cytotoxicity in MDA-MB-435, MDA-MB-231, and OVCAR3 cells by a factor of ~40, ~90, and ~30, respectively.
Table 5:
Cytotoxic activities of compounds 11–13.
| compounda | IC50 values in nMb | ||
|---|---|---|---|
| MDA-MB-435 | Ovcar3 | MDA-MB-231 | |
| 11 | 12 | 23 | 95 |
| 12 | 21 | 77 | 113 |
| 13 | 800 | 2230 | 9940 |
| taxolc | 0.1 | 1.45 | 171 |
Compounds 1–10 were inactive, IC50 values >25 μM.
IC50 is the concentration to inhibit 50% of growth with a 72 h incubation.
Positive control.
Supplementary Material
Acknowledgments
This research was supported, in part, by the Deanship of Research, Jordan University of Science and Technology, Irbid, Jordan (Grant No. 258/2017) and the National Cancer Institute/National Institutes of Health, Bethesda, MD, USA via P01 CA125066. We thank Dr. L. Flores Bocanegra, J. M. Gallagher, Z. Y. Al Subeh, and N. D. Paguigan from UNCG for technical help and valuable suggestions. This work was performed in part at the Joint School of Nanoscience and Nanoengineering, a member of the Southeastern Nanotechnology Infrastructure Corridor (SENIC) and National Nanotechnology Coordinated Infrastructure (NNCI), which is supported by the National Science Foundation (Grant ECCS-1542174).
Footnotes
Conflict of interest
All the authors have no conflict of interest.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.fitote.2020.104706
References
- [1].Harvey AL, Natural products in drug discovery, Drug Discovery Today 13 (2008) 894–901. [DOI] [PubMed] [Google Scholar]
- [2].Newman DJ, Cragg GM, Natural products as sources of new drugs from 1981 to 2014, J. Nat. Prod 79 (2016) 629–661. [DOI] [PubMed] [Google Scholar]
- [3].Dias DA, Urban S, Roessner U, A historical overview of natural products in drug discovery, Metabolites 2 (2012) 303–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Newman DJ, Cragg GM, Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019, J. Nat. Prod 83 (2020) 770–803. [DOI] [PubMed] [Google Scholar]
- [5].Feinbrun-Dothan N, Flora Palestina. The Israel Academy of Sciences and Humanities, Jeruselum, 1986. [Google Scholar]
- [6].Al-Eisawi DM, Field Guide to Wild Flowers of Jordan and Neighbouring Countries. Jordan Press Foundation Al Rai, Amman, 1998. [Google Scholar]
- [7].Al-Khalil S, A survey of plants used in Jordanian traditional medicine, Int. J. Pharmacogn 33 (1995) 317–323. [Google Scholar]
- [8].Abu-Irmaileh B, Afifi FU, Treatment with medicinal plants in Jordan, Dirasat: Med. Biol. Sci. (in Arabic) 27 (2000) 53–74. [Google Scholar]
- [9].Online DatabaseRoskov Y. AL, Orrell T, Nicolson D, Flann C, Bailly N, Kirk P, Bourgoin T, DeWalt RE, Decock W, De Wever A, Species 2000 & ITIS Catalogue of Life, 2016 Annual Checklist. Digital resource at http://www.catalogueoflife.org/col/, Species 2000: Naturalis, Leiden, the Netherlands: ISSN 2405–884X, 2017. [Google Scholar]
- [10].Alali FQ, El-Elimat T, Li C, Qandil A, Alkofahi A, Tawaha K, Burgess JP, Nakanishi Y, Kroll DJ, Navarro HA, Falkinham JO, Wani MC, Oberlies NH, New Colchicinoids from a Native Jordanian Meadow Saffron, Colchicum brachyphyllum: Isolation of the First Naturally Occurring Dextrorotatory Colchicinoid, J. Nat. Prod 68 (2005) 173–178. [DOI] [PubMed] [Google Scholar]
- [11].Alali FQ, Ma’aya’h AS, Alkofahi A, Qandil A, Li C, Burgess J, Wani MC, Oberlies NH, A New Colchicinoid from Colchicum tauri, an Unexplored Meadow Saffron Native to Jordan, Natural Product Communications 1 (2006) 1934578X0600100203. [Google Scholar]
- [12].Al-Mahmoud MS, Alali FQ, Tawaha K, Qasaymeh RM, Phytochemical study and cytotoxicity evaluation of Colchicum stevenii Kunth (Colchicaceae): A Jordanian meadow saffron, Natural Product Research 20 (2006) 153–160. [DOI] [PubMed] [Google Scholar]
- [13].Alali FQ, Tawaha K, El-Elimat T, Qasaymeh R, Li C, Burgess J, Nakanishi Y, Kroll DJ, Wani MC, Oberlies NH, Phytochemical studies and cytotoxicity evaluations of Colchicum tunicatum Feinbr and Colchicum hierosolymitanum Feinbr (Colchicaceae): two native Jordanian meadow saffrons, Natural Product Research 20 (2006) 558–566. [DOI] [PubMed] [Google Scholar]
- [14].Alali FQ, Tahboub YR, Al-Daraysih IS, El-Elimat T, LC-MS and LC-PDA vs. phytochemical analysis of Colchicum brachyphyllum, Die Pharmazie - An International Journal of Pharmaceutical Sciences 63 (2008) 860–865. [PubMed] [Google Scholar]
- [15].Alali FQ, Gharaibeh AA, Ghawanmeh A, Tawaha K, Qandil A, Burgess JP, Sy A, Nakanishi Y, Kroll DJ, Oberlies NH, Colchicinoids from Colchicum crocifolium Boiss. (Colchicaceae), Natural Product Research 24 (2010) 152–159. [DOI] [PubMed] [Google Scholar]
- [16].Membrives N, Pedrola-Monfort J, Caujapé-Castells J, Relative influence of biological versus historical factors on isozyme variation of the genus Androcymbium (Colchicaceae) in Africa, Plant Syst. Evol 229 (2001) 237–260. [Google Scholar]
- [17].Watt JM, Breyer-Brandwijk MG, The medicinal and poisonous plants of southern and eastern Africa: being an account of their medicinal and other uses, chemical composition, pharmacological effects and toxicology in man and animal. E. & S. Livingstone 1962. [Google Scholar]
- [18].Tojo E, The homoaporphine alkaloids J Nat. Prod 52 (1989) 909–921. [Google Scholar]
- [19].Tojo E, Zarga MHA, Freyer AJ, Shamma M, The dibenzocycloheptylamine alkaloids J Nat. Prod 52 (1989) 1163–1166. [Google Scholar]
- [20].Ellington E, Bastida J, Viladomat F, V. Šimánek, C. Codina, Occurrence of colchicine derivatives in plants of the genus Androcymbium, Biochem. Syst. Ecol 31 (2003) 715–722. [Google Scholar]
- [21].Potěšilová H, Sedmera P, Guénard D, Šimánek V, Alkaloids of Androcymbium melanthioides var. stricta, Planta Med 51 (1985) 344–345. [DOI] [PubMed] [Google Scholar]
- [22].Tojo E, Zarga MHA, Sabri SS, Freyer AJ, Shamma M, The homoaporphine alkaloids of Androcymbium palaestinum, J. Nat. Prod 52 (1989) 1055–1059. [Google Scholar]
- [23].Battersby AR, Herbert RB, Pijewska L, Santavy F, The constitution of androcymbine, Chem. Commun. (London) (1965) 228–230. [Google Scholar]
- [24].Alali FQ, El-Elimat T, Li C, Qandil A, Alkofahi A, Tawaha K, Burgess JP, Nakanishi Y, Kroll DJ, Navarro HA, New colchicinoids from a native jordanian meadow saffron, Colchicum brachyphyllum: Isolation of the first naturally occurring dextrorotatory colchicinoid, J. Nat. Prod 68 (2005) 173–178. [DOI] [PubMed] [Google Scholar]
- [25].Rae J, Creighton C, Meck J, Haddad B, Johnson M, MDA-MB-435 cells are derived from M14 melanoma cells–a loss for breast cancer, but a boon for melanoma research, Breast Cancer Res. Treat 104 (2007) 13–19. [DOI] [PubMed] [Google Scholar]
- [26].El-Elimat T, Figueroa M, Raja HA, Swanson SM, Falkinham JO, Lucas DM, Grever MR, Wani MC, Pearce CJ, Oberlies NH, Sorbicillinoid analogues with cytotoxic and selective Anti-Aspergillus Activities from Scytalidium album, J. Antibiot 68 (2015) 191–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].El-Elimat T, Raja HA, Falkinham JO, Day CS, Oberlies NH, Greensporones: Resorcylic acid lactones from an aquatic Halenospora sp., J. Nat. Prod 77 (2014) 2088–2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Hufford CD, Capraro H-G, Brossi A, 13C- and 1H-NMR. Assignments for colchicine derivatives, Helv. Chim. Acta 63 (1980) 50–56. [Google Scholar]
- [29].Hufford CD, Collins CC, Clark AM, Microbial transformations and 13C-NMR analysis of colchicine, J. Pharm. Sci 68 (1979) 1239–1243. [DOI] [PubMed] [Google Scholar]
- [30].Meksuriyen D, Lin L-J, Cordell GA, Mukhopadhyay S, Banerjee SK, NMR studies of colchicine and its photoisomers, β- and λ-lumicolchicines, J. Nat. Prod 51 (1988) 88–93. [DOI] [PubMed] [Google Scholar]
- [31].Yusupov M, Chommadov BC, Aslanov KA, N-oxides of homoaporphine alkaloids from Merendera raddeana, Chem. Nat. Compd 27 (1991) 75–79. [Google Scholar]
- [32].Brossi A, O’Brien J, Teitel S, Totalsynthese und absolute konfiguration von natürlichem multifloramin, Helv. Chim. Acta 52 (1969) 678–689. [Google Scholar]
- [33].Yusupov M, Aslanov KA, Structure of szovitsidine, Chem. Nat. Compd 11 (1975) 289–290. [Google Scholar]
- [34].Hoye TR, Jeffrey CS, Shao F, Mosher ester analysis for the determination of absolute configuration of stereogenic (chiral) carbinol carbons, Nat. Protoc 2 (2007) 2451–2458. [DOI] [PubMed] [Google Scholar]
- [35].Battersby AR, Herbert RB, McDonald E, Ramage R, Clements JH, Alkaloid biosynthesis. Part XVIII. Biosynthesis of colchicine from the 1-phenethylisoquinoline system, J. Chem. Soc., Perkin Trans 1 (1972) 1741–1746. [DOI] [PubMed] [Google Scholar]
- [36].Aladesanmi AJ, Kelley CJ, Leary JD, The constituents of Dysoxylum lenticellare. I. Phenylethylisoquinoline, homoerythrina, and dibenzazecine alkaloids, J. Nat. Prod 46 (1983) 127–131. [Google Scholar]
- [37].Czarnocki Z, MacLean DB, Szarek WA, Enantioselective synthesis of isoquinoline alkaloids: phenylethylisoquinoline and aporphine alkaloids, Biochem. Syst. Ecol 65 (1987) 2356–2361. [Google Scholar]
- [38].Itoh T, Nagata K, Yokoya M, Miyazaki M, Kameoka K, Nakamura S, Ohsawa A, The synthesis of isoquinoline alkaloid and its related compounds using alanine derivatives as chiral auxiliaries, Chem. Pharm. Bull 51 (2003) 951–955. [DOI] [PubMed] [Google Scholar]
- [39].Bussotti L, D’Auria M, Foggi P, Lesma G, Righini R, Silvani A, The photochemical behavior of colchicone and thiocolchicone, Photochem. Photobiol 71 (2000) 29–34. [DOI] [PubMed] [Google Scholar]
- [40].Jaromír Hrbek J, Hruban L, Šimánek V, Šantavý F, Snatzke G, Yemul SS, Circular dichroism of alkaloids of colchicine type and their derivatives, Collect. Czech. Chem. Commun 47 (1982) 2258–2279. [Google Scholar]
- [41].Vilanova Gallén C, Díaz Oltra S, Murga Clausell J, Falomir Ventura E, Carda Usó M, Redondo Horcajo M, Fernando Díaz J, Barasoain I, Alberto Marco J, Design and synthesis of pironetin analogue/colchicine hybrids and study of their cytotoxic activity and mechanisms of interaction with tubulin, J. Med. Chem 57 (2014) 10391–10403. [DOI] [PubMed] [Google Scholar]
- [42].Zhang X, Kong Y, Zhang J, Su M, Zhou Y, Zang Y, Li J, Chen Y, Fang Y, Zhang X, Design, synthesis and biological evaluation of colchicine derivatives as novel tubulin and histone deacetylase dual inhibitors, Eur. J. Med. Chem 95 (2015) 127–135. [DOI] [PubMed] [Google Scholar]
- [43].Huczyński A, Rutkowski J, Popiel K, Maj E, Wietrzyk J, Stefańska J, Majcher U, Bartl F, Synthesis, antiproliferative and antibacterial evaluation of C-ring modified colchicine analogues, Eur. J. Med. Chem 90 (2015) 296–301. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.