

Abstract
Congenital hydrocephalus (CH), characterized by enlarged brain ventricles, is considered a disease of excessive cerebrospinal fluid (CSF) accumulation and thereby treated with neurosurgical CSF diversion with high morbidity and failure rates. The poor neurodevelopmental outcomes and persistence of ventriculomegaly in some post-surgical patients highlight our limited knowledge of disease mechanisms. Through whole-exome sequencing of 381 patients (232 trios) with sporadic, neurosurgically treated CH, we found that damaging de novo mutations account for >17% of cases, with five different genes exhibiting a significant de novo mutation burden. In all, rare, damaging mutations with large effect contributed to ~22% of sporadic CH cases. Multiple CH genes are key regulators of neural stem cell biology and converge in human transcriptional networks and cell types pertinent for fetal neuro-gliogenesis. These data implicate genetic disruption of early brain development, not impaired CSF dynamics, as the primary pathomechanism of a significant number of patients with sporadic CH.
Hydrocephalus has been classically defined as cerebral ventricular enlargement (‘ventriculomegaly’), resulting from progressive accumulation of CSF1. This mechanism is most often observed in acquired (‘secondary’) hydrocephalus associated with brain hemorrhage, tumor or infection. In these cases, an increase in CSF production relative to CSF reabsorption leads to an increase in intracranial pressure (ICP), which causes tissue damage, neurological impairment and death if untreated. In this context, neurosurgical CSF diversion (via ventricular shunting or brain endoscopy) can acutely decrease ICP and ventricular size and dramatically restore neurological function. Nonetheless, these neurosurgeries have high morbidity and failure rates2.
In contrast, many neonatal or infantile cases of CH are classified as developmental (‘primary’) hydrocephalus3, because they lack a known antecedent. CH can occur in the setting of no obvious intraventricular obstruction to CSF flow (communicating hydrocephalus) or exhibit partial or complete intraventricular obstruction, usually from aqueductal stenosis. CH may be associated with motor deficits, intellectual disability and epilepsy that can persist despite neurosurgical intervention4. These observations suggest that CH is not simply a disorder of impaired ‘brain plumbing’ treatable by CSF diversion, but a complex neurodevelopmental disorder with associated functional deficits referable to the brain parenchyma.
While some communicating forms of CH are associated with increased ICPs, other cases can have documented ICPs in the borderline-high, normal or even low range and can be associated with severe thinning of the cortical mantle1,5. Neurosurgical CSF diversion when undertaken in this context can fail to reduce ventriculomegaly or improve neurodevelopmental outcomes6. The determination of which patients with CH may benefit from neurosurgical intervention can be a complex clinical challenge, as their clinical and neuroradiographic presentations may be similar1. A molecular classification of CH could improve prognostication and clinical decision-making for caregivers.
Epidemiological studies and reports of familial CH suggest genetic etiologies for up to 40% of cases7, but few causal mutations have been identified. Traditional linkage and targeted sequencing approaches have identified mutations in L1CAM (OMIM no. 307000), MPDZ (OMIM no. 615219), CCDC88C (OMIM no. 236600) and AP1S2 (OMIM no. 300629)8. Other genes have been linked with CH in Mendelian syndromes (for example, ciliopathies) characterized by severe systemic (such as respiratory, cardiac and renal) abnormalities8, but >95% of CH cases are sporadic and of unknown cause8.
Next-generation sequencing has revolutionized the identification of genetic causes of human disease. We recently used whole-exome sequencing (WES) to identify four new genes not previously implicated in human CH9. We now present the largest WES study to date of sporadic, neurosurgically treated CH, integrated with transcriptomics of human brain development. Our data implicate genetically encoded neural stem cell (NSC) dysregulation and an associated impairment of fetal neuro-gliogenesis as primary pathophysiological events in a significant number of CH cases.
Results
We recruited 381 genetically undiagnosed probands (including 232 parent–offspring trios) with sporadic, neurosurgically treated, primary (developmental) CH (excluding myelomeningocele) (Supplementary Table 1), including 169 previously reported CH probands with 125 trios9. Studies were Institutional Review Board (IRB)-approved by Yale’s Human Research Protection Program (Methods). DNA was isolated and WES was performed9. A total of 1,798 control trios (comprising unaffected siblings and parents of patients with autism spectrum disorder (ASD) from the Simons Simplex Collection (SSC) cohort) were analyzed in parallel (Supplementary Tables 2 and 3). Overall, 8.7% of probands were from consanguineous union, versus 1.3% ASD sibling controls (Supplementary Dataset 1 and Supplementary Table 2; Methods for sequence variant calling, calibration, annotation and validation). Mutations in known familial CH genes8 accounted for ~2.1% of cases, including mutations in L1CAM (OMIM no. 307000), MPDZ (OMIM no. 615219), FLNA (OMIM no. 300049) and CRB2 (OMIM no. 219730) described in Supplementary Table 4. Removal of the eight patients from further analyses yielded 373 CH probands, including 225 trios.
Protein-damaging de novo mutations account for a large fraction of sporadic CH.
The average de novo mutation (DNM) rate of 1.307 per subject (Supplementary Dataset 2) resembled previous results with the identical sequencing platform10 and followed a Poisson distribution (Supplementary Fig. 1). Protein-damaging DNMs were significantly enriched among all genes (enrichment of 1.72, P = 6.6 × 10−7; Supplementary Table 5), with greater enrichment among genes intolerant of loss-of-function (LoF) mutations (pLI ≥ 0.9 in gnomAD v.2.1.1) and among genes in the top quartile of mouse brain bulk RNA-sequencing (RNA-seq) expression (Methods). Enrichment was greatest among genes meeting both criteria (3.71-fold, P = 5.0 × 10−9; Supplementary Table 5). We estimated that damaging DNMs can account for 17.7% of cases in this cohort (Supplementary Table 5).
Twelve genes had ≥2 protein-altering DNMs (Table 1a) versus 2.7 genes expected by chance (4.5-fold enrichment; P = 8.0 × 10−6 by 1 million permutations; Table 1b). Greater enrichment of recurrent genes was observed in LoF-intolerant genes with multiple DNMs (8.9-fold enrichment; P = 1.0 × 10−5; Table 1c), supporting these as causal CH disease genes. Five genes (TRIM71, SMARCC1, PTEN, PIK3CA and FOXJ1) had significantly more protein-altering DNMs than expected by chance (P value threshold of 8.6 × 10−7 after correction for testing 19,347 RefSeq genes in triplicate using a one-tailed Poisson test; Table 1a). Three other genes that are highly intolerant of LoF mutations exhibited ≥2 protein-altering DNMs: MTOR, PTCH1 and FMN2.
Table 1 |.
Genes with multiple DNMs are candidate CH risk genes
| (a) Genes with ≥2 protein-altering DNMs | ||||||
|---|---|---|---|---|---|---|
| Gene | No. LoF | No. D-Mis | No. T-Mis | Poisson P value | pLI | mis_z |
| TRIM71 | 0 | 6 | 0 | 2.4 × 10−16 | 1.00 | 3.28 |
| PTEN | 2 | 1 | 0 | 1.9 × 10−8 | 0.26 | 3.49 |
| SMARCC1 | 2 | 1 | 0 | 2.0 × 10−8 | 1.00 | 2.45 |
| FOXJ1 | 2 | 0 | 0 | 1.4 × 10−7 | 0.97 | 0.70 |
| PIK3CA | 0 | 1 | 2 | 4.9 × 10−7 | 1.00 | 5.60 |
| PTCH1 | 2 | 0 | 0 | 3.0 × 10−6 | 1.00 | 1.68 |
| PLOD2 | 0 | 2 | 0 | 1.6 × 10−5 | 0.00 | 0.56 |
| SGSM3 | 0 | 0 | 2 | 1.0 × 10−4 | 0.00 | 0.16 |
| LRIG1 | 1 | 0 | 1 | 1.7 × 10−4 | 0.04 | −1.18 |
| FMN2 | 1 | 0 | 1 | 4.4 × 10−4 | 1.00 | 0.32 |
| MTOR | 0 | 1 | 1 | 9.1 × 10−4 | 1.00 | 7.02 |
| MUC17 | 0 | 0 | 2 | 1.3 × 10−3 | 0.00 | −7.83 |
| (b) Genes with multiple DNMs in 225 cases (observed versus expected) | ||||
|---|---|---|---|---|
| Observed | Expected | Enrichment | P value | |
| Syn | 0 | 0.18 | 0 | 1 |
| Missense | 6 | 1.86 | 3.23 | 0.01 |
| D-Mis | 2 | 0.29 | 7.01 | 0.03 |
| LoF | 4 | 0.08 | 48.38 | 3.0 × 10−6 |
| Protein-damaging | 6 | 0.85 | 7.08 | 1.3 × 10−4 |
| Protein-altering | 12 | 2.66 | 4.5 | 8.0 × 10−6 |
| (c) LoF-intolerant genes with multiple DNMs in 225 cases (observed versus expected) | ||||
| Syn | 0 | 0.05 | 0 | 1 |
| Missense | 3 | 0.54 | 5.52 | 0.02 |
| D-Mis | 1 | 0.12 | 8.2 | 0.12 |
| LoF | 3 | 0.02 | 121.02 | 2.0 × 10−6 |
| Protein-damaging | 4 | 0.36 | 11.03 | 4.5 × 10−4 |
| Protein-altering | 7 | 0.79 | 8.9 | 1.0 × 10−5 |
| (d) LoF-tolerant genes with multiple DNMs in 225 cases (observed versus expected) | ||||
| Syn | 0 | 0.13 | 0 | 1 |
| Missense | 3 | 1.31 | 2.28 | 0.14 |
| D-Mis | 1 | 0.16 | 6.12 | 0.15 |
| LoF | 1 | 0.06 | 17.27 | 0.06 |
| Protein-damaging | 2 | 0.48 | 4.12 | 0.08 |
| Protein-altering | 5 | 1.88 | 2.66 | 0.04 |
Genes that surpassed the bonferroni multiple-testing threshold are shown in bold type. a, Twelve genes with >1 protein-altering DNm found in cases. P values are calculated using a one-tailed Poisson test comparing the observed number of DNms for each gene versus expected. As separate tests were performed for protein-altering, protein-damaging and LoF DNms, the bonferroni multiple-testing threshold is equal to 8.6 × 10−7 (= 0.05 / (3 tests × 19,347 genes)). The most significant P value of the three tests was reported. pLI and mis-z values are based on gnomAD v.2.1.1. b, more genes with multiple DNms were detected in 225 case trios than expected by chance, as shown by the observed numbers of genes with >1 DNm in each variant category. One million simulations were performed, based on the per-base probability of mutations in each category, to determine the likelihood and the expected number of genes with >1 DNm. c, Greater enrichment than expected by chance was observed when restricting analysis to LoF-intolerant genes (n = 3,049) with multiple DNms in 225 case trios. d, restricting analysis to genes tolerant to LoF mutations showed marginal enrichment for genes with multiple protein-altering mutations. T-mis, tolerated missense mutations; Protein-altering, missense + LoF; Protein-damaging, D-mis + LoF.
TRIM71 and SMARCC1 are bona fide CH risk genes that likely define new syndromes.
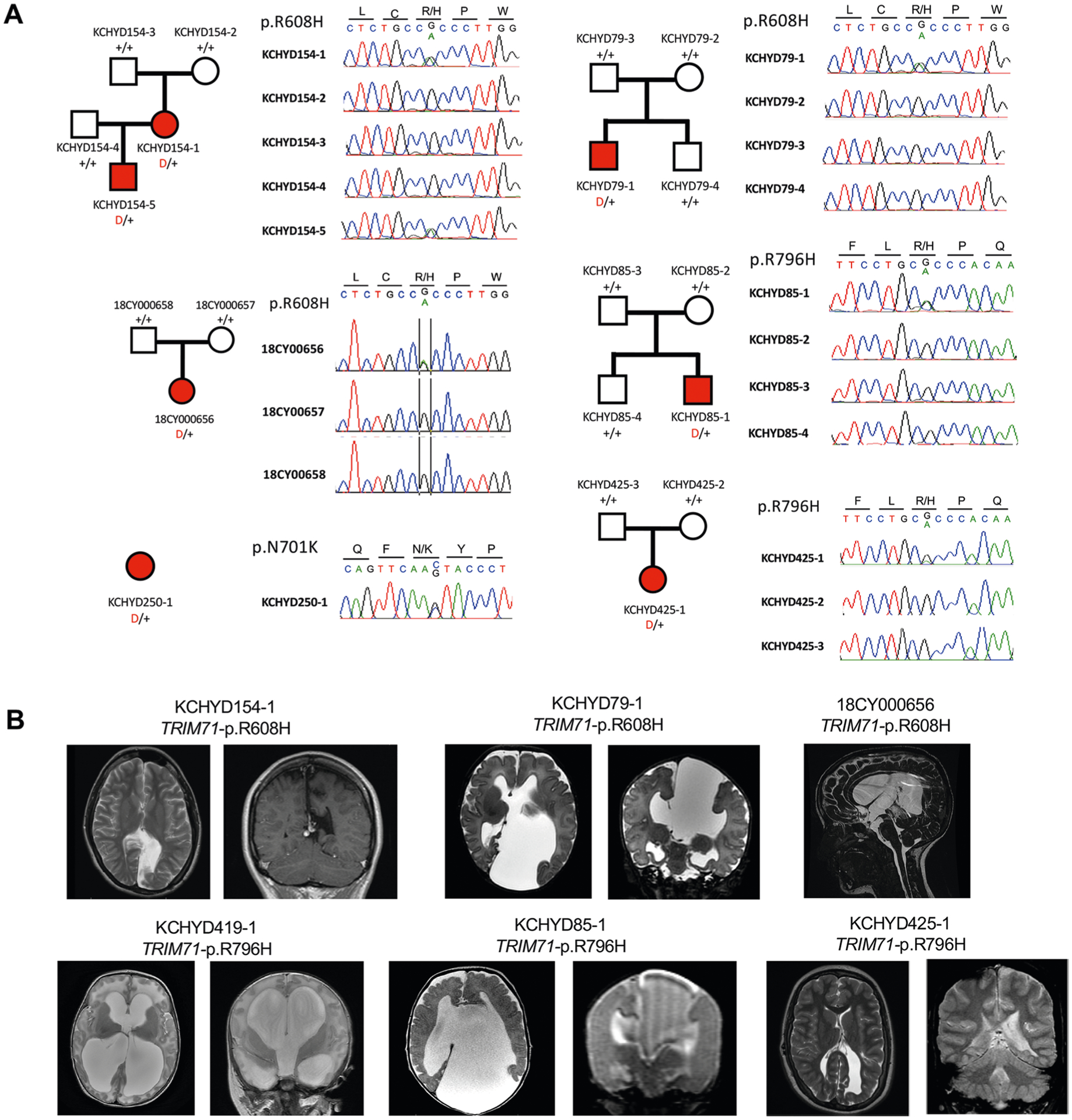
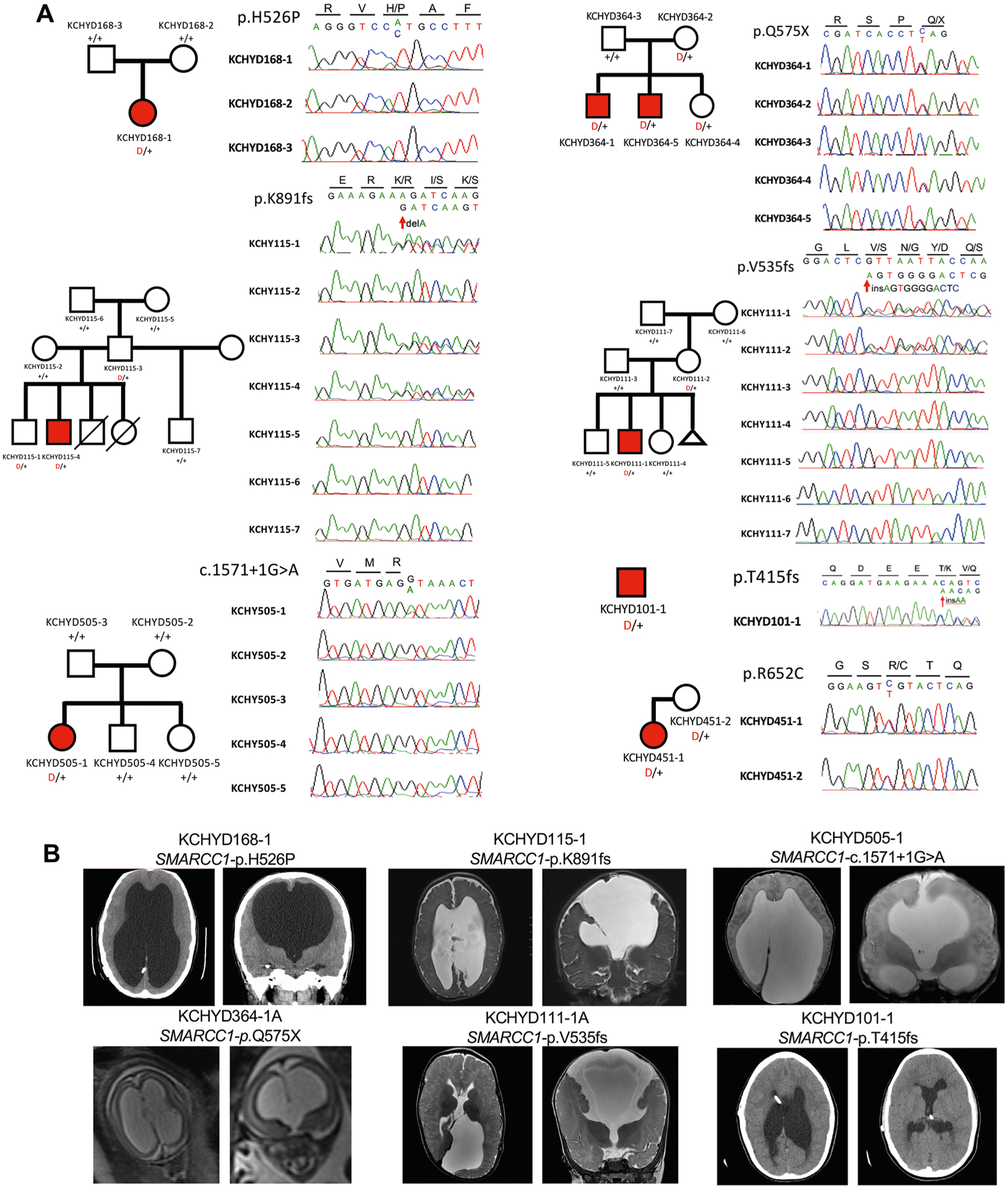
By comparing observed and expected DNMs among CH cases, we previously9 identified enrichment in CH cases of protein-altering DNMs in TRIM71, encoding the RNA-binding protein Tripartite Motif Containing 71, homolog of let-7 (lethal 7) microRNA target lin-41, and in SMARCC1, encoding BAF155 subunit of BRG1/BRM-associated factor (BAF; Saccharomyces cerevisiae SWI/SNF) chromatin remodeling complex. Our expanded cohort included three new damaging TRIM71 DNMs and one in SMARCC1 (c.1571+1G>A) (Fig. 1, Supplementary Tables 6–7, Extended Data Figs. 1–2 and Supplementary Figs. 2–3). Both genes surpassed genome-wide significance thresholds in DNM enrichment and case–control tests (Table 1a, Fig. 1a and Supplementary Table 8).
Fig. 1 |. TRIM71 and SMARCC1 are bona fide CH risk genes.
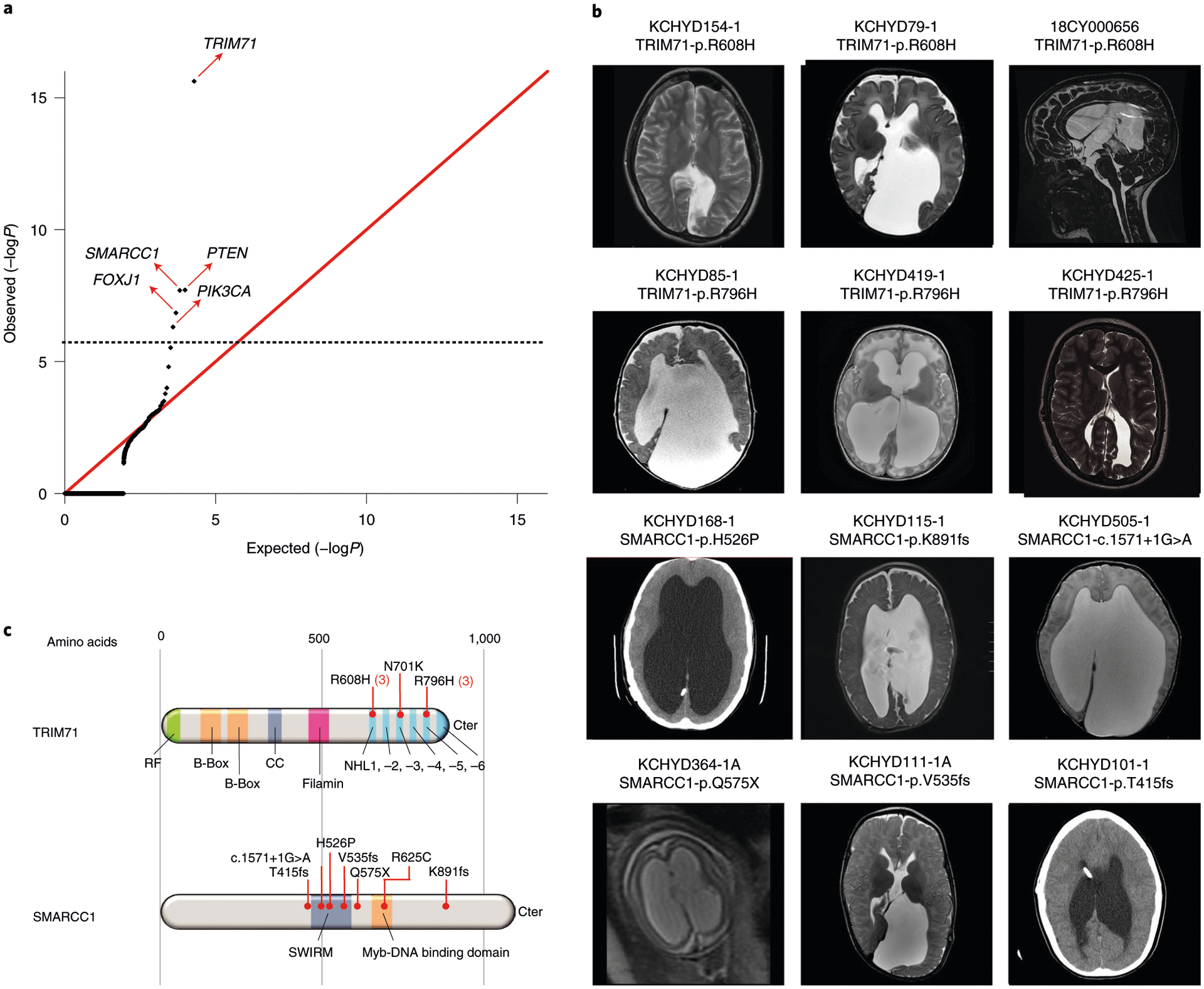
a, Quantile-quantile plot comparing observed versus expected P values for DNMs in each gene in 225 cases. TRIM71 and SMARCC1 exhibit genome-wide significant enrichment of DNMs in CH cases and TRIM71 is the overall most commonly mutated CH gene. b, Representative T1- or T2-weighted axial and sagittal brain magnetic resonance images (MRIs) or head computed tomography (CT) images of neurosurgically treated CH probands with the indicated TRIM71 and SMARCC1 mutations (Supplementary Tables 6 and 7 and Extended Data Figs. 1 and 2 contain clinical and neuroradiographic details for available patients). c, Locations of identified TRIM71 and SMARCC1 mutations in relation to critical functional domains. The recurrent TRIM71 p.Arg608His and p.Arg796His mutations impact conserved residues in the 16th position of the respective first and fifth blades of NHL domain, which mediates the binding to target mRNAs (Supplementary Fig. 2). p.Asn701Lys localizes to the third NHL domain and is predicted to destabilize protein–RNA interactions (Supplementary Fig. 2). RF, ring finger domain; CC, coiled-coil domain. The identified SMARCC1 mutations were mapped in relation to its SWIRM and Myb-DNA-binding domains, which mediate SMARCC1 interaction with BAF47 and its in vivo function in the SWI-SNF and ADA complexes, respectively.
TRIM71 maintains stem cell pluripotency by the post-transcriptional silencing of target mRNAs via interactions with its RNA-binding NHL domain (reviewed previously11). Six DNMs in TRIM71 included three p.Arg608His mutations and three p.Arg796His mutations, mapping respectively to homologous positions in the conserved first and fifth blades of TRIM71’s RNA-binding NHL domain (Fig. 1c and Supplementary Table 6a). The probability of three occurrences of a single DNM in this cohort is low (P = 7.7 × 10−9) and vanishingly small for two sites in the same gene (Supplementary Information). A previously unidentified, unphased damaging TRIM71 mutation (p.Asn701Lys) in NHL domain’s third blade was also predicted to destabilize protein–RNA interactions (Fig. 1c). Mutations p.Arg608His and p.Arg796His impair TRIM71’s degradation of specific target RNAs12. Trim71 deletion in mice results in exencephaly and embryonic lethality by decreasing NSC proliferation (reviewed previously11).
SMARCC1 is an ATP-dependent chromatin remodeler that regulates gene expression required for NSC proliferation, differentiation and survival during telencephalon development13. SMARCC1 harbored three DNMs (p.His526Pro, p.Lys891fs and c.1571+1G>A), two transmitted LoF mutations (p.Gln575X and p.Val535fs), one unphased rare LoF variant (p.Thr415fs) and one transmitted rare damaging missense (D-Mis) variant (p.Arg652Cys), with five of seven likely LoF mutations (Fig. 1c and Supplementary Table 7a). The damaging SMARCC1 DNM, p.His526Pro, is predicted to abolish interaction with the backbone carbonyl oxygen of p.Leu505 at the end of an adjacent helix in the SWIRM domain mediating BAF complex subunit interactions14 (Fig. 1c). Another rare transmitted D-Mis SMARCC1 p.Arg652Cys variant alters a conserved residue in the Myb-like DNA-binding catalytic domain. All other variants are absent in gnomAD and Bravo databases. Approximately 80% of mice homozygous for Smarcc1 missense allele Baf155msp/msp exhibit exencephaly similar to Trim71 mutant mice as a result of defective NSC proliferation and increased apoptosis15. These data suggest that SMARCC1 haploinsufficiency increases CH risk.
CH probands with recurrent TRIM71 DNMs each had significant white matter volume loss and corpus callosum abnormalities, with cranial nerve deficits (n = 4), nonobstructive interhemispheric cysts (n = 3), hearing loss (n = 3) and skeletal abnormalities (n = 2) (Supplementary Table 6b). In contrast, all six patients with SMARCC1 mutations with available brain imaging data had aqueductal stenosis and corpus callosum abnormalities, along with cardiac (n = 3) and skeletal abnormalities (n = 2) (Supplementary Table 7b). Complete septal agenesis or septal abnormalities, cerebellar tonsillar ectopia, developmental delay and epilepsy were additional common features in both TRIM71 and SMARCC1 mutant probands. We conclude that TRIM71 and SMARCC1 are new bona fide CH risk genes whose mutation likely defines new Mendelian syndromes with variable expressivity of associated phenotypes.
PI3K signaling genes PIK3CA, PTEN and MTOR are frequently mutated in sporadic CH.
PI3K pathway genes regulate cell growth, proliferation and differentiation in multiple tissues16, including NSCs in developing ventricular zone17 (Fig. 2a). Somatic PIK3CA or MTOR gain-of-function (GoF) mutations and PTEN LoF mutations drive tumorigenesis by increasing PIP3 levels18. Related germline or mosaic mutations have been identified in multiple brain and body overgrowth syndromes that also predispose to cancer19.
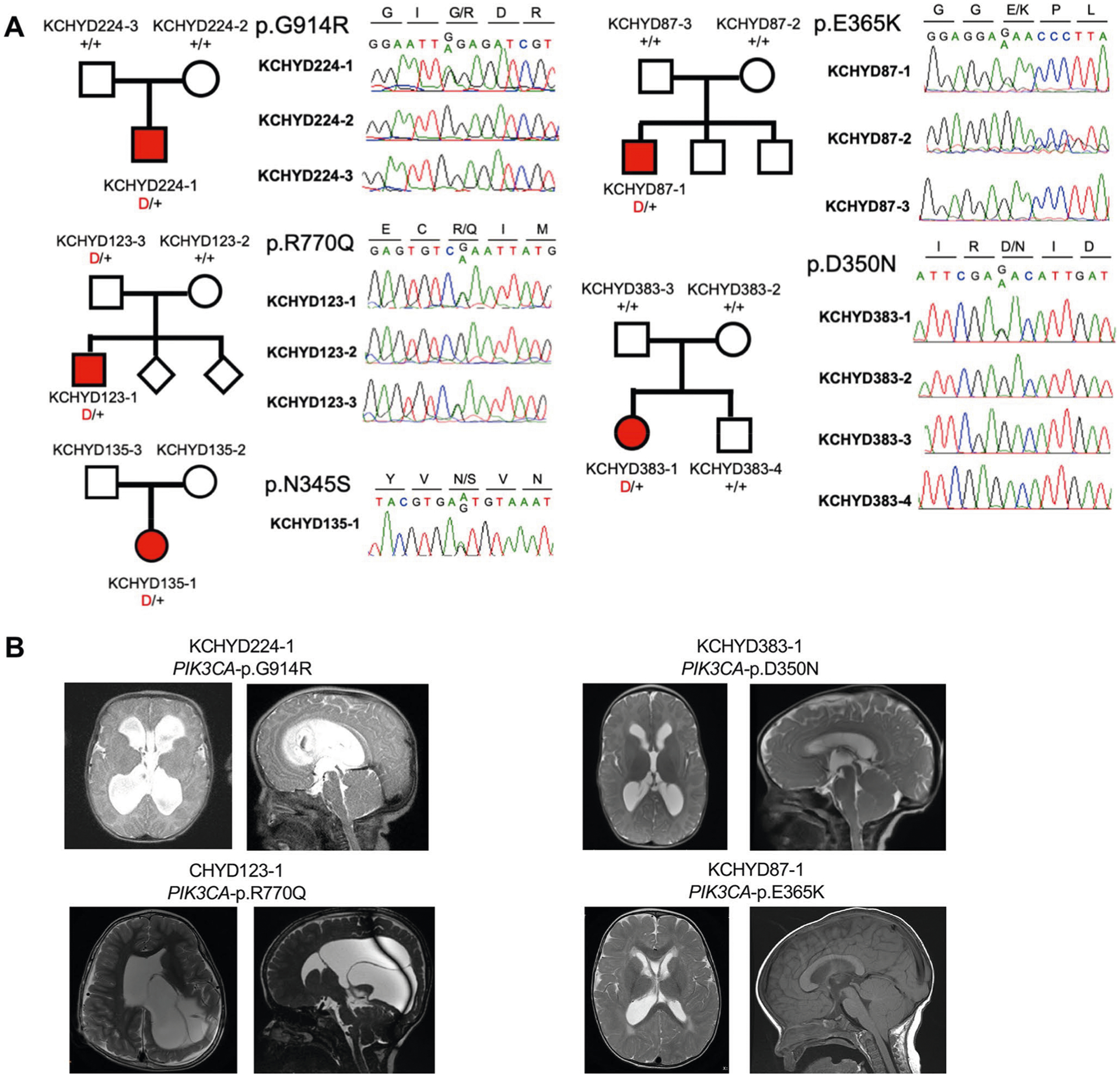
Fig. 2 |. PI3K signaling genes PIK3CA, PTEN and MTOR are frequently mutated in sporadic CH.
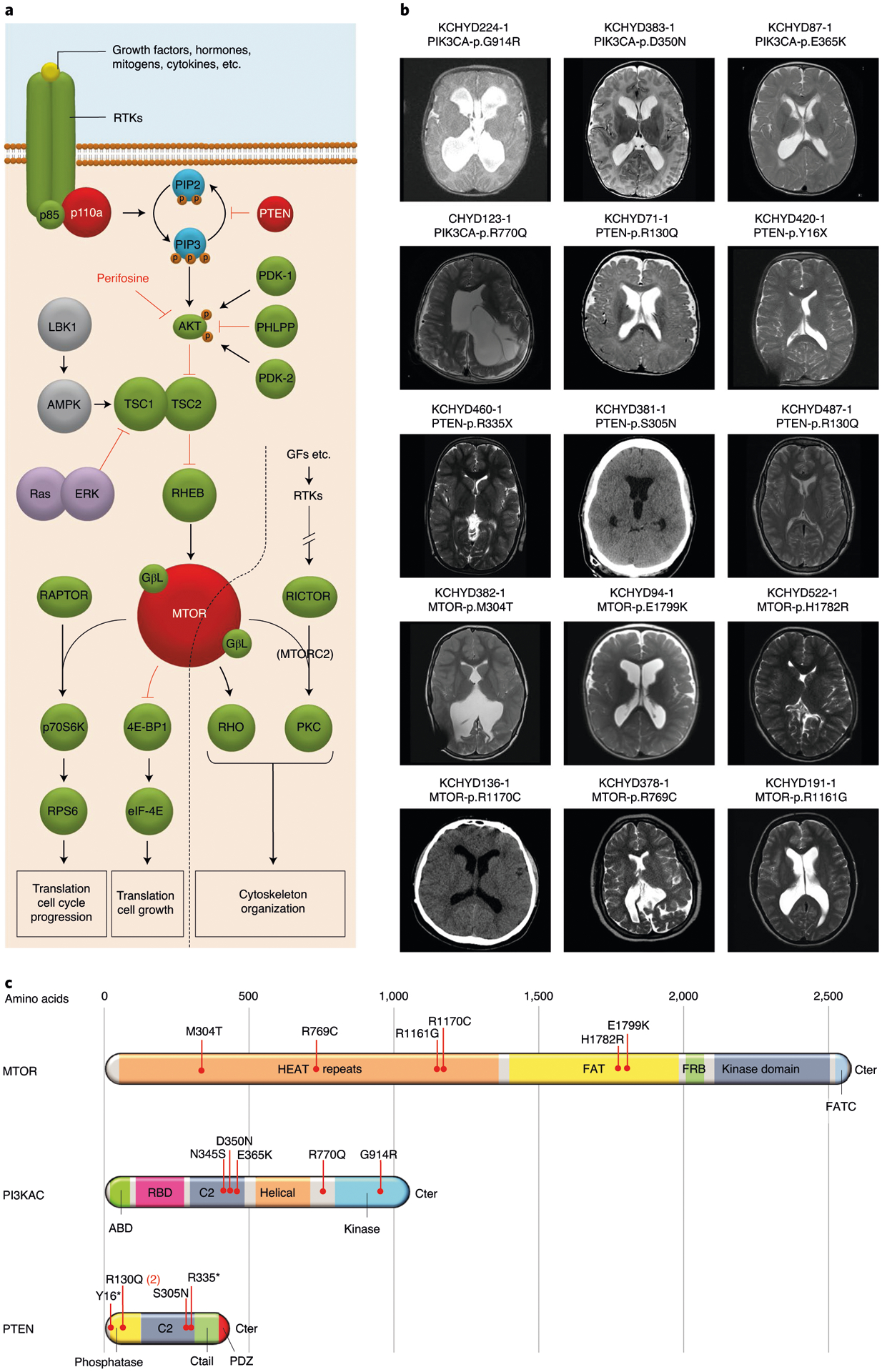
a, Depiction of the PI3K signaling pathway with genes mutated in sporadic CH indicated by a red-filled circle. PIK3CA and PTEN mutations are anticipated to lead to increased PIP3 production and mTOr activation, effects mimicked by CH-associated MTOR mutations. PI3K signaling regulates growth, proliferation and differentiation of embryonic and early postnatal NSCs. GF, growth factor; rTK, receptor tyrosine kinase. b, representative T1 or T2-weighted axial brain mrIs or head CT images of neurosurgically treated CH probands with the indicated PIK3CA, PTEN and MTOR mutations (Supplementary Table 11 and extended Data Figs. 3–5 contain clinical and neuroradiographic details for each patient). c, mutation mapping of PIK3CA, PTEN and MTOR mutations in relation to critical functional domains in each molecule. Frb, FKbP12-rapamycin-binding domain; FATC, C-terminal FAT domain. AbD, adaptor-binding domain; rbD, ras-binding domain; PDZ, PSD95, DLG1 and ZO1 domain.
PIK3CA harbored three DNMs (p.Asp350Asn, p.Glu365Lys and p.Gly914Arg) in unrelated probands with shunted CH, macrocephaly, megalencephaly, polymicrogyria and craniofacial abnormalities (Fig. 2, Supplementary Tables 9–11 and Extended Data Fig. 3). This DNM burden surpassed thresholds for genome-wide significance (P = 4.9 × 10−7; Table 1a). All three DNMs were previously linked to megalencephaly-capillary malformation-polymicrogyria syndrome (MCAP) (OMIM no. 602501; Supplementary Table 9);20,21 localize to sites of recurrent mutation in cancer;22 and are predicted to alter catalytic subunit structure (Supplementary Fig. 4). The two variants p.Glu365Lys and p.Gly914Arg constitutively increase PI3K activity and mTOR phosphorylation20,21, whereas the biochemical activity of p.Asp350Asn remains unevaluated. The fraction of mutant allele reads (43–53%) provided no evidence of somatic mosaicism (Supplementary Table 10). Consistent with a GoF effect of these PIK3CA DNMs, NSC-specific conditional expression of a Pik3ca activating allele during mouse embryogenesis induced 100% penetrant, severe nonobstructive murine hydrocephalus with focally increased NSC proliferation and disruption of cell adhesion at the neural-ependymal transition zone23.
PIK3CA also harbored two rare, unphased D-Mis mutations, p.Arg770Gln and p.Asn345Ser, both less frequent somatic targets in cancer (Supplementary Table 9 and Fig. 2). Similar to CH probands with PIK3CA DNMs, the CH proband with p.Arg770Gln exhibited macrocephaly, megalencephaly and polymicrogyria, supporting a similar functional effect, whereas the patient with p.Asn345Ser shared no syndromic features.
Notably, these PI3K pathway mutant CH probands carried no clinical or genetic diagnosis at time of study recruitment (Supplementary Table 11), despite previous neurosurgical treatment. Nonetheless, four CH probands with PIK3CA mutations retrospectively meet Martinez-Glez’s clinical criteria24 for MCAP, rarely associated with treated hydrocephalus24.
PTEN contained three DNMs (p.Tyr16X, p.Arg130Gln and p.Arg335X) in unrelated CH probands (P = 1.9 × 10−8; Supplementary Table 9 and Extended Data Fig. 4). Probands had macrocephaly (without megalencephaly) and cerebellar tonsillar ectopia. Two probands had polymicrogyria (Fig. 2, Extended Data Fig. 4 and Supplementary Table 11). All three DNMs are linked to PTEN hamartoma tumor syndrome25–27 and are sites of recurrent somatic mutation in cancer (Supplementary Table 9). Consistent with a LoF mechanism for PTEN CH DNMs, Pten conditional deletion in mouse NSCs causes increased PIP3 signaling and severe obstructive hydrocephalus due to increased ventricular zone NSC proliferation and cell size, with associated cerebral aqueduct obliteration28.
PTEN p.Arg130Gln (unphased) was detected in an unrelated CH proband with macrocephaly, cerebellar tonsillar ectopia and neurodevelopmental delay (Fig. 2). Another CH proband with macrocephaly, cerebellar tonsillar ectopia and neurodevelopmental delay carried the rare, inherited D-Mis PTEN VUS, p.Ser305Asn (Supplementary Tables 9 and 11). Both probands had aqueductal stenosis. p.Ser305Asn is predicted to disrupt PTEN C2 domain structure (Supplementary Fig. 5), but is not recurrently mutated in cancer.
These PTEN-mutated probands had no previous genetic or clinical diagnosis of PTEN hamartoma tumor syndrome (PHTS) (OMIM no. 158350), including Cowden, Bannayan–Riley–Ruvalcaba and autism-macrocephaly syndrome subtypes). However, one patient with PTEN p.Arg335X DNM retrospectively met diagnostic criteria29.
MTOR harbored two DNMs (p.Glu1799Lys and p.Met304Thr) in unrelated CH probands with macrocephaly, craniofacial abnormalities and skeletal defects (Fig. 2, Supplementary Tables 9 and 11 and Extended Data Fig. 5). A site of recurrent cancer mutation30, p.Glu1799Lys has also been implicated in ASD30, megalencephaly30 and Smith–Kingsmore (or MINDS) syndrome (OMIM no. 616638)30. However, the patient carrying this mutation did not meet the criteria for Smith–Kingsmore syndrome. The p.Glu1799Lys DNM increases mTORC1 kinase activity30 and is predicted to alter mTOR helix positioning at the FAT domain interface that binds negative regulators of mTOR kinase activity (Supplementary Fig. 6)31. The new p.Met304Thr DNM (also a site of recurrent mutation) alters a highly conserved amino acid residue predicted to alter structure of the mTOR HEAT domain required for interaction with inhibitory regulators (Supplementary Fig. 6). p.Met304Thr has not been implicated in Smith–Kingsmore syndrome. Consistent with a GoF mechanism for MTOR CH DNMs, mTOR inhibitor rapamycin can rescue the severe neonatal hydrocephalus associated with constitutive mTORC1 hyperactivation in NSCs due to primary cilia ablation23.
MTOR contained other inherited or unphased rare, D-Mis variants of uncertain significance (p.Arg769Cys, p.Arg1161Gly, p.Arg1170Cys and p.His1782Arg) (Supplementary Table 9 and Extended Data Fig. 5). Notably, similar to the CH proband with de novo MTOR p.Glu1799Lys, the CH proband with the nearby transmitted p.His1782Arg variant had macrocephaly, craniofacial abnormalities and skeletal abnormalities (Supplementary Table 11).
FOXJ1, FMN2 and PTCH1 harbor multiple DNMs and other inherited damaging variants.
Three other LoF-intolerant genes harbored ≥2 damaging DNMs (Fig. 3, Supplementary Table 12 and Extended Data Figs. 6–8). The forkhead family transcription factor FOXJ1 (Forkhead Box J1, pLI = 0.97) contained two LoF DNMs (p.Gln276X and p.Glu323fs) surpassing thresholds for genome-wide significance (P = 1.4 × 10−7; Fig. 3, Table 1a and Supplementary Table 12) and one inherited rare D-Mis mutation of uncertain significance (p.Thr96Arg). All FOXJ1-mutated CH probands exhibit obstructive hydrocephalus with aqueductal stenosis (Fig. 3, Extended Data Fig. 6 and Supplementary Table 13). Consistent with these results, Foxj1 depletion in mice causes obstructive hydrocephalus and aqueductal stenosis and disrupts a transcriptional network required for the differentiation of radial glial NSC into multiciliated ependymal cells32.
Fig. 3 |. Multiple damaging DNMs in FOXJ1, FMN2, PTCH1 and an excess burden of rare LoF heterozygous mutations in FXYD2 in sporadic CH.
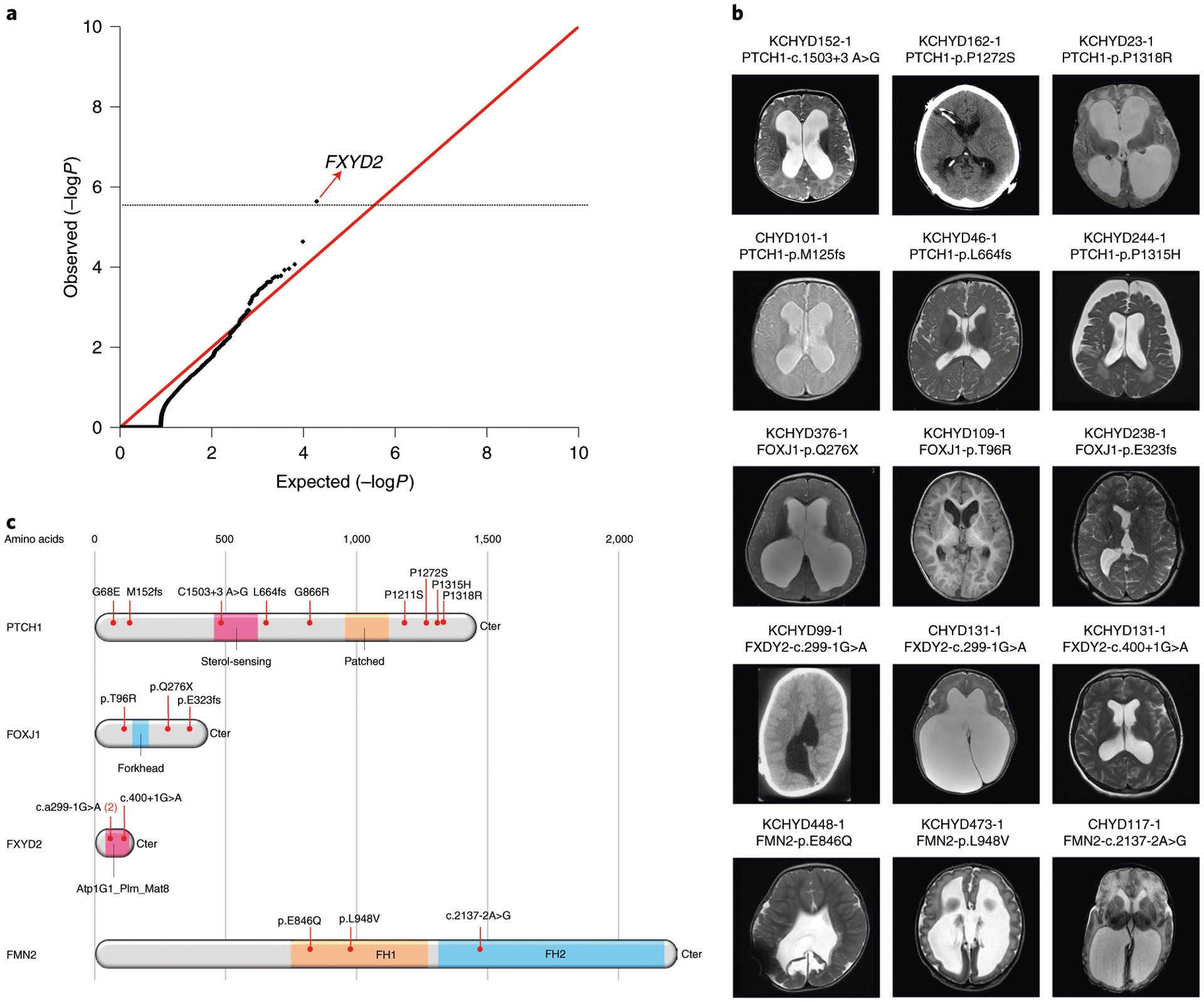
a, Quantile-quantile plot of observed versus expected P value of rare LoF heterozygous mutations. A one-tailed binomial test was conducted by comparing the observed number of LoF heterozygous mutations to the expected count. The genome-wide significant cutoff was 2.6 × 10−6 (0.05 of 19,347). FXYD2 was the single gene showing enrichment of rare, LoF heterozygous mutations. b, representative T1- or T2-weighted axial brain mrIs or axial head CT images of neurosurgically treated CH probands with the indicated FOXJ1, FMN2, PTCH1 and FXYD2 mutations (Supplementary Table 13 and extended Data Figs. 6–9 contain clinical and neuroradiographical details for each patient). c, mutation mapping of the FOXJ1, FMN2, PTCH1 and FXYD2 mutations in relation to critical functional domains in each molecule. FH, formin homology; Atp1G1_plm_mat8, ATP1G1/PLm/mAT8 family domain.
FMN2 (formin-2; pLI = 1.0) contained two DNMs (c.2137−2A>G and p.Glu846Gln; P = 4.4 × 10−4) and one inherited D-Mis variant of uncertain significance (p.Leu948Val) (Fig. 3, Extended Data Fig. 7 and Supplementary Table 12) in CH probands with obstructive hydrocephalus and aqueductal stenosis (Supplementary Table 13). None of the FMN2 mutations are present in gnomAD and Bravo. The CRYP-SKIP algorithm33 suggests that the canonical splice-site mutation c.2137−2A>G likely causes exon skipping (PCR-E = 0.30; Extended Data Fig. 7). Both p.Glu846Gln and p.Leu948Val map to the proline-rich FH1 domain of formin-2 (Fig. 3)34. Fmn2 overexpression disrupts neuroepithelial integrity and impairs NSC proliferation and neuronal migration in mouse embryos35. Fmn2 and FlnA double knockout mice show significantly thinned cortices and microcephaly associated with NSC proliferation36.
We previously identified two LoF DNMs (c.1503+3A>G and p.Met152fs) in PTCH1 (Patched 1) among CH probands9. In our expanded cohort, we identified a total of seven (including six new) rare, damaging transmitted or unphased PTCH1 variants in unrelated CH probands (Extended Data Fig. 8 and Supplementary Table 12), including an inherited PTCH1 p.Leu664fs mutation in a CH proband who, like his transmitting mother, had Gorlin syndrome. Two inherited D-Mis mutations (p.Pro1315His and p.Gly68Glu) and four unphased D-Mis mutations (p.Gly866Arg, p.Pro1211Ser, p.Pro1272Ser and p.Pro1318Arg) were of unknown significance (Supplementary Table 12). All D-Mis mutations in PTCH1 were rare and altered conserved residues with evident clustering of four of six altering proline residues within a 107 amino acid segment of the carboxy terminus (Fig. 3). In support of their pathogenicity, several CH probands with inherited or unphased PTCH1 D-Mis variants had phenotypes similar to patients with Gorlin syndrome with PTCH1 LoF mutations, but did not meet formal criteria for Gorlin syndrome (Supplementary Table 13 and Extended Data Fig. 8). Consistent with these results, Ptch1+/− mice develop hydrocephalus with incomplete penetrance and variable expressivity37. Primary cilia sense gradients of Sonic Hedgehog via PTCH1, which transduces these signals to regulate growth and differentiation of hindbrain NSCs38,39.
FXYD2 contains a significant burden of inherited dominant mutations, including a recurrent splice-site mutation.
To identify additional haplo-insufficient genes associated with CH otherwise not revealed by DNM analysis, we compared the observed and expected number of rare (minor allele frequency (MAF) ≤ 5.0 × 10−5) heterozygous LoF mutations in each gene using a one-tailed binomial test while adjusting for gene mutability (Methods). FXYD2 (pLI = 0.24), encoding the regulatory γ-subunit of the Na+/K+-ATPase, surpassed genome-wide significance thresholds (123.5-fold enrichment, P = 2.3 × 10−6; Fig. 3 and Extended Data Fig. 9). No DNMs or recessive mutations were observed in FXYD2. Case–control burden analysis for rare LoF mutations in all probands versus gnomAD controls also identified FXYD2 as having high mutational burden in CH probands (odds ratio = 49.3, one-sided Fisher’s exact test, P = 4.8 × 10−5). Three unrelated CH probands exhibited two identical transmitted canonical splice-site mutations in FXYD2 (c.299−1G>A) and one unphased FXYD2 splice-site mutation (c.410+1G>A) predicted by the CRYP-SKIP algorithm33 to cause exon skipping (Extended Data Fig. 9). The maximum haplotype shared by the two kindreds (~548 kb) suggests a remote common ancestor (Supplementary Table 14 and Supplementary Fig. 7). Recurrent heterozygous missense mutations in FXYD2 (p.Gly41Arg) underlie defective Na+/K+-ATPase plasma membrane expression and function in autosomal dominant type 2 renal hypomagnesemia (OMIM no. 154020). All FXYD2 mutant CH probands shared normal serum magnesium levels, and the majority displayed corpus callosum abnormalities and cerebellar tonsillar ectopia (Supplementary Table 13).
Recessive genotypes in homologs of mouse hydrocephalus genes are enriched in consanguineous CH cases.
The 8.7% consanguinity of our CH cohort (Supplementary Table 2 and Supplementary Fig. 8) prompted evaluation for enrichment in CH probands of damaging recessive genotypes (RGs) in homologs of 189 mouse hydrocephalus (mH) genes8,40 (Supplementary Datasets 3 and 4; Methods). Among 90 damaging RGs among probands, six occurred in the mH gene set, (P = 3.7 × 10−3) (Supplementary Table 15a). Enrichment of RGs in the mH gene set was greater for LoF mutations (P = 4.9 × 10−4; Supplementary Table 16). Homozygous RGs new for CH included one each in POMGNT1 (c.1111–1G>A), FKRP (D-Mis p.Gly354Glu), RHPN1 (p.Met281fs), CEP290 (c.6012−2A>G), KCNG4 (p.Gly442Arg) and KIF19 (p.Gly859fs) (Supplementary Table 15b and Extended Data Fig. 10). All probands were products of consanguineous union except the RHPN1 proband, P = 1.9 × 10−3; Supplementary Table 17), revealing a substantial contribution of RGs among probands from consanguineous union (15.6%). Homozygous loss of each of these genes causes severe postnatal hydrocephalus8,40.
POMGNT1 and FKRP mutations cause human muscular dystrophy-dystroglycanopathy, characterized by hypotonia, seizures, retinal degeneration, cobblestone lissencephaly and, rarely, ventriculomegaly41. A set of 12 human muscular dystrophy-dystroglycanopathy genes (Supplementary Dataset 5) was enriched among CH probands (P = 8.5 × 10−5; Supplementary Table 15a) and included POMGNT2, a gene with a homozygous (consanguineous) LoF mutation (p.Tyr367X) whose depletion causes hydrocephalus in humans and zebrafish (Supplementary Table 15b)42. Other pathway gene sets implicated in syndromic hydrocephalus8, including cilia structure and function (Supplementary Dataset 6), cell adhesion (Supplementary Dataset 7), synaptic vesicle biology (Supplementary Dataset 8), planar cell polarity (Supplementary Dataset 9), Ras signaling (Supplementary Dataset 10), Wnt signaling (Supplementary Dataset 11), PI3K-AKT-mTOR signaling (Supplementary Dataset 12) and lysosomal storage (Supplementary Dataset 13) were not enriched among CH probands (Supplementary Table 15a).
CH risk genes converge in fetal human coexpression networks and cell types relevant for fetal neurogenesis.
Because animal and pre-clinical evidence suggests that many CH mutations disrupt NSC regulation (Supplementary Table 18), we tested whether high-confidence, probable and/or known human CH risk genes (Supplementary Dataset 14) converge in gene coexpression networks of the midgestational human cortex (Methods)43. Notably, CH risk genes converged in a single transcriptional network (‘yellow’ module; P = 1.19 × 10−3; Fig. 4a), previously associated with ASD (Supplementary Dataset 15) and other undiagnosed developmental disorders (DDs) (Supplementary Dataset 16)43. The top enriched Gene Ontology (GO) biological process terms for the yellow module (Fig. 4b) include neuronal differentiation and RNA processing (for example, GO: 0000904 and GO: 0048667). The top enriched human phenotype (HP) ontology terms (Fig. 4b) describe several congenital defects of craniofacial development and behavioral abnormalities, including ‘autistic behavior’ (for example, HP: 0000252 and HP: 0000729).
Fig. 4 |. CH risk genes are enriched in a coexpression network pertinent to other neurodevelopmental disorders and in cell types of early fetal neurogenesis.
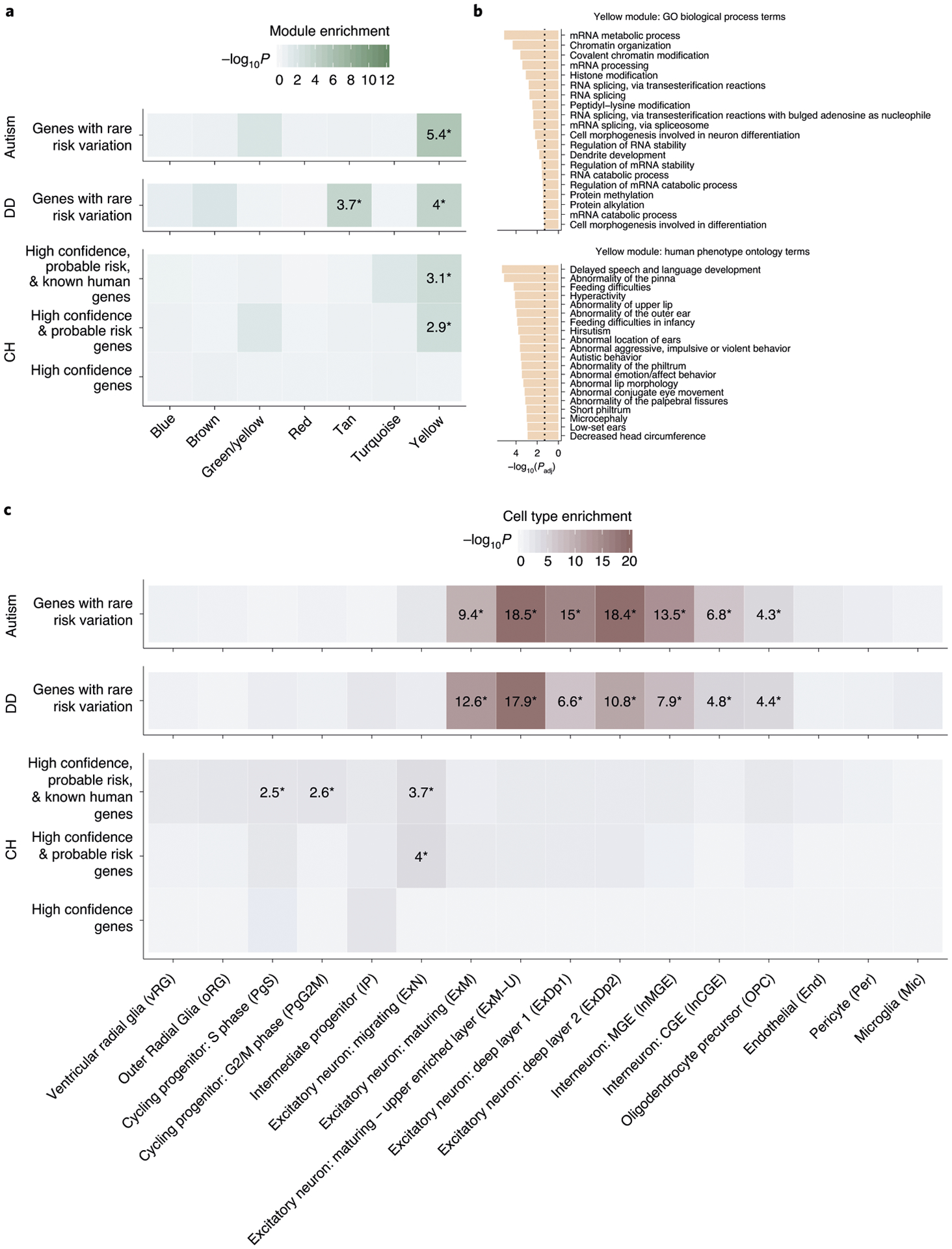
a, enrichment analysis across weighted gene coexpression network analysis (WGCNA) modules of the midgestational human brain for genes with rare risk variation in CH (high confidence, probable risk and known human genes), ASD and DD (methods contains details of gene set definitions). Only seven modules are shown (labeled by color in line with Walker et al.43); other modules demonstrated no significant enrichment for tested gene sets. Tiles labeled with −log10(P value) and an asterisk represent statistically significant enrichment at the bonferroni multiple-testing cutoff (α = 0.05/17 = 2.94 × 10−3). b, Top 20 GO biological process terms and top 20 HP ontology terms enriched for the yellow module. The x axis depicts −log (adjusted P value) and the dotted line represents the α = 0.05 significance threshold. (P values are adjustted according to the g:SCS algorithm from g:Profiler59). c, enrichment analysis across cell type markers of the midgestational human brain44 for genes with rare risk variation in CH (high confidence, probable risk and known human genes), ASD and DD. Tiles labeled with −log10(P value) and an asterisk represent significant enrichment at the bonferroni multiple-testing cutoff (α = 0.05/16 = 3.13 × 10−3).
We also examined potential enrichment of CH risk genes in cell type markers of the largest available single-cell (sc)RNA transcriptomic atlas of midgestational brain development44 (spanning 17–18 gestational weeks; Fig. 4c). High confidence and probable CH genes were enriched in nascent migrating excitatory neurons (P = 9.98 × 10−5). Adding known human genes to our cohort’s risk genes led to additional enrichment in mitotic progenitors PgS (P = 2.85 × 10−3) and PgG2M (P = 2.44 × 10−3). These data suggest that mutations in biologically pleiotropic CH genes disrupt pathways that regulate neurogenesis in the developing human brain.
CH shares genetic risk factors with other neurodevelopmental disorders.
The transcriptional overlap of risk genes for CH, ASD and DD during brain development (Fig. 4a); the frequent presence of other neurodevelopmental phenotypes in patients with CH;45 and the association of ventriculomegaly with ASD46 and other neurodevelopmental conditions47 prompted our hypothesis that sporadic CH may share common genetic risk factors with ASD and other neurodevelopmental conditions. Indeed, CH and ASD exhibited significant overlap, with 7 genes harboring LoF DNMs and 20 genes harboring damaging DNMs in both cohorts (Supplementary Table 19). CH and other DDs also exhibited significant overlap, with 6 genes harboring LoF DNMs and 22 harboring damaging DNMs in both cohorts (Supplementary Table 20). The data suggest partial overlap of genetic risk factors among CH, ASD and other severe neurodevelopmental disorders.
Discussion
Our WES study of the largest cohort of sporadic, neurosurgically treated CH to date has coupled integrative genomics with deep clinical and neuroradiographic phenotyping to uncover new insights into CH genetic architecture and biology with potential implications for patient care. We show rare mutations with large effect contributed to 22.2% of CH cases (17.7% damaging DNMs, 1.6% RGs, 0.8% transmitted heterozygous LoF variants). Overall, 2.1% of CH cases represented known familial CH mutations. Insertion-deletions, rearrangements, noncoding variants and intronic splice mutations, also likely contribute to genetic risk for CH and will be subjects of future studies. Additional CH cases may arise from complex interactions between genetic and environmental risk factors.
We estimate from the distribution of protein-altering DNMs in LoF-intolerant genes that 34 genes contribute to CH via a DNM mechanism (Supplementary Fig. 9a; Methods). This estimate is relatively low compared to the ~400 genes contributing to ASD and CHD, respectively48,49. Simulations suggest that sequencing of 2,500 or 5,000 WES trios will yield 90.3% or 97.6% saturation, respectively for CH (Supplementary Fig. 9b; Methods). Sequencing of additional trios and isolated probands will therefore detect additional rare mutations with a large effect on disease risk.
These results corroborate and significantly extend our previous work9, with discovery of new DNMs in TRIM71 and SMARCC1 as likely bona fide CH risk genes. We also provide evidence that PIK3CA, PTEN, MTOR, FOXJ1, FMN2, PTCH1 and FXYD2 are new high-confidence sporadic CH genes, collectively accounting for ~7.3% of CH cases. The phenotypes associated with each orthologous gene in corresponding zebrafish and/or murine disease models support their roles in embryonic neurogenesis and CH pathogenesis (Supplementary Table 18).
Clinical and neuroradiographic phenotyping of CH cases provides evidence for genotype-specific subtypes of CH. For example, TRIM71 and SMARCC1 likely define new Mendelian CH syndromes based on clustering of distinctive features associated with each gene (for example, cranial nerve deficits, nonobstructive interhemispheric cysts and hearing loss associated with TRIM71, and aqueductal stenosis, cardiac and skeletal abnormalities with SMARCC1). These observations support the pathogenicity of identified mutations and suggest that phenotypic subsets of CH are influenced by specific genetic determinants. As ongoing WES of deeply phenotyped trios continues, we anticipate emergence among CH cases of different genetic disorders with predictable clinical histories and distinctive neuroradiologic features. The phenotypic spectra of TRIM71, SMARCC1 and other CH risk variants will also be better defined.
Several of the identified CH risk genes harboring damaging DNMs and inherited mutations have been implicated in other Mendelian diseases, sometimes producing quite different phenotypes. For example, three CH probands carried mutations in PTEN previously implicated in PTEN hamartoma tumor syndrome (OMIM no. 607174), but none met criteria for this or related PTEN disorders25–27. The same is true of a CH proband harboring an MTOR mutation previously implicated in Smith–Kingsmore syndrome (OMIM no. 616638) that did not meet criteria for this disorder30. Similarly, although the identical FOXJ1 DNMs in our CH probands were recently identified in patients with type 43 primary ciliary dyskinesia (OMIM no. 618699, associated with bronchiectasis and situs inversus)50, none of our FOXJ1 mutant patients exhibit these pulmonary or cardiac phenotypes. These observations highlight the phenotypic heterogeneity and variable expressivity associated with these gene mutations, which could arise from environmental modifiers, working in concert with the identified rare mutations and/or specific genetic modifiers, including mosaicism and other somatic mutations.
Our study also highlights how distinctive features of known Mendelian syndromes in sporadic CH probands can be overlooked by clinical caregivers. For example, although four out of five PIK3CA-mutated probands (all treated at different institutions across the country) retrospectively met clinical criteria for MCAP, none carried a diagnosis before involvement in our study. Similarly, we identified four new L1CAM mutations in men with CH with aqueductal stenosis and classic stigmata of L1 syndrome (OMIM no. 307000) undiagnosed due to unrecognized syndromic findings, along with lack of genetic testing. Our study’s structure, including patient recruitment from a multitude of domestic and international institutions, enabled a ‘real-life’ snapshot of CH care and diagnosis, with important implications for genetic screening of newly diagnosed children with CH.
Much hydrocephalus research has centered on understanding the production, circulation and reabsorption mechanisms of CSF. While these mechanisms are important for acquired hydrocephalus in children and adults or in elderly patients with normal pressure hydrocephalus, our data and much murine data40 implicate earlier, more fundamental genetic insults in CH. Notably, each high confidence CH gene harboring DNMs is highly expressed in the neuroepithelium lining embryonic neural tube and/or ventricular (VZ) and subventricular (SVZ) zones, where they regulate proliferation, differentiation and/or fate specification of multipotent NSCs or rapidly proliferative neural precursors (Supplementary Dataset 17). Genetic disruption of embryonic and fetal brain development is therefore the primary event underlying CH pathogenesis in a significant subset of patients.
In this NSC model of CH pathogenesis (Fig. 5), nonobstructive ventriculomegaly can result from impaired neurogenesis due to dysregulation in NSC pluripotency, leading to decreased cortical cell mass and a thinned cortical mantle51. Obstructive ventriculomegaly can arise from progressive CSF accumulation due to aqueductal obstruction from maldevelopment52 or to peri-aqueductal NSC hyperproliferation53. Other potential mechanisms include impaired growth or size regulation of the ventricular apical domain of primary cilia-containing radial glia NSCs54 or impaired differentiation of radial glia NSCs into multiciliated ependymal cells32. These primary genetic events impairing neuro-gliogenesis could then secondarily disrupt CSF homeostasis by altering normal multiciliated ependymal or possibly glia-lymphatic structure and function. Notably, germinal matrix hemorrhage in premature neonates, the most common cause of acquired pediatric hydrocephalus, is associated with impaired neurogenesis due to ependymal denudation and NSC damage in the VZ-SVZ55. An NSC model could thus provide a ‘unified’ mechanism explaining multiple forms of neonatal hydrocephalus, both congenital and acquired.
Fig. 5 |. A neural stem cell model of sporadic CH.
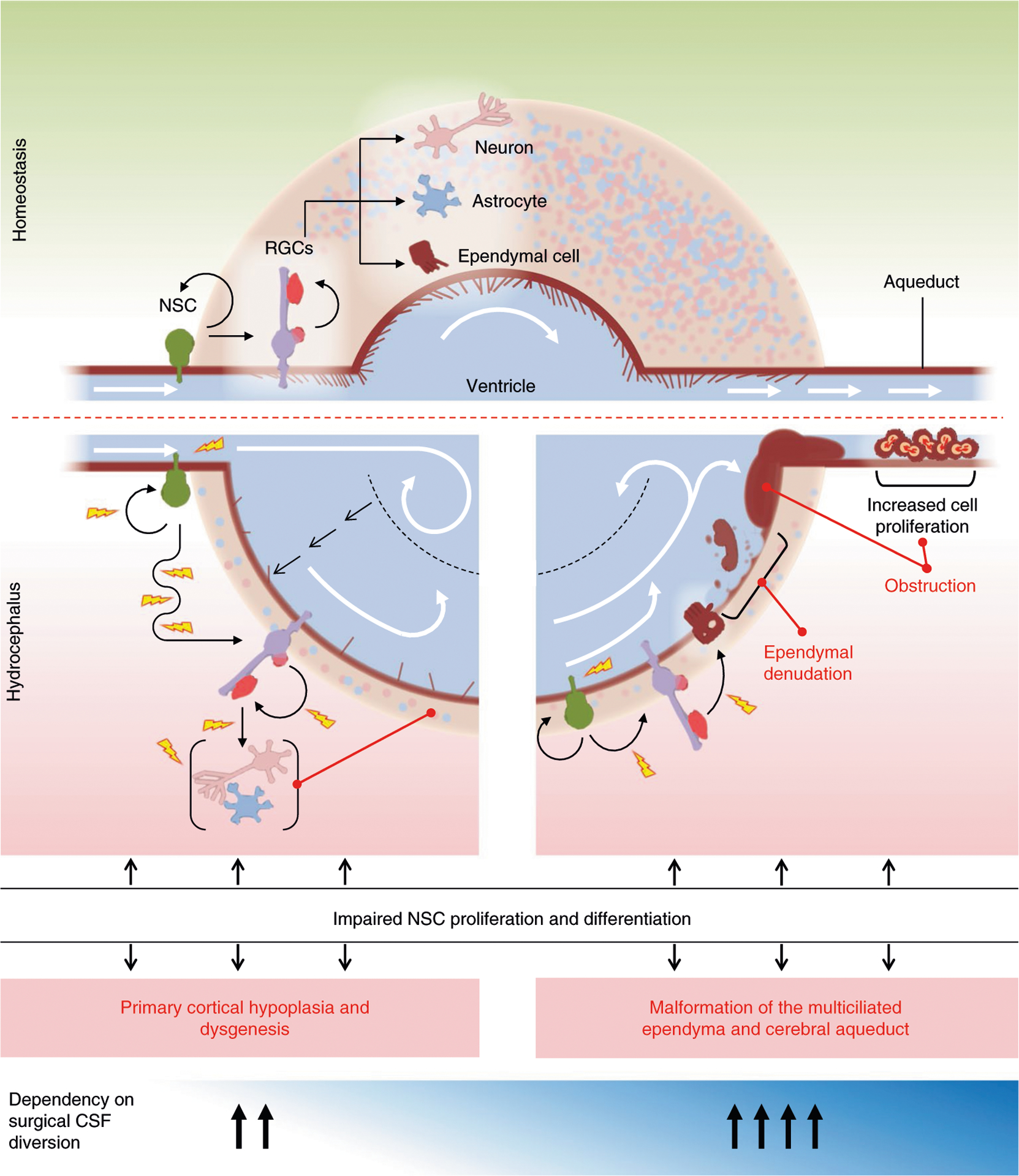
Schematic of the normal developing brain with the ventricular system surrounded by parenchyma consisting of neurons, astrocytes and components of neurogenesis at the cellular level (top). embryonic and fetal NSC populations, including neuroepithelial cells and radial glia cells (rGCs), together generate virtually all neuronal and glial cells that populate the brain, including multiciliated ependymal cells that line the ventricular system thought to participate in CSF circulation and maintenance of ventricular integrity. Defects in embryonic and fetal NSCs secondary to genetic mutations can thus drive CH via multiple pathogenic mechanisms that impact development and maturation of different cell types. Schematic of two possible developmental mechanisms of NSC alteration that may lead to ventriculomegaly (bottom). In one hypothesized scenario (left), ventriculomegaly results from impaired neurogenesis and an associated decrease in cortical cell mass that reflects a reduction in NSC proliferation. Continued CSF production from the unaffected choroid plexus would further expand the already enlarged ventricular compartment and even at low hydrostatic pressure push the thin, low-resistance cortical ribbon to the dural–bone interface. Ventricular enlargement and dysmorphology could then promote further ventricular expansion through secondary disruption of normal linear CSF laminar flow, eliciting fluid turbulence and current reversal. In another hypothesized scenario (right) that is not necessarily mutually exclusive from the former, altered NSC regulation leads to malformation of ependymal cells and their motile cilia, leading to impaired intraventricular CSF circulation and attendant CSF accumulation responsible for progressive ventricular dilation. Furthermore, defects in cilia-related genes may cause hydrocephalus not only by impairing motile cilia-driven CSF flow, but also by affecting development of primary cilia, which are nonmotile sensory organelles present on embryonic and fetal NSCs, crucial for multiple developmental processes, including patterning, neurogenesis, migration and survival. A combination of defects in NSC patterning and/or the proliferation–differentiation balance can also introduce anatomical defects, resulting in physical obstruction to CSF flow, such as aqueductal stenosis.
Consistent with mutations impacting fundamental aspects of fetal brain development, associated phenotypes such as intellectual disability, neurodevelopmental delay, epilepsy and autistic-like features are not infrequent findings among patients with CH4, including those of our cohort. In addition, ventricular enlargement in low-birth-weight infants is a risk factor for ASD56, including those with de novo PTEN mutations. We found enriched overlap of genetic risk factors between CH and ASD and DDs, along with CH risk gene enrichment in coexpression networks previously implicated in these conditions. However, analysis showed convergence of CH risk genes in neural precursors of relatively earlier origin than those of ASD and DDs57, perhaps accounting for the increased frequency of structural brain abnormalities in CH probands relative to these other disorders. The power of integrative genomics to identify specific cell types and developmental pathways impacted by CH genes will be increased as more high-confidence CH risk genes are discovered.
The diversity of genetic etiologies and underlying biochemical pathways in CH supports implementation of routine clinical WES for newly diagnosed patients. Current recommendations for workup of fetal/neonatal ventriculomegaly include rapid testing for known chromosomal and copy-number abnormalities58. However, this strategy does not address CH cases explained by known mutations. Application of routine WES or whole genome sequencing would provide improved diagnosis and management of children with CH. WES or whole genome sequencing could also aid prognostication, increase vigilance for medical screening of mutation-associated conditions (such as cancer surveillance for patients with CH with PIK3CA or PTEN) and provide recurrence rates to restore reproductive confidence.
In the longer term, we speculate that WES of patients with CH, coupled with deep clinical and neuroradiographical phenotyping, might improve precision of classification schemes to prognosticate neurocognitive outcomes and stratify patients to specific treatments (such as endoscopy versus CSF shunting versus pharmacological therapies). For example, in some nonobstructive CH with excessively thinned cortical mantles from disrupted neurogenesis and normal or even borderline moderately elevated ICPs, surgical CSF shunting may merely expose patients with CH to surgical morbidity without addressing disease pathogenesis. Surgical intervention in these contexts is unlikely to improve associated neurodevelopmental phenotypes such as seizures, motor impairment or intellectual function, more likely arising from genetic disruptions of embryonic neurogenesis than from reversible sequelae of CSF accumulation. These observations should raise thresholds for surgical intervention (or subsequent shunt revision) in patients with CH without radiographical obstruction, high ICPs or high-pressure-associated symptoms.
Our data explain ~20% of CH cases; however, most sporadic CH cases remain unexplained. Our current sample size still lacks statistical power adequate to detect the many rare, inherited or sporadic CH-associated risk genes. Although our patients are mostly of European origin, international collaborative studies will soon overcome our current limitations of small cohort size and limited ethnic diversity. Moreover, mechanistic insights into newly identified CH causal genes and core pathways will arise from in vivo experiments in model organisms. Our current work identifying new human gene targets and CH-specific mutations will serve as entry points for these functional studies. Successful pursuit of these next steps will refine current heuristics for clinical decision-making and render personalized treatments for patients with CH, including nonsurgical targeted therapies, a realistic goal.
Methods
Patients.
All study procedures and protocols comply with Yale University’s Human Investigation Committee and Human Research Protection Program. Written informed consent for genetic studies was obtained from all participants. Inclusion criteria included patients with primary CH who did not carry a genetic diagnosis before surgical treatment or inclusion in the study. Subjects with either a known chromosomal aneuploidy or a copy-number variation with known association to CH were also excluded. Hydrocephalus cases with secondarily acquired etiologies such as intraventricular hemorrhage, meningitis or other central nervous system infection, obstruction due to tumors or cysts and stroke were excluded. Children with hydranencephaly, large cysts and cephaloceles, myelomeningocele (Chiari II malformation) or benign extra-axial CSF accumulation (benign external hydrocephalus) were also excluded. Sequenced trios were composed of 381 primary CH probands including 232 parent–offspring trios and 149 singletons (Supplementary Tables 1 and 2). All probands had undergone surgery for therapeutic CSF diversion (shunt placement and/or endoscopic third ventriculostomy). Patients and participating family members provided buccal swab samples (Isohelix SK-2S DNA buccal swab kits), medical records, neuroimaging studies, operative reports and CH phenotype data.
Controls consisted of 1,798 unaffected siblings of people with ASD and unaffected parents from SSC60. Only the unaffected siblings and parents, as designated by SSC, were included in the analysis and served as controls for this study. Permission to access to the genomic data in the SSC on the National Institute of Mental Health Data Repository was obtained. Written informed consent for all participants was provided by the Simons Foundation Autism Research Initiative.
Whole-exome sequencing and variant calling.
Exon capture was performed on genomic DNA samples derived from saliva or blood using Roche SeqCap EZ MedExome Target Enrichment kit or IDT xGen target capture kit followed by 101 or 148 base-paired-end sequencing on the Illumina platforms as described previously9,10. Sequence reads were aligned to the human reference genome GRCh37/hg19 using BWA-MEM. Single-nucleotide variants and small indels were called using a combination of GATK HaplotypeCaller61,62 and Freebayes63 and annotated using ANNOVAR64. Allele frequencies were annotated in the Exome Aggregation Consortium, gnomAD (v.2.1.1) and Bravo databases65,66. MetaSVM and MPC algorithms were used to predict deleteriousness of missense variants (D-Mis, defined as MetaSVM-deleterious or MPC-score ≥2)67,68. Inferred LoF variants consisted of stop-gain, stop-loss, frameshift insertions/deletions, canonical splice site and start-loss. LoF and D-Mis mutations were considered ‘damaging’. PCR amplicons containing the mutation verified mutations in genes of interest.
DNMs were called using TrioDeNovo69. Candidate DNMs were further filtered based on the following criteria: (1) exonic or splice-site variants; (2) read depth (DP) of 10 in the proband and both parents; (3) minimum proband alternative read depth of 5; (4) proband alternative allele ratio ≥28% if having <10 alternative reads or ≥20% if having ≥10 alternative reads; (5) alternative allele ratio in both parents ≤3.5%; and (6) global MAF ≤ 4 × 10−4 in the Exome Aggregation Consortium database.
For recessive variant analysis, we filtered for rare (MAF ≤ 1 × 10−3 in Bravo and in-cohort MAF ≤ 5 × 10−3) homozygous and compound heterozygous variants that exhibited high-quality sequence reads (pass GATK variant quality score recalibration, ≥4 total reads total for homozygous and ≥8 reads for compound heterozygous variants, genotype quality (GQ) score ≥10 for homozygous and GQ score ≥20 for compound heterozygous variants). Only LoF, D-Mis and nonframeshift indels were considered potentially damaging to the disease. For probands whose parents’ WES data were not available, only homozygous variants were analyzed.
For rare heterozygous variants, only LoF and D-Mis mutations were considered to be potentially disease associated and were filtered using the following criteria: (1) pass GATK variant quality score recalibration; (2) MAF ≤ 5 × 10−5 in Bravo and in-cohort MAF ≤5 × 10−3; (3) DP ≥8 independent reads; and (4) GQ score ≥20. RGs and DNMs were excluded.
After filtering using the aforementioned criteria for each type of mutation, in silico visualization was performed to remove false-positive calls. Variants in the top candidate genes were further confirmed by Sanger sequencing.
Quantification and statistical analysis.
DNM expectation model.
Because the CH trios were captured by two different reagents (MedExome and IDT), we took the union of all bases covered by different capture reagents and generated a Browser Extensible Data file representing a unified capture for all trios. We used bedtools (v.2.27.1) to extract sequences from the Browser Extensible Data file70. We then applied a sequence context-based method to calculate the probability of observing a DNM for each base in the coding region, adjusting for sequencing depth in each gene as described previously71. Briefly, for each base in the exome, the probability of observing every trinucleotide mutating to other trinucleotides was determined. ANNOVAR (v2015Mar22) was used to annotate the consequence of each possible substitution. RefSeq was used to annotate variants (based on the file ‘hg19_refGene.txt’ provided by ANNOVAR). For each gene, the coding consequence of each potential substitution was summed for each functional class (synonymous, missense, canonical splice site, frameshift insertions/deletions, stop-gain, stop-loss and start-lost) to determine gene-specific mutation probabilities71. The probability of a frameshift mutation was determined by multiplying the probability of a stop-gain mutation by 1.25, as described previously71. In-frame insertions or deletions are not accounted for by the model and were not considered in the downstream statistical analyses. To align with ANNOVAR annotations, analysis was limited to variants that were located in the exonic or canonical splice site regions and were not annotated as ‘unknown’ by ANNOVAR. Following the inclusion criteria, we identified potential coding mutations and generated gene-specific mutation probabilities for 19,347 unique genes. Owing to the difference in exome capture kits, DNA sequencing platforms and variable sequencing coverage between case and control cohorts, separate de novo probability tables were generated for cases and controls, respectively.
Estimation of expected number of rare transmitted variants.
We implemented a multivariate regression model to quantify the enrichment of rare transmitted variants in a specific gene or gene set in cases, independent of controls. Additional details about the modeling of the distribution of recessive and transmitted heterozygous variant counts are described in our recent study48.
De novo enrichment analysis.
The burden of DNMs in CH cases and unaffected ASD controls was determined using the denovolyzeR package72 as previously described48. Briefly, the expected number of DNMs in case and control cohorts across each functional class was calculated by taking the sum of each functional class-specific probability multiplied by the number of probands in the study 2× (diploid genomes). Then, the expected number of DNMs across functional classes was compared to the observed number in each study using a one-tailed Poisson test71. Gene set enrichment analyses only considered mutations observed or expected in genes within the specified gene set (high brain-expressed, LoF-intolerant).
To examine whether any individual gene contains more protein-altering DNMs than expected, the expected number of protein-altering DNMs was calculated from the corresponding probability adjusting for cohort size. A one-tailed Poisson test was then used to compare the observed DNMs for each gene versus expected. As separate tests were performed for protein-altering, protein-damaging and LoF DNMs, the Bonferroni multiple-testing threshold is, therefore, equal to 8.6 × 10−7 (= 0.05 / (3 tests × 19,347 genes)).
To estimate the number of genes with multiple DNMs, one million permutations were performed to derive the empirical distribution of the number of genes with multiple DNMs. For each permutation, the number of DNMs observed in each functional class was randomly distributed across the genome adjusting for gene mutability. The empirical P value was calculated as the proportion of times that the number of recurrent genes from the permutation equals or exceeds the observed number of recurrent genes as follows:
Enrichment analysis for dominant and recessive variants.
We implemented a polynomial regression model coupled with a one-tailed binomial test to quantify the enrichment of damaging RGs in a specific gene or gene set in cases and controls, separately as described previously48. The expectation of the RG count for each gene was calculated by the formula below:
where ‘i’ denotes the ‘ith’ gene and ‘N’ denotes the total number of RGs. For a given gene set, the expected RG count was based on the sum of fitted values for the gene set.
For rare LoF heterozygous variants, we found that the number of rare LoF heterozygous variants in a gene was inversely correlated with the pLI score obtained from the gnomAD database. To control for the potential confounding effect due to the pLI score, we stratified genes into five subsets by pLI quartiles: (1) those with a pLI score between 0 and the first quantile (6.4 × 10−8); (2) those with a pLI score between the first quantile and the second quantile (pLI = 1.9 × 10−3); (3) those with a pLI score between the second quantile and the third quantile (pLI = 0.48); (4) those with a pLI score between third quantile and 1; and (5) those without a pLI score. For each set, the expected number of LoF heterozygous variants for a gene was estimated by the following formula:
where ‘j’ denotes the ‘jth’ gene, ‘k’ denotes the ‘kth’ set, and ‘L’ denotes the total number of rare LoF heterozygous variants.
Case–control burden analysis.
Case and control cohorts were processed using the same pipeline and filtered with the same criteria. A one-sided Fisher’s exact test was used to compare the observed number of total alternative alleles, regardless of the transmission pattern in cases to controls in the gnomAD (without disease-enriched TOPMed samples) database.
Determining gene lists.
The gene lists used for recessive enrichment analysis were curated as below. The mH genes were compiled by the association of their disease model, disease ortholog or phenotype with hydrocephalus per MGI (http://www.informatics.jax.org/) (Supplementary Dataset 4). The dystroglycanopathies genes (Supplementary Dataset 5) and ciliopathies genes (Supplementary Dataset 6) were compiled by Kousi and Katsanis8. Cell adhesion molecules (Supplementary Dataset 7), synaptic vesicle cycle (Supplementary Dataset 8), Ras signaling pathway (Supplementary Dataset 10), Wnt signaling (Supplementary Dataset 11), PI3K–ATK–mTOR pathway (Supplementary Dataset 12) and lysosomal storage disorder (Supplementary Dataset 13) gene sets were curated based on KEGG and pathway database and the HUGO Gene Nomenclature Committee. A planar cell polarity gene list (Supplementary Dataset 9) was curated based on Wang et al.73 and Tissir and Goffinet74.
Gene lists from transcriptomic analyses were curated as below. Risk genes from our CH cohort were defined as genes that harbored ≥1 inherited heterozygous LoF mutation of genome-wide significance, genes intolerant to LoF mutations (pLI > 0.9) with ≥1 LoF DNM and genes intolerant to missense mutations (mis-Z > 2) with ≥1 missense DNM. These genes were categorized as high confidence if they harbored ≥1 inherited heterozygous LoF mutation of genome-wide significance or ≥2 protein-altering DNMs; and as probable risk if they harbored 1 protein-altering DNM. This yielded a high confidence set of 9 hydrocephalus genes (TRIM71, PTEN, PIK3CA, SMARCC1, FMN2, MTOR, FOXJ1, PTCH1 and FXYD2) and a probable set of 55 genes.
We assembled lists of genes previously known to cause isolated and syndromic forms of hydrocephalus in humans (Supplementary Dataset 14) from three publications: Kousi and Katsanis8 summarized over 100 genes described in known hydrocephalus syndromes8, Furey et. al. outlined new genes implicated in CH through WES9 and Shaheen et. al. summarized genes with recessive mutations linked to familial forms of CH75.
We compiled a list of genes with rare risk variation in ASD from two papers: Ruzzo et. al.76, which describes genes harboring rare inherited variants and Satterstrom et. al.77, which describes genes with de novo variants and case–control variation (Supplementary Dataset 15). We compiled a list of developmental disorder (DD) risk genes from DDD 2017 (ref.78), which describes genes enriched in damaging DNMs (Supplementary Dataset 16).
Module enrichment.
Module gene lists were obtained from a bulk RNA-seq atlas from of the midgestational human prenatal cortex (14–21 gestational weeks)43. WGCNA79 of this atlas identified modules (labeled by color) of genes that share highly similar expression patterns during midgestational cortical development43. In a background set of all genes categorized in coexpression modules, we used a logistic regression for an indicator-based enrichment: is.disease ~ is.module + gene covariates (GC content, gene length and mean expression in bulk RNA-seq atlas), as described previously43. Of the 18 WGCNA modules, the gray module, by WGCNA convention80, contains all genes that do not coexpress and are consequently unassigned to a coexpression network. Thus, the gray module was excluded from enrichment testing and enrichment significance was defined at the Bonferroni multiple-testing cutoff (α = 0.05 / 17 = 2.94 × 10−3).
Module GO and HP profiling.
We used g:GOSt from g:Profiler, a tool for functional profiling of gene lists, to obtain descriptive terms for enriched modules59. We used all annotated genes as the statistical domain scope, the g:SCS algorithm to address multiple testing and P = 0.05 as a user-defined threshold for statistical significance. For each gene list, we retained terms of 100–1,000 genes and we plotted the top 20 enriched terms from GO biological process annotations and the top 20 enriched terms from HP ontology annotations.
Cell type enrichment.
Cell-type-enriched genes (cell type markers), were obtained from a scRNA-seq atlas that maps the human midgestational cortex (17–18 gestational weeks)44. In a background set of all genes expressed in ≥3 cells of the scRNA-seq atlas, we used a logistic regression for indicator-based enrichment: is.cell.type ~ is.disease + gene covariates (GC content, gene length). All P values were adjusted with Bonferroni correction. Enrichment significance was defined at the Bonferroni multiple-testing cutoff (α = 0.05 / 16 = 3.13 × 10−3).
Overlap analysis.
As described previously48, the permutation test was performed to assess the enrichment of overlapping genes with either damaging (D-Mis + LoF) or LoF DNMs shared between CH and two other trio-based cohorts: autism and developmental disorder. Given the observed numbers of genes with DNMs in the CH and other cohorts as N1 and N2, respectively and the observed number of overlapping genes as M, we sampled N1 genes from all genes in the CH cohort and N2 genes from all genes in the autism cohorts without replacement using the probability of observing at least one DNM as weight. The number of overlapping genes, G, was determined in each interaction of the simulation. A total of 1,000,000 iterations were conducted to construct the empirical distribution. The empirical number of overlapping genes was calculated by taking the average of the number of overlapping genes across all iterations. The empirical P value was calculated as follows:
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The sequencing data for all CH parent–offspring trios and singletons reported in this study have been deposited in the NCBI Database of Genotypes and Phenotypes under accession number phs000744.v4.p2. Our in-house R and Python pipelines and codes are available upon request.
Code availability
Our in-house Python and R pipelines are available from the corresponding author on request.
Extended Data
Extended Data Fig. 1 |. De novo, transmitted, and unphased mutations in TRIM71.
a, Pedigrees and sequencing electropherograms of Sanger sequencing depict all TRIM71 mutations in genomic DNA from CH probands. b, representative T1 or T2-weighted brain magnetic resonance images for all available probands.
Extended Data Fig. 2 |. De novo, transmitted, and unphased mutations in SMARCC1.
a, Pedigrees and sequencing electropherograms of Sanger sequencing depict all SMARCC1 mutations in genomic DNA from CH probands. b, representative T1 or T2-weighted brain magnetic resonance images or head CTs for all available probands.
Extended Data Fig. 3 |. De novo, transmitted, and unphased mutations in PIK3CA.
a, Pedigrees and sequencing electropherograms of Sanger sequencing depict all PIK3CA mutations in genomic DNA from CH probands. b, representative T1 or T2-weighted brain magnetic resonance images for all available probands.
Extended Data Fig. 4 |. De novo, transmitted, and unphased mutations in PTEN.
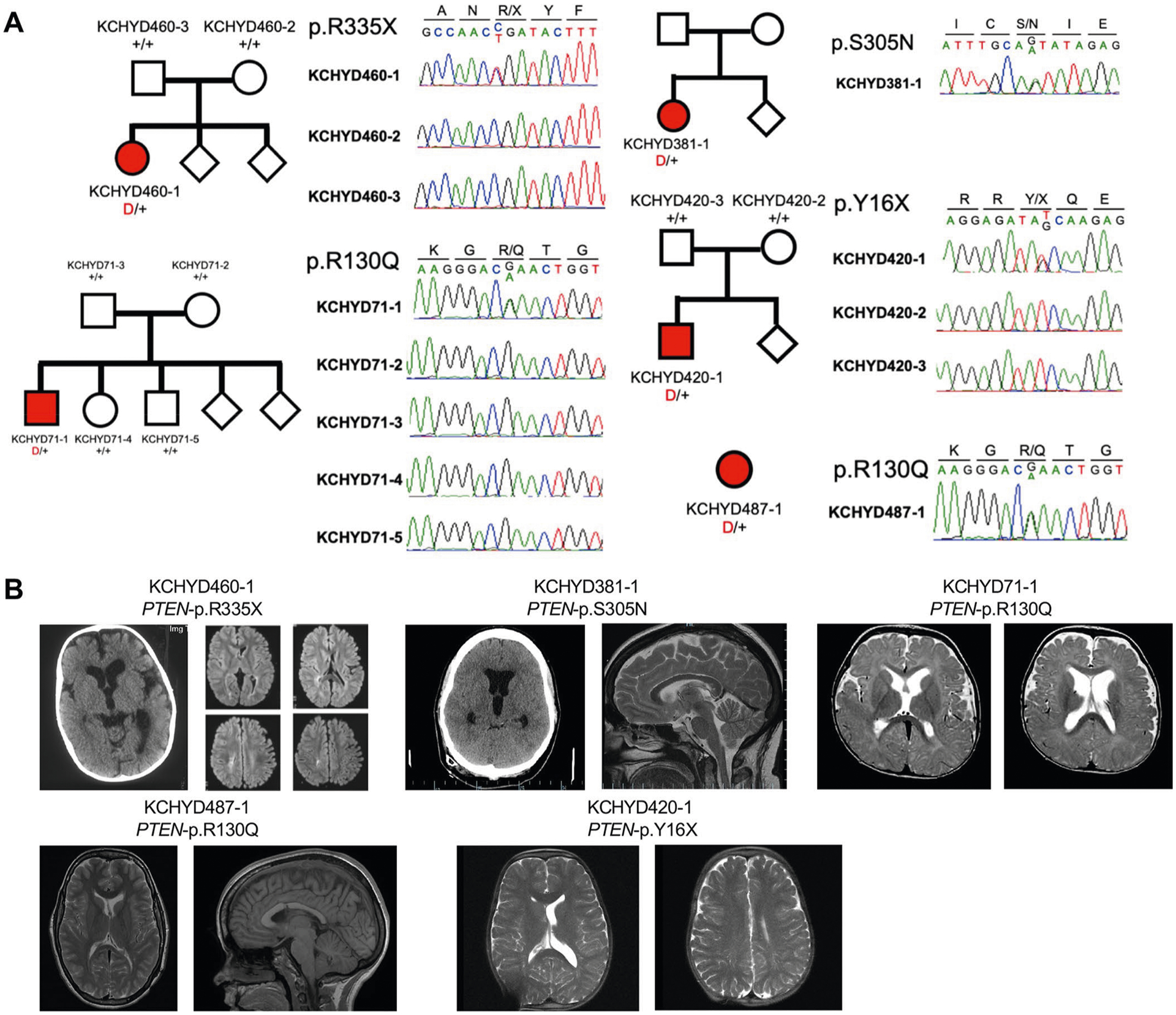
a, Pedigrees and sequencing electropherograms of Sanger sequencing depict all PTEN mutations in genomic DNA from CH probands. b, representative T1 or T2-weighted brain magnetic resonance images or head CTs for all available probands.
Extended Data Fig. 5 |. De novo, transmitted, and unphased mutations in MTOR.
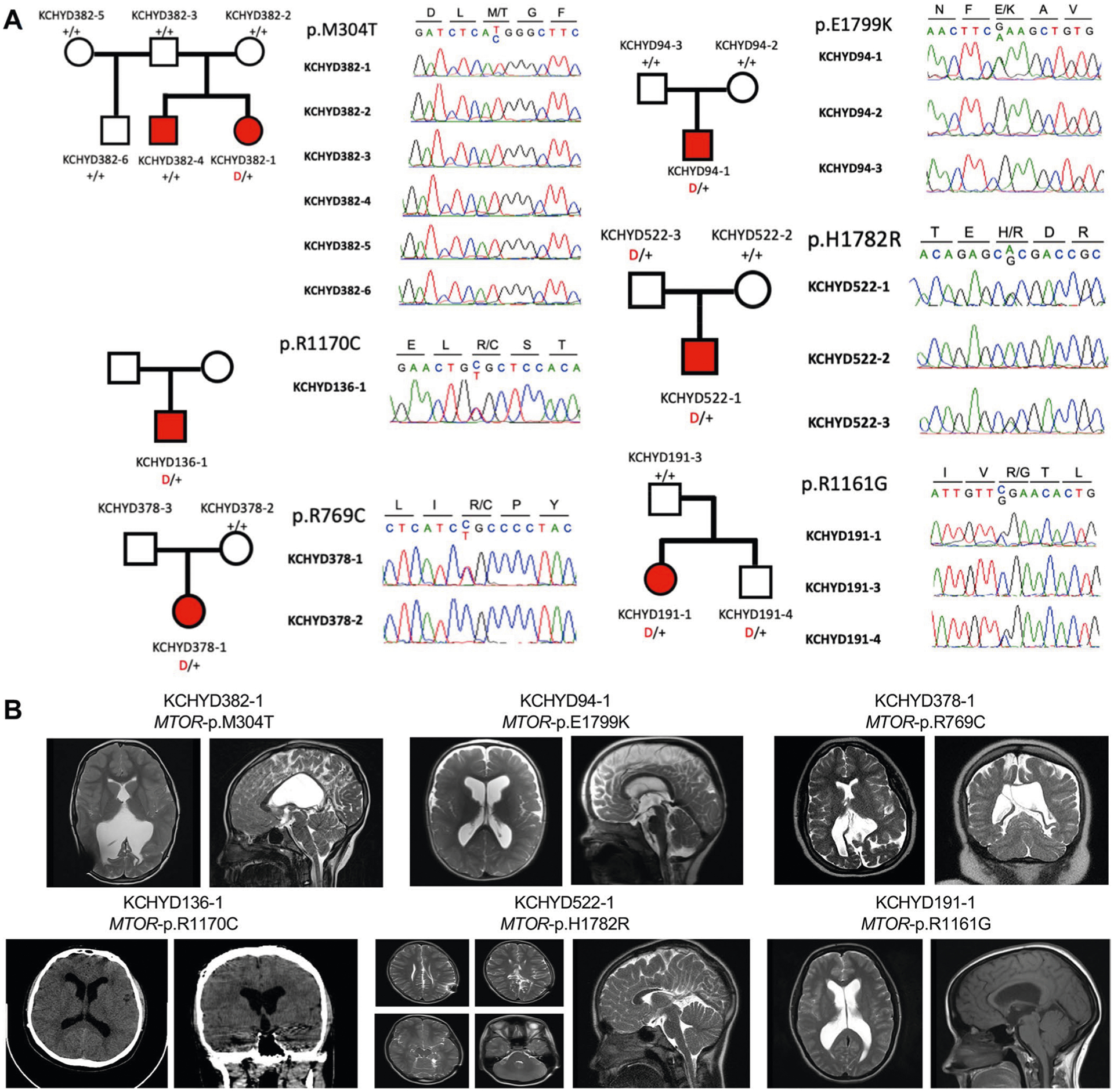
a, Pedigrees and sequencing electropherograms of Sanger sequencing depict all MTOR mutations in genomic DNA from CH probands. b, representative T1 or T2-weighted brain magnetic resonance images or head CTs for all available probands.
Extended Data Fig. 6 |. De novo and transmitted mutations in FOXJ1.
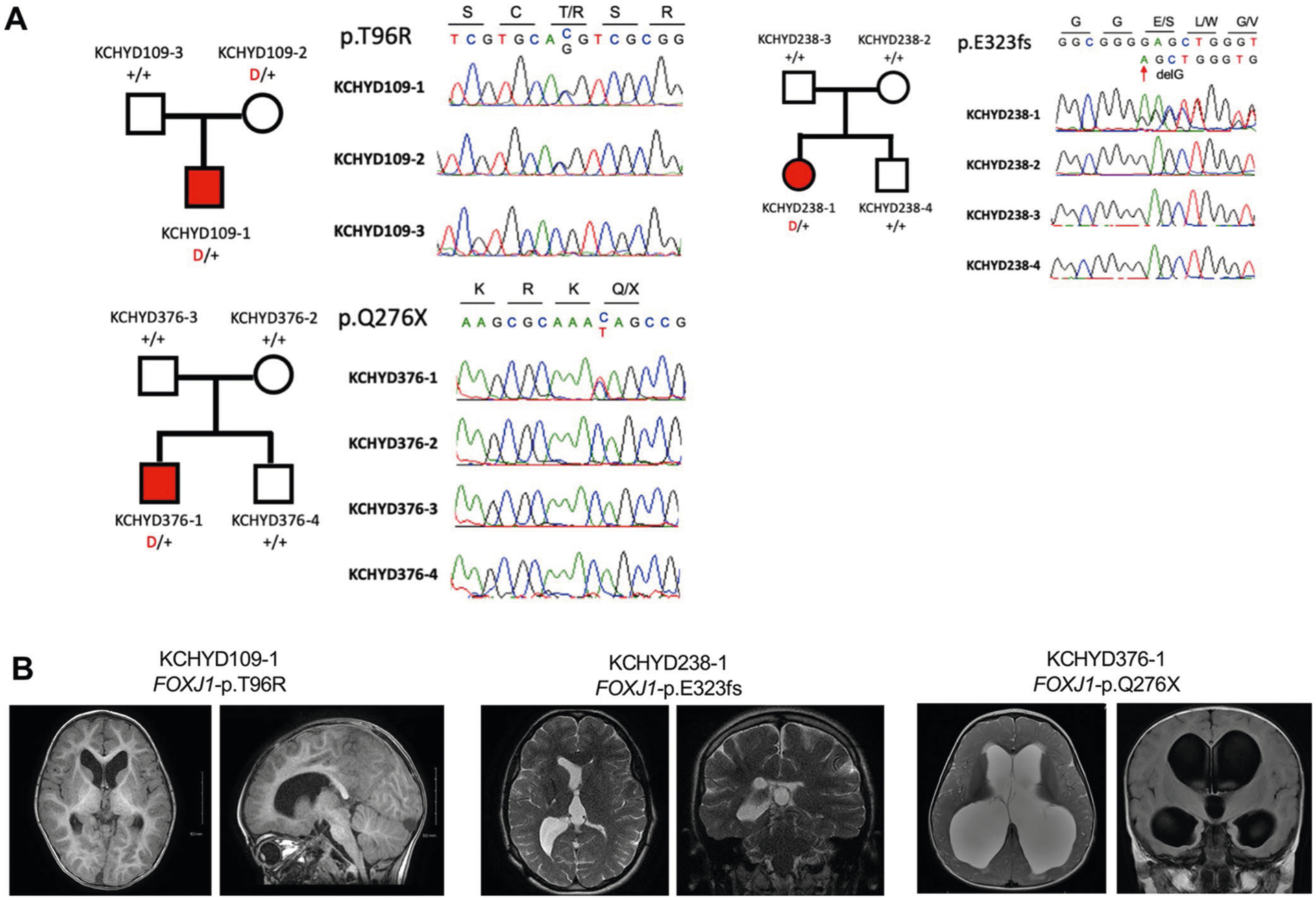
a, Pedigrees and sequencing electropherograms of Sanger sequencing depict all FOXJ1 mutations in genomic DNA from CH probands. b, representative T1 or T2-weighted brain magnetic resonance images for all available probands.
Extended Data Fig. 7 |. De novo and transmitted mutations in FMN2.
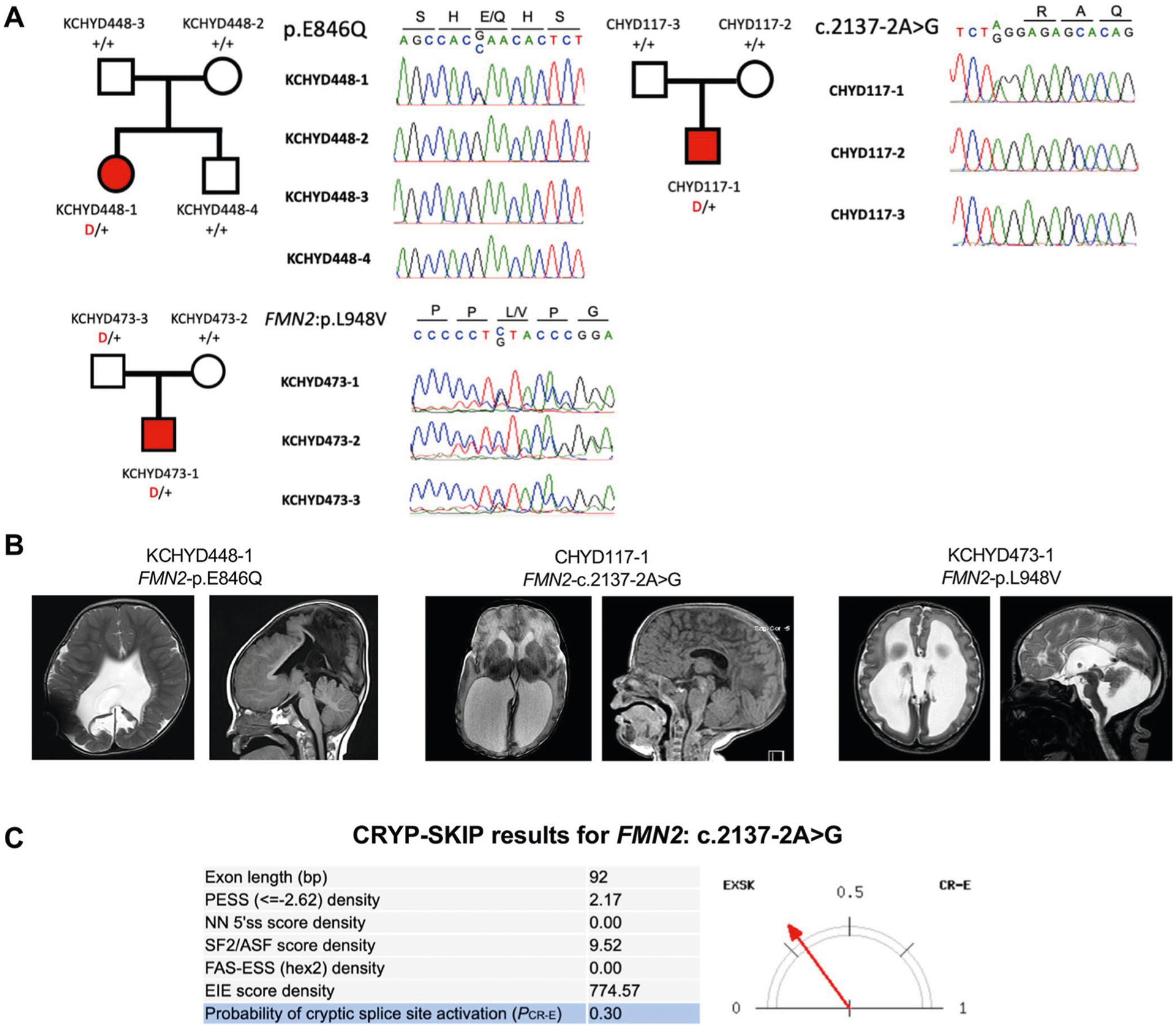
a, Pedigrees and sequencing electropherograms of Sanger sequencing depict all FMN2 mutations in genomic DNA from CH probands. b, representative T1 or T2-weighted brain magnetic resonance images for all available probands. c, The CrYP-SKIP algorithm prediction on splicing defects for FMN2: c.2137−2 A > G.
Extended Data Fig. 8 |. De novo, transmitted, and unphased mutations in PTCH1.
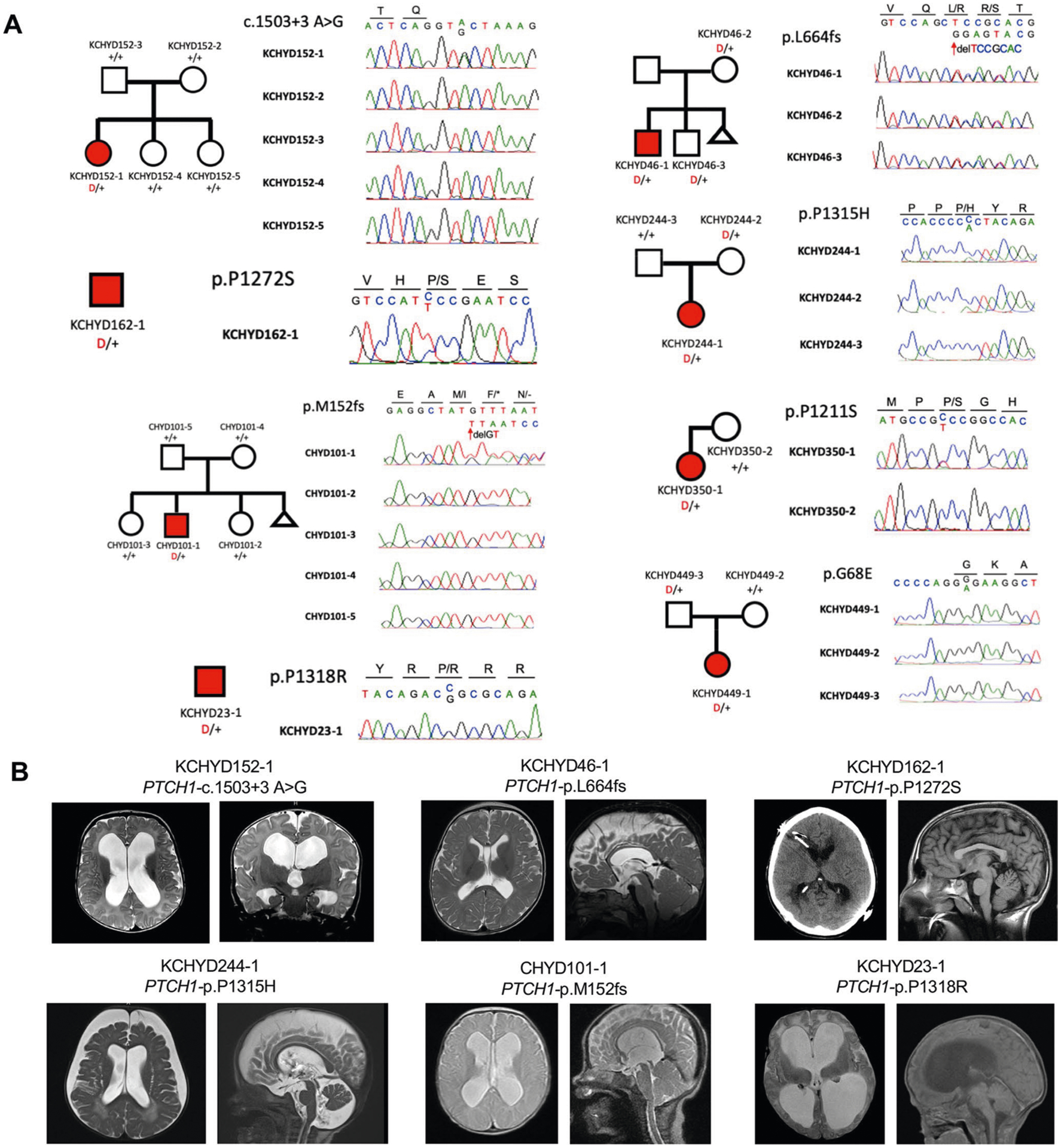
a, Pedigrees and sequencing electropherograms of Sanger sequencing depict all PTCH1 mutations in genomic DNA from CH probands. b, representative T1 or T2-weighted brain magnetic resonance images or head CTs for all available probands.
Extended Data Fig. 9 |. Transmitted and unphased mutations in FXYD2.
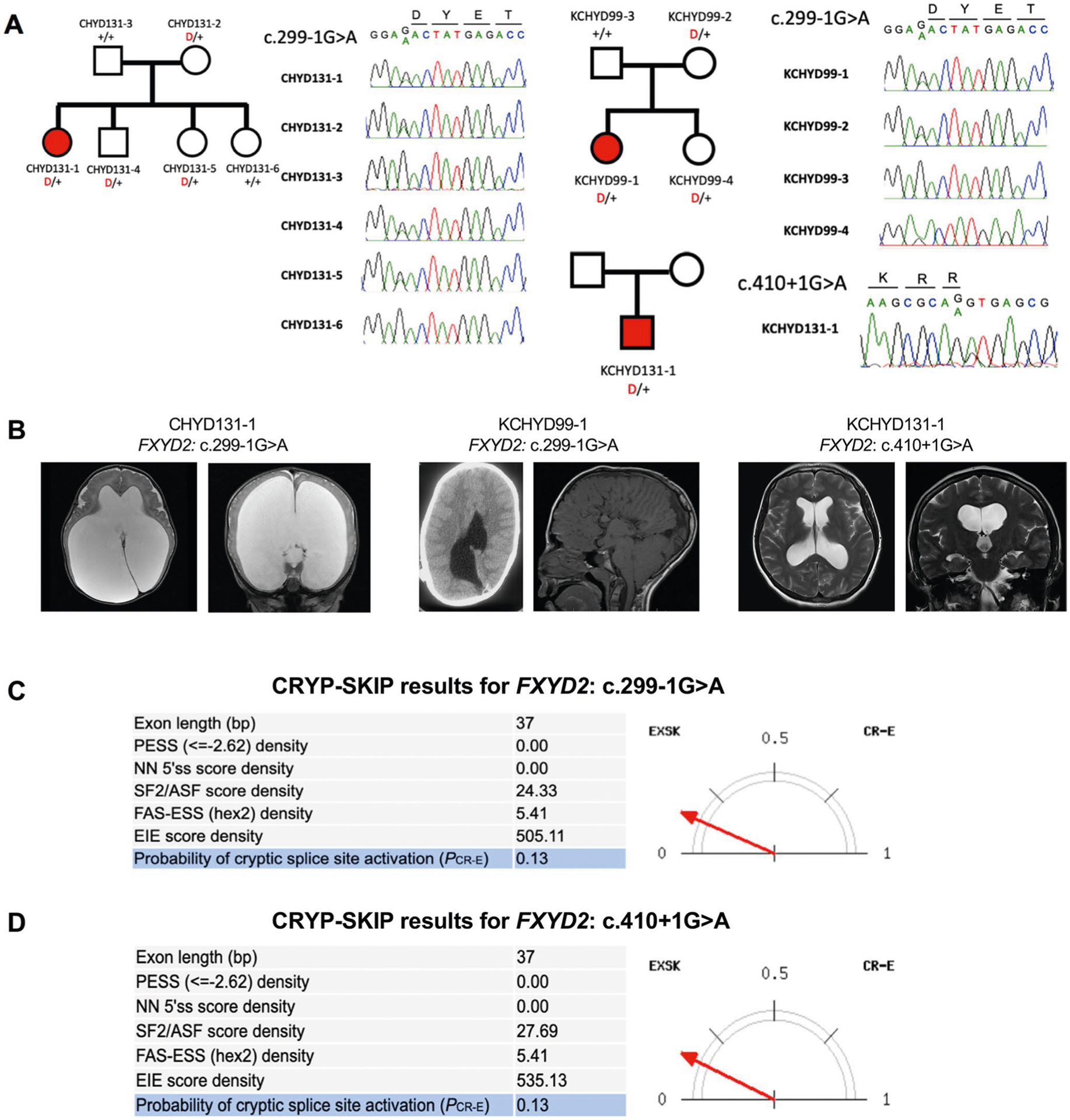
a, Pedigrees and sequencing electropherograms of Sanger sequencing depict all FXYD2 mutations in genomic DNA from CH probands. b, representative T1 or T2-weighted brain magnetic resonance images for all available probands. c, The CrYP-SKIP algorithm prediction on splicing defects for FXYD2: c.299−1 G > A. d, The CrYP-SKIP algorithm prediction on splicing defects for FXYD2: c.410 + 1 G > A.
Extended Data Fig. 10 |. Damaging recessive genotypes in human dystroglycanopathy genes and homologs of mouse hydrocephalus genes.
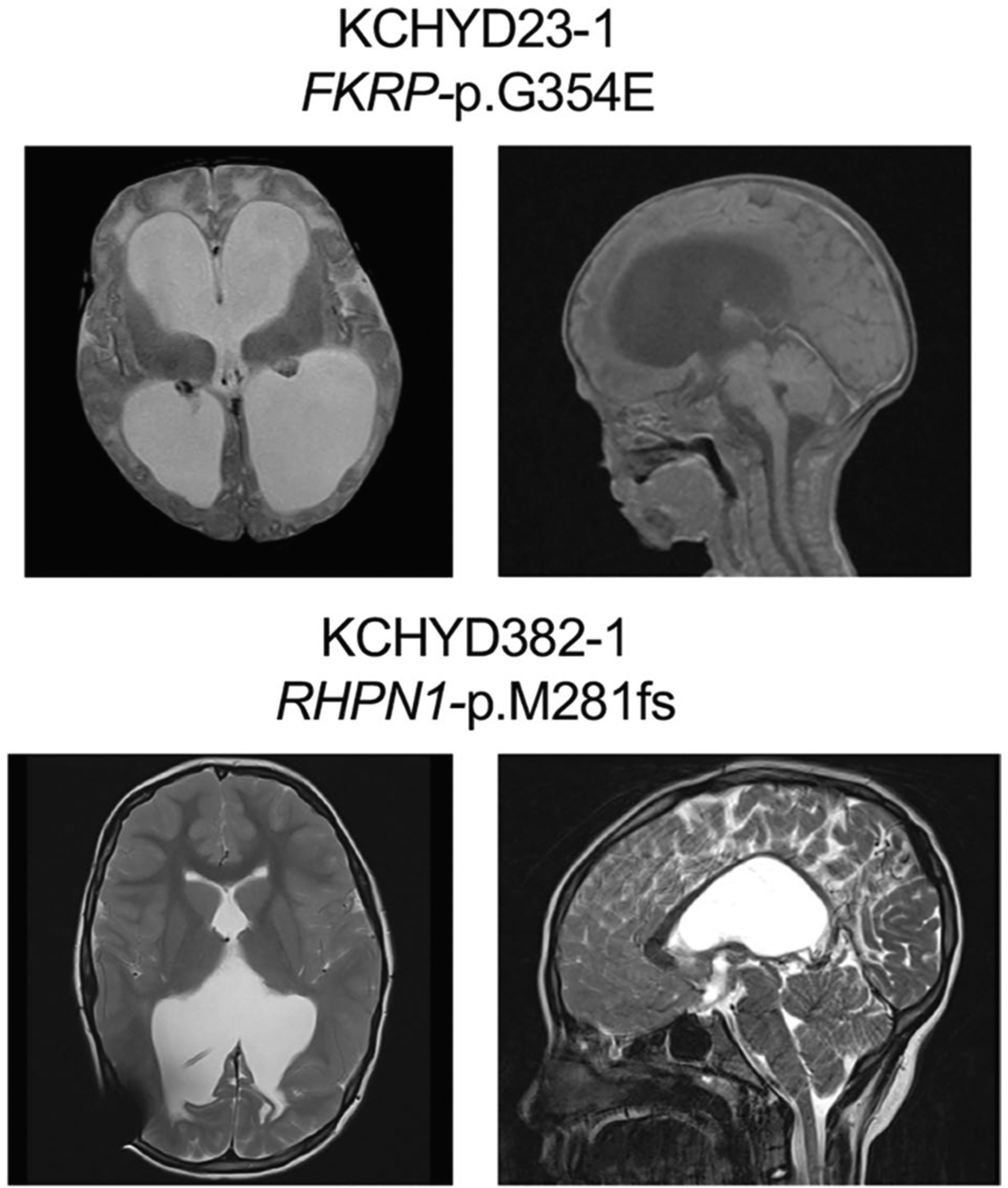
Available clinical-neuroimaging phenotypes of CH probands with damaging recessive mutations.
Supplementary Material
Acknowledgements
We are grateful to the patients and their families who participated in this research. We thank the Hydrocephalus Association (HA) for their support. We also thank J. Koschnitzky (HA), J. Rockefeller (Yale), J. Freeman (Yale) and J. Nicolleli (Yale) for their help and support. This work is supported by the Yale–National Institutes of Health (NIH) Center for Mendelian Genomics (5U54HG006504); NIH Director’s Pioneer Award DP1HD086071 and NIH Director’s Transformative Award 1R01AI145057 (S.J.S.); R01 NS111029-01A1, R01 NS109358, K12 228168 and the Rudi Schulte Research Institute (K.K.); NIH Medical Scientist Training Program (NIH/National Institute of General Medical Sciences Grant T32GM007205); NIH Clinical and Translational Science Award from the National Center for Advancing Translational Science (TL1 TR001864); James Hudson Brown – Alexander B. Coxe Fellowship at Yale School of Medicine, the American Heart Association Postdoctoral Fellowship (18POST34060008), the K99/R00 Pathway to Independence Award (K99HL143036 and R00HL143036-02) (S.C.J.); the American Heart Association Predoctoral Fellowship (19PRE34380842, W.D.); the Pediatric Hydrocephalus Foundation (P.H.F.). We thank M. C. Kruer at Phoenix Children’s Hospital and H. Zhao at Yale School of Public Health for critical discussion.
Footnotes
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41591-020-1090-2.
Competing interests
The authors declare no competing interests.
Extended data is available for this paper at https://doi.org/10.1038/s41591-020-1090-2.
Supplementary information is available for this paper at https://doi.org/10.1038/s41591-020-1090-2.
References
- 1.Albright AL, Adelson PD & Pollack IF Principles and Practice of Pediatric Neurosurgery (Thieme, 2008). [Google Scholar]
- 2.Bondurant CP & Jimenez DF Epidemiology of cerebrospinal fluid shunting. Pediatr. Neurosurg 23, 254–258 (1995). [DOI] [PubMed] [Google Scholar]
- 3.Tully HM & Dobyns WB Infantile hydrocephalus: a review of epidemiology, classification and causes. Eur. J. Med. Genet 57, 359–368 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lindquist B, Carlsson G, Persson EK & Uvebrant P Behavioural problems and autism in children with hydrocephalus: a population-based study. Eur. Child Adolesc. Psychiatry 15, 214–219 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Kahle KT, Kulkarni AV, Limbrick DD Jr. & Warf BC Hydrocephalus in children. Lancet 387, 788–799 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Chervenak FA et al. Outcome of fetal ventriculomegaly. Lancet 2, 179–181 (1984). [DOI] [PubMed] [Google Scholar]
- 7.Haverkamp F et al. Congenital hydrocephalus internus and aqueduct stenosis: aetiology and implications for genetic counselling. Eur. J. Pediatrics 158, 474–478 (1999). [DOI] [PubMed] [Google Scholar]
- 8.Kousi M & Katsanis N The genetic basis of hydrocephalus. Annu Rev. Neurosci 39, 409–435 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Furey CG et al. De novo mutation in genes regulating neural stem cell fate in human congenital hydrocephalus. Neuron 99, 302–314 e304 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duran D et al. Mutations in chromatin modifier and ephrin signaling genes in vein of Galen malformation. Neuron 101, 429–443 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Duy PQ, Furey CG & Kahle KT Trim71/lin-41 links an ancient miRNA pathway to human congenital hydrocephalus. Trends Mol. Med 25, 467–469 (2019). [DOI] [PubMed] [Google Scholar]
- 12.Welte T et al. The RNA hairpin binder TRIM71 modulates alternative splicing by repressing MBNL1. Genes Dev. 33, 1221–1235 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Narayanan R et al. Loss of BAF (mSWI/SNF) complexes causes global transcriptional and chromatin state changes in forebrain development. Cell Rep. 13, 1842–1854 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Da G et al. Structure and function of the SWIRM domain, a conserved protein module found in chromatin regulatory complexes. Proc. Natl Acad. Sci. USA 103, 2057–2062 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harmacek L et al. A unique missense allele of BAF155, a Core BAF chromatin remodeling complex protein, causes neural tube closure defects in mice. Developmental Neurobiol. 74, 483–497 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu P, Cheng H, Roberts TM & Zhao JJ Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Disco 8, 627–644 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li L, Liu F & Ross AH PTEN regulation of neural development and CNS stem cells. J. Cell Biochem 88, 24–28 (2003). [DOI] [PubMed] [Google Scholar]
- 18.Chalhoub N & Baker SJ PTEN and the PI3-kinase pathway in cancer. Annu Rev. Pathol 4, 127–150 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Keppler-Noreuil KM, Parker VE, Darling TN & Martinez-Agosto JA Somatic overgrowth disorders of the PI3K/AKT/mTOR pathway & therapeutic strategies. Am. J. Med. Genet. C. Semin. Med. Genet 172, 402–421 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Riviere JB et al. De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nat. Genet 44, 934–940 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oda K et al. PIK3CA cooperates with other phosphatidylinositol 3′-kinase pathway mutations to effect oncogenic transformation. Cancer Res. 68, 8127–8136 (2008). [DOI] [PubMed] [Google Scholar]
- 22.Dogruluk T et al. Identification of variant-specific functions of PIK3CA by rapid phenotyping of rare mutations. Cancer Res. 75, 5341–5354 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Foerster P et al. mTORC1 signaling and primary cilia are required for brain ventricle morphogenesis. Development 144, 201–210 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martinez-Glez V et al. Macrocephaly-capillary malformation: analysis of 13 patients and review of the diagnostic criteria. Am. J. Med. Genet. A 152A, 3101–3106 (2010). [DOI] [PubMed] [Google Scholar]
- 25.O’Rourke DJ, Twomey E, Lynch SA & King MD Cortical dysplasia associated with the PTEN mutation in Bannayan–Riley–Ruvalcaba syndrome: a rare finding. Clin. Dysmorphol 21, 91–92 (2012). [DOI] [PubMed] [Google Scholar]
- 26.Chen HH et al. Immune dysregulation in patients with PTEN hamartoma tumor syndrome: analysis of FOXP3 regulatory T cells. J. Allergy Clin. Immunol 139, 607–620 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sarquis MS et al. Distinct expression profiles for PTEN transcript and its splice variants in Cowden syndrome and Bannayan–Riley–Ruvalcaba syndrome. Am. J. Hum. Genet 79, 23–30 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Groszer M et al. Negative regulation of neural stem/progenitor cell proliferation by the Pten tumor suppressor gene in vivo. Science 294, 2186–2189 (2001). [DOI] [PubMed] [Google Scholar]
- 29.Pilarski R & Eng C Will the real Cowden syndrome please stand up (again)? Expanding mutational and clinical spectra of the PTEN hamartoma tumour syndrome. J. Med. Genet 41, 323–326 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mirzaa GM et al. Association of MTOR mutations with developmental brain disorders, including megalencephaly, focal cortical dysplasia, and pigmentary mosaicism. JAMA Neurol. 73, 836–845 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baynam G et al. A germline MTOR mutation in aboriginal Australian siblings with intellectual disability, dysmorphism, macrocephaly, and small thoraces. Am. J. Med. Genet. A 167, 1659–1667 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Jacquet BV et al. FoxJ1-dependent gene expression is required for differentiation of radial glia into ependymal cells and a subset of astrocytes in the postnatal brain. Development 136, 4021–4031 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Divina P, Kvitkovicova A, Buratti E & Vorechovsky I Ab initio prediction of mutation-induced cryptic splice-site activation and exon skipping. Eur. J. Hum. Genet 17, 759–765 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schonichen A & Geyer M Fifteen formins for an actin filament: a molecular view on the regulation of human formins. Biochim. Biophys. Acta 1803, 152–163 (2010). [DOI] [PubMed] [Google Scholar]
- 35.Lian G, Chenn A, Ekuta V, Kanaujia S & Sheen V Formin 2 regulates lysosomal degradation of wnt-associated β-catenin in neural progenitors. Cereb. Cortex 29, 1938–1952 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lian G et al. Filamin A- and formin 2-dependent endocytosis regulates proliferation via the canonical Wnt pathway. Development 143, 4509–4520 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gavino C & Richard S Patched1 haploinsufficiency impairs ependymal cilia function of the quaking viable mice, leading to fatal hydrocephalus. Mol. Cell. Neurosci 47, 100–107 (2011). [DOI] [PubMed] [Google Scholar]
- 38.Palma V et al. Sonic hedgehog controls stem cell behavior in the postnatal and adult brain. Dev. (Camb., Engl.) 132, 335–344 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Palma V & Ruiz i Altaba A Hedgehog-GLI signaling regulates the behavior of cells with stem cell properties in the developing neocortex. Development 131, 337–345 (2004). [DOI] [PubMed] [Google Scholar]
- 40.Bult CJ et al. Mouse genome database (MGD) 2019. Nucleic Acids Res. 47, D801–D806 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hehr U et al. Novel POMGnT1 mutations define broader phenotypic spectrum of muscle-eye-brain disease. Neurogenetics 8, 279–288 (2007). [DOI] [PubMed] [Google Scholar]
- 42.Manzini MC et al. Exome sequencing and functional validation in zebrafish identify GTDC2 mutations as a cause of Walker-Warburg syndrome. Am. J. Hum. Genet 91, 541–547 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Walker RL et al. Genetic control of expression and splicing in developing human brain informs disease mechanisms. Cell 179, 750–771 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Polioudakis D et al. A single-cell transcriptomic atlas of human neocortical development during mid-gestation. Neuron 103, 785–801 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kurata H et al. Neurodevelopmental disorders in children with macrocephaly: a prevalence study and PTEN gene analysis. Brain Dev. 40, 36–41 (2018). [DOI] [PubMed] [Google Scholar]
- 46.Palmen SJ et al. Increased gray-matter volume in medication-naive high-functioning children with autism spectrum disorder. Psychol. Med 35, 561–570 (2005). [DOI] [PubMed] [Google Scholar]
- 47.Gilmore JH et al. Outcome in children with fetal mild ventriculomegaly: a case series. Schizophrenia Res. 48, 219–226 (2001). [DOI] [PubMed] [Google Scholar]
- 48.Jin SC et al. Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat. Genet 49, 1593–1601 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Iossifov I et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wallmeier J et al. De novo mutations in FOXJ1 result in a motile ciliopathy with hydrocephalus and randomization of left/right body asymmetry. Am. J. Hum. Genet 105, 1030–1039 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guerra MM et al. Cell junction pathology of neural stem cells is associated with ventricular zone disruption, hydrocephalus, and abnormal neurogenesis. J. Neuropathol. Exp. Neurol 74, 653–671 (2015). [DOI] [PubMed] [Google Scholar]
- 52.Wagner C et al. Cellular mechanisms involved in the stenosis and obliteration of the cerebral aqueduct of hyh mutant mice developing congenital hydrocephalus. J. Neuropathol. Exp. Neurol 62, 1019–1040 (2003). [DOI] [PubMed] [Google Scholar]
- 53.Zega K et al. Dusp16 deficiency causes congenital obstructive hydrocephalus and brain overgrowth by expansion of the neural progenitor pool. Front Mol. Neurosci 10, 372 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Henzi R et al. Neural stem cell therapy of foetal onset hydrocephalus using the HTx rat as experimental model. Cell Tissue Res. 381, 141–161 (2020). [DOI] [PubMed] [Google Scholar]
- 55.McAllister JP et al. Ventricular zone disruption in human neonates with intraventricular hemorrhage. J. Neuropathol. Exp. Neurol 76, 358–375 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Movsas TZ et al. Autism spectrum disorder is associated with ventricular enlargement in a low birth weight population. J. Pediatrics 163, 73–78 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li M et al. Integrative functional genomic analysis of human brain development and neuropsychiatric risks. Science 362, eaat7615 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Etchegaray A, Juarez-Penalva S, Petracchi F & Igarzabal L Prenatal genetic considerations in congenital ventriculomegaly and hydrocephalus. Childs Nerv. Syst 36, 1645–1660 (2020). [DOI] [PubMed] [Google Scholar]
- 59.Raudvere U et al. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 47, W191–W198 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Krumm N et al. Excess of rare, inherited truncating mutations in autism. Nat. Genet 47, 582–588 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McKenna A et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Van der Auwera GA et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinforma 43, 11–33 (2013). 11 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Garrison EMG Haplotype-based variant detection from short-read sequencing. Preprint at arXivhttps://arxiv.org/abs/1207.3907 (2012).
- 64.Wang K, Li M & Hakonarson H ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Karczewski KJ et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Taliun D et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Preprint at bioRxiv 10.1101/563866 (2019). [DOI] [PMC free article] [PubMed]
- 67.Samocha K et al. Regional missense constraint improves variant deleteriousness prediction. Preprint at bioRxiv 10.1101/148353 (2017). [DOI]
- 68.Dong C et al. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum. Mol. Genet 24, 2125–2137 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wei Q et al. A Bayesian framework for de novo mutation calling in parents-offspring trios. Bioinformatics 31, 1375–1381 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Quinlan AR & Hall IM BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Samocha KE et al. A framework for the interpretation of de novo mutation in human disease. Nat. Genet 46, 944–950 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ware JS, Samocha KE, Homsy J & Daly MJ Interpreting de novo variation in human disease using denovolyzeR. Curr. Protoc. Hum. Genet 87, 21–15 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang M, Marco P, Capra V & Kibar Z Update on the role of the non-canonical wnt/planar cell polarity pathway in neural tube defects. Cells 8, 1198 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tissir F & Goffinet AM Shaping the nervous system: role of the core planar cell polarity genes. Nat. Rev. Neurosci 14, 525–535 (2013). [DOI] [PubMed] [Google Scholar]
- 75.Shaheen R et al. The genetic landscape of familial congenital hydrocephalus. Ann. Neurol 81, 890–897 (2017). [DOI] [PubMed] [Google Scholar]
- 76.Ruzzo EK et al. Inherited and de novo genetic risk for autism impacts shared networks. Cell 178, 850–866 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Satterstrom FK et al. Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell 180, 568–584 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Deciphering Developmental Disorders Study. Prevalence and architecture of de novo mutations in developmental disorders. Nature 542, 433–438 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Langfelder P & Horvath S WGCNA: an R package for weighted correlation network analysis. BMC Bioinforma. 9, 559 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li J et al. Application of weighted gene co-expression network analysis for data from paired design. Sci. Rep 8, 622 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequencing data for all CH parent–offspring trios and singletons reported in this study have been deposited in the NCBI Database of Genotypes and Phenotypes under accession number phs000744.v4.p2. Our in-house R and Python pipelines and codes are available upon request.