

Abstract
Sensors that detect double stranded RNA stimulate IFN-responses as a defense against viral infection. IFN-responses are also well documented in a variety of human autoimmune diseases including relapsing remitting multiple sclerosis in which increased IFN-responses result from increased levels of double-stranded endogenous Alu RNAs. Mechanisms underlying increases in double-stranded Alu RNAs in multiple sclerosis are obscure. We find widespread loss of A-to-I editing of Alu RNAs in multiple sclerosis. Unedited Alu RNAs are potent activators of both IFN- and NF-κB responses via the dsRNA sensors, RIG-I and TLR3. Minor editing of highly active Alu elements abrogates ability to activate both transcriptional responses. Thus, A-to-I editing may also represent an important defense against autoimmune diseases, such as multiple sclerosis.
Introduction
A major host defense by multicellular organisms against viral infection is expression of the interferon (IFN) class of cytokines (1-3). IFNs are sub-divided into type 1 IFNs, IFN-α or leukocyte IFN, and IFN-β or fibroblast IFN, type 2 IFN, IFN-γ or immune interferon, and type 3 IFN, IFN-λ (also named IL-29, IL28A and IL28B, respectively). IFN-α, IFN-β and IFN-γ activate transcription factors, termed interferon regulatory factors, IRFs, that bind to conserved transcriptional enhancer elements, termed interferon-stimulated response elements, ISREs, and induce similar but not entirely overlapping gene sets, termed IFN-stimulated genes (ISGs) with antiviral properties (4-6). IFN-γ also plays a critical role orchestrating both innate and adaptive arms of the immune system (7). Induction of type 1 IFNs by viruses is mediated via pattern recognition receptors that recognize viral components, such as cytosolic double-stranded RNA (dsRNA) (8). These include TLR3 and the DExD/H-box helicases, RIG-I and MDA5 (9-12). A general view is that RIG-I recognizes short (< 400 nucleotides) 5’ triphosphate uncapped dsRNA or ssRNA while MDA5 and TLR3 recognize dsRNAs of greater nucleotide (nt) length (10, 13, 14). Activation of these sensors induces IFN-α or IFN-β, and in turn, ISGs, as well as activation of the pro-inflammatory transcription factor, NF-κB.
Increased expression of ISGs in the absence of apparent viral infection is also a hallmark of many human autoimmune diseases, most notably systemic lupus erythematosus, but also relapsing remitting multiple sclerosis (MS), rheumatoid arthritis, Sjogren’s syndrome, systemic sclerosis, myositis and others (15-22). Sources of stimuli that lead to induction of ISGs in these human autoimmune diseases are incompletely understood. Aicardi-Goutieres syndrome is another example of a human disease associated with increased expression of type 1 IFNs and ISGs and results in severe early onset progressive encephalopathy (23, 24). Of note, Aicardi-Goutieres syndrome is a rare genetic disorder that can result from activating mutations in IFIH1, the gene that encodes the cytosolic dsRNA sensor, MDA5. One activating mutation in IFIH1 found in Aicardi-Goutieres syndrome causes MDA5 loss of tolerance to endogenous cytosolic dsRNA structures and MDA5-mediated induction of IFNs and ISGs (13). A second class of mutations found in Aicardi-Goutieres syndrome is an inactivating mutation in ADAR1, the gene that encodes the enzyme double stranded RNA specific adenosine deaminase, ADAR (24). ADAR deaminates adenosines in RNA resulting in conversion of adenosine to inosine, termed A-to-I editing, a process thought to destabilize dsRNA structures (25-28). In the case of the ADAR1 mutation in Aicardi-Goutieres syndrome, loss of ADAR function also results in increased expression of IFNs and ISGs. The general view is that loss of ADAR1 results in accumulation of endogenous cytoplasmic dsRNAs and activation of dsRNA sensors, such as RIG-I, MDA5 or TLR3 (29-36). Thus, it is possible that alterations in the sensing of endogenous dsRNAs or in A-to-I editing of endogenous dsRNAs may contribute to increased induction of ISGs observed in these more common autoimmune diseases.
The primary sources of dsRNA structures that exist in human leukocytes are RNAs produced by the retrotransposon class of genetic elements, approximately 80% of these dsRNAs are derived from Alu elements, which are short interspersed elements (SINE) of ~300 nt in length unique to primates and approximately 10% of these dsRNAs are long interspersed elements, LINE (37), of up to about 6,000 nt in length (29, 35, 38-40). Approximately 1,000,000 Alu elements and 1,000,000 LINE elements exist in human genomes and make up about 30% of the human genome. Alu elements can be transcribed by RNA polymerase III as part of their normal life cycle. Alu elements are also embedded within protein-coding genes, predominantly within introns and 3’ untranslated regions (3’UTR), and are also transcribed by RNA polymerase II as pre-mRNAs. Importantly, studies show that most A-to-I editing sites are found within these Alu RNAs and given their repetitive nature can form dsRNA structures. As such, Alu dsRNAs can be recognized by endogenous dsRNA sensors resulting in stimulation of downstream IFN- and NF-κB-responses. ADAR-mediated A-to-I editing of these endogenous Alu dsRNAs may decrease their dsRNA character resulting in loss of their ability to be recognized by dsRNA sensors (12, 30, 31). Thus, A-to-I editing of these highly prevalent Alu and Line RNAs likely represents a critical cellular function of ADAR.
In leukocytes from patients with MS, induction of ISGs is mediated, at least in part, by increases in the levels of endogenous Alu dsRNAs (41). As such, increases in levels of Alu dsRNAs in leukocytes from patients with MS may result from defective A-to-I editing of Alu RNAs. To test this hypothesis, we examined levels of A-to-I editing using whole genome RNA-sequencing data. Here, we show that marked defects in A-to-I editing exist in leukocytes from MS patients compared to healthy controls (HC). We also find that native Alu RNAs highly expressed in leukocytes from patients with MS are recognized by RIG-I and TLR3 dsRNA sensors and induce potent IFN- and NF-κB responses. Minor editing (A-to-G nucleotide changes) of these native Alu elements results in marked loss of this activity. Thus, defects in A-to-I editing of Alu RNAs may mediate the observed increased IFN- and NF-κB responses in this disease. It seems possible that similar defects in A-to-I editing of Alu RNAs may also contribute to increased IFN- and NF-κB responses and pathogenesis associated with other autoimmune diseases and syndromes.
Materials and Methods
Whole genome RNA-sequencing and genome-wide analysis of A-to-I editing
Whole genome RNA-sequencing files employed here are deposited in the NCBI Gene Expression Omnibus (42). Accession numbers are GSE92472 and GSE126427 (https://www.ncbi.nlm.nih.gov/geo). We employed SPRINT, a toolkit that identifies RNA editing sites without the need for filtering single nucleotide polymorphisms using whole genome RNA sequencing data (43). We used the following workflow to identify RNA editing sites from paired FASTQ sequencing files. The main identification tool is a python-based package called the SPRINT toolkit for identifying RNA editing sites. The multithread toolkit accepts sequence files and produces a text file that contains information about each editing site in the sequence data. This information includes the following for each edit site: (i) genomic location; (ii) type of edit (e.g., A-to-G or T-to-C); strand (“+” or “-“); (iii) number of edits reads at site); and (iv) total number of reads at site. This analysis is performed on each sample of the data groups (healthy control, HC, MS patients prior to therapeutic intervention, MS-N, and MS patients with established disease on therapy, MS-E, Table 1 (42). Mathematica (Wolfram Research, Inc.) programs were developed to synthesis the data for each group. Mainly, we were interested in the number of samples in groups that share editing sites, mean numbers of total reads and edits for each editing site across the group, and the editing sites that are common and distinct to group pairs (e.g., HC versus MS-E). Lastly, the editing site information was tied to an Alu database which annotated each site with respect to nearby genes, type of location (intronic, ncRNA, intergenic, 3UTR), if the site was indeed an Alu or if the site was a non-Alu repetitive site.
TABLE I.
Subject demographics.
| # | agea | % Fb | Therapyc | CD14d | CD4 | CD8A | CD19 | CD56 | FCGR3A | |
|---|---|---|---|---|---|---|---|---|---|---|
| MS-N | 8 | 34 ±3 | 83 | − | 1.26 | 1.04 | 0.88 | 1.16 | 0.89 | 1.13 |
| MS-E | 8 | 39 ±3 | 67 | + | 1.08 | 0.84 | 0.86 | 0.80 | 1.09 | 1.05 |
| HC | 8 | 38 ± 11 | 75 | − | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
Average age ± standard deviation, P > 0.05 compared to HC.
P > 0.05 compared to HC, all subjects were Caucasian.
Present therapy for multiple sclerosis
From RNA-seq data, ratios of expression of the different cell-type specific cell surface markers in each disease cohort compared to HC, P > 0.05 compared to HC; CD14, monocytes; CD4, helper T cells; CD8A, cytotoxic T cells; CD19, B cells; CD56, NK cells; FCGR3A (CD16) neutrophils.
Determination of A-to-I editing enriched locations, EELs, and EEL indices
We employed the following scheme to classify EELs. First, genomic locations of A-to-I editing sites present in ≥ 2 HC samples or ≥ 2 MS-E samples were pooled into a single list. Second, distances between neighboring A-to-I editing sites in the genome were calculated. Third, EEL boundaries were formed if the next neighboring common A-to-I editing site in either 5’ or 3’ directions in the genome was > 5,000 bp in distance. Fourth, we employed a rather arbitrary cutoff of presence of ≥5 A-to-I editing sites within EEL boundaries for inclusion in the analysis as this cutoff still captured the vast majority of common A-to-I editing sites across the genome. For example, the length of the CSAD gene EEL is 4,576 bp and contains 51 common A-to-I editing sites for an average distance between editing sites of 90 bp (range of 1-1,676 bp). The next nearest A-to-I editing site in the 5’ direction is 1,874,544 bp away and the next nearest A-to-I editing site in the 3’ direction is 118,150 bp away, thus establishing the EEL boundary.
To create an EEL editing index, we identified all A-to-I editing sites within an EEL and separated them into those present in ≥2 HC samples or ≥2 MS-E samples. We summed the A-to-I edit/total read ratios for all sites for the HC class or the MS-E class and multiplied this value by the total number of HC or MS-E samples with these edits. These calculations formed an EEL editing index to help gauge if individual EELS were preferentially edited in the HC class, the MS-E class or exhibited similar EEL editing levels between classes.
Production of Alu RNAs from dsDNA templates
Reference DNA sequences of Alu and Line elements were obtained from the UCSC genome browser and a SP6 bacteriophage promoter was added to the 5’ end of the sequence and synthetic dsDNAs were obtained from Integrated DNA Technologies (44). RNA transcription was performed using the Megascript SP6 kit (Invitrogen, cat. no. AM1330) per manufacturer’s instructions. Briefly, 8μl of DNA was placed in a 1.5 ml microfuge tube and heated for 5 minutes at 65 °C and cooled. ATP, CTP, GTP, UTP, 10X reaction buffer and SP6 RNA polymerase were added and the reaction was allowed to incubate overnight at 37°C. Turbo DNAse was added and incubated for 30 minutes and the reaction was stopped with the addition of 30μl of dH20 and 30μl of lithium chloride precipitation solution and incubated for 30 minutes at −20 °C, followed by centrifugation at 4 °C at 20,000xg for 15 minutes. The supernatant was removed and pellet washed with 70% ethanol. The centrifugation step was repeated under the same conditions. The 70% ethanol was removed and the pellet briefly air-dried prior to suspension in 50μl nuclease free water. Absorbance was determined at 260 nm to quantitate yields of RNA and solutions stored at −80 °C. Yields typically were 10-70μg RNA
Cell culture
THP-1 and HEK-293 reporter cell lines were obtained from Invivogen. THP-1 reporter cells contain a stably integrated luciferase gene under the control of either an interferon-stimulated response element (ISRE) or a NF-κB response element. HEK-293 reporter cells contain a stably integrated luciferase gene under the control of the ISRE element. A second line of HEK-293 reporter cells, HEK-RIG-I also contained a stably integrated DDX58 gene encoding RIG-I. Cells were cultured in RPMI 1640 media (Gibco, cat. no. 21870) supplemented with 10% (vol/vol) fetal calf serum (Atlanta Biologicals cat. no. S12450), 100 U/ml penicillin, 100 μg/ml streptomycin and 2mM L-Glutamine (final concentration, Gibco, cat. no. 15140). Cells were cultured at 37°C in 5% CO2 in humidified air. TLR3, IFIH1 (MDA5), and DDX58 (RIG-I) expression plasmids were obtained from Invivogen.
Cell transfections
Cell transfections were performed using Lipofectamine ® RNAiMAX transfection reagent (ThermoFisher Scientific, cat. no. 13778150) according to manufacturer’s instructions (45). All dilutions were performed in Opti-MEM medium (ThermoFisher Scientific, cat. no. 31985070). Briefly, on day 0, cells were plated at 100,000 cell/ml and in 96 well plates at volumes of 100μl. On day 1, 1.5 μl of RNAiMAX was diluted into 25μl of Opti-MEM. This was scaled depending upon the number of samples, dilutions and culture volumes. RNAs were diluted in Opti-MEM at varying concentrations before addition to cell cultures. Culture after transfection was typically 24 hr. Levels of secreted luciferase in culture fluids were determined using a luciferin substrate (Invivogen) and light emission determined using a TD20/20 luminometer.
Results
Defective A-to-I editing in MS
We examined our whole-genome RNA-sequencing data using the Sprint software package to identify differences in A-to-I editing in leukocytes between HC and patients with MS. For these initial analyses, we used two criteria to identify A-to-I editing sites. First, we required the proportion of A-to-I edits to exceed 5% of total reads at individual nucleotide sites to guard against possible sequencing errors and total read count >10. Second, we required that individual candidate A-to-I editing sites were edited in ≥25% of the HC cohort, ≥25% of the MS-E cohort, or ≥25% of both HC and MS-E cohorts. Overall, >90% of editing events we identified occurred within Alu elements and these were mainly distributed among introns, 3’ untranslated regions of poly (A)+ protein-coding genes and intergenic space (Fig. 1A). Of note, using the above criteria, we identified three distinct patterns of differential A-to-I editing of Alu elements. One pattern showed extensive editing of individual Alu elements in the HC group with the absence of editing in the MS group, two examples include the AluSx1 element in the MAVS gene and the AluSz element in the TPM4 gene (Fig. 1B). The converse pattern also existed showing editing of individual Alu elements in the MS group but not the HC group, examples include the AluSx1 element in the PLEKHG2 gene and the AluSz6 element in the COTL1 gene (Fig. 1C). The third pattern we identified showed extensive editing at identical adenosines within individual Alu elements in both HC and MS-E groups, examples include the AluSq element in the NUP43 gene and the AluSp element in the SLC12A6 gene (Fig. 1D).
FIGURE 1.

A-to-I editing profiles in MS. (A) Total number of unique A-to-I editing sites determined using the following criteria: present in at least 2 HC or 2 MS-E samples, >5% edit/read ratios and >10 total reads. All unique A-to-I editing sites were present in Alu elements. Number of A-to-I editing sites found in introns, exons, 5’ UTRs, and 3’UTRs are also shown. (B) Alu elements within MAVS and TPM4 genes with editing in HC but not MS-E. (C) Alu elements within PLEKHG2 and COTL1 genes with editing in MS-E but not HC. (D) Alu elements within NUP43 and SLC12A6 genes with editing in both HC and MS-E. Y-axis is the average proportion of edits to total reads in HC, N=8, (open black circles) or MS-E, N=8, (open green circles); note that A-to-I edit/read ratios <0.05 were excluded to guard against sequencing errors so Y-axis lower limit is set at 0.05. Standard deviations of the average editing proportions were <10%, error bars not shown.
Next, we examined A-to-I editing sites genome-wide. Using the above criteria, we found 2,724 nucleotide sites that were A-to-I edited in genomes of the HC cohort but were not A-to-I edited in genomes from the MS-E cohort. In sharp contrast, we identified only 665 nucleotide sites that were A-to-I edited in genomes from the MS cohort and not edited in genomes from the HC cohort (P<0.0001, χ2 analysis) (Fig. 2A). We also found 2243 nucleotide sites that were A-to-I edited in both HC and MS cohorts (Fig 2B). Average proportion of edits to total reads of these shared A-to-I editing sites was similar, between 35-40%, and the proportion of edits to reads ranged from 5% to 100%. We next identified those A-to-I edits present in 100% of HC or 100% of MS samples or 100% of both. We identified 280 editing sites present in 100% of HC samples and absent from MS samples and only 64 editing sites present in 100% of MS samples and absent from HC samples (P<0.0001, χ2 analysis) (Fig. 2C). As above, we identified 174 nucleotide sites edited in all HC and all MS samples (Fig. 2D). Average proportion of edits to total reads of these A-to-I editing sites was also similar, between 35-40%, and the proportion of edits to reads ranged from 5% to 100%.
FIGURE 2.

Loss of genome-wide A-to-I editing in MS. (A) Unique A-to-I editing sites present in ≥ 2 HC samples and 0 MS-E samples ranked according to edit/read ratio (Y-axis, black open circles) or present in ≥ 2 MS-E samples and 0 HC samples (open green circles). (B) Number of unique sites that are A-to-I edited in both ≥ 2 HC and ≥ 2 MS-E samples ranked according to HC edit/read ratios (open black circles; corresponding MS-E edit/read ratios at the identical nucleotide sites are also shown (open green circles) (C) Number of unique A-to-I editing sites present in all 8 HC samples and 0 MS-E samples ranked according to edit/read ratios (Y-axis, black open circles) or present in all 8 MS-E samples and 0 HC samples (open green circles). (D) Number of unique sites that are A-to-I edited in all 8 HC and all 8 MS-E samples ranked according to HC edit/read ratios (open black circles; corresponding MS-E edit/read ratios at the identical nucleotide sites are also shown (open green circles). (E) Unique A-to-I editing sites present in all 8 HC samples and 0 MS-N samples ranked according to edits/read ratios (Y-axis, black open circles) or present in all 8 MS-N samples and 0 HC samples (open magenta circles). (F) Number of unique sites that are A-to-I edited in all 8 HC and all 8 MS-N samples ranked according to HC edit/read ratios (open black circles); corresponding MS-E edit/read ratios at the identical nucleotide sites are also shown (open magenta circles).
We next examined A-to-I editing in a second group of MS patients using RNA-seq data. Blood samples from these patients were obtained at initial diagnosis of MS but before onset of therapies so were treatment naïve (MS-N). We identified A-to-I edits present in 100% of HC or 100% of MS-N samples or 100% of both. We identified 271 A-to-I edits present in 100% of HC samples and absent from MS-N samples but only 96 editing sites present in MS-N samples and absent from HC samples (P<0.0001, χ2 analysis) (Fig. 2E). We also identified 220 sites that were edited in all HC and all MS samples (Fig. 2F). Average proportion of edits to total reads of these editing sites was similar, between 35-40%, and the proportion of edits to reads ranged from 5% to 100%. Thus, we identified A-to-I editing sites that were shared between both MS-E patient samples and HC as well as A-to-I editing sites that were unique to either the MS patient groups or the HC group. At these editing sites, the average proportion of edited reads to total reads was similar across all samples. The major differences we found were in edits that were unique to either the HC cohort, which were high in number, or the MS-E and MS-N cohorts, which were much lower in number. Thus, leukocytes from MS patients showed an overall deficit in A-to-I editing.
A-to-I editing enriched locations (EELs) in the genome
We also enumerated numbers of A-to-I editing sites within protein-coding genes counting those in exons, introns, 5’ untranslated regions (5’UTRs) and 3’ UTRs by including editing sites present in either ≥2 HC samples, ≥2 MS-E samples, or those shared between ≥2 HC samples and ≥2 MS-E samples. For this comparison, we focused on genes with the greatest number of A-to-I editing sites. Two classes of editing patterns emerged. We identified genes with approximately equivalent numbers of total A-to-I editing sites across the gene in both HC and MS-E classes (Fig. 3A) and these differences were not statistically significant. We determined expression levels of these genes from RNA-seq data and expression levels of these genes were not significantly different between HC and MS-E classes (Fig. 3B). We also identified genes with relatively greater numbers of editing sites in the HC class compared to the MS-E class (Fig. 3C), which is similar to what we observed genome-wide (Figure 2 B, C). We determined expression levels of these genes from the RNA-seq analysis of these same samples. We found that those genes with reduced numbers of A-to-I editing sites in the MS-E class compared to the HC class also demonstrated overall reduced levels of mRNA expression in the MS-E class compared to the HC class (Fig. 3D). Thus, genes with reduced levels of edits in MS-E compared to HC also exhibit reduced levels of expression compared to HC while genes with similar numbers of edits between HC and MS-E classes exhibited similar levels of expression between the two classes. These results do not prove causality but suggest that further experimentation is warranted to better understand the relationship between A-to-I editing within protein-coding genes and mRNA expression levels of these genes.
FIGURE 3.
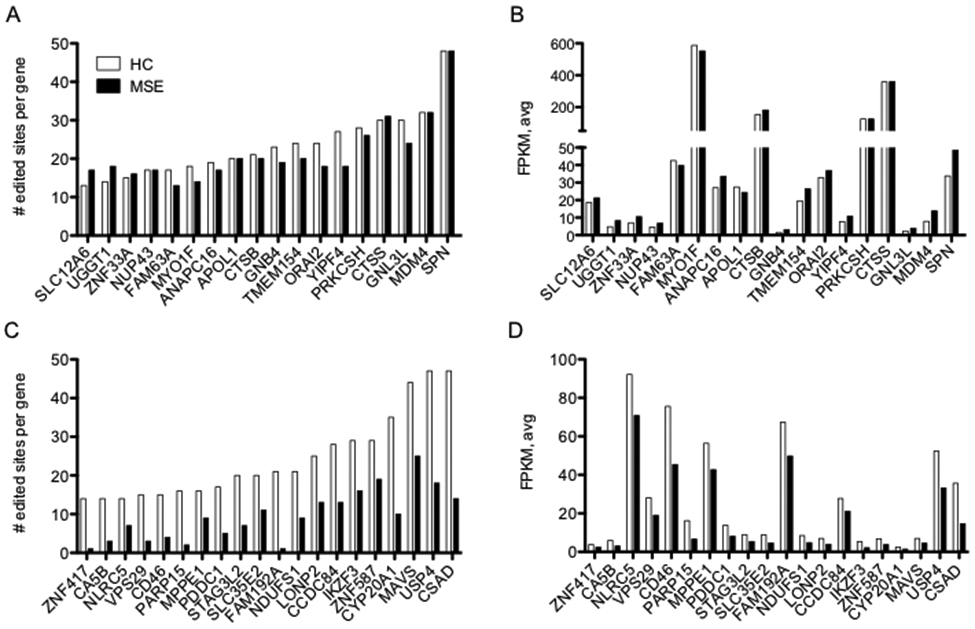
Correlation between levels of A-to-I editing and gene expression between HC and MS-E. (A) Total numbers of A-to-I editing sites present in individual genes in HC or MS-E cohorts. A-to-I editing sites were counted if edited sites were present in ≥ 2 of 8 HC or ≥ 2 of 8 MS-E samples with edit/read ratios > 0.05 and expression levels > 10 read counts, P>0.05, 2-way ANOVA. (B) Average expression levels (FPKM) of genes identified in (A) determined by RNA-seq, P>0.05, 2-way ANOVA. (C) As in (A), except genes with reduced levels of A-to-I editing sites in MS-E compared to HC are shown, P < 0.0001, 2-way ANOVA. (D) As in B except expression levels of genes in (C) are shown, P<0.0001, 2-way ANOVA.
We selected two of these genes for closer inspection, TMEM154, and CSAD, and compared locations of A-to-I editing sites to gene organization and underlying Alu elements (Fig. 4 A-B). We used the same criteria as above and only included editing sites present in ≥2 HC samples, ≥2 MS-E samples or shared between ≥2 HC and ≥2 MS-E samples. The inset to the left of each gene shows the overall average edit/read ratios at each editing site for the HC and MS-E samples. To the right of the inset is the genomic location of TMEM154 and CSAD genes along with positions of UTRs, exons, and introns. Below that are the positions of all the Alu elements across each gene. The green bars indicate the approximate locations of all A-to-I editing sites within each gene. We found that A-to-I editing sites were not evenly distributed across the gene but were confined to relatively small locations or editing enriched locations (EELs). Thus, the TMEM154 EEL is <1,000 bp while the TMEM154 gene is ~65,000 bp. Similarly, each gene contained multiple Alu elements and many of these were transcribed as RNA (not shown), but only a small number of Alu elements within the EEL were A-to-I edited. Patterns of editing in the different EELs were mixed as the CSAD EEL was predominately edited in HC samples but not MS-E samples while the TMEM154 EELs was edited similarly in both HC and MS-E samples. Thus, only very small fractions of potential editing sites within Alu elements are actually recognized as dsRNA and A-to-I edited by ADAR.
FIGURE 4.

Examples of gene A-to-I editing enriched locations, EELs. (A) The inset shows genomic locations of all A-to-I editing sites found in the TMEM154 gene (X-axis) defined by presence in ≥ 2 HC or ≥ 2 MS-E samples with proportion of edits/reads >0.05 and total read density > 10. Y-axis is the average edit/read ratio. To the right, approximate location of the EEL is labeled with a green bar. Genomic locations, exon-intron structure of TMEM154 and Alu content (SINE) are shown in the tracks. (B) As in (A) except the CSAD gene with genomic EEL location is shown.
To explore this idea further, we pooled all editing sites present in ≥2 HC samples, ≥2 MS-E samples, or shared between ≥2 HC samples and ≥2 MS-E samples into a single list of common A-to-I editing sites. We determined the genomic distance between one editing site and its nearest neighbor in the genome. We found that >60% of these common editing sites were within 100 bp of its nearest neighbor and almost 80% were within 1,000 bp of its nearest neighbor (Fig. 5A). Next, we defined EEL length as described in Methods. Using these criteria, we found that the length of EELs ranged from 5-~4,500 bp with a mean length of ~ 800 bp (Fig. 5B). Numbers of common A-to-I editing sites within an EEL ranged from 6-~65 sites with a mean number of editing sites per EEL of ~15 (Fig. 5C). Thus, as above, we found that the majority of genome-wide common editing sites are not evenly distributed but are clustered within discrete EELs.
FIGURE 5.
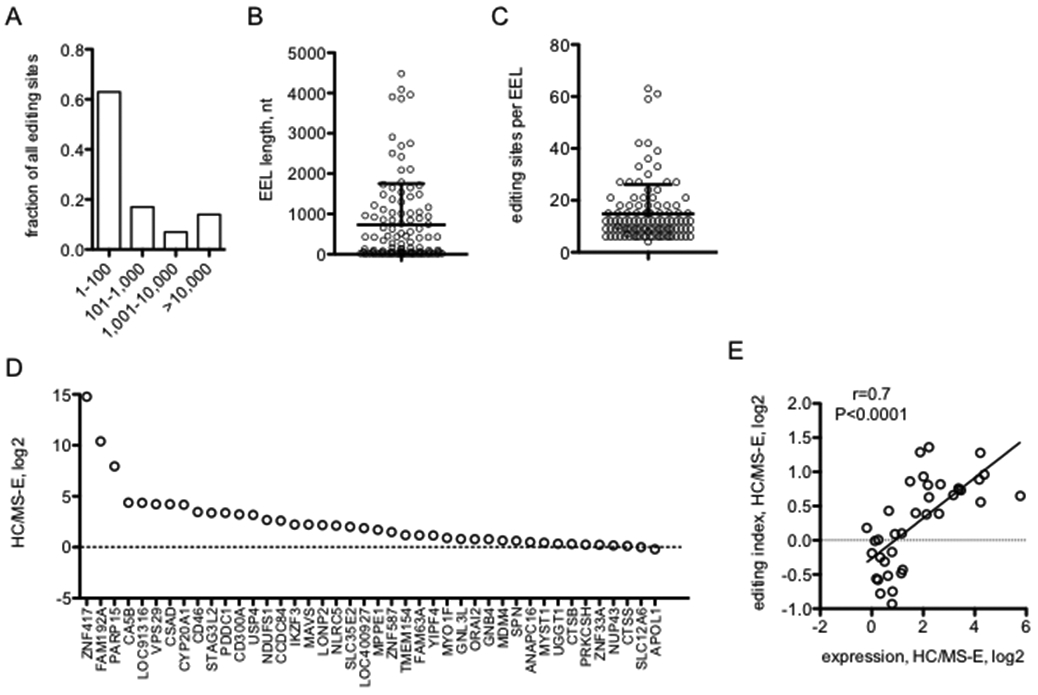
Genome-wide A-to-I editing enriched locations, EELs. (A) Genomic distances between common A-to-I editing sites. Fraction of common A-to-I editing sites (present in ≥ 2 HC or ≥ 2 MS-E samples) within the indicated genomic distance in nucleotides to the nearest neighboring common A-to-I editing site. (B) Average nucleotide length +/− SD of all EELs in the genome. (C) Average +/− SD number of common A-to-I editing sites within EELs. (D) EEL editing index. Y-axis is the HC editing index/MS-E editing index, log2, for individual EELs within the indicated genes, X-axis. (E) Correlation between A-to-I editing indices from (D) and gene expression (shown in Figure 3 A,C).
Using the EEL definition, we next examined editing profiles of individual EELs rather than individual A-to-I editing sites. To do so, we created an EEL editing index (see Methods) to gauge if all editing sites within an EEL may be preferentially edited in the HC class, the MS-E class or if there was no difference in the overall level of EEL editing between the HC and MS-E classes. This index showed that editing indices of individual EELs were fairly diverse with certain EELs displaying similar editing index values between HC and MS-E classes while other EELs were preferentially edited in the HC class compared to the MS-E class (Fig. 5D). We also found that differences between EEL indices in HC and MS-E classes were highly correlated with differences in expression of the genes in which these EELs resided between HC and MS-E classes (Fig. 5E).
Interferon, NF-κB responses and Alu and Line RNAs
Given the above results we wanted to determine the ability of individual Alu and Line RNAs to activate dsRNA sensors and stimulate downstream transcriptional responses as well as the effect of editing on their biologic activity. To do so, we identified several Alu elements present at high levels in leukocytes (RNA-seq analysis) with elevated levels of A-to-I editing in HC compared to MS-E samples for further analysis. We in vitro transcribed these Alu elements and Line elements also highly expressed as dsRNAs in MS leukocytes and tested their ability to stimulate luciferase reporter constructs under the control of either an ISRE or NF-κB response element stably integrated into the human monocyte cell line, THP-1 (41). We identified two Alu RNAs of the AluJb and AluY2 classes that were potent stimulators of both the ISRE and NF-κB transcriptional responses, while a third Alu RNA of the AluYK4 class was a weak stimulator of these response elements (Fig. 6 A, B). Similarly, we identified Line dsRNA elements that were very potent inducers of both the ISRE and NF-κB responses (Fig. 6 C, D). For comparison, we also stimulated THP-1 reporter cells with varying amounts of IFN-alpha. We found that maximal activation of the ISRE response by IFN-alpha was similar in magnitude to activation by Alu or Line dsRNA elements. In contrast, activation of the NF-κB response by IFN-alpha was somewhat less in magnitude that that observed for the Alu or Line dsRNA elements. To actually demonstrate that these Alu and Line dsRNA elements induced changes in gene expression, we measured induction of RELB and IL6, two known NF-κB responsive genes, and DDX58 and IFIT5, two known ISRE-responsive genes. We compared two cell lines of myeloid origin, THP-1 and HMC3, a human microglial cell line (46), and found these genes were induced >20-fold after stimulation by a representative Alu or Line dsRNA element (Fig. 6F). Gene induction in response to IFN-alpha was also determined. We also found that the more potent Alu and Line dsRNA elements induced ~5-fold greater ISRE- and NF-κB-transcriptional responses than poly IC at amounts ~10-fold less than poly IC. Alu and Line dsRNA elements also induced higher levels of gene expression at lower amounts of RNA than poly IC. Thus, we found that both Alu and Line dsRNA elements that were highly expressed in MS patient leukocytes were potent inducers of both ISRE- and NF-κB transcriptional responses measured using either reporter assays or induction of known ISRE- and NF-κB responsive genes in two distinct myeloid-derived cell lines.
FIGURE 6.

Activation of interferon and NF-κB transcriptional responses by Alu and Line RNAs. (A-D) Cultures of THP-1 ISRE- or NF-κB-reporter cells were transfected with the indicated amounts of the indicated Alu RNAs or polyIC (A,B) or the indicated Line RNAs (C,D). Results are expressed as the increase in luciferase activity in cultures relative to mock transfected cultures. (E) THP-1 ISRE- or NF-κB reporter cells were treated with the indicated amounts of IFN-alpha, results are expressed as the increase in luciferase activity relative to untreated cultures. (F) Induction of IFN- and NF-κB response genes by Alu and Line RNAs. THP-1 or HMC3 cells (3 ml cultures) were transfected with the indicated amounts of AluJb dsRNA, L5 dsRNA, or poly IC or were treated with the 10 ng/ml IFN-alpha. After 24 hr, expression levels of the indicated genes were determined by PCR. Results are expressed as the fold increase in expression activity relative to mock-treated cultures.
We in vitro transcribed additional Alu and Line RNA elements that were highly expressed as dsRNAs in the MS cohort and tested their ability to induce ISRE- and NF-κB transcriptional responses using the reporter assays. We tested induction of these responses at RNA amounts ranging from 0.1-100 ng/culture and data are expressed as the effective concentration required for 10x-induction of either ISRE- or NF-κB-responses over backgrounds observed in cultures transfected without added exogenous RNA, EC10x. We identified both potent Alu and Line RNAs that stimulated ISRE- and NF-κB-responses by >10 fold at amounts of ≤1 ng RNA/culture, Alu and Line RNAs with moderate activity stimulating ISRE- and NF-κB -responses by >10 fold at RNA amounts of 1-10 ng/culture RNA, and those that were inactive and failed to stimulate ISRE- and NF-κB-responses at amounts of RNA > 100 ng/culture (TABLE II). Both Alu and Line elements are sub-divided into classes, e.g. AluJ, AluY, AluS, L1-L5. We tested representatives of these different classes and found highly active Alu and Line RNAs represented by multiple classes. Also, we found representatives from identical classes that were both highly active and completely inactive. Thus, it seems that ability to activate dsRNA sensors and ISRE- and NF-κB-responses was not dependent upon belonging to a specific Alu or Line class. Thus, these experiments demonstrated that multiple Alu and Line RNAs are potent activators of both ISRE- and NF-κB-responses.
TABLE II.
Diverse Alu and Line dsRNAs stimulate IFN- and NF-κB responses
| Alu/Line class | genomic location | ISREa | NF-κBa |
|---|---|---|---|
| AluJb | chr17:76,418,582-76,418,856 | 0.3 | 1 |
| AluY2 | chr5:175,126,575-175,126,866 | 0.5 | 1 |
| AluJo | chr9:68,416,267-68,416,574 | 3 | 30 |
| AluJb | chr1:36,944,168-36,944,489 | 4 | >100 |
| AluSx1 | chr1:36,943,848-36,944,158 | 5 | >100 |
| AluSx3 | chr1:36,942,858-36,943,160 | 8 | >100 |
| AluY | chr3:71,827,084-71,827,387 | 20 | >100 |
| AluSp | chr15:74,893,758-74,894,053 | >100 | >100 |
| AluYk4 | chr12:51,711,490-51,711,800 | >100 | >100 |
| L5 | chr3:122,593,731-122,594,215 | 0.3 | 0.5 |
| L5-2 | chr2:197,877,529-197,878,005 | 0.5 | 0.5 |
| L1P1 | chr8:12,399,322-12,400,289 | 0.3 | 1 |
| L1PB1 | chr5:77462505-77463456 | 0.3 | 2 |
| L4 | chr7:36,559,566-36,560,029 | 0.3 | 3 |
| L4 | chr7:105,910,258-105,910,706 | 0.5 | 3 |
| L1MA4A | chr20:1,464,664-1,465,526 | 0.5 | 4 |
| L1PA3 | chr1:174,431,977-174,432,809 | 0.3 | 8 |
| L2a | chr6:24,920,947-24,921,396 | 0.5 | 10 |
| L2 | chr12:62,712,029-62,712,774 | 0.3 | 20 |
| L1P1 | chr16:31,910,131-31,910,962 | 1 | 30 |
| L1MA8 | chr6:11,274,877-11,275,769 | 2 | 40 |
| L3 | chrX:30,509,147-30,509,509 | 3 | 10 |
| L1PA4 | chr2:149,419,501-149,420,396 | 3 | 20 |
| L1PA2 | chr2:144,395,314-144,396,233 | 4 | 33 |
| L3 | chr13:100,034,217-100,034,854 | >100 | >100 |
| poly ICb | >100 | >100 |
Amount of the indicated RNAs required to stimulate a 10-fold increase in reporter activity compared to mock-transfected cell cultures.
Poly IC did not stimulate a 10-fold increase in secreted luciferase over unstimulated cell cultures at RNA amounts up to 100 ng.
Activation of dsRNA sensors by Alu and Line RNAs
We employed the human embryonic kidney 293 cell line (HEK293) to determine which dsRNA sensors were activated by representative Alu and Line RNAs. HEK293 cells do not express dsRNA sensors RIG-I and MDA5 but do express TLR3 (37). We compared responses in HEK293 cells (HEK-TLR3), HEK293 cells with a stably integrated RIG-I (DDX58) expression construct (HEK-TLR3 + RIG-I), and HEK293 cells transiently transfected with a MDA5 expression construct (HEK-TLR3 + MDA5). Each of these cell lines also contained a stably integrated ISRE-luciferase reporter construct. We found that transfection of poly IC activated ISRE responses almost equivalently in the three HEK lines suggesting that under these conditions, poly IC only activates TLR3 (Fig. 7A). We found that AluJb dsRNA, one of the more potent inducers of ISRE- and NF-κB-responses in THP1 cells (genomic location-chr17:76,418,582-76,418,856), was a potent inducer of ISRE-responses in HEK293-TLR3 + RIG-I cells compared to HEK-TLR3 and HEK-TLR3 + MDA5 cells (Fig. 7B). ISRE-responses were also detected in HEK-TLR3 cells indicating that AluJb is recognized by RIG-I and TLR3. We did not find an increase in ISRE-responses in HEK-TLR3 + MDA5 cells compared to HEK-TLR3 cells suggesting that AluJb does not activate MDA5. We performed a similar analysis using one of the Line dsRNA elements, L1P1 (genomic location-chr8:12,399,322-12,400,289). We found that this L1P1 dsRNA element activated ISRE-responses in HEK-TLR3, HEK-TLR3 + RIG-I, and HEK-TLR3 + MDA5 reporter cells (Fig. 7C). Activation of ISRE responses was strongest in the HEK-TLR3 + RIG-I cells. ISRE responses were higher in HEK-TLR3 + MDA5 cells compared to HEK-TLR3 cells suggesting that the L1P1 dsRNA element also activated ISRE-responses via MDA5. Thus, these results are consistent with the notion that the AluJb dsRNA element activated ISRE-responses through RIG-I and TLR3 while the L1P1 dsRNA element activated ISRE-responses via RIG-I, TLR3, and MDA5 sensors.
FIGURE 7.
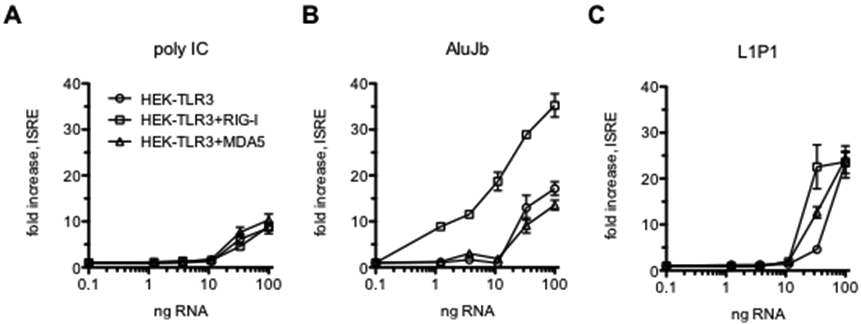
Activation of ISRE-responses via TLR3, RIG-I or MDA5 dsRNA sensors. (A-C) HEK293-TLR3, HEK293-TLR3+RIG-I, or HEK293-TLR3+MDA5 cells were transfected with the indicated amounts of polyIC, AluJb, or L1P1 RNAs. Luciferase activity was determined after 24 hrs. Results are expressed as fold increase in luciferase activity compared to mock-transfected controls.
Structure-function relationships
An Alu element is composed of a left arm of ~100 nucleotides (nt) and a right arm of ~200 nt (39). Certain models predict that, if transcribed, an Alu RNA forms two dsRNA structures, one composed of the left arm and one composed of the right arm with a linker sequence in between (47). We prepared new AluJb elements one consisting of the right arm and one consisting of the left arm, in vitro transcribed each new element, and tested them for activation of ISRE-responses in HEK-TLR3+RIG-I reporter cells. We found that activity of the AluJb left arm dsRNA was almost equivalent to activity of the intact AluJb dsRNA while the AluJb right arm possessed almost no activity (Fig. 8A). These results seem to support the model predicting that Alu RNAs contain two independent arms, each with its own dsRNA structure.
FIGURE 8.
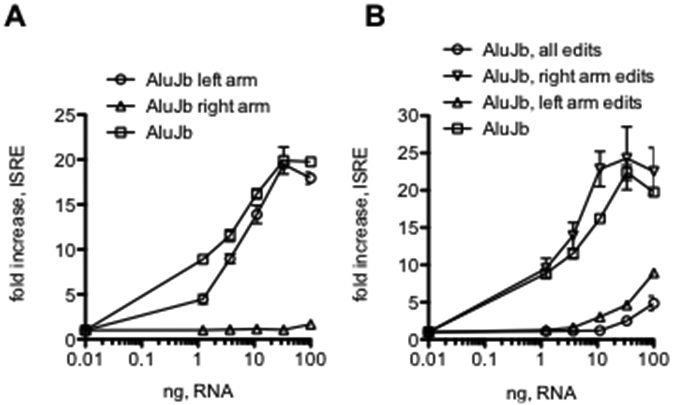
Structural analysis of the functional activity of AluJb dsRNA. A, HEK293-TLR3+RIG-I reporter cells were transfected with the indicated amounts of intact AluJb RNA, AluJb left arm (nt 1-137) or AluJb right arm (nt 138-275). Results are expressed as the fold increase in luciferase activity relative to mock transfected controls. B, HEK293-TLR3+RIG-I reporter cells were transfected with the indicated amounts of the following dsRNAs: intact AluJb, AluJB, all edits (all A nts changed to G nts), AluJb, right arm edits (all A nts changed to G nts in the right arm only) or AluJb, left arm edits (all A nts changed to G nts in the left arm only). Results are expressed as the fold increase in luciferase activity relative to mock transfected controls.
Further, ADAR catalyzed A-to-I editing of Alu dsRNA elements is thought to result in loss of dsRNA character and the ability of these RNA elements to be recognized by dsRNA sensors and stimulate downstream signaling cascades resulting in activation of ISRE- and NF-κB-responses. Our results, as well as those of others, indicate that not all A’s are equal targets for A-to-I editing. Our results comparing editing of Alu elements in HC and MS showed that only a portion of total A’s within an Alu RNA element are edited to I’s and edit/read ratios at a given editing site were generally <100%. However, it is generally unknown how efficient or extensive A-to-I editing needs to be to convert an active Alu RNA recognized by dsRNA sensors into an inactive Alu RNA incapable of triggering interferon-responses. To explore this idea further, we prepared new AluJb DNA elements with all A’s changed to G’s in the entire element, all A’s changed to G’s in the AluJb left arm and all A’s changed to G’s in the AluJb right arm, in vitro transcribed these new elements and tested them for activity in HEK-TLR3+RIG-I reporter cells containing the ISRE-luciferase construct. We found that changing all A’s to G’s in the entire AluJb element or only changing A’s to G’s in the left arm of the intact AluJb element resulted in an almost complete loss of ability to activate ISRE-responses (Fig. 8B). In contrast, changing A’s to G’s only in the AluJb right arm resulted in a small increase in activity.
Given the above results, we next determined how changes in AluJb dsRNA nucleotide sequence altered ISRE- and NF-κB-responses in THP-1 reporter cells. As above, we found that the AluJb left arm dsRNA activated both ISRE-responses and NF-κB responses approximately as well as the full-length AluJb dsRNA while the AluJb right arm did not activate ISRE- and NF-κB responses (Fig. 9A). We also found that changing either all A’s to G’s in the entire AluJb element or changing only A’s to G’s in the left arm resulted in almost complete loss of activity while changing A’s to G’s only in the right arm resulted in a small increase in the ability of this modified AluJb dsRNA to activate either ISRE- or NF-κB-responses in THP-1 reporter cells (Fig. 9B).
FIGURE 9.
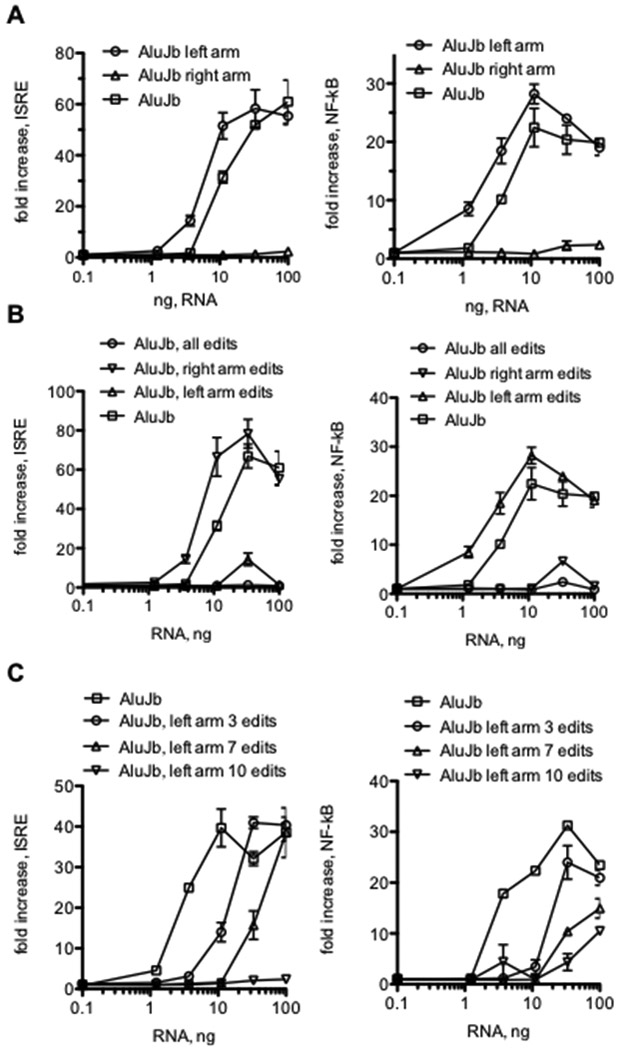
AluJb dsRNA structure-function studies in THP-1 cells. (A) AluJb, AluJb left arm, or AluJb right arm RNAs were transfected into THP-1 ISRE- (left) or NF-κB (right) reporter cells. Results are expressed as the fold increase in luciferase activity relative to mock transfected controls. B, THP-1 reporter cells were transfected with the indicated amounts of intact AluJb RNA, AluJB, all edits RNA (all A nts changed to G nts) AluJb, right arm edits RNA (all A nts changed to G nts in the right arm only) or AluJb, left arm edits (all A nts changed to G nts in the left arm only. Results are expressed as the fold increase in luciferase activity relative to mock transfected controls (left ISRE, right NF-κB). C, THP-1 reporter cells were transfected with the indicated amounts of intact AluJb RNA, AluJB with 3, 7, or 10 A nt to G nt left arm. Results are expressed as the fold increase in luciferase activity relative to mock transfected controls (left ISRE, right NF-κB).
The AluJb element is 274 nt in length and changing all A’s to G’s in the left arm results in 15 nucleotide changes. To pursue this notion further, we asked if fewer A to G changes in the AluJb left arm affected ability to activate ISRE- and NF-κB-responses in THP-1 reporter cells. We created three new constructs containing 10, 7 or 3 A-to-G changes in the AluJb left arm and tested the ability of these RNAs to stimulate ISRE- and NF-κB-responses in THP-1 reporter cells. We found that 10 A-to-G changes resulted in almost complete loss of the ability to stimulate ISRE- or NF-κB responses (Fig. 9C). Making 7 A-to-G changes in the AluJb left arm also resulted in substantial loss of the ability to activate ISRE- and NF-κB responses while even 3 A-to-G changes in the AluJb left arm resulted in partial loss of the ability to activate ISRE- and NF-κB responses by this modified dsRNA. Thus, small changes in nucleotide sequence can markedly affect ability of Alu dsRNAs to stimulate dsRNA sensors and downstream signaling paths leading to activation of ISRE- and NF-κB transcriptional responses.
Discussion
Our results document substantial differences in A-to-I editing between HC and MS patient leukocytes. Perhaps the most striking difference is complete absence of editing of certain Alu RNA sequences in MS leukocytes and presence of substantial editing at these same Alu RNA elements in HC leukocytes. Importantly, these differences are seen early in MS disease and prior to onset of treatments. In contrast, certain Alu RNA elements are A-to-I edited at approximately equivalent levels in both HC and MS leukocytes. We also find that A-to-I editing sites within Alu elements not evenly distributed across the genome but are clustered in the genome in, what we term, editing enriched locations or EELs. Interestingly, loss of EEL editing in MS is associated with loss of expression of genes in which the EELs are embedded. We also find that unedited Alu and Line dsRNAs are potent activators of dsRNA sensors resulting in induction of ISRE- and NF-κB-transcriptional responses and known ISRE- and NF-κB-regulated genes. The unedited Alu dsRNAs stimulate ISRE-responses via both RIG-I and TLR3, but not MDA5, while the longer unedited Line dsRNAs stimulate ISRE-responses via all three dsRNA sensors examined, RIG-I, TLR3, and MDA5. Our results show that for the AluJb dsRNA element, one half or arm possesses all the ability to activate these inflammatory transcriptional responses while the other half or arm is completely inactive and we have replicated this finding by analyzing a second Alu RNA element (not shown). Further, converting A’s in the active arm to G’s, as a model of A-to-I editing, results in almost complete loss of activity while converting A’s to G’s in the inactive arm results in a small gain in overall activity. Finally, replacing a small number of A’s with G’s as a model of A-to-I editing has profound effects on the ability of these dsRNAs to activate ISRE- and NF-κB responses. In summary, our results are consistent with a model where deficiencies of A-to-I editing of Alu RNAs in MS patients increase the dsRNA character of these abundant Alu and Line RNAs giving rise to increased activation of both interferon and NF-κB signaling paths.
The concept of editing-enriched locations or regions in the genome is not entirely new (48). Using similar strategies as outlined here, previous studies have referred to these regions as ‘editing-enriched regions’ or EERs and have documented presence of EERs in C. elegans, mice and humans. Since our methodology is not identical, we chose to refer to these regions as EELs to avoid any confusion. Interestingly, presence of EERs within a gene versus absence of EERs within a gene is associated with overall higher expression levels compared to genes that lack EERs. We show here that loss of A-to-I editing in gene-associated EELs, which is seen in MS-E, is also associated with loss of gene expression suggesting perhaps that one function of A-to-I editing in EELs may be to enhance expression of genes with embedded EELs. EELs probably have additional functions and understanding the basic functions of EELs will contribute to a better understanding of the critical importance of A-to-I editing for development and survival.
One example of human disease resulting from persistent activation of the interferon system is Aicardi-Goutieres Syndrome (23, 24). One form of this syndrome results from loss of function mutations in ADAR1 and deficiencies in A-to-I editing. A second form results from gain of function mutations in IFIH1, the gene that encodes MDA5, resulting in loss of tolerance to endogenous dsRNAs and elevated interferon responses. Age-related macular degeneration is another disorder in which accumulation of Alu dsRNA elements contributes to activation of interferon-responses and cell death (49, 50). In this disease, accumulation of Alu dsRNAs appears to result from loss of DICER1 and subsequent failure of DICER1-mediated degradation of Alu dsRNAs. Our results show there is dramatic loss of A-to-I editing at certain editing sites in MS patient leukocytes. Several factors may contribute to selective loss of A-to-I editing depending upon nucleotide site. First, our studies were performed using whole blood which contains multiple leukocyte lineages and A-to-I editing processes in unique leukocyte lineages may be affected differently in MS. Second, besides ADAR and DICER, multiple other RNA binding proteins (RBPs) can both positively and negatively influence A-to-I editing and these effects seem to vary depending upon the specific nucleotide editing site in the genome (51). For example, depletion of TROVE2 in cell lines, the gene that encodes the RBP, Ro60, results in both increases and decreases of A-to-I editing depending upon the editing site. Ro60 also plays an important role in RNA surveillance or quality control and we have previously shown that both TROVE2 mRNA and Ro60 protein are highly under-expressed in MS leukocytes, which may influence A-to-I editing (52-54). Several other genes that encode RBPs shown to affect genome-wide A-to-I editing are also differentially expressed in MS compared to HC leukocytes and differences in expression of these RBPs may also contribute to the observed differences in A-to-I editing in MS. Thus, there may be multiple molecular mechanisms contributing to the observed deficiencies in A-to-I editing in leukocytes in MS.
In contrast to diseases or syndromes where increased production of type 1 interferons is considered to be pathogenic, commonly referred to as ‘interferonopathies’, including Aicardi-Goutieres Syndrome and certain autoimmune diseases, such as systemic lupus erythematosus, therapeutic versions of IFN-β are mainstays for treatment of MS reducing both rates of disease relapse and long-term disability (55-57). Similarly, type 1 interferons reduce severity of disease in animal models of MS. Thus, one might speculate that in contrast to the prevailing views of ‘interferonopathies’, decreased A-to-I editing and increased accumulation of un-edited Alu dsRNA elements and elevated expression of ISGs may have positive impacts on MS. A better understanding of mechanisms that regulate levels of A-to-I editing in MS may improve disease management and perhaps point to new therapeutic targets.
In mouse models, early embryonic lethality and interferon responses caused by ADAR deletion are prevented by concomitant deletion of the dsRNA sensor, MDA5/IFIH1, but not by deletion of RIG-I, suggesting that MDA5, but not RIG-I, is the key sensor of endogenous dsRNAs (31, 58). These interpretations are complicated by the fact that the RIG-I/DDX58 knockout is also embryonic lethal so it may not be possible to rescue embryonic lethality observed by ADAR deletion with the DDX58 knockout since it also causes embryonic lethality and employ this experimental approach to exclude the role for RIG-I as a key sensor of endogenous dsRNAs. However, studies of inactivating human ADAR mutations in cell lines further support that MDA5 is a key sensor of endogenous dsRNAs (13, 59). In contrast, studies in certain stress models such as ischemia/reperfusion injury show that ADAR plays a critical role in this injury response, such that deletion of ADAR greatly exacerbates the interferon response associated with this injury model as well as resulting pathology (60). In this case, injury caused by ADAR loss is reversed by concomitant depletion of RIG-I suggesting that in this model, RIG-I is a key sensor of endogenous dsRNAs. Thus, in one experimental setting, MDA5 appears to be the major sensor of endogenous dsRNAs while in a second experimental setting, RIG-I appears to be the major of endogenous dsRNAs. Two key differences between these two experimental systems are that those showing MDA5 rescue of ADAR deletion are seen early in development and result from ADAR inactivating mutations or gene knockouts while systems showing RIG-I rescue of ADAR depletion employ knockdown strategies that do not result in complete inactivation of ADAR and employ fully developed liver tissue from adult mice. Thus, it seems there may be different contributions of MDA5 and RIG-I as endogenous dsRNA sensors depending upon either developmental status, cell lineage or response to external or internal stimuli. In addition, we do not see total loss of A-to-I editing in MS as might result from either ADAR deletion or presence of ADAR inactivating mutations. These differences may suggest that loss of A-to-I editing observed in MS-E may be a kind of stress response to exogenous or endogenous stimuli.
Further, loss of the ADAR p150 isoform disrupts intestinal homeostasis and B cell development while loss of the ADAR p110 isoform disrupts kidney and brain function(58). These developmental defects are independent of induction of interferon responses via MDA5 activation. Thus, A-to-I editing is also essential for multiple developmental processes. Whether selective loss of A-to-I editing in EELs as observed in MS-E may contribute to MS pathogenesis via mechanisms independent of activation of interferon responses is unknown.
In MS leukocytes, the endogenous dsRNA fraction capable of stimulating interferon responses is composed primarily of Alu RNAs and Line RNAs (41). We in vitro transcribed and tested different Alu and Line RNAs that are highly expressed in MS leukocytes to assess their ability to activate ISRE- and NF-κB responses. Even though RNAs derived from Alu elements have a great deal of sequence similarity, they display very different abilities to activate ISRE- and NF-κB responses where some activate maximal responses at very low RNA amounts, 1 ng/culture, while others are completely inactive. Line elements exhibit similar properties with some having very high activity and others exhibiting no activity. Sensors that recognize dsRNA include RIG-I, MDA5 and TLR3. A general view is that RIG-I activation involves sensing of a free 5’ triphosphate short dsRNA while MDA5 and TLR3 are activated by longer dsRNA structures. The contribution of double-stranded structure or perhaps higher-order structure to the ability to activate these dsRNA sensors is incompletely understood. Our results show that Alu RNA elements activate both RIG-I and TLR3 dsRNA sensors but not MDA5, while the longer Line RNA elements activate all three dsRNA sensors to induce potent ISRE- and NF-κB responses. The fact that certain Alu RNAs activate potent ISRE- and NF-κB responses while others are without activity and all possess a 5’ triphosphate suggests that either the double-stranded structure or perhaps higher order structure also contributes to recognition of these Alu RNAs by dsRNA sensors.
Alu retrotransposons inserted in the genome are composed of a left arm and a right arm and linker sequence. Certain models predict that both left and right arms may form independent dsRNA structures while other models predict that the total Alu RNA sequence is involved in forming only one continuous dsRNA structure (47). Our results show, that at least for one highly active Alu RNA, all the ISRE- and NF-κB inducing activity lies in the left arm while the right arm is completely inactive. In addition, A-to-G nucleotide editing in the inactive arm results in an increase in biologic activity while A-to-G editing in the active arm completely abrogates activity. These results lend further support to this general model of a left arm-right arm structure of Alu RNAs. At this point, we do not know if all active Alu RNAs can be sub-divided into an active arm-inactive arm structure or if in some cases, both arms possess biologic activity.
The general view is that A-to I editing is a critical process to ‘de-toxify’ endogenous RNAs to prevent their recognition by dsRNA sensors. Our sequencing analyses demonstrate that only a fraction of possible A-to-I editing sites in an Alu RNA are actually edited raising the question of how many edits are actually necessary to de-toxify an Alu RNA and prevent it from activating dsRNA sensors and downstream signaling paths. Our A-to-G editing model of Alu RNA editing indicates that as few as three A-to-G changes in a ~275 nt Alu element significantly impacts activity while 5-10 A-to-G changes almost completely abrogates biologic activity. This small number of editing changes is similar in magnitude to the A-to-I editing differences found in individual Alu RNAs elements when HC and MS are compared. Thus, we propose that loss of A-to-I editing observed in MS contributes to increased dsRNA character of Alu RNAs and increased activation of ISRE- and NF-κB transcriptional responses and downstream inflammatory responses.
Key Points.
A-to-I RNA editing of Alu dsRNAs is markedly decreased in MS
endogenous Alu dsRNAs are potent activators of IFN and NF-κB responses
minor RNA editing of active Alu dsRNAs abrogates this activity
Acknowledgments
This work was supported by grants from the NIH, R01AI044924 and R21AI144193 (TMA) Whole genome RNA-sequencing files are deposited in the NCBI Gene Expression Omnibus. Accession numbers are GSE92472 and GSE126427 (https://www.ncbi.nlm.nih.gov/geo/).
Abbreviations used in this article:
- 3’UTR
3’ untranslated region
- dsRNA
double-stranded RNA
- EEL
edited enriched location
- EER
editing enriched region
- HC
healthy control
- ISRE
interferon-stimulated response element
- ISGs
interferon stimulated genes
- LINE
long interspersed element
- MS
relapsing remitting multiple sclerosis
- RBP
RNA binding protein
- SINE
short interspersed element
Footnotes
Disclosures
The authors declare no competing interests
References
- 1.Samuel CE 2001. Antiviral actions of interferons. Clin Microbiol Rev 14: 778–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Theofilopoulos AN, Baccala R, Beutler B, and Kono DH. 2005. Type I interferons (alpha/beta) in immunity and autoimmunity. Annual Review of Immunology 23: 307–336. [DOI] [PubMed] [Google Scholar]
- 3.Trinchieri G 2010. Type I interferon: friend or foe? Journal of Experimental Medicine 207: 2053–2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Billiau A 2006. Anti-inflammatory properties of Type I interferons. Antivir Res 71: 108–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Der SD, Zhou AM, Williams BRG, and Silverman RH. 1998. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. P Natl Acad Sci USA 95: 15623–15628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, and Rice CM. 2011. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472: 481–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schoenborn JR, and Wilson CB. 2007. Regulation of interferon-gamma during innate and adaptive immune responses. Adv Immunol 96: 41–101. [DOI] [PubMed] [Google Scholar]
- 8.Janeway CA Jr., and Medzhitov R. 2002. Innate immune recognition. Annu Rev Immunol 20: 197–216. [DOI] [PubMed] [Google Scholar]
- 9.Sharma S, Fitzgerald KA, Cancro MP, and Marshak-Rothstein A. 2015. Nucleic Acid-Sensing Receptors: Rheostats of Autoimmunity and Autoinflammation. J Immunol 195: 3507–3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dias AG, Sampaio NG, and Rehwinkel J. 2019. A Balancing Act: MDA5 in Antiviral Immunity and Autoinflammation. Trends Microbiol 27: 75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim Y, Lee JH, Park JE, Cho J, Yi H, and Kim VN. 2014. PKR is activated by cellular dsRNAs during mitosis and acts as a mitotic regulator. Gene Dev 28: 1310–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lassig C, and Hopfner KP. 2017. Discrimination of cytosolic self and non-self RNA by RIG-I-like receptors. J Biol Chem 292: 9000–9009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ahmad S, Mu X, Yang F, Greenwald E, Park JW, Jacob E, Zhang CZ, and Hur S. 2018. Breaching Self-Tolerance to Alu Duplex RNA Underlies MDA5-Mediated Inflammation. Cell 172: 797–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goubau D, Schlee M, Deddouche S, Pruijssers AJ, Zillinger T, Goldeck M, Schuberth C, Van der Veen AG, Fujimura T, Rehwinkel J, Iskarpatyoti JA, Barchet W, Ludwig J, Dermody TS, Hartmann G, and Sousa CRE. 2014. Antiviral immunity via RIG-I-mediated recognition of RNA bearing 5 ‘-diphosphates. Nature 514: 372–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, Shark KB, Grande WJ, Hughes KM, Kapur V, Gregersen PK, and Behrens TW. 2003. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci U S A 100: 2610–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, and Pascual V. 2003. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med 197: 711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chiappinelli KB, Strissel PL, Desrichard A, Li HL, Henke C, Akman B, Hein A, Rote NS, Cope LM, Snyder A, Makarov V, Buhu S, Slamon DJ, Wolchok JD, Pardoll DM, Beckmann MW, Zahnow CA, Mergoub T, Chan TA, Baylin SB, and Strick R. 2015. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell 162: 974–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Crow MK 2014. Type I Interferon in the Pathogenesis of Lupus. Journal of Immunology 192: 5459–5468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lubbers J, Brink M, de Stadt LAV, Vosslamber S, Wesseling JG, van Schaardenburg D, Rantapaa-Dahlqvist S, and Verweij CL. 2013. The type I IFN signature as a biomarker of preclinical rheumatoid arthritis. Ann Rheum Dis 72: 776–780. [DOI] [PubMed] [Google Scholar]
- 20.Nezos A, Gravani F, Tassidou A, Kapsogeorgou EK, Voulgarelis M, Koutsilieris M, Crow MK, and Mavragani CP. 2015. Type I and II interferon signatures in Sjogren’s syndrome pathogenesis: Contributions in distinct clinical phenotypes and Sjogren’s related lymphomagenesis. J Autoimmun 63: 47–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ranoa DR, Parekh AD, Pitroda SP, Huang X, Darga T, Wong AC, Huang L, Andrade J, Staley JP, Satoh T, Akira S, Weichselbaum RR, and Khodarev NN. 2016. Cancer therapies activate RIG-I-like receptor pathway through endogenous non-coding RNAs. Oncotarget 7: 26496–26515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Verweij CL, and Vosslamber S. 2013. Relevance of the type I interferon signature in multiple sclerosis towards a personalized medicine approach for interferon-beta therapy. Discov Med 15: 51–60. [PubMed] [Google Scholar]
- 23.Oda H, Nakagawa K, Abe J, Awaya T, Funabiki M, Hijikata A, Nishikomori R, Funatsuka M, Ohshima Y, Sugawara Y, Yasumi T, Kato H, Shirai T, Ohara O, Fujita T, and Heike T. 2014. Aicardi-Goutieres Syndrome Is Caused by IFIH1 Mutations. Am J Hum Genet 95: 121–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rice GI, Kasher PR, Forte GMA, Mannion NM, Greenwood SM, Szynkiewicz M, Dickerson JE, Bhaskar SS, Zampini M, Briggs TA, Jenkinson EM, Bacino CA, Battini R, Bertini E, Brogan PA, Brueton LA, Carpanelli M, De Laet C, de Lonlay P, del Toro M, Desguerre I, Fazzi E, Garcia-Cazorla A, Heiberg A, Kawaguchi M, Kumar R, Lin JPSM, Lourenco CM, Male AM, Marques W, Mignot C, Olivieri I, Orcesi S, Prabhakar P, Rasmussen M, Robinson RA, Rozenberg F, Schmidt JL, Steindl K, Tan TY, van der Merwe WG, Vanderver A, Vassallo G, Wakeling EL, Wassmer E, Whittaker E, Livingston JH, Lebon P, Suzuki T, McLaughlin PJ, Keegan LP, O’Connell MA, Lovell SC, and Crow YJ. 2012. Mutations in ADAR1 cause Aicardi-Goutieres syndrome associated with a type I interferon signature. Nat Genet 44: 1243–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.George CX, Ramaswami G, Li JB, and Samuel CE. 2016. Editing of Cellular Self-RNAs by Adenosine Deaminase ADAR1 Suppresses Innate Immune Stress Responses. J Biol Chem 291: 6158–6168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Samuel CE 2019. Adenosine deaminase acting on RNA (ADAR1), a suppressor of double-stranded RNA-triggered innate immune responses. J Biol Chem 294: 1710–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yablonovitch AL, Deng P, Jacobson D, and Li JB. 2017. The evolution and adaptation of A-to-I RNA editing. Plos Genet 13(11):e1007064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Walkley CR, and Li JB. 2017. Rewriting the transcriptome: adenosine-to-inosine RNA editing by ADARs. Genome Biol 18:205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eisenberg E, and Levanon EY. 2018. A-to-I RNA editing - immune protector and transcriptome diversifier. Nat Rev Genet 19: 473–490. [DOI] [PubMed] [Google Scholar]
- 30.Lamers MM, van den Hoogen BG, and Haagmans BL. 2019. ADAR1: “Editor-in-Chief” of Cytoplasmic Innate Immunity. Front Immunol 10:1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liddicoat BJ, Piskol R, Chalk AM, Ramaswami G, Higuchi M, Hartner JC, Li JB, Seeburg PH, and Walkley CR. 2015. RNA EDITING RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 349: 1115–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nishikura K 2016. A-to-I editing of coding and non-coding RNAs by ADARs. Nat Rev Mol Cell Bio 17: 83–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pfaller CK, Donohue RC, Nersisyan S, Brodsky L, and Cattaneo R. 2018. Extensive editing of cellular and viral double-stranded RNA structures accounts for innate immunity suppression and the proviral activity of ADAR1(p150). Plos Biol 16(11):e2006577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Porath HT, Knisbacher BA, Eisenberg E, and Levanon EY. 2017. Massive A-to-I RNA editing is common across the Metazoa and correlates with dsRNA abundance. Genome Biol 18:185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Portal MM, Pavet V, Erb C, and Gronemeyer H. 2015. Human cells contain natural double-stranded RNAs with potential regulatory functions. Nat Struct Mol Biol 22: 89–97. [DOI] [PubMed] [Google Scholar]
- 36.Yang SY, Deng P, Zhu ZW, Zhu JZ, Wang GL, Zhang LY, Chen AF, Wang T, Sarkar SN, Billiar TR, and Wang QD. 2014. Adenosine Deaminase Acting on RNA 1 Limits RIG-I RNA Detection and Suppresses IFN Production Responding to Viral and Endogenous RNAs. Journal of Immunology 193: 3436–3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Linehan MM, Dickey TH, Molinari ES, Fitzgerald ME, Potapova O, Iwasaki A, and Pyle AM. 2018. A minimal RNA ligand for potent RIG-I activation in living mice. Sci Adv 4: e1701854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen LL, and Yang L. 2017. ALUternative Regulation for Gene Expression. Trends Cell Biol 27: 480–490. [DOI] [PubMed] [Google Scholar]
- 39.Deininger P 2011. Alu elements: know the SINEs. Genome Biol 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mighell AJ, Markham AF, and Robinson PA. 1997. Alu sequences. FEBS Lett 417: 1–5. [DOI] [PubMed] [Google Scholar]
- 41.Heinrich MJ, Purcell CA, Pruijssers AJ, Zhao Y, Spurlock CF, Sriram S, Ogden KM, Dermody TS, Scholz MB, Crooke PS, Karijolich J, and Aune TM. 2019. Endogenous double-stranded Alu RNA elements stimulate IFN-responses in relapsing remitting multiple sclerosis. J Autoimmun 100: 40–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aune TM, Crooke PS, Patrick AE, Tossberg JT, Olsen NJ, and Spurlock CF. 2017. Expression of long non-coding RNAs in autoimmunity and linkage to enhancer function and autoimmune disease risk genetic variants. J Autoimmun 81: 99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang F, Lu YL, Yan SJ, Xing QH, and Tian WD. 2017. SPRINT: an SNP-free toolkit for identifying RNA editing sites. Bioinformatics 33: 3538–3548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brown JE, Klement JF, and McAllister WT. 1986. Sequences of three promoters for the bacteriophage SP6 RNA polymerase. Nucleic Acids Res 14: 3521–3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gibbons HR, Shaginurova G, Kim LC, Chapman N, Spurlock CF 3rd, and Aune TM. 2018. Divergent lncRNA GATA3-AS1 Regulates GATA3 Transcription in T-Helper 2 Cells. Front Immunol 9: 2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dello Russo C, Cappoli N, Coletta I, Mezzogori D, Paciello F, Pozzoli G, Navarra P, and Battaglia A. 2018. The human microglial HMC3 cell line: where do we stand? A systematic literature review. J Neuroinflamm 15: 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jady BE, Ketele A, and Kiss T. 2012. Human intron-encoded Alu RNAs are processed and packaged into Wdr79-associated nucleoplasmic box H/ACA RNPs. Gene Dev 26: 1897–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reich DP, and Bass BL. 2019. Mapping the dsRNA World. Cold Spring Harb Perspect Biol 11: a035352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kaneko H, Dridi S, Tarallo V, Gelfand BD, Fowler BJ, Cho WG, Kleinman ME, Ponicsan SL, Hauswirth WW, Chiodo VA, Kariko K, Yoo JW, Lee DK, Hadziahmetovic M, Song Y, Misra S, Chaudhuri G, Buaas FW, Braun RE, Hinton DR, Zhang Q, Grossniklaus HE, Provis JM, Madigan MC, Milam AH, Justice NL, Albuquerque RJC, Blandford AD, Bogdanovich S, Hirano Y, Witta J, Fuchs E, Littman DR, Ambati BK, Rudin CM, Chong MMW, Provost P, Kugel JF, Goodrich JA, Dunaief JL, Baffi JZ, and Ambati J. 2011. DICER1 deficit induces Alu RNA toxicity in age-related macular degeneration. Nature 471: 325–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tarallo V, Hirano Y, Gelfand BD, Dridi S, Kerur N, Kim Y, Cho WG, Kaneko H, Fowler BJ, Bogdanovich S, Albuquerque RJC, Hauswirth WW, Chiodo VA, Kugel JF, Goodrich JA, Ponicsan SL, Chaudhuri G, Murphy MP, Dunaief JL, Ambati BK, Ogura Y, Yoo JW, Lee DK, Provost P, Hinton DR, Nunez G, Baffi JZ, Kleinman ME, and Ambati J. 2012. DICER1 Loss and Alu RNA Induce Age-Related Macular Degeneration via the NLRP3 Inflammasome and MyD88. Cell 149: 847–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Quinones-Valdez G, Tran SS, Jun HI, Bahn JH, Yang EW, Zhan LJ, Brummer A, Wei XT, Van Nostrand EL, Pratt GA, Yeo GW, Graveley BR, and Xiao XS. 2019. Regulation of RNA editing by RNA-binding proteins in human cells. Commun Biol 2: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Belair C, Sim S, and Wolin SL. 2017. Noncoding RNA Surveillance: The Ends Justify the Means. Chem Rev 118: 4422–4447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Spurlock CF, Tossberg JT, Guo Y, Sriram S, Crooke PS, and Aune TM. 2015. Defective structural RNA processing in relapsing-remitting multiple sclerosis. Genome Biol 16: 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hung T, Pratt GA, Sundararaman B, Townsend MJ, Chaivorapol C, Bhangale T, Graham RR, Ortmann W, Criswell LA, Yeo GW, and Behrens TW. 2015. The Ro60 autoantigen binds endogenous retroelements and regulates inflammatory gene expression. Science 350: 455–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Goodkin DE 1994. Interferon Beta-1b. Lancet 344: 1702–1703. [DOI] [PubMed] [Google Scholar]
- 56.Polman CH, and Hartung HP. 1995. The Treatment of Multiple-Sclerosis - Current and Future. Curr Opin Neurol 8: 200–209. [DOI] [PubMed] [Google Scholar]
- 57.Nortvedt MW, Riise T, Myhr KM, Nyland HI, and Hanestad BR. 1999. Type I interferons and the quality of life of multiple sclerosis patients. Results from a clinical trial on interferon alfa-2a. Mult Scler 5: 317–322. [DOI] [PubMed] [Google Scholar]
- 58.Pestal K, Funk CC, Snyder JM, Price ND, Treuting PM, and Stetson DB. 2015. Isoforms of RNA-Editing Enzyme ADAR1 Independently Control Nucleic Acid Sensor MDA5-Driven Autoimmunity and Multi-organ Development. Immunity 43: 933–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chung HC, Calis JJA, Wu XF, Sun T, Yu YP, Sarbanes SL, Thi VLD, Shilvock AR, Hoffmann HH, Rosenberg BR, and Rice CM. 2018. Human ADAR1 Prevents Endogenous RNA from Triggering Translational Shutdown. Cell 172: 811–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang H, Wang GL, Zhang LY, Zhang JB, Zhang JX, Wang QD, and Billiar TR. 2016. ADAR1 Suppresses the Activation of Cytosolic RNA-Sensing Signaling Pathways to Protect the Liver from Ischemia/Reperfusion Injury. Sci Rep-Uk 6:20248. [DOI] [PMC free article] [PubMed] [Google Scholar]