

Abstract
Strategies to engineer surfaces that can enable the selective inhibition of bacterial pathogens while preserving beneficial microbes can serve as tools to precisely edit the microbiome. In the oral microbiome, this selectivity is crucial in preventing the proliferation of cariogenic species such as Streptococcus mutans (S. mutans). In this communication, coatings consisting of a covalently tethered hydroxylated azobenzene (OH-AAZO) on glassy acrylic resins are studied and characterized for their ability to selectively prevent the attachment and growth of oral Streptococci biofilms. The coating applied on the surface of glassy resins inhibits the growth and proliferation of cariogenic S. mutans and S. oralis biofilms while A. actinomycetemcomitans, S. aureus, and E. coli biofilms are unaffected by the coating . The antibacterial effect is characterized as a function of both the OH-AAZO concentration in the coatings (≥50 mg mL−1) and the structure of the monomer in the coating. Preliminary mechanistic results suggest that the targeted bactericidal effect against Streptococci species is caused by a disruption of membrane ion potential, inducing cell death.
Keywords: antibacterial coatings, biofilms, biomaterials, polymers
Graphical Abstract
The rise in multidrug resistance toward pathogenic bacteria has severe implications for future treatments against infectious disease and calls for the urgent development of alternative biotechnologies and strategies to adequately address this crisis.[1] Bacterial biofilms that grow in difficult-to-reach areas limit the access of antibiotics that can be delivered, reducing the antibiotic’s effectiveness and worsening antibiotic resistance.[2,3] Antifouling coatings applied on medical devices such as catheters and endoscopes are one available option to prevent pathogenic biofilm growth while not exacerbating antibiotic resistance.[4–7] Several emerging technologies in this field include novel antibiotic drug delivery systems,[8–10] nanofabricated patterns,[5,6,11] silver nanoparticles,[12–14] quaternary ammonium compounds,[13,15–20] and anti-microbial peptides.[21–25] The challenges faced by emerging technologies such as toxicity and growing antibiotic resistance (silver nanoparticles and quaternary ammonium compounds),[26,27] enzymatic degradation and cost (AMPs),[28] and a reliance on a limited amount of chemical agent (antibiotic release) hinder the widespread adaptation of these technologies on a clinical level. Therefore, approaches that seek to minimize bacterial attachment and growth via the incorporation of inherently antibacterial coatings have gained attention.
Previously, our group was successful in designing an acrylated azobenzene (AAZO) coating that could be optomechanically induced to detach bacterial biofilms.[29] Azobenzenes are a class of photoisomeric compounds that consist of a N=N double bond connecting two benzene rings with the ability to transition between trans and cis isomers upon exposure to specific wavelengths of light. Upon simultaneous irradiation of multiple wavelengths of light, azobenzenes undergo photofluidization via rapid and transient trans–cis–trans isomerization. By covalently tethering the AAZO as coatings on polymers, a nonpathogen specific, antibiofilm coating that used light-induced mechanical motion to disrupt biofilms was developed. The successful disruption of bacterial biofilms from multiple species such as Escherichia coli (E. coli), Pseudomonas aeruginosa (P. aeruginosa), and Staphylococcus aureus (S. aureus) was demonstrated.[29]
However, a significant limitation of using mechanical forces to disrupt biofilms is that the success of this approach is dependent solely on the ability of the specific AAZO molecule to generate optimal mechanical forces to detach the biofilm. This limitation was highlighted when the AAZO molecule from the previous study failed to detach the robust Streptococcus mutans biofilm grown under sucrose-dependent conditions at 24 h. Unlike other bacteria, which saw 3-log reductions in colony forming units (CFU), S. mutans grown under sucrose-rich conditions saw only a minimal reduction in total CFUs that were attached to the coating. Within the oral cavity, S. mutans is a known cariogenic pathogen that plays a central role in the etiology of dental caries, the most prevalent biofilm-associated disease in the world.[30] S. mutans biofilms are difficult to remove using conventional dental hygiene practices following the biofilm’s adhesion to the surface of a tooth and/or a dental restoration.[31–35] Our continuing work in this area is part of a growing interest in materials science to develop novel strategies to prevent attachment and proliferation of cariogenic bacteria in the mouth.[28,36,37]
Recent studies of the oral microbiome have emphasized that the ability to preserve the beneficial microbial flora is almost as imperative as the need to eliminate pathogenic microbes.[38,39] Therefore, strategies that aim to inhibit or eliminate the growth of targeted bacteria while preserving beneficial species can be utilized to alter the microbiome in a targeted manner. Toward this end, we hypothesized that in addition to enabling the optomechanical disruption of biofilms, it would be beneficial to design azobenzene coatings that inhibit bacterial growth and proliferation. Therefore, a cytocompatible phenolic acrylated azobenzene (OH-AAZO) molecule was developed and covalently bonded onto substrates. Based on previous studies that explored the antibacterial properties of both natural and synthetic small molecule (poly)phenols,[40–43] the OH-AAZO molecule with a single hydroxyl group has been designed as part of library of acrylated molecules to be evaluated for their antibacterial properties. In contrast to prior studies on polyphenols, which focus primarily on free-floating molecules, the OH-AAZO molecule used in this study is covalently tethered on to the surface of the substrate and is severely limited in mobility. It is on the surprising pathogen-specific impacts of the covalently tethered OH-AAZO molecule that we wish to report in this communication.
OH-AAZO was synthesized from a commercially available azobenzene using conventional acylation methods (Figure S1, Supporting Information). For the in vitro studies, methyl methacrylate (MMA), triethylene glycol dimethacrylate (TEGDMA), and polymethyl methacrylate (PMMA) formulations (hereafter referred to as the substrate) were mixed in a ratio of 56:30:14 by weight and thermally cast in molds (80 °C for 1.5 h).The methacrylic C=C bond was monitored to ensure >95% double bond conversion via Fourier-transformed infrared spectroscopy.
The coating was prepared with a mixture of OH-AAZO in tetrahydrofuran (THF), azobisisobutyronitrile (AIBN, 2 wt%, thermal initiator), and acrylated Rhodamine B (0.2 wt% for imaging). THF was selected for more efficient packing and aggregation of the pendant azobenzene groups in the coating solution.[44] The coating solutions were applied to the substrate via a spin-coating protocol and cured thermally at 100 °C. The coated and uncoated substrates were subsequently extracted in MilliQ H2O and wet-autoclaved at 120 °C and 15 psi. The comprehensive extraction protocol of samples was implemented to ensure that there was complete absence of diffusible bactericidal materials from both the OH-AAZO coated and uncoated substrates. The coated and uncoated substrates were analyzed via UV–vis to ensure the uniformity of the coating prior to use (Figure S2, Supporting Information).
The antibacterial activity of OH-AAZO was observed to be concentration dependent. Figure 1 compares OH-AAZO coatings prepared from coating solution concentrations of 100, 50, 25, and 5 mg mL−1 in THF, which correspond to OH-AAZO surface concentrations of 12.6, 6.3, 3.2, and 0.6 μg mm−2, respectively. After 24 h of biofilm growth, and in the absence of any light treatment, the 100 and the 50 mg mL−1 OH-AAZO substrates had no observable CFUs detected on their surfaces, which indicated that both substrates were completely sterile (which we define here as 0 CFUs). At 25 mg mL−1 OH-AAZO, there was a 4-log reduction in CFUs for the S. mutans biofilm relative to the uncoated control. At 5 mg mL−1 OH-AAZO, there was no significant difference in CFUs between the coated and uncoated substrates (≈1.0 × 108 CFUs for both). In the case of the uncoated control, 5, and 25 mg mL−1 coated substrates, the concentration of bacterial biofilms growing on the surface of the substrates and the surrounding media was comparable. However, when complete biofilm inhibition was observed on the substrates, as in the case of the 50 and 100 mg mL−1 coated substrates, bacterial growth in the surrounding media was absent as well. Because the substrates and surrounding media reached complete sterility at concentrations at or above 50 mg mL−1 OH-AAZO, 50 mg mL−1 substrates were selected for all subsequent studies unless stated otherwise. Cytotoxicity studies (ISO9993 protocol) confirmed that the 50 mg mL−1 OH-AAZO substrates were cytocompatible with L929 cells (Figure S3, Supporting Information). To confirm the absence of a biofilm, a crystal violet assay was performed on 50 mg mL−1 coated and uncoated substrates, and the absence of a biofilm was observed on the coated sample (Figure S4, Supporting Information).
Figure 1.
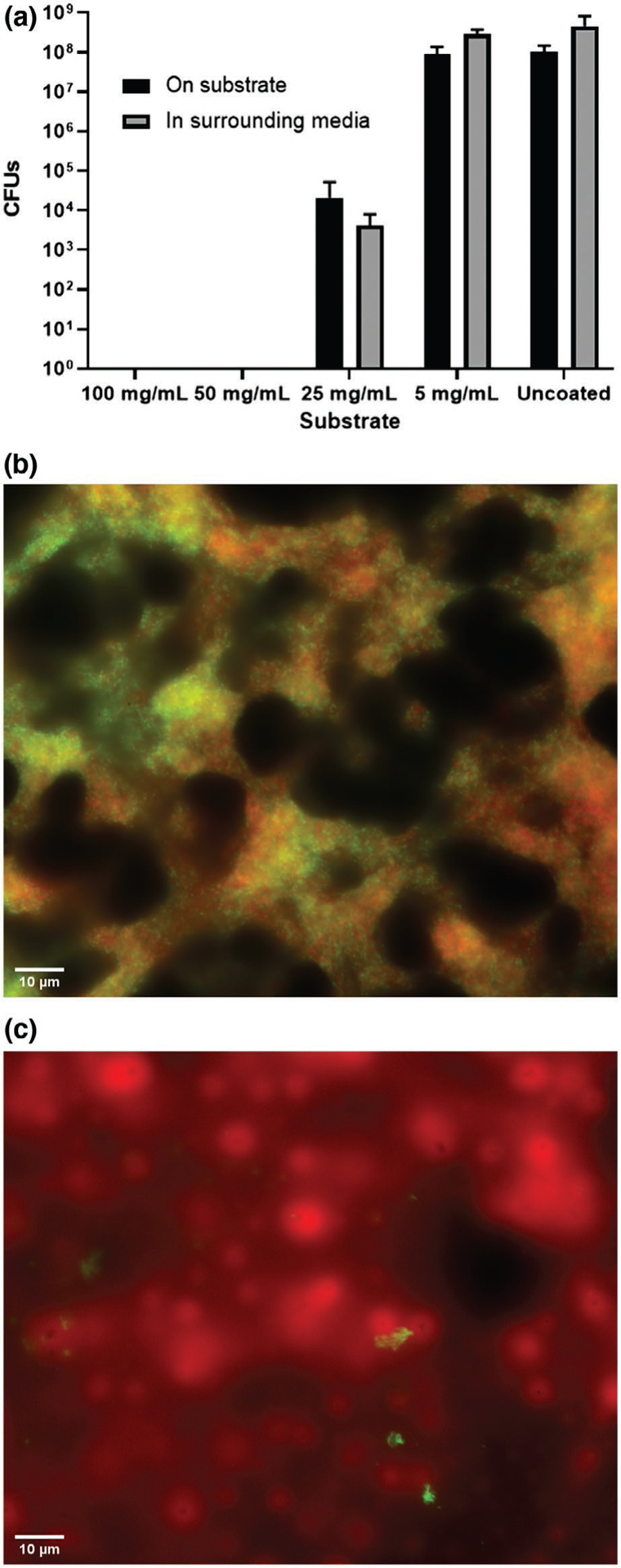
a) S. mutans biofilms growth as a function of OH-AAZO concentration within the coating formulation (all n ≥ 3). Digital microscope images (630X magnification) of S. mutans bacteria growing on both b) uncoated and c) coated substrates.
Microscopic images (Zeiss Axioplane II digital microscope) of the coated OH-AAZO coatings confirmed visually the prevention of S. mutans biofilm growth. Following 24 h of biofilm growth, the substrates were stained with BacLight live-dead stains and imaged using the appropriate fluorescence channels (Figure 1). The coated substrates (Figure 1c) contained high amounts of dead bacteria (red) and little to no live bacteria (green) when compared to the uncoated control substrate (Figure 1b).
The efficacy of the coating to inhibit the growth and proliferation of other oral bacterial strains was also studied. The commensal bacteria S. oralis is often seen as a component of the normal human oral microbiota, but is a Gram-positive species that is capable of opportunistic pathogenicity in immunocompromised individuals and is implicated bacterial endocarditis and respiratory diseases.[45] A. actinomycetemcomitans is a Gram-negative, oral disease-causing pathogen that is often known to cause aggressive periodontal disease. Additionally, the OH-AAZO coatings were also tested against biofilm-forming nonoral pathogenic bacterial strains such as S. aureus and E. coli which are Gram-positive and Gram-negative, respectively.
As seen in Figure 2, neither S. mutans nor S. oralis grew on the OH-AAZO substrates nor in the media in the well containing the substrate over the time required for the biofilms to reach stationary phase. However, robust biofilm growth was observed on the uncoated control substrates, indicating that the presence of the coating prevented the growth and proliferation of the Streptococci biofilms. Interestingly, there was no discernable inhibitory effect or reduction in CFUs observed for the A. actinomycetemcomitans, S. aureus, or E. coli biofilms grown on the substrates in comparison to the controls, indicating that the OH-AAZO selectively inhibited the growth and proliferation of Streptococci biofilms. The exciting findings from our initial studies imply that an in-depth understanding of the mechanism by which the OH-AAZO-coated substrate can prevent the growth and proliferation of Streptococci biofilms can allow us to design coatings that can be then used to alter the microbiome and eliminate pathogenic species in a targeted manner. To further elucidate the mechanism by which biofilm inhibition was achieved, sucrose-dependent S. mutans biofilms were utilized as model biofilms for the remainder of this study. S. mutans biofilms have long been utilized as model biofilms due their ability to generate a robust extracellular polysaccharide matrix.[30]
Figure 2.
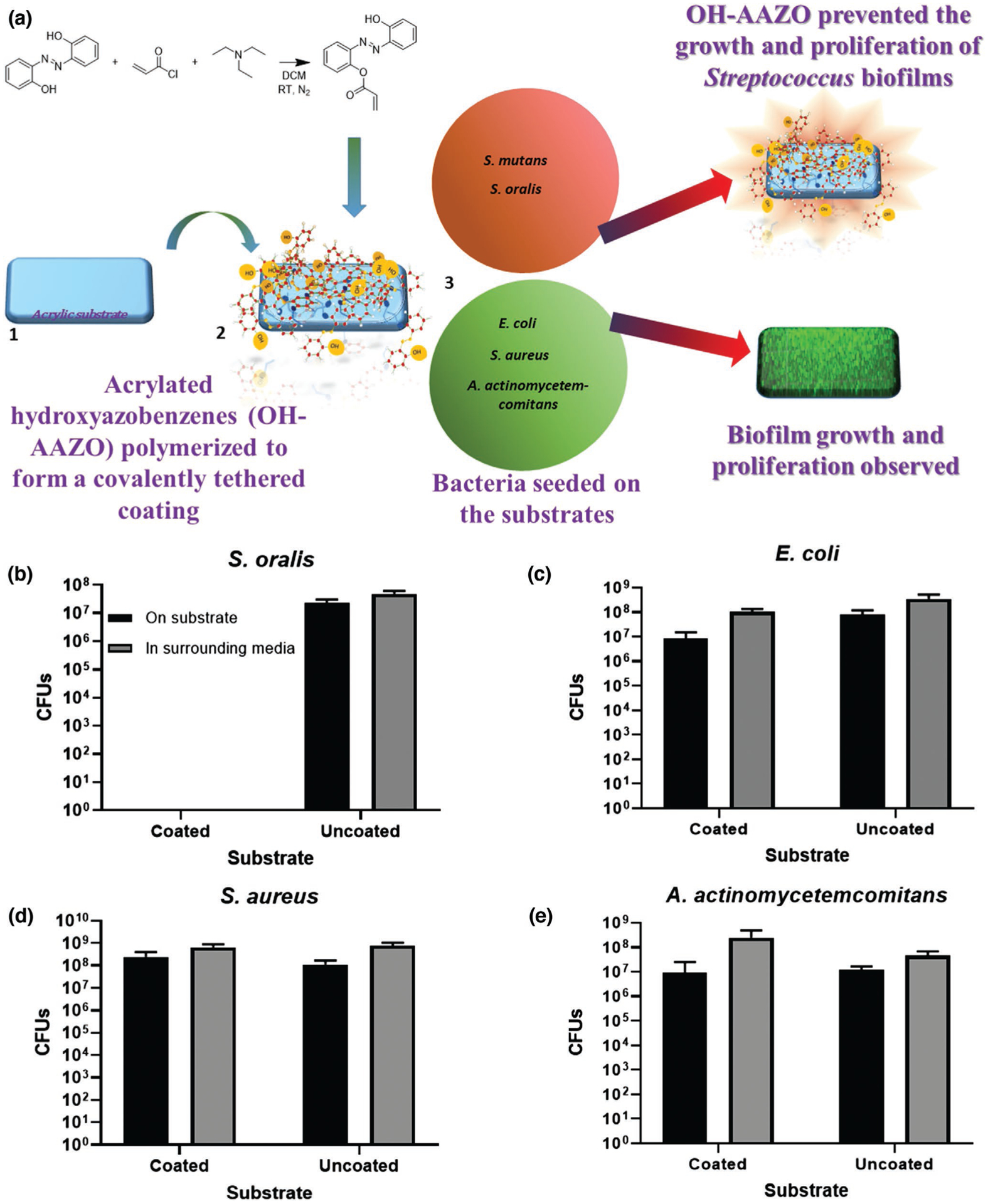
a) Biofilms grown on OH-AAZO coatings and the observed selective inhibitory effect on Streptococcus biofilms. Bacterial biofilms of b) S. oralis indicate selective inhibition of Streptococci biofilms on OH-AAZO coated-substrates, whereas bacterial biofilms of c) E. coli, d) S. aureus, and e) A. actinomycetemcomitans were not inhibited. All biofilms grown on coated substrates were compared to those grown on uncoated substrates.
Toward developing a mechanistic understanding as to what specific functionality within the OH-AAZO molecule is responsible for the selective inhibition of Streptococci, several other polymerized coatings containing similar structural moieties to OH-AAZO were studied. The three monomers tested were AAZO (an acrylic azobenzene), 4-hydroxybenzyl acrylate (HBA, an acrylic phenol), and hydroxyethyl methacrylate (HEMA, an aliphatic hydroxyl methacrylate). Robust biofilms on the surfaces of AAZO, HBA, and HEMA-coated substrates were observed at 24 h (107–108 CFUs). Only the OH-AAZO coating was successful in inhibiting S. mutans growth. The structure–property implication of these results shows that the combination of azobenzenes and phenols give rise to the anti-Streptococci biofilm effect, which will be of great consideration for us as we design more molecules for antibacterial applications (Figure 3a).
Figure 3.
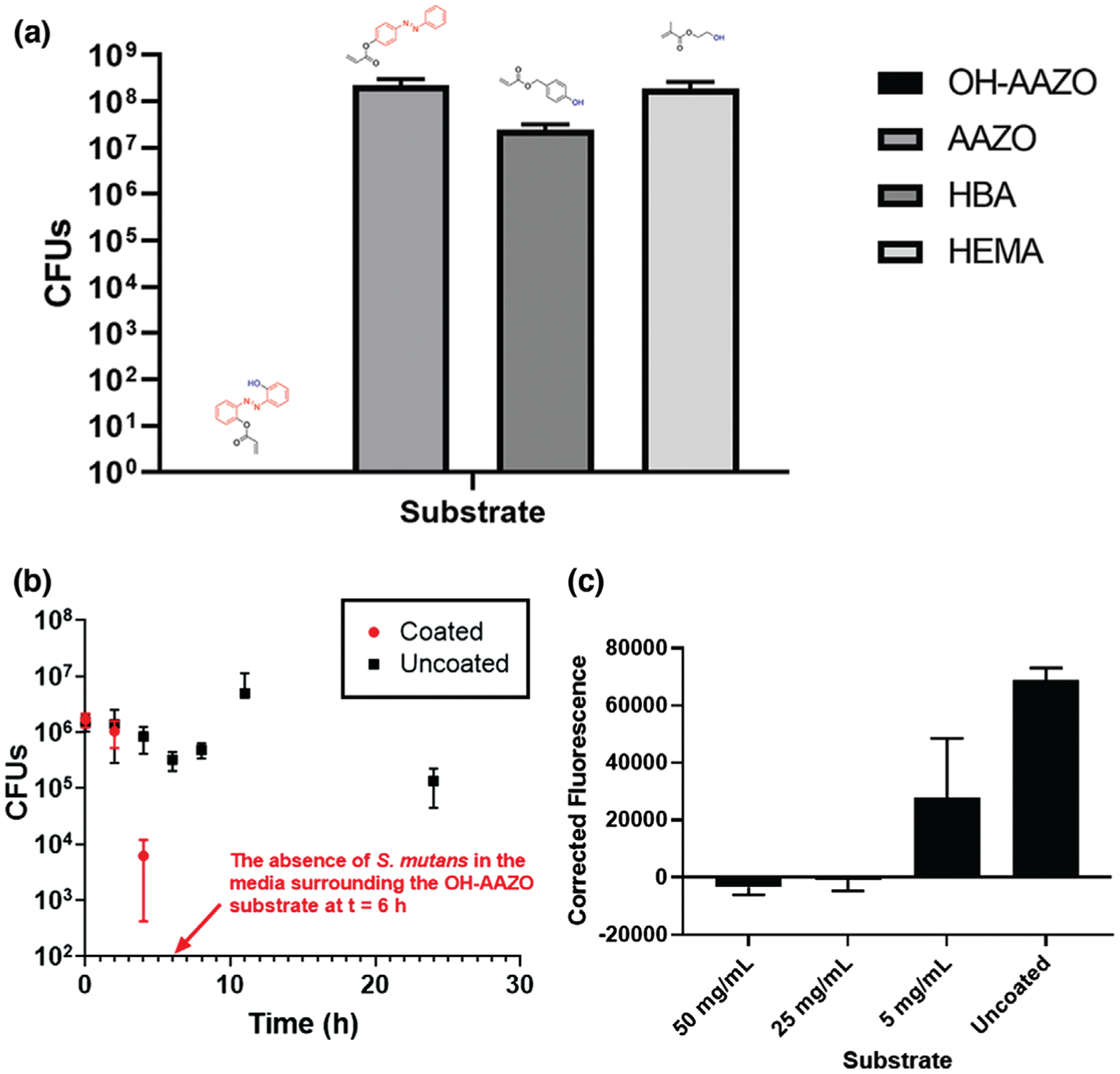
a) Coatings generated from monomers with different functional groups to study the inhibitory effect. CFU values listed are from biofilms growing on the substrate. S. mutans biofilm growth quantified at 24 h indicates that the antibacterial properties are specific to the OH-AAZO coatings. b) Kill curve of S. mutans in media containing OH-AAZO coated substrates in comparison with an uncoated substrate over time. c) The membrane potential assay results at different concentrations of coated OH-AAZO substrates. Raw data was corrected against the fluorescence values of the corresponding substrate (all raw values n = 3).
Interestingly, the presence of OH-AAZO prevented S. mutans biofilm formation not only on the substrate but also in the surrounding media, a phenomenon that was observed in all studies reported here. To ensure the absence of leachables (small molecules, trace solvents, etc.) from the substrates, all substrates used in this study were exhaustively extracted to ensure that the molecules that formed the coating were covalently linked to the underlying substrate. To this end, an additional study was performed in which an OH-AAZO coated substrate was exposed to the aqueous extraction process and placed in a well containing media, which was then sampled in a Kirby-Bauer assay over the course of 12 h on plates streaked with S. mutans. The outcome of this process, described in Table S1 in the Supporting Information, indicates that no small molecules were leaching from the coating and inducing cell death to a significant degree. Therefore, it can be concluded that the tethered OH-AAZO coating was responsible for the absence of biofilms in both the substrate and in the surrounding media. To further confirm the OH-AAZO coating’s ability to induce cell death in the surrounding area as a function of time, a kinetic study to observe bacteria growth in the surrounding media was designed. Toward this end, aliquots of the media surrounding the OH-AAZO substrate with S. mutans were sampled at different time points (Figure 3b). At t = 0, S. mutans colonies (1 × 106 CFUs) were introduced to the well containing the OH-AAZO coated substrate. At t = 4 h, the CFU count in the well saw a 2-log reduction, and by t = 6 h, the CFU count had reduced to zero, indicating that the OH-AAZO substrate was successful in eliminating S. mutans not just from the surface of the OH-AAZO substrate but from the well as a whole in a relatively short amount of time. In a separate study, the entire volume of the media in the well plated onto a brain heart infusion (BHI) agar plate yielded no discernable CFU counts, further confirming that the OH-AAZO coating was successful in eliminating the S. mutans colonies from the surrounding media (Figure S5, Supporting Information).
Currently ongoing studies in our lab are aimed at identifying the specific pathway by which the OH-AAZO coating is able to inhibit Streptococci biofilm growth and proliferation. For our preliminary studies on identifying the mechanism by which Streptococci biofilms are impacted by the coating, a DIBAC4(3) membrane potential assay was selected to quantify any decrease in metabolism in the biofilm cells.[41] The DIBAC4(3) assay results on S. mutans exposed to OH-AAZO showed a drop in fluorescence at higher concentrations of OH-AAZO, which is a sign of hyperpolarization of the membrane (Figure 3c). These results imply a disruption of ion homeostasis, which may ultimately be leading to cell death. Clearly, more detailed studies are required to establish a pathway via which the tethered OH-AAZO coatings are able to inhibit biofilm growth both on the substrate and in the surrounding media.
Our initial results indicate that the OH-AAZO has the potential to form a Streptococci-selective antibacterial coating. Future work will focus on the further the understanding the biochemical mechanism behind the observed biofilm inhibition effect, such as monitoring reactive oxygen species generation, a likely cause of the observed ion homeostasis disruption, upon exposure to OH-AAZO (specifically for S. mutans biofilms).[40,46–49] Establishing biochemical pathways that are affected by the selective inhibition observed here will be utilized to design more potent, cytocompatible antibacterial coatings that can selectively inhibit multiple species and provide a toolkit to tailor and edit microbiomes.
Experimental Section
Materials:
Unless otherwise stated, all compounds and solvents were used as received. 4-hydroxyazobenzene, 2’-dihydroxyazobenzene, and AIBN were acquired from Aldrich. AIBN was recrystallized in methanol prior to use. HEMA, MMA, and PMMA (≈35 k) were acquired from Acros Organics. TEGDMA, 2-hydroxybenzyl alcohol, and acyl chloride were acquired from Tokyo Chemical Industry. N,N-dimethylformamide (DMF), triethylamine, and silica gel were acquired from Fisher. THF was acquired from VWR. Dichloromethane (DCM) was acquired from Millipore. Rhodamine B was acquired from PolySciences.
S. mutans American Type Culture Collection (ATCC 25175), S. aureus (Herbert, 2010 HG001 or AH2183 or RN1HG), S. oralis (ATCC 9811), and A. actinomycetemcomitans (ATCC 43718), and E. coli (Mysorekar, 2013) were provided from the sources listed. BBL BHI media and Difco agar were acquired from Becton, Dickinson and Company. Sucrose for cell growth was acquired from MP Biomedicals, LLC. Phosphate-buffered saline (PBS) was acquired from Bioland Scientific. Dulbecco’s modified eagle medium (DMEM) media and fetal bovine serum (FBS) was acquired from Fisher. Penicillin-streptomycin and trypsin solution 10X (2.5%) was acquired from Aldrich.
Synthesis of AAZO:
Acrylated azobenzene (AAZO) was synthesized as described by Kehe et al.[29] 1H NMR: (500 MHz, CDCl3, δ) 7.97 (d, 2H), 7.91 (d, 2H), 7.51 (m, 3H), 7.30 (d, 2H), 6.63 (d, 1H), 6.35 (dd, 1H), 6.05 (d, 1H). The outline for the synthesis is described in Figure S1 in the Supporting Information.
Synthesis of OH-AAZO:
The phenolic acrylated azobenzene (OH-AAZO) was synthesized as described by Kehe et al. with several modifications. Briefly, 2,2’-dihydroxyazobenzene (0.5261 g, 2.46 mmol), trimethylamine (0.41 mL, 2.95 mmol), and DCM (25 mL) were combined in a flame-dried Schlenk flask under N2 and stirred for 0.5 h at 0 °C. Acyl chloride (0.20 mL, 2.46 mmol) was added dropwise and the solution was stirred overnight at room temperature. The organic layer was washed with water three times (15 mL per wash) and dried over MgSO4. The crude product was purified via column chromatography using DCM as an eluent (Rf = 0.77) and yielded OH-AAZO as a red-orange crystal. Yield: 0.41007 g (62%). 1H NMR (500 MHz, CDCl3, δ) 12.75 (s, 1H), 7.97 (d, 2H), 7.55 (t, 1H), 7.38 (m, 3H), 7.09 (m, 2H), 6.75 (d, 1H), 6.46 (dd, 1H), 6.16 (d, 1H). The outline for the synthesis is described in Figure S1 in the Supporting Information.
Synthesis of HBA Control Monomer:
HBA was prepared by the method described previously.[50] Briefly, 4-hydroxybenzyl alcohol (1.2637 g, 10.2 mmol) was combined with triethylamine (1.70 mL, 12.2 mmol) in THF (12 mL) in a flame-dried Schlenk flask under N2. The solution was mixed for 0.5 h at room temperature and then cooled to 0 °C. Acyl chloride (0.82 mL, 10.2 mmol) was added slowly dropwise and allowed to stir for 24 h. The THF was removed via rotovap, and the residue was extracted with a 20 mL DCM wash twice with 0.1 m HCl, then twice with DI water. The DCM layer was run through a silica plug to yield an opaque slightly yellow oil. Yield: 0.8132 g (45%). 1H NMR (500 MHz, dimethylsulfoxide-d6 (DMSO-d6), δ) 9.56 (s, 1H), 7.47 (d, 2H), 7.21 (d, 2H), 6.76 (d, 1H), 6.52 (d, 1H), 6.37 (m, 2H), 6.16 (m, 2H), 5.99 (dd, 1H). The outline for the synthesis is described in Figure S1 in the Supporting Information.
Making Polymer Substrates:
Formulations of MMA:TEGDMA:PMMA base polymers were prepared using 56 wt% MMA, 30 wt% tetraethylene glycol dimethacrylate (TEGDMA), and 14 wt% PMMA (MW ≈35 k). The components were stirred at 70 °C for 2 h. 1 wt% AIBN (relative to the total mass of the mixture) was added and the formulation was stirred for an additional 0.5 h.
Circular holes (d = 5.5 mm) were punched in a rubber spacer (0.8 mm thickness) placed on a glass microscope slide. The formulations were pipetted into each hole and sandwiched between an additional glass slide with binder clips. The substrates were cured at 80 °C for 1.5 h. Conversion rates were observed via Fourier-transformed infrared spectroscopy for the integration under the acrylate peak (6250–6096 cm−1) (Figure S6, Supporting Information).
OH-AAZO Coatings:
Coating formulations of OH-AAZO were prepared in THF, all others were prepared in DMF. The THF volume was kept constant (1 mL) and the mass of OH-AAZO monomer was varied to attain the desired concentration. In addition to the OH-AAZO monomer, the coating also consisted of AIBN (2 wt% relative to the mass of the monomer) and Rhodamine B (0.2 wt% relative to the mass of the monomer).
Once cured, the MMA:TEGDMA:PMMA base polymers were placed into a mount on a square glass microscope slide. 3 μL of the coating formulation was added via pipette to one side of the substrate and spin coated (1000 rpm for 1 min, 6000 rpm for 3 s) then the process was repeated on the other side. Once both sides were coated, they were placed in an oven for 3 h at 100 °C, then under vacuum for an additional 0.5 h.
Extraction Protocol:
Following curing, residual monomer was aqueously extracted by autoclaving (15 psi, 120 °C) for 1 h in MilliQ water (≈5 mL water per substrate). The MilliQ water was replaced with fresh water (≈5 mL water per substrate) and placed in an 80 °C oven for 2 h. The substrates were blotted dry and analyzed via UV–vis spectroscopy to confirm the relative amount of the coating (Figure S2, Supporting Information). All substrates were autoclaved prior to their use with both mammalian and bacterial cells.
Cytotoxicity:
Cytotoxicity studies were performed as described previously.[51] Briefly, L929 mouse epithelial cells were suspended in cell culture media (DMEM, 10% FBS, 1% penicillin streptomycin) to a final concentration of 1.06 × 105 cells mL−1. 1 mL of the suspension was added to each well in a 24-well plate containing the substrates and allowed to grow to confluence. Cell growth was monitored via a phase contrast inverted microscope (Leica, WLD MPS32, Germany). Once confluent, the media was aspirated off, and 500 μL of a 1 mg mL−1 solution of MTT reagent in DMEM media was added to each well. The plate was incubated for 4 h, and 500 μL of 20% sodium dodecyl sulfate in 50% DMF was added to each well containing the MTT reagent. The plate was incubated for an additional 24 h, then removed and analyzed by an absorbance reading at λ = 570 nm.
Culturing S. Mutans:
S. mutans biofilms were grown from a 24 h BHI liquid culture inoculated with an isolated S. mutans colony. The liquid culture was diluted 1:50 in BHI media containing 1 wt% sucrose in each well in a 96-well plate containing a substrate of interest. The plate was incubated in the dark at 37 °C and 5% CO2 for t = 24 h. The growth curve on all substrates tested in this study is outlined in Figure S7 in the Supporting Information.
Culturing S. Oralis, A. Actinomycetemcomitans, S. Aureus, E. Coli Bacterial Strains:
S. oralis and A. actinomycetemcomitans were streaked on a BHI agar plate and incubated at 37 °C and 5% CO2 for 48 h. An isolated colony was inoculated in 5 mL BHI media for 12 h under the same incubation conditions, followed by a 1:10 dilution in BHI media in a 96-well plate containing the substrates of interest. S. oralis biofilms were allowed to grow for 9 h and A. actinomycetemcomitans biofilms were allowed to grow for 30 h.
Methicillin-susceptible S. aureus biofilms were grown from a 20 h BHI liquid culture inoculated with an S. aureus colony. The liquid culture was diluted 103 to a stock solution in BHI, then diluted 1:10 in BHI media containing 0.4 wt% glucose in a 96-well plate. The plate was incubated in the dark at 37 °C for 24 h.
E. coli biofilms were grown from a 20 h BHI inoculated with an E. coli colony. The liquid culture was diluted 103 to a stock solution in BHI, then diluted 1:10 in BHI media in a 96-well plate. The plate was incubated in the dark at 37 °C for 24 h.
Biofilm Inhibition Assays:
Following biofilm growth in the 96-well plate, the substrates were aseptically removed from the 96-well plate and placed in a vial containing 5 mL of 1X PBS solution. The vials were sonicated in a water bath for 10 min at 45 °C and the wells that previously contained the substrates were briefly sonicated using a probe sonicator (3 W root mean square output) for 10 s per well. 200 μL aliquots of the sonicated PBS solution were placed in a 96-well plate and diluted 1:10 in each subsequent row. 20 μL aliquots of the sonicated media wells were placed in a 96-well plate containing 180 μL of PBS solution and diluted 1:10 in each subsequent row. Each column was then plated at a volume of 10 μL on a BHI agar plate in triplicate. The plates were then incubated at 37 °C and 5% CO2 for 48 h. The CFUs on each plate were counted in order to quantify the amount of biofilm remaining on the surface of the substrate and the bacteria remaining in the media after the removal of the substrate using the following equations
| (1) |
| (2) |
where CFUsubstrate and CFUmedia are the total CFU counts on the substrate and in the media well, respectively, CFUspot is the number of CFUs counted in a single spot on the plate, and Row is the corresponding numerical row number for the spot counted.
Microscopy:
Microscope images were obtained using a Zeiss digital microscope at 630X magnification using the fluorescein isothiocyanate and CY3 channels. Following biofilm growth, the substrates were placed in a solution of live-dead stains (Invitrogen BacLight) containing 1.5 μL Component A (SYTO 9, green fluorescent nucleic acid stain) and 1.5 μL Component B (propidium iodide, red fluorescent nucleic acid stain) in 200 μL of sterile water. The substrates were soaked for 3 min, then gently washed in sterile water for 10 s, dried, and fixed to a glass slide prior to imaging.
Membrane Potential Assay:
Membrane potential assays were performed as described by Wu et al. with minor modifications.[41] S. mutans bacteria grown for 24 h in BHI broth were inoculated in a 96-well plate as described above. The plate was incubated for 8 h at 37 °C and 5% CO2. 2 μL of 0.1 mg mL−1 DIBAC4(3) solution in DMSO was added to each well. The plate was then incubated in the dark overnight. The fluorescence reading was taken in a plate reader at λexcitation = 492 nm and λemission = 515 nm. Fluorescence values in bacteria-containing wells were normalized against fluorescence values containing the same substrate but no bacteria.
Supplementary Material
Acknowledgements
D.P.N. acknowledges financial support from National Institutes of Health-National Institute of Dental and Craniofacial Research (Grant No. K25DE027418). The authors thank Dr. Alexander Horswill (University of Colorado Department of Immunology and Microbiology) and Dr. Indira Mysorekar (Washington University Department of Obstetrics & Gynecology) for the S. aureus and E. coli bacterial strains, respectively. The authors thank Gannon Kehe for providing the AAZO control monomer.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
The ORCID identification number(s) for the author(s) of this article can be found under https://doi.org/10.1002/admi.201902149.
Contributor Information
Dylan I. Mori, Department of Craniofacial Biology, University of Colorado Anschutz Medical Campus, Aurora, CO 80045, USA
Michael J. Schurr, Department of Immunology and Microbiology, University of Colorado Anschutz Medical Campus, Aurora, CO 80045, USA
References
- [1].Davies J, Microbiologia 1996, 12, 9. [PubMed] [Google Scholar]
- [2].Mah T-FC, O’Toole GA, Trends Microbiol. 2001, 9, 34. [DOI] [PubMed] [Google Scholar]
- [3].Baptista PV, McCusker MP, Carvalho A, Ferreira DA, Mohan NM, Martins M, Fernandes AR, Front. Microbiol 2018, 9, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Li X, Bai H, Yang Y, Yoon J, Wang S, Zhang X, Adv. Mater 2019, 31, 1805092. [DOI] [PubMed] [Google Scholar]
- [5].Mann EE, Manna D, Mettetal MR, May RM, Dannemiller EM, Chung KK, Brennan AB, Reddy ST, Antimicrob. Resist. Infect. Control 2014, 3, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].May RM, Hoffman MG, Sogo MJ, Parker AE, O’Toole GA, Brennan AB, Reddy ST, Clin. Transl. Med 2014, 3, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Gu H, Lee SW, Buffington SL, Henderson JH, Ren D, ACS Appl. Mater. Interfaces 2016, 8, 21140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Guo J-H, Skinner G, Harcum W, Barnum P, Pharm. Sci. Technolo. Today 1998, 1, 254. [Google Scholar]
- [9].Oh JK, Drumright R, Siegwart DJ, Matyjaszewski K, Prog. Polym. Sci 2008, 33, 448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Billings N, Birjiniuk A, Samad TS, Doyle PS, Ribbeck K, Rep. Prog. Phys 2015, 78, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Mann EE, Mettetal MR, May RM, Drinker MC, Stevenson BC, Baiamonte VL, Marso JM, Dannemiller EA, Parker AE, Reddy ST, Sande MK, J. Microbiol. Exp 2014, 1, 1. [Google Scholar]
- [12].Wang S, Huang Q, Liu X, Li Z, Yang H, Lu Z, ACS Biomater. Sci. Eng 2019, 5, 2030. [DOI] [PubMed] [Google Scholar]
- [13].Cocco AR, De Oliveira Da Rosa WL, Da Silva AF, Lund RG, Piva E, Dent. Mater 2015, 31, 1345. [DOI] [PubMed] [Google Scholar]
- [14].Zhao L, Wang H, Huo K, Cui L, Zhang W, Ni H, Zhang Y, Wu Z, Chu PK, Biomaterials 2011, 32, 5706. [DOI] [PubMed] [Google Scholar]
- [15].Shadmon H, Basu A, Eckhard LH, Domb AJ, Beyth N, New J. Chem. 2018, 42, 15427. [Google Scholar]
- [16].Venault A, Yang HS, Chiang YC, Lee BS, Ruaan RC, Chang Y, ACS Appl. Mater. Interfaces 2014, 6, 3201. [DOI] [PubMed] [Google Scholar]
- [17].Chou YN, Venault A, Cho CH, Sin MC, Yeh LC, Jhong JF, Chinnathambi A, Chang Y, Chang Y, Langmuir 2017, 33, 9822. [DOI] [PubMed] [Google Scholar]
- [18].Venault A, Chang Y, Langmuir 2019, 35, 1714. [DOI] [PubMed] [Google Scholar]
- [19].Rzhepishevska O, Hakobyan S, Ruhal R, Gautrot J, Barbero D, Ramstedt M, Biomater. Sci 2013, 1, 589. [DOI] [PubMed] [Google Scholar]
- [20].Magennis EP, Francini N, Mastrotto F, Catania R, Redhead M, Fernandez-Trillo F, Bradshaw D, Churchley D, Winzer K, Alexander C, Mantovani G, PLoS One 2017, 12, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].He J, Eckert R, Pharm T, Simanian MD, Hu C, Yarbrough DK, Qi F, Anderson MH, Shi W, Antimicrob. Agents Chemother 2007, 51, 1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Li L, Guo L, Lux R, Eckert R, Yarbrough D, He J, Anderson M, Shi W, Int. J. Oral Sci 2010, 66. [DOI] [PMC free article] [PubMed]
- [23].Murugesh J, Sameera ASRS, Aysha S, Int. J. Oral Heal. Dent 2015, 1, 81. [Google Scholar]
- [24].Liang D, Li H, Xu X, Liang J, Dai X, Zhao W, Chem. Biol. Drug Des 2019, 94, 1768. [DOI] [PubMed] [Google Scholar]
- [25].Zhang L, Fang Z, Li Q, Cao CY, J. Mater. Sci. Mater. Med 2019, 30, 1. [DOI] [PubMed] [Google Scholar]
- [26].Salas-Orozco M, Niño-Martínez N, Martínez-Castañón G-A, Méndez FT, Jasso MEC, Ruiz F, J. Nanomater 2019, 2019, 1. [Google Scholar]
- [27].Zhang C, Cui F, Zeng G, Jiang M, Yang Z, Yu Z, Zhu M, Shen L, Sci. Total Environ 2015, 518.–, 352. [DOI] [PubMed]
- [28].Ding X, Duan S, Ding X, Liu R, Xu F-J, Adv. Funct. Mater 2018, 28, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kehe GM, Mori DI, Schurr MJ, Nair DP, ACS Appl. Mater. Interfaces 2019, 11, 1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Colombo APV, do Souto RM, da Silva-Boghossian CM, Miranda R, Lourenço TGB, Curr. Oral Heal. Rep 2015, 37.
- [31].Kidd EAM, J. Dent. Educ 2001, 65, 997. [PubMed] [Google Scholar]
- [32].Lai G, Li M, in Contemporary Approach to Dental Caries, InTech Open, London, UK: 2012, pp. 403–422. [Google Scholar]
- [33].Jokstad A, Dent. Mater 2016, 32, 11. [DOI] [PubMed] [Google Scholar]
- [34].Kidd EAM, Beighton D, J. Dent. Res 1996, 75, 1942. [DOI] [PubMed] [Google Scholar]
- [35].Cugini MA, Warren PR, J. Can. Dent. Assoc. (Tor) 2006, 72, 323. [PubMed] [Google Scholar]
- [36].Siedenbiedel F, Tiller JC, Polymers (Basel, Switz) 2012, 4, 46. [Google Scholar]
- [37].Hasan J, Crawford RJ, Ivanova EP, Trends Biotechnol. 2013, 31, 295. [DOI] [PubMed] [Google Scholar]
- [38].Kreth QF, Merritt J, Shi W,J, J. Bacteriol 2005, 187, 7193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Abranches J, Zeng L, Kajfasz JK, Palmer SR, Chakraborty B, Wen ZT, Richards VP, Brady LJ, Lemos JA, Microbiol. Spectr 2018, 6, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Rempe CS, Burris KP, Lenaghan SC, Stewart CN, Front. Microbiol 2017, 8, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Wu Y, Bai J, Zhong K, Huang Y, Qi H, Jiang Y, Gao H, Molecules 2016, 21, 1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Piotto S, Concilio S, Sessa L, Diana R, Torrens G, Juan C, Caruso U, Iannelli P, Molecules 2017, 22, 1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Nijampatnam B, Zhang H, Cai X, Michalek SM, Wu H, Velu SE, ACS Omega 2018, 3, 8378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Jeong SP, Renna LA, Boyle CJ, Kwak HS, Harder E, Damm W, Venkataraman D, Sci. Rep 2017, 7, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Do T, Jolley KA, Maiden CJ, Gilbert SC, Clark D, Wade WG, Beighton D, Microbiology 2009, 155, 2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Nijampatnam B, Casals L, Zheng R, Wu H, Velu SE, Bioorg, Med. Chem. Lett 2016, 26, 3508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Akagawa M, Shigemitsu T, Suyama K, Biosci. Biotechnol. Biochem 2003, 67, 2632. [DOI] [PubMed] [Google Scholar]
- [48].Nakamura K, Ishiyama K, Sheng H, Ikai H, Kanno T, Niwano Y, J. Agric Food Chem 2015, 63, 7707. [DOI] [PubMed] [Google Scholar]
- [49].Matsumoto M, Hamada S, Ooshima T, FEMS Microbiol. Lett 2003, 228, 73. [DOI] [PubMed] [Google Scholar]
- [50].Seo S, Lee DW, Ahn JS, Cunha K, Filippidi E, Ju SW, Shin E, Kim BS, Levine ZA, Lins RD, Israelachvili JN, Waite JH, Valentine MT, Shea JE, Ahn BK, Adv. Mater 2017, 29, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Riss TL, Moravec RA, Niles AL, Duellman S, Benink HA, Worzella TJ, Minor L, Cell Viability Assays (Promega) 2016, 1.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.