

SUMMARY
Radiation sensitive 52 (RAD52) is an important factor for double-strand break repair (DSBR). However, deficiency in vertebrate/mammalian Rad52 has no apparent phenotype. The underlying mechanism remains elusive. Here, we report that RAD52 deficiency increased cell survival after camptothecin (CPT) treatment. CPT generates single-strand breaks (SSBs) that further convert to double-strand breaks (DSBs) if they are not repaired. RAD52 inhibits SSB repair (SSBR) through strong single-strand DNA (ssDNA) and/or poly(ADP-ribose) (PAR) binding affinity to reduce DNA-damage-promoted X-Ray Repair Cross Complementing 1 (XRCC1)/ligase IIIα (LIG3α) co-localization. The inhibitory effects of RAD52 on SSBR neutralize the role of RAD52 in DSBR, suggesting that RAD52 may maintain a balance between cell survival and genomic integrity. Furthermore, we demonstrate that blocking RAD52 oligomerization that disrupts RAD52’s DSBR, while retaining its ssDNA binding capacity that is required for RAD52’s inhibitory effects on SSBR, sensitizes cells to different DNA-damaging agents. This discovery provides guidance for developing efficient RAD52 inhibitors in cancer therapy.
In Brief
Wang et al. show that vertebrate/mammalian RAD52 promotes CPT-induced cell death via inhibition of PARP-mediated SSBR, which involves RAD52’s strong ssDNA/PAR binding affinity that reduces DNA-damage-promoted XRCC1-LIG3a interaction. Blocking of RAD52 oligomerization, while retaining the ssDNA binding capacity of RAD52, efficiently sensitizes cells to different DNA-damaging agents.
Graphical Abstract
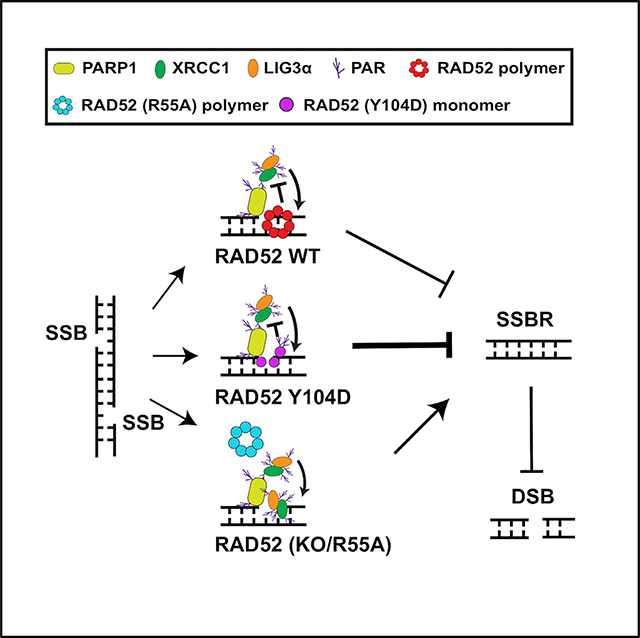
INTRODUCTION
DNA strand breaks include single-strand breaks (SSBs) and double-strand breaks (DSBs). SSB repair (SSBR) is a much more efficient process compared to DSB repair (DSBR): approximately 70% of SSBs but only ~30% of DSBs can be repaired within a 10-min time frame in mammalian cells (Schipler and Iliakis, 2013). Although SSBs do not directly result in cell death and DSBs are a much severer threat to cell survival, SSBs closely located on opposite strands easily form lethal DSBs if they are not repaired properly in a timely manner (Sutherland et al., 2000).
SSBR in vertebrate/mammalian cells depends mainly on the Poly (ADP-ribose) polymerase 1 (PARP1)/polyADP-ribose glycohydrolase (PARG)-mediated pathway. Efficient SSBR requires polyADP-ribosylation (PARylation) of XRCC1 and ligase IIIα (LIG3α) (Jungmichel et al., 2013; Li et al., 2013) to promote the recruitment of the XRCC1/LIG3α complex to SSB sites (Caldecott, 2008; Hu et al., 2018; London, 2015) and requires PARG to quickly remove the PARylation modification of XRCC1 (Gravells et al., 2017; Wei et al., 2013). XRCC1 does not bind double-strand DNA (dsDNA) but binds single-strand DNA (ssDNA) and PAR (to be PARylated by PARP1; Jungmichel et al., 2013; Li et al., 2013). PAR is able to compete with DNA for common histone binding sites (Panzeter et al., 1992). Accumulated evidence supports that PAR exhibits similarities to ssDNA due to its anionic composition and flexibility, resulting in its consequent capability to conform to variously structured ssDNA binding sites (London, 2015).
DSBs in vertebrate/mammalian cells are mainly repaired by KU-dependent non-homologous end-joining (NHEJ), RAD51-dependent homologous recombination (HR), and a back-up pathway, PARP-dependent alternative end-joining (a-EJ). Both HR and a-EJ require end resection and are efficient only during S/G2 phase. Differently, NHEJ is a much more efficient DSBR pathway that does not require end resection and is independent of cell cycle (Scully et al., 2019). As a HR factor, RAD52 is conserved from yeast to mammals (McIlwraith et al., 2000; Van Dyck et al., 1999). RAD52 has strong binding affinity for both ssDNA and dsDNA (Kagawa et al., 2002; Saotome et al., 2018). Combining deficiency in RAD52 with another major HR factor, such as XRCC3 or Breast cancer gene 2 (BRCA2), causes synthetic lethality in vertebrate/mammalian cells (Feng et al., 2011; Fujimori et al., 2001). However, deficiency in vertebrate/mammalian Rad52 alone has no apparent phenotype in cell response to general DNA-damaging agents (Rijkers et al., 1998; Yamaguchi-Iwai et al., 1998); the mechanism remains unclear.
Camptothecin (CPT), a topoisomerase I (Top1) inhibitor, induces SSBs by trapping the Top1-cleavage complex during replication, further resulting in single-ended DSBs (Pommier et al., 2016b) that are strongly inhibited by KU-dependent NHEJ (Adachi et al., 2004; Chanut et al., 2016; Foster et al., 2011; Hochegger et al., 2006). KU readily and stably binds to single-ended DSBs (without other ends for joining), which prevents the binding of additional repair factors for end resection, thus preventing DSBR and promoting cell death (Chanut et al., 2016). Considering that RAD52 has an essentially synthetic role with other HR repair proteins, CPT is an ideal agent to be used to initially study the role of vertebrate/mammalian RAD52 in DNA repair.
RESULTS
Vertebrate/Mammalian RAD52 Reduces Cell Survival after CPT Treatment
CPT-induced DNA damage interferes with both transcription and DNA replication (Pommier et al., 2016b). Treatment with aphidicolin (an inhibitor of DNA polymerase α) to block cells from G1 to S phase completely abolished CPT-induced cell killing (Figures S1A–S1C), suggesting that CPT-induced cell death mainly depends on active DNA replication and is independent of transcription.
Unexpectedly, Rad52-deficient chicken B (DT40) cells were more resistant to CPT than their wild-type (WT) counterparts (Figures S2A and 1A, left panel). Similarly, RAD52 inhibitor (RAD52i) treatment (which interrupts RAD52’s binding to DNA) also rendered WT cells resistant to CPT (note: in order to show clear difference in compared samples, we present data in bar charts that are derived from the curve plots in the corresponding supplemental figures throughout the manuscript). Both Rad54 and Atm knockout (KO) DT40 cells were more sensitive to CPT than WT controls (Adachi et al., 2004; Chanut et al., 2016; Hochegger et al., 2006), confirming that RAD54/ATM associated with RAD51-mediated HR contributes to repair of CPT-induced DSBs. RAD52i treatment also rendered Rad54 or Atm KO DT40 cells more resistant to CPT (Figures S2B and 1A, right panel), suggesting that RAD52’s inhibitory effect on repair of CPT-induced DNA damage is independent of HR. Knocking out Ku70 from Rad52-deficient DT40 cells using CRISPR-Cas9 (Figures S2C and S2D) resulted in greater resistance to CPT, and similar phenotypes were observed in Ku70-deficient cells treated with RAD52i (Figure S2D). These results also exclude the RAD52-associated inhibitory effects linked to KU-dependent NHEJ.
Figure 1. RAD52 Promotes CPT-Induced Vertebrate/Mammalian Cell Killing.

(A) WT, Rad52−/−, Rad54−/−, or Atm−/− DT40 cell sensitivities to CPT were measured using the Cell-Titer-Glo Luminescent Cell Viability Assay Kit following our modified protocol and confirmed by clonogenic assay. RAD52 inhibitor (RAD52i) CD1, 2.5 μM, was added to cell culture 1.5 h before adding CPT at 20 nM. Data are mean ± SEM from three independent experiments. **p < 0.01.
(B) RAD52 levels were detected by western blotting in WT (mock) and Rad52 knockout MEFs generated using CRISPR-Cas9 (2 from targeting exon 3 and 2 from targeting exon 5).
(C) WT, Rad52-deficient (Rad52 d1 targeting exon 3–1; Rad52 d2 targeting exon 3–2), or Ku70-deficient (Ku70 d) MEF sensitivities to CPT using a clonogenic assay. Data are mean ± SEM from three independent experiments. **p < 0.01.
(D) RAD52 levels were detected by western blotting in U2OS cells treated with control RNA (Ct RNA) or Rad52 siRNA (for 48 h).
(E) U2OS cell sensitivity to CPT after treatment with Ct RNA, Rad52 siRNA, or RAD52i. Data are mean ± SEM from three independent experiments. **p < 0.01.
Similar to DT40 cells, Ku70-deficient mouse embryonic fibroblasts (MEFs) were more resistant and Rad54-deficient MEFs were more sensitive to CPT than their WT counterparts (Figures S2G and 1C). Knocking Rad52 out in MEFs (Figures S2E and 1B), or treating WT MEFs with RAD52i, resulted in increased resistance to CPT (Figures S2G and 1C). Knocking down Rad52 with small interfering RNA (siRNA) in human U2OS cells (Figure 1D) or treating the cells with RAD52i also resulted in greater resistance of the cells to CPT (Figures S2H and 1E). These results confirm that vertebrate/mouse/human cells shared a similar RAD52-dependent inhibitory effect on repair of CPT-induced damage. To elucidate the underlying mechanism, we used different cell types (chicken, MEF, or human) based on the cell line availability for each particular experiment.
RAD52 Suppresses PARP-Mediated Repair of CPT-Induced SSBs
RAD52 foci (reflecting RAD52 binding to damage DNA) appeared in most MEFs within 5 min of CPT treatment and maintained to 6 h post-treatment (Figure 2A), indicating that RAD52 rapidly binds to CPT-damaged DNA. Rad52-deficient MEFs showed fewer γ-H2AX foci-positive (DSB-detecting marker) cells than their WT counterparts (Figure 2B), indicating the reduced generation of DSBs in Rad52-deficient cells after CPT treatment. Considering that RAD52 is a DSBR protein, possible explanation for fewer DSBs in Rad52-deficient cells is a negative effect of RAD52 on SSBR. To test this hypothesis, we first examined the chromatin-bound levels of different repair proteins at different times after CPT treatment because SSBR is a fast process. The increased chromatin-bound repair proteins reflect whether the specific protein efficiently involves the repair, and they also reflect unrepaired DNA damage level at the examined time points, which should be explained by combining the cell biological phenotypes. Rad52-expressing MEFs showed higher chromatin-bound levels of all examined proteins compared with Rad52-deficient MEFs (vector control), suggesting a faster repair rate in CPT-treated Rad52-deficient MEFs. Notably, these MEFs showed high chromatin-bound levels of PARP1 at 5 min after CPT treatment control cells, which occurred much earlier than the increased levels of RAD51 and RPA70, the key HR factors for DSBR (Figure 2C). RAD52 showed a similar chromatin-bound pattern to PARP1. These results suggest that RAD52 might interfere with PARP-mediated SSBR.
Figure 2. RAD52 Suppresses PARP-Mediated Repair of CPT-Induced SSBs.

(A) Top: images of RAD52 foci in Rad52-expressing MEFs following 50 nM CPT treatment using a RAD52 antibody (Ochs et al., 2016; scale bar represents 4 μM). Bottom: quantification of RAD52 foci per cell at indicated times after CPT treatment is shown. Data are mean ± SEM (n = 50 cells). ***p < 0.001.
(B) Left: images of CPT-induced γ-H2AX foci in WT or Rad52-deficient (Rad52−/−) MEFs treated with CPT for 30 min. The cells were collected for immunostaining with an anti-γ-H2AX antibody DAPI (scale bar represents 8 μM). Right: percentage of cells with γ-H2AX foci was analyzed. Data are mean ± SEM (n = 50 cells). **p < 0.01.
(C) Left: chromatin-bound “C” and whole-cell “W” fractions of different DNA repair proteins from vector or Rad52-expressing Rad52-deficient MEFs were evaluated using western blotting after 50 nM CPT treatment. Right: chromatin-bound protein quantification was based on western blotting image data from 3 independent experiments of 2 clones of Rad52-deficient cells. Data are mean ± SEM from biological triplicates. **p < 0.01.
(D) WT, Parp1−/−, Rad52−/−, DT40 cell sensitivities to CPT with or without PARPi (olaparib, 1 μM for 1.5 h before CPT treatment). ***p < 0.001.
(E) WT, Rad52 d1 (R52–1), or Rad52 d2 (R52–2) MEF sensitivities to CPT. Data are mean ± SEM from three independent experiments. **p < 0.01; ***p < 0.001.
(F) U2OS cell sensitivities to CPT after treatment with Ct RNA or Rad52 siRNA. Data are mean ± SEM from three independent experiments. **p < 0.01; ***p < 0.001.
(G) Left: WT or Parp1-deficient DT40 cells were treated with CPT for 30 min and then DSBs in these cells were assessed by neutral comet assays (scale bar represents 8 μM). Right: percentage of tail DNA in total DNA (from 100 cells) was analyzed using the CometScore software. Data are mean ± SEM from three independent experiments. ****p < 0.0001.
PARP inhibitor (PARPi) treatment rendered all of the tested cells (DT40, MEF, and human) more sensitive to CPT (Figure S3A), indicating that PARP-mediated pathway plays an important role in repair of CPT-induced damage. Notably, treatment of Rad52-deficient cells with PARPi increased the cell sensitivity to CPT, although Rad52-deficient cells without PARPi were more resistant to CPT (Figures S3A and 2D–2F). Combining PARPi and CPT treatment induced more γ-H2AX foci in Rad52-deficient MEFs versus (versus) in their WT counterparts (Figure S3B), although CPT treatment alone without PARPi induced fewer γ-H2AX foci in Rad52-deficient MEFs (Figure 2B). These results support that RAD52-mediated inhibitory effects are associated with PARP-dependent repair. In addition, the results suggest that RAD52 contributes to repair of CPT-induced DSBs, which might be inhibited by PARP.
DT40 cells have no PARP2, and PARP1 is essential for PARP-mediated repair. MEFs have PARP2 that plays an important complementary role for PARP1 because deficiency in both PARP1 and PARP2 is lethal (Boehler et al., 2011) and deficiency in PARP1 or PARP2 alone is viable. Parp1 KO MEFs did not show the same sensitivity to CPT as the PARPi-treated cells (Patel et al., 2012); however, knocking down Parp2 in Parp1-deficient MEFs (~70%) increased the sensitivity to CPT (~2-fold; Figures S3C and S3D), supporting a back-up role of PARP2 in Parp1-deficient MEFs. PARPi is involved in either trapping PARP protein or inhibiting PARP activity (Pommier et al., 2016a). Niraparib, a PARPi, generated greater trapping effects than olaparib, another PARPi (Murai et al., 2012). Although niraparib increased CPT sensitization to a greater level than olaparib in WT MEFs, the increased sensitization ratio was also observed in Rad52-deficient MEFs (Figure S3E). If RAD52 affecting PARP-mediated repair is through trapping PARP protein, compared to olaparib, niraparib treatment should not increase CPT sensitivity in Rad52-deficient MEFs. Therefore, these data exclude the possibility that the inhibitory effects of RAD52 on repair of CPT-induced damage mainly depend on PARP trapping.
PARP1 is involved in both SSBR and DSBR that is via the a-EJ pathway (Ray Chaudhuri and Nussenzweig, 2017). DNA Polymerase Theta (POLQ) is a key factor in DSBR by PARP1-mediated a-EJ (Wood and Doublié, 2016). Polq-deficient DT40 cells showed similar CPT sensitivity to WT cells, with or without PARPi treatment (Figure S3F), suggesting that repair of CPT-induced DNA damage is independent of PARP1-mediated DSBR. Furthermore, Parp1-deficient DT40 cells were highly sensitive although Rad52-deficient DT40 cells were more resistant to H2O2 (a SSB inducer), compared to WT cells (Figure S3F), providing additional evidence to support that RAD52 may interfere with PARP1-mediated SSBR. A neutral comet assay to detect DSBs showed that Parp1-deficient versus WT DT40 cells significantly increased tail movement (reflecting DSBs) at 30 min following a low dose of CPT (10 nM) treatment (Figure 2G), further supporting that PARP-mediated SSBR may play a major role in preventing DSB generation, thus promoting cell survival after CPT treatment.
PARP-mediated PARylation is a rapid cellular response to DNA damage; this dynamic change reflects DNA damage and repair status. We then tested the possibility whether RAD52 competed with PARP1 for DNA binding and thus affected the PARylation activity because both PARP1 and RAD52 showed binding affinities to ssDNA ends and dsDNA ends (Figures S4A and S4B). Unexpectedly, RAD52 did not affect PARP1 DNA binding to ssDNA or dsDNA ends (Figure S4C). In addition, RAD52 did not directly affect PARP1 PARylation activity, which was measured by fluorescence signal of a PARP-HSA-enzyme combined with western blotting of co-immunoprecipitation (IP) assay (Figures S4D and S4E). These results exclude the possibility that RAD52 directly affects PARP1 activity but may affect PARP1’s partners to interfere with SSBR in CPT-treated cells.
RAD52 Suppresses XRCC1/LIG3α Co-localization in CPT-Treated Cells
Continually PARylated signals indirectly indicate unrepaired damage in the cells. CPT treatment stimulated whole-cell PAR levels (PARP-mediated PARylation) at 5 min in both WT and Rad52-deficient MEFs, but PAR levels rapidly decreased and remained low in Rad52-deficient MEF; whereas WT cells sustained a relatively high PAR level until 1 h after CPT treatment (Figure 3A). Together with the survival data (Figure 1), these results suggest that, without RAD52, the cells more efficiently repaired CPT-induced damage (showing reduced PAR signals), possibly through the PARP-mediated SSBR.
Figure 3. RAD52 Suppression of PARP-Mediated SSBR Is through Interference with XRCC1/LIG3a Co-localization.

(A) Whole-cell PAR levels were measured in WT or Rad52-deficient MEFs after 50 nM CPT treatment.
(B) Western blot analysis of lysates and IP from HEK293T cells transfected with the indicated genes (GFP-XRCC1 or FLAG-LIG3α) for 27 h and then treated with CPT (20 nM) for the indicated times.
(C) PLA plots show the percentage of cells with various numbers of XRCC1/LIG3α foci in vector or WT Rad52 expressing in Rad52−/− MEFs after CPT or MMS treatment. Data from 5 to 6 randomly selected fields (n = 50 cells) in each group were quantified using ImageJ.
(D) PAR polymer dot blot analysis. Incremental amounts of each protein (1, 2, 4, and 8 pMol) were spotted onto a nitrocellulose membrane (Bio-Rad), incubated in PBS-T containing 50 nM pADPr polymer (Trevigen), and subsequently subjected to western blotting using a PAR antibody.
(E) IP of PAR polymer incubated with RAD52 and glutathione S-transferase (GST)-XRCC1 proteins at different ratios (12:0, 12:6, 12:12, 0:12, 6:12, and 12:12 [pMol:pMol]) using a PAR antibody.
(F) WT and Xrcc1-deficient MEFs were treated with DMSO, RAD52i, or PARPi for 1.5 h and then treated with CPT for 24 h. The survival results were obtained using the clonogenic assay. Data are mean ± SEM from three independent experiments. *p < 0.05. **p < 0.01. ***p < 0.001.
XRCC1 is one of the most important factors in PARP-mediated SSBR. XRCC1 focus formation is an index for SSBR (Das et al., 2014), which indirectly indicates XRCC1/LIG3α function at SSB sites (London, 2015). When endogenous Rad52 was knocked down in U2OS cells (Figure 1D), more XRCC1 foci-positive cells were observed at 5 min after CPT (10 nM) treatment, although the levels returned to the background (similar to that shown in the control RNA-treated cells) at 30 min after CPT treatment (Figure S5A). These results suggest that RAD52 may interfere with XRCC1 function in the PARP-dependent SSBR. The XRCC1/LIG3α complex was initially identified without DNA damage (Caldecott et al., 1995; Whitehouse et al., 2001), and it was believed that the heterodimer occurs naturally. Later, it has been known that LIG3 has XRCC1-independent form in mitochondria and XRCC1-dependent form in nucleus (Gao et al., 2011; Simsek et al., 2011). Therefore, it is possible that DNA damage might stimulate the nuclear XRCC1/LIG3a complex formation for DNA repair. To test this hypothesis, we detected the complex level using an IP assay with GFP-XRCC1 and FLAG-LIG3α co-transfected HEK293T cells at different times after CPT treatment. Without CPT treatment, there was a very low level of interaction between XRCC1 and LIG3α. However, at 5 min after DNA damage (20 nM CPT treatment), the complex level significantly increased and then gradually decreased (Figure 3B).
To confirm the inhibitory effects of RAD52 on DNA-damage-stimulated XRCC1/LIG3α complex formation, we used a proximity ligation assay (PLA) to detect XRCC1/LIG3α complex foci (Figure S5B). There were no positive PLA signals in Xrcc1 KO cell nuclei (Figure S5C), indicating the signal specificity. There were a few XRCC1/LIG3α complex foci without DNA-damaging agent treatment, indicating a background SSBR level in normal cells and confirming those shown from the IP complex (Figure 3B). Efficient SSBR requires initially rapid PARP activity followed by PARG activity to remove PARylation of XRCC1 (Wei et al., 2013). PARPi treatment reduced but PARGi increased the number of foci-positive cells without DNA damage (Figure S5C), confirming the involvement of PARP/PARG in SSBR. Notably, at 5 min after CPT (20 nM) treatment, more XRCC1/LIG3α foci-positive cells were detected in vector control (Rad52 deficient) than in Rad52-expressing MEFs (Figure 3C), suggesting more-efficient SSBR in Rad52-deficient cells. To confirm the inhibitory effects of RAD52 on XRCC1/LIG3α foci were involved in SSBR, we detected the PLA foci in methyl methanesulfonate (MMS) treated these cells because MMS mainly induces SSBs (Lundin et al., 2005). Similarly, more XRCC1/LIG3α foci-positive cells appeared in vector control (Rad52 deficient) than in Rad52-expressing MEFs after MMS treatment (Figure 3C). These results support that RAD52 directly interferes with SSB-stimulated XRCC1/LIG3α complex formation.
To examine whether RAD52 has a stronger ssDNA/PAR binding activity than XRCC1, we compared PAR binding activities between purified RAD52 and XRCC1 proteins in an in vitro polymer blot assay. Such assay does not need PARP activity and directly detects the protein’s PAR-binding ability, which indirectly reflects the protein’s ssDNA-binding affinity. RAD52 showed a much stronger PAR-binding affinity than XRCC1 (Figure 3D). This was further verified by the competition of purified RAD52 and XRCC1 proteins in a PAR-binding complex, where RAD52 significantly inhibited PAR-bound XRCC1 levels (Figure 3E). These results support that RAD52 has a stronger binding affinity for ssDNA and/or PAR than XRCC1. We then found that, similar to XRCC1, RAD52 was PARylated by PARP1 after DNA damage (Figures S6A–S6C), suggesting that RAD52 might compete with XRCC1 for PARylation modification and thus inhibit SSBR. Unexpectedly, the XRCC1 PARylation levels in cells were not affected by RAD52 expression at different times after CPT treatment (data not shown). These results suggest that RAD52’s inhibitory effects on DNA-damage-promoted XRCC1/LIG3α foci formation may not directly affect through XRCC1 PARylation but through ssDNA binding affinity, although to confirm the conclusion requires more experiments in the future.
To verify the inhibitory effects of RAD52 on XRCC1-dependent SSBR, we tested the sensitivity of Xrcc1-deficient MEFs to CPT. Xrcc1-deficient MEFs without PARP1 treatment showed similar CPT sensitivities to WT cells treated with PARPi (Figures S6D and 3F), indicating that XRCC1 mainly functions in PARP-dependent SSBR. PARPi treatment rendered Xrcc1-deficient MEFs more sensitive to CPT (Figures S6D and 3F). This can be explained by the previous finding that PARP1-mediated replication fork reversal (RFR) also contributes to repair of CPT-induced damage but is XRCC1 independent (Ray Chaudhuri et al., 2012; Ray Chaudhuri and Nussenzweig, 2017). It is possible that Xrcc1-deficient MEFs depend more on PARP-mediated RFR to repair CPT-induced damage than their WT counterparts. RAD52i had no effects on the sensitivity of Xrcc1-deficient MEFs to CPT treatment at low doses (5 and 10 nM), although RAD52i increased the resistance of WT MEFs to CPT at the same doses (Figure S6D). These results suggest that low doses (5 and 10 nM) of CPT-induced damage are mainly repaired by PARP1/XRCC1-mediated SSBR, which is inhibited by RAD52. RAD52i significantly sensitized Xrcc1-deficient MEFs to a high dose (20 nM) of CPT treatment, more significantly than PARPi treatment (Figures S6D and 3F), providing additional evidence that RAD52 contributes to repair of CPT-induced DSBs, which might be inhibited by PARP activity.
A Strategy to Enhance the Inhibitory Effects of RAD52 on SSBR and Sensitize Cells to Different DNA-Damaging Agents
RAD52 is known to participate in DSBR via HR functions (Chang et al., 2017; Costantino et al., 2014; Kan et al., 2017; Sotiriou et al., 2016; Verma and Greenberg, 2016). We have discovered in this study a negative effect of RAD52 on SSBR. To study whether we can utilize these characteristics of RAD52 to enhance cell sensitivity to DNA-damaging agents, we generated R55A and Y104D RAD52 mutants based on RAD52’s structure (Figures 4A and 4B), because these mutants can separate RAD52’s inhibitory effects on SSBR and promoting effects on DSBR. R55A significantly reduces ssDNA, but not dsDNA, binding activity (Kagawa et al., 2002; Saotome et al., 2018), which is similar to WT cells treated with an available RAD52i. Y104D, a mimetic of phospho-serine, retained RAD52 DNA binding activity (Honda et al., 2011) but lost RAD52 self-interaction (oligomerization; Figure S6A) and thus lost its DSBR ability. c-ABL phosphorylation of Y104 was disrupted by mutation to F104 (Honda et al., 2011; Kitao and Yuan, 2002) but still maintained RAD52 oligomerization (Figure S7A); thus, Y104F was used as a control for Y104D.
Figure 4. A Strategy to Enhance the Inhibitory Effects of RAD52 on SSBR and Sensitize Cells to DNA-Damaging Agents.

(A and B) Description of key RAD52 mutants (R55A and Y104D) tested in this study. Y104 is conserved across variant species. Y104F (abolished Abelson tyrosine kinase (c-ABL) phosphorylation) was used as a control for Y104D.
(C) Expression of FLAG-tagged WT or mutant mouse Rad52 in Rad52-deficient MEFs was measured using western blotting.
(D) Sensitivity of vector, WT, or mutant Rad52-expressing cells to CPT (40 nM). Data are mean ± SEM from three independent experiments. **p < 0.01.
(E) Left: images of XRCC1/LIG3α foci in R55A or Y104 mutant Rasd52-expressing Rad52-deficient MEFs on slides treated with or without CPT (20 nM) for 5 min and then fixed for PLA (scale bar represents 5 μM). Right: the plots show the percentage of XRCC1/LIG3α foci in R55A or Y104 mutant Rad52-expressing Rad52-deficient MEFs with different numbers of foci/cell after CPT treatment from 5 to 6 randomly selected fields (n = 50 cells) in each group, quantified using ImageJ.
(F) Sensitivity of MEFs expressing vector, WT, or mutant Rad52 (R55A or Y104D) to different DNA damage inducers: IR; MMS; or etoposide (Top II inhibitor). Data are mean ± SEM from three independent experiments. For IR- and MMS-treated cells, as compared to empty-vector-expressing cells, **p < 0.01 and ***p < 0.001; for etoposide-treated cells, as compared to WT Rad52-expressing cells, **p < 0.01.
(G) Comparison of CPT-induced PARylation levels between WT and mutant Rad52-expressing (R55A or Y104D) HEK293T cells under the same conditions as described in Figure S7A.
Y104F mutant RAD52-expressing cells showed similar sensitivity to WT RAD52-expressing cells (Figures 4C, 4D, and S7B), excluding the possibility that c-ABL phosphorylation is critical for cell sensitivity to CPT. Notably, Y104D expression showed most sensitive to CPT among all tested cells (Figures 4C, 4D, and S7B). PARPi abolished the difference in sensitivity between R55A and Y104D cells (Figure S7B) because PARPi abolished SSBR, the inhibitory effects of RAD52 depended on intact SSBR, and both R55A (lost ssDNA binding) and Y104D (lost RAD52 oligomerization) lost RAD52-mediated DSBR. This was further supported by CPT-induced XRCC1/LIG3α complex focus data (Figure 4E). Y104D expression significantly reduced CPT-promoted XRCC1/LIG3α foci compared to R55A expression (Figure 4E), supporting that ssDNA binding is essential for RAD52’s inhibitory effects on XRCC1/LIG3α foci. Notably, R55A-expressing cells showed similar levels of CPT-induced XRCC1/LIG3α foci to those Rad52-deficient cells (Figures 3C and 4E), providing additional evidence to support that the inhibitory effects of RAD52 on SSBR depend on RAD52’s DNA-binding ability.
To study whether inhibitory effects of RAD52 on SSBR are not limited to CPT treatment, we examined the sensitivity of WT and Rad52-deficient DT40 cells to different DNA-damaging agents, including MMS (generates SSBs), ionizing radiation (IR) (generates SSBs and DSBs), and etoposide (topoisomerase [Top] II inhibitor, mainly generates DSBs; Pommier et al., 2016b). Although Rad52-deficient cells were more resistant to MMS than WT cells, PARPi treatment abolished the different MMS sensitivities between these cells (Figure S7C). These results further support that RAD52-mediated inhibitory effects on SSBR depend on PARP-mediated SSBR and that RAD52-mediated DSBR does not contribute to the repair of MMS-induced damage. In contrast, there was no clear difference in sensitivity to IR between Rad52-deficient versus WT cells with or without PARPi treatment (Figure S7C), supporting that RAD52-mediated inhibitory effects on SSBR neutralize RAD52’s DSBR effects. Rad52-deficient cells were more sensitive than WT cells to etoposide (Figure S7C), suggesting that RAD52 contributes to repair of etoposide-induced DSBs. These phenotypes provide evidence to support the inhibitory effects of RAD52 on SSBR commonly occurring in cell response to DNA damage.
In irradiated MEFs, vector and R55A mutant showed the same sensitivity as WT Rad52-expressing cells, although the Y104D mutant exhibited significant sensitivity to IR (Figure 4F). Thus, cells became more sensitive to IR only when they lost RAD52’s DSBR but retained inhibition of SSBR. WT RAD52-expressing cells were relatively sensitive to MMS compared to vector control cells, although Y104D-expressing cells were more sensitive to MMS (Figure 4F) because Y104D had stronger ssDNA/PAR-binding activity. This is supported by the data shown in Figure 4G. Expression of RAD52 R55A showed no PARylation although Y104D expression significantly increased PARylation after CPT treatment compared to expression of their WT RAD52 counterparts (Figure 4G). Without RAD52-mediated DSBR (vector alone, R55A, and Y104D), cells were more sensitive to etoposide than WT RAD52-expressing cells (Figure 4F). These results, as summarized in the graphic abstract, describe that the inhibitory effect of RAD52 on SSBR is a common phenotype in cells in response to DNA-damaging agents.
DISCUSSION
In this study, we have shown an inhibitory effect of RAD52 on PARP1-mediated SSBR, which may occur through RAD52’s strong ssDNA/PAR-binding affinity to interfere with XRCC1/LIG3α-complex formation. CPT and IR generate both SSBs and DSBs, but compared to WT cells, RAD52-deficient cells showed more resistant only to CPT treatment, but not to IR exposure. This might be due to the different effects of CPT and IR on DNA damage generation. CPT traps Top1-cleavage complex to induce SSBs during replication, resulting in DSBs with replication fork movement (Pommier et al., 2016b). Therefore, efficient PARP-dependent SSBR plays a key role in preventing CPT-induced lethal DSBs that are inhibited by KU-dependent NHEJ (Chanut et al., 2016) but mainly repaired by HR (a relatively slow repair process). RAD52’s inhibitory effects on SSBR could promote the conversion of SSBs to lethal DSBs. In contrast, IR generates SSBs and DSBs at the same time, and IR-generated DSBs are mainly repaired by NHEJ, a more-efficient DSBR pathway than HR and a-EJ, because most cells, in general, are in G1 phase. Thus, it is possible that the opposing effects of RAD52 on SSBR and DSBR neutralize the phenotypes of irradiated cells. Thus, the major role of mammalian RAD52 in response to DNA damage is a mediator to adjust the balance of cell death and genomic integrity. We reported here that RAD52 interferes with SSBR, which promotes DSB generation and cell death. This identified function of RAD52 might play an important role in facilitating genomic integrity because RAD52 binding to ssDNA prevents ssDNA degradation and genomic instability (Malacaria et al., 2019).
RAD52 forms an 11-mer ring structure, which is essential for its role in DSBR (Chandramouly et al., 2015). However, RAD52’s DNA-binding affinity does not depend on the ring structure because it occurs mainly through the outside positive charge of the oligomer ring and the positively charged surface of the RAD52 monomer binds DNA efficiently (Kagawa et al.; 2002; Saotome et al., 2018). Because the Y104D mutant loses RAD52’s oligomerization and is defective in ring formation, this results in more RAD52 monomers having an opportunity to directly reach and bind ssDNA/PAR, as we observed (Figure 4G). This may also explain why Y104D mutant RAD52-expressing cells showed greater sensitivity to DNA-damaging agents because such cells lost RAD52’s DSBR ability and displayed more inhibition of SSBR with the stronger ssDNA/PAR-binding affinity of RAD52 monomers. Disruption of RAD52’s DNA binding with currently available RAD52i sensitized only BRCA1/2 mutant tumor cells to PARPi (Hengel et al., 2016; Huang et al., 2016; Sullivan-Reed et al., 2018). Although such RAD52i reduced RAD52-mediated DSBR, they lose the inhibitory effects on SSBR, thus limiting their usage. Our results provide an opportunity to disrupt RAD52’s oligomerization (ablating RAD52-mediated DSBR) but retain RAD52’s DNA-binding activity (maintaining RAD52’s inhibitory effects on SSBR), which would be able to efficiently sensitize BRCA1/2 WT tumor cells to DNA-damaging agents.
Yeast has no Parp1 and Lig3 genes and mainly depends on DNA polymerase to repair SSBs, and Rad52 has no effects on such repair (Ogorek and Bryant, 1985a, 1985b). In contrast, vertebrate/mammalian cells use a PARP1/XRCC1/LIG3α-mediated pathway to repair SSBs (Caldecott, 2008), which is affected by RAD52. The biggest difference in SSBR between yeast and mammalian cells may depend on their different growth rates. Most vertebrate/mammalian cells are not in S phase, so they cannot depend on DNA synthesis processes but require the PARP1/XRCC1/LIG3α pathway to repair SSBs. Meanwhile, RAD52-mediated DSBR might be limited by PARP activity because PARylation reduced RAD52 DNA-binding activity (Figure S7D). These explanations are summarized in Figure S7E.
RAD52-mediated single-strand annealing (SSA) is a process associated with RAD52’s other HR functions (Kan et al., 2017). It is known that SSA is an error-prone repair pathway by generating large deletion of DNA during the annealing process (Bhargava et al., 2016). PARylation reduced RAD52 DNA-binding activity, supporting that PARP may inhibit RAD52-mediated DSBR as described in Figure 2 to maintain genomic integrity. PARP1 has many substrates (Jungmichel et al., 2013), which suggests that any DNA damage response protein might be able to be PARylated if it could bind DNA. PARylation may adjust a protein’s function through reducing its DNA-binding affinity, such as the effects of PARP on RAD51 and BRCA1 (Hu et al., 2014; Schultz et al., 2003). In this way, PARP may maintain a balance between different types of repair pathways based on cellular requirements. Further studies are required to explore this prediction.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and request for resources should be directed to and will be fulfilled by the Lead Contact, Ya Wang (ywang94@emory.edu).
Materials Availability
Materials, including plasmids and cell lines, are available from the authors upon request.
Data and Code Availability
The published article includes data generated in this study, no unique software or code were generated for this study.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines and Cell Culture
Different DT40 (chicken B), MEF and human cell lines were used in this study. DT40 cells were grown in RPMI1640 supplemented with 10% fetal calf serum, 1% chicken serum, and 10−5 M β-mercaptoethanol at 39°C. MEF and human cell lines were grown in DMEM medium supplemented with 10% fetal calf serum. Detail information of cell lines used in this study is included in Key Resources Table.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-Beta-actin | Santa Cruz | Cat #: sc-47778; RRID: AB_2714189 |
| Mouse anti-Flag M5 | Sigma Aldrich Inc | Cat #: F4042; RRID: AB_439686 |
| Rabbit anti-Gamma-H2AX | Cell Signaling Technology (CST) | Cat #: 2577; RRID: AB_2118010 |
| Rabbit anti-GAPDH | CST | Cat #: 5174; RRID: AB_10622025 |
| Rabbit anti-GFP | Biomol | Cat #: 600-401-215S; RRID: AB_2612814 |
| Rabbit anti-Histone 3 | CST | Cat #: 4499; RRID: AB_10544537 |
| Mouse anti-KU70 | Santa Cruz | Cat #: sc-17789; RRID: AB_628454 |
| Rabbit anti-MRE11 | CST | Cat #: 4895; RRID: AB_2145100 |
| Mouse anti-PAR | Abcam | Cat #: 14459; RRID: AB_301239 |
| Rabbit anti-PAR | Millipore Sigma | Cat #: MAB3192; RRID: AB_94743 |
| Rabbit anti-PARP | CST | Cat #: 9532; RRID: AB_659884 |
| Rabbit anti-RAD51 | Santa Cruz | Cat #: sc-8349; RRID: AB_2253533 |
| Goat anti-RAD52 | A gift from Dr. Jiri Lukas’s lab | (Ochs et al., 2016) |
| Rabbit anti-TurboGFP (tGFP) | Thermo Fisher | Cat #: PA5-22688; RRID: AB_2540616 |
| Rabbit anti-XRCC1 | CST | Cat #: 2735; RRID: AB_2218471 |
| Mouse anti-XRCC1 | Abcam | Cat #: Ab1838; RRID: AB_302636 |
| Mouse anti-LIG3α | Thermo Fisher Scientific Inc. | Cat #: PA5-21480; RRID: AB_11153448 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| CPT | VWR | 101757-582 |
| etoposide | VWR | ss419-42-0 |
| RAD52 inhibitor, CD1 | ChemDiv Inc. | C267-0065 |
| PARP inhibitor, niraparib | SELLECK | S7625 |
| PARP inhibitor, olaparib | SELLECK | S1060 |
| PARG inhibitor | Millipore Sigma | 118415 |
| methyl methanesulfonate (MMS) | Sigma-Aldrich Inc | 129925-5G |
| H2O2 | Fisher Scientific | H325-100 |
| aphidicolin | Sigma-Aldrich Inc | A4487 |
| nocodazole | CST | 2190 |
| DAPI | Vector Laboratory | H-1200 |
| Critical Commercial Assays | ||
| CellTiter-Glo Luminescent Cell Viability Assay Kit | Promega Corp | G9241 |
| Chromatin Extraction Kit | Abcam | ab117152 |
| Single cell electrophoresis (comet assay) Kit | Trivigen | 4250-050-K |
| Duolink In Situ Orange Starter (Mouse/Rabbit) Kit | Sigma-Aldrich Inc | DUO92102-1KT |
| PARP Inhibition Assay Kit | Trevigen | 4690-096-K |
| Experimental Models: Cell Lines | ||
| DT40 WT, Rad52-deficient | Dr. Shinichi Takeda’s Lab | (Yamaguchi-Iwai et al., 1998) |
| DT40 Rad52/Ku70-deficient | This study | N/A |
| DT40 Rad54, Ku70, Rad54/Ku70-deficient | Dr. Shinichi Takeda’s Lab | (Takata et al., 1998) |
| DT40 Atm, Atm/Ku70-deficient | Dr. Shinichi Takeda’s Lab | (Morrison et al., 2000) |
| DT40 Parp1 or Parp11/Ku70 deficient | Dr. Shinichi Takeda’s Lab | (Hochegger et al., 2006) |
| DT40 Polq (theta)-deficient | Dr. Shinichi Takeda’s Lab | (Kohzaki et al., 2010) |
| MEF WT and Ku70-deficient | Dr. David Chen’s Lab | (Li et al., 1998) |
| MEF Parp1-deficient | Dr. Zhaoqi Wang’s Lab | (Wang et al., 1995) |
| MEF Rad54-deficient | Dr. George Iliakis’s Lab with Dr. Kevin Mill’s permission | (Mills et al., 2004) |
| MEF Xrcc1-deficient | Dr. Li Lan’s lab | (Wei et al., 2013) |
| MEF Rad52-deficient or Rad52 mutants including Rad52R55A |
In this study | N/A |
| Rad52Y104D and Rad52Y104F | ||
| HEK293T | ATCC | ACS-4500 |
| U2OS | ATCC | HTB96 |
| Oligonucleotides | ||
| KO cXrcc6-Ex3, F: 5′-CACCAAGCGTTGTGTTCTACGGCA-3′ | IDT | N/A |
| KO cXrcc6-Ex3, R: 5′-AAACTGCCGTAGAACACAACGCTT-3′ | IDT | N/A |
| KO cXrcc6-Ex4, F: 5′-CACCGGCATGAGGGTTATCCTCGT-3′ | IDT | N/A |
| KO cXrcc6-Ex4, R: 5′-AAACGACTGATTACCTTGTGACCC-3′ | IDT | N/A |
| KO mRad52-Ex3, F: 5′-CACCATTATAGAGCCAGTATACAG-3′ | IDT | N/A |
| KO mRad52-Ex3, F: 5′-CACCATTATAGAGCCAGTATACAG-3′ | IDT | N/A |
| KO mRad52-Ex3, R: 5′-AAACCTGTATACTGGCTCTATAAT-3′ | IDT | N/A |
| KO mRad52-Ex5,F: 5′-CACCAACAATGGCAAGTTCTACGT-3′ | IDT | N/A |
| KO mRad52-Ex5, R: 5′-CACCTTAGTTAAATCCACATCAAG-3′ | IDT | N/A |
| Primers for mutations of mRad52 R55A, Y104F and Y104D are as described in Table S1 | IDT | N/A |
| A biotin-labeled DNA oligo Top: GGCCGCACGCGTCCACCATGGGGTAC AA [BioTEGi] GCACGCGTCCACCATGGGGTACAA; | Eurofins | N/A |
| A biotin-labeled DNA oligo Bottom: GTAGTTGTACCCCATGGTGGACGCGTGCTTGTACCCCATGGTGGACGCGTGC | Eurofins | N/A |
| Recombinant DNA | ||
| pX330-U6-Chimeric_BB-CBh-hSpCas9 | Addgene | (Cong et al., 2013) |
| p3XFLAG-CMV | Sigma-Aldrich Inc | E7533 |
| 3xFLAG-Rad52 | In this study | N/A |
| 3xFLAG-Rad52 R55A | In this study | N/A |
| 3xFLAG-Rad52Y104F | In this study | N/A |
| 3xFLAG-Rad52Y104D | In this study | N/A |
| Software and Algorithms | ||
| ImageJ | NIH | https://imagej.nih.gov/ |
| Comet Score analysis software | TriTek Corp | N/A |
| SoftWorx 5.5 | Applied Precision | N/A |
| Other | ||
| mParp2 siRNA 5′-CTACCTTATTCAGCTGTTA-3′ | Santa Cruz | N/A |
| mParp2 siRNA 5′-GTTGAAGCGTGCAATGAAT-3′ | Santa Cruz | N/A |
| mParp2 siRNA 5′-CCATCACAGGTTATATGTT-3′ | Santa Cruz | N/A |
| hRad52 siRNA 5′-GGAGUGACUCAAGAAUUAATT-3′ | QIAGEN | SI02629865 |
| hRad52 siRNA 5′-GGCCCAGAAUACAUAAGUATT-3′ | QIAGEN | SI03020794 |
METHOD DETAILS
Gene Knockout, Mutation, and Cell Cultures
Ku70-knockout in Rad52−deficient DT40 cells or Rad52-knockout in MEFs was accomplished with the CRISPR-Cas9 plasmid (pX330-U6-Chimeric_BB-CBh-hSpCas9) following the published protocol with the primers targeting different genes shown in the KEY RESOURCES TABLE. The following WT or mutant mouse Rad52 constructs were generated using vector p3XFLAG-CMV: 3xFLAG-Rad52, 3xFLAG-Rad52Y104D, 3xFLAG-Rad52Y104F, or 3xFLAG-Rad52R55A. The primers for WT and mutant mouse Rad52 are listed in the Table S1. None of the cell lines used were listed as commonly misidentified by the International Cell Line Authentication Committee. Ku70-deficient DT40 cells were transfected with vector pAneoKu70 cDNA by using Nucleofector (Amaxa) according to the manufacturer’s instructions. G418 resistant colonies were selected and developed into cell lines. Rad52-deficient MEFs were transfected with vector containing Flag or GFP-tagged WT or mutant mouse Rad52.
Cell Synchrony
DT40 cells were seeded into 100 mm dishes (2 × 106 cells with 10 mL medium/dish). Fourteen hours later, 10 mL pre-warmed fresh medium was added to each dish and nocodazole was added to the culture at a final concentration of 0.4 μg/mL for 4.5 h. The cells were then collected, centrifuged, and put into new dishes. Aphidicolin was added to the cell cultures at a final concentration of 2.5 μg/mL and CPT was added to the same culture 15 min later. Cells were collected at the required times and were stained with a solution of 62 μg/mL RNase A, 40 μg/mL propidium iodide, and 0.1% Triton X-100 in phosphate buffered saline (PBS) at room temperature for 1 h. The distribution of cells in the cell cycle was measured in a flow cytometer (Beckman Coulter CytoFLEX).
Cell Sensitivity Assay
MEF and human cell survival was measured using a traditional clonogenic assay as described previously (Wang et al., 2010) with minor modifications. MEFs or human cells were plated for 24 h then treated with or without inhibitors of RAD52 (CD1, 2.5 μM) or PARP (olaparib, 1 μM) for 1 h followed by different concentrations of CPT for an additional 24 h. The cells were then collected and plated with different dilutions aimed at forming 20–150 colonies per dish and left in an incubator for 7–10 days until colonies formed. DT40 cell survival was assessed using clonogenic assay and compared with a modified CellTiter-Glo Luminescent Cell Viability Assay Kit to measure the ATP enzyme activity (an indicator of viability) in CPT-treated cells. The results using traditional clonogenic assay (Wang et al., 2010) and the modified kit approach were comparable and all key survival data using the modified kit approach were verified by the clonogenic assay in this study. Since using a clonogenic assay to measure survival of suspension growth cells (such as DT40 cells) required more steps, we introduced the modified kit method here. Briefly, we plated 10,000 WT cells in each well of a 24-well plate with 1 mL medium/well as untreated controls (numbers of other cell types were adjusted depending on the plating efficiency ratio to WT cells), with different concentrations of CPT. At the end of 6 doubling times, 100 μL of cultured cells were taken from each well and added to each individual well of a 96-well plate. The plates were kept at room temperature for approximately 30 min, and 100 μL of CellTiter-Glo reagent was added and mixed for 2 min to lyse the cells. The plate was then incubated at room temperature for 10 min to stabilize the luminescence signal. Luminescence signals were measured using a Synergy H1 Multi-Mode Reader (BIOTEK, Winooski).
Neutral Comet Assay
DNA DSBs were evaluated by single cell electrophoresis (comet assay) using a reagent kit. Briefly, after 30 min at different concentrations of CPT, WT or Parp1-deficient DT40 cells (1×105/mL) were mixed with molten LMAgarose (at 37°C) at a ratio of 1:10 (v/v) and 50 μL were immediately pipetted onto a CometSlide. The slides were placed flat at 4°C in the dark for 10 min, and then immersed in lysate solution overnight at 4°C. The slides were changed to 50 mL neutral electrophoresis (1X TBE) buffer for 30 min at 4°C, placed in an electrophoresis tray of the CometAssay® ES unit with ~850 mL 1X TBE buffer, and 21 V were applied for 45 min at 4°C. The slides were then immersed in DNA precipitation solution for 30 min, then in 70% ethanol at room temperature for 30 min, and dried for 15 min at 37°C. The dried agarose on each slide was stained with 100 μL of diluted SYBR® Gold for 30 min at room temperature in the dark. The slides were rinsed briefly in water and dried completely at 37°C. The slides were observed using an epifluorescence microscope (SYBR® Gold’s maximum excitation/emission is 496 nm/522 nm) and the percentage of tail DNA in total DNA for each cell was analyzed using Comet Score analysis software (TriTek Corp).
Proteins Competitively Binding to DNA Using an in vitro Assay
A biotin-labeled DNA oligo was synthesized by Eurofins as shown in the KEY RESOURCES TABLE. Annealing of the top with the bottom oligo resulted in dsDNA molecules with a 4 nt overhang at the 3′ end. Proteins were incubated with ssDNA or dsDNA in a binding buffer containing 10 mM Tris–HCl (pH 7.5), 1 mM EDTA, 0.5% glycerol, 1 mM DTT, 0.05 mg/mL BSA, and 150 mM NaCl at 25°C for 20 min. Dynabeads streptavidin beads were added to the mixture and incubated at 25°C for 30 min. The beads were washed with binding buffer 4 times, and then re-suspended in SDS loading buffer for western blotting.
Western Blotting
Cells were collected and counted. A total of 1 × 106 cells were lysed in 50 μL of RIPA buffer (50 mM Tris-HCl, pH 7.4; 1% NP-40; 0.25% sodium deoxycholate; 150 mM NaCl; 1mM EGTA; 1 mM PMSF; 1 μg/mL each of aprotinin, leupeptin, pepstatin; 1 mM Na3VO4; 1 mM NaF) and mixed with 50 μL of 2 x protein loading buffer. After boiling for 5 min, 20 μL of whole cell lysates were loaded onto a 10% polyacrylamide gel. After the first antibody reaction, the membrane was washed with PBS and stained with IRDye 680 or 800 anti-mouse/rabbit secondary antibody (VWR International LLC) and visualized with the Li-Cor Odyssey system.
Detection of Foci in Cells Using Immunofluorescence
For γ-H2AX or RAD52 foci assays, MEFs were seeded on glass coverslips that were coated with poly-D-lysine. At 24 h later, the cells were treated with CPT for different times. Cells were fixed with 2% paraformaldehyde in culture media for 15 min at room temperature, rinsed with PBS twice, and permeabilized with 0.3% Triton X-100 in PBS for 10 min. The cell coverslips were rinsed 3 times with PBS, each time for 5 min, incubated in 3% normal goat serum in PBS for 30 min at room temperature, and then incubated with a primary antibody in 3% normal goat serum overnight at 4°C. The cell coverslips were rinsed three times with PBS for 5 min each and incubated with a fluorochrome-conjugated secondary antibody using an appropriate dilution in 3% normal goat serum for 1 h at 37°C in the dark. After the cell coverslips were rinsed 3 times in PBS for 5 min each in the dark, they were mounted with a drop of VECTASHIELD Antifade Mounting Medium with DAPI (H-1200, from Vector Laboratory) and sealed with nail polish oil. The image was observed with a Leica SP8 confocal microscope system. For XRCC1 foci, U2OS cells were transfected with control or Rad52 siRNA and incubated for 48 h, then treated with 10 nM CPT for various times. Cells were then fixed in 4% paraformaldehyde for 10 min at room temperature, permeabilized by incubation with 0.2% Triton X-100 for 15 min at room temperature, and incubated in blocking solution (10% goat serum) for 1 h at room temperature. After blocking, cells were incubated with mouse monoclonal anti-XRCC1 antibody (1:200 dilution in blocking solution) overnight at 4°C, followed by incubation with Alexa Fluor 488-conjugated goat anti-mouse antibody at a dilution of 1:500 for 1 h at room temperature. Cells were then mounted with VECTASHIELD medium with DAPI (Vector Laboratories). Images were taken using a DeltaVision microscope and deconvolved and projected using SoftWorx 5.5 software. Foci in cells from 4 to 5 randomly selected fields (40 to 63 cells) in each group were quantified using ImageJ.
Detection of Protein Bound to Chromatin DNA in Cells
Chromatin binding proteins were prepared using a chromatin extraction kit purchased from ABCAM, Inc., according to the manufacturer’s instructions. Briefly, 107 cell pellets were washed with PBS, added to working lysis buffer and transferred to a 1.5 mL vial on ice for 10 min. The samples were vortexed vigorously for 10 s and centrifuged at 5000 rpm for 5 min. The supernatants were carefully removed and mixed with 500 μL (1×106 cells/50 μL) of working extraction buffer on ice for 10 min and vortexed occasionally. The samples were sonicated 2 × 20 s to increase chromatin extraction. The samples were cooled on ice between sonication pulses for 30 s, and centrifuged at 12,000 rpm at 4°C for 10 min. The supernatants were transferred to a new vial, and chromatin buffer was added at a ratio of 1:1. The proteins were then separated by SDS-PAGE. Detection with western blots was done using the proper antibodies.
In vitro PARP Activity Assay
The Homogeneous PARP Inhibition Assay Kit from Trevigen was used in this assay and performed in 2 successive steps requiring only the consecutive addition of reaction components. A PARP reaction was first performed, followed by a detection step. PARP activity was identified according to the recommendations of the manufacturer by a fluorescence signal when PARP mediated NAD+ depletion. In our protocol, purified RAD52 protein was incubated with 200 ng of high specific activity (HSA) PARP enzyme prior to the addition of the PARP substrate cocktail and activated DNA. The fluorescence signals (excitation at 544 nm and emission at 590 nm) were measured using a Synergy H1 Microplate Fluorescence Reader (BioTek Instruments Inc.).
Immunoprecipitation (IP) Assay
Cells were lysed in RIPA buffer with protease and phosphatase inhibitors. The cell lysates were then incubated with anti-Flag M2 agarose (Sigma-Aldrich) at 4°C overnight according to the manufacturer’s instructions (for tagged proteins). The beads were washed 4 times (5 min each) with the same buffer used for cell lysate and boiled in 2 X SDS sample buffer for 5 min. Samples were then analyzed by SDS-PAGE followed by immunoblotting.
Proximity Ligation Assay (PLA)
This method is used to detect two proteins that are interacting within close proximity (≤40 nM). The assay was performed using a Duolink In Situ Orange Starter (Mouse/Rabbit) Kit according to the manufacturer’s protocol. Briefly, CPT-treated MEFs on a slide were fixed in 4% paraformaldehyde and permeabilized in 0.5% Triton X-100. Cells were washed and incubated in blocking solution for 1 h at 37°C, then incubated with a mouse anti-XRCC1 antibody and a rabbit anti-LIG3α antibody overnight at 4°C. The next day, cells were washed and incubated with the anti-rabbit PLUS and anti-mouse MINUS oligonucleotide-conjugated secondary antibodies for 1 h at 37°C. Cells were washed again and then incubated for 30 min for ligation and 100 min for amplification. Fluorochrome-coupled oligonucleotides were then hybridized to the amplified substrates. After a final wash, slides were mounted with DAPI containing Duolink in situ mounting medium.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis of survival assays were performed using the Student’s t test (unpaired, two-tailed) on Excel, foci as well as comet assays were performed using two-way ANOVA analysis, and chromatin bound level measurement was performed using Odyssey V3.0 software. Error bars, p value, and sample sizes are indicated in figure legends. Significance was accepted as p < 0.05.
Supplementary Material
Highlights.
Vertebrate/mammalian RAD52 as a DSBR factor promotes CPT-induced cell killing
RAD52 interferes with PARP-mediated SSBR via reducing XRCC1-LIG3a interaction
Blocking of RAD52 oligomerization alone sensitizes cells to DNA-damaging agents
ACKNOWLEDGMENTS
We thank Drs. George Iliakis, Zhaoqi Wang, Kevin Mill, David Chen, and Li Lan for cell lines; Dr. Jiri Lukas for the RAD52 antibody; and Dr. Alexander Mazin for providing the information on RAD52 inhibitor sources. This work was supported by NIH grants CA76203, CA186129, CA185882, and P30CA138292, as well as by pilot grants of Winship Cancer Institute, Emory University, United States. This work was directly supported by a Grant-in-Aid from the Ministry of Education, Science, Sport and Culture (KAKENHI 19K22561 and 16H06306) and the Japan Society for the Promotion of Science Core-to-Core Program, Advanced Research Networks.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108625.
REFERENCES
- Adachi N, So S, and Koyama H (2004). Loss of nonhomologous end joining confers camptothecin resistance in DT40 cells. Implications for the repair of topoisomerase I-mediated DNA damage. J. Biol. Chem 279, 37343–37348. [DOI] [PubMed] [Google Scholar]
- Bhargava R, Onyango DO, and Stark JM (2016). Regulation of single-strand annealing and its role in genome maintenance. Trends Genet. 32, 566–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehler C, Gauthier L, Yelamos J, Noll A, Schreiber V, and Dantzer F (2011). Phenotypic characterization of Parp-1 and Parp-2 deficient mice and cells In Poly(ADP-ribose) Polymerase: Methods and Protocols, Tulin AV, ed. (Humana; ), pp. 313–336. [DOI] [PubMed] [Google Scholar]
- Caldecott KW (2008). Single-strand break repair and genetic disease. Nat. Rev. Genet. 9, 619–631. [DOI] [PubMed] [Google Scholar]
- Caldecott KW, Tucker JD, Stanker LH, and Thompson LH (1995). Characterization of the XRCC1-DNA ligase III complex in vitro and its absence from mutant hamster cells. Nucleic Acids Res. 23, 4836–4843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandramouly G, McDevitt S, Sullivan K, Kent T, Luz A, Glickman JF, Andrake M, Skorski T, and Pomerantz RT (2015). Small-molecule disruption of RAD52 rings as a mechanism for precision medicine in BRCA-deficient cancers. Chem. Biol 22, 1491–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HHY, Pannunzio NR, Adachi N, and Lieber MR (2017). Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol 18, 495–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanut P, Britton S, Coates J, Jackson SP, and Calsou P (2016). Coordinated nuclease activities counteract Ku at single-ended DNA double-strand breaks. Nat. Commun 7, 12889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, and Zhang F (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantino L, Sotiriou SK, Rantala JK, Magin S, Mladenov E, Helleday T, Haber JE, Iliakis G, Kallioniemi OP, and Halazonetis TD (2014). Break-induced replication repair of damaged forks induces genomic duplications in human cells. Science 343, 88–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das BB, Huang SY, Murai J, Rehman I, Amé J-C, Sengupta S, Das SK, Majumdar P, Zhang H, Biard D, et al. (2014). PARP1-TDP1 coupling for the repair of topoisomerase I-induced DNA damage. Nucleic Acids Res. 42, 4435–4449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z, Scott SP, Bussen W, Sharma GG, Guo G, Pandita TK, and Powell SN (2011). Rad52 inactivation is synthetically lethal with BRCA2 deficiency. Proc. Natl. Acad. Sci. USA 108, 686–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster SS, Balestrini A, and Petrini JHJ (2011). Functional interplay of the Mre11 nuclease and Ku in the response to replication-associated DNA damage. Mol. Cell. Biol 31, 4379–4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimori A, Tachiiri S, Sonoda E, Thompson LH, Dhar PK, Hiraoka M, Takeda S, Zhang Y, Reth M, and Takata M (2001). Rad52 partially substitutes for the Rad51 paralog XRCC3 in maintaining chromosomal integrity in vertebrate cells. EMBO J. 20, 5513–5520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Katyal S, Lee Y, Zhao J, Rehg JE, Russell HR, and McKinnon PJ (2011). DNA ligase III is critical for mtDNA integrity but not Xrcc1-mediated nuclear DNA repair. Nature 471, 240–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravells P, Grant E, Smith KM, James DI, and Bryant HE (2017). Specific killing of DNA damage-response deficient cells with inhibitors of poly(ADP-ribose) glycohydrolase. DNA Repair (Amst.) 52, 81–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengel SR, Malacaria E, Folly da Silva Constantino L, Bain FE, Diaz A, Koch BG, Yu L, Wu M, Pichierri P, Spies MA, and Spies M (2016). Small-molecule inhibitors identify the RAD52-ssDNA interaction as critical for recovery from replication stress and for survival of BRCA2 deficient cells. eLife 5, e14740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochegger H, Dejsuphong D, Fukushima T, Morrison C, Sonoda E, Schreiber V, Zhao GY, Saberi A, Masutani M, Adachi N, et al. (2006). Parp-1 protects homologous recombination from interference by Ku and Ligase IV in vertebrate cells. EMBO J. 25, 1305–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda M, Okuno Y, Yoo J, Ha T, and Spies M (2011). Tyrosine phosphorylation enhances RAD52-mediated annealing by modulating its DNA binding. EMBO J. 30, 3368–3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Petit SA, Ficarro SB, Toomire KJ, Xie A, Lim E, Cao SA, Park E, Eck MJ, Scully R, et al. (2014). PARP1-driven poly-ADP-ribosylation regulates BRCA1 function in homologous recombination-mediated DNA repair. Cancer Discov. 4, 1430–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu L-Y, Chang C-C, Huang Y-S, Chou W-C, Lin Y-M, Ho C-C, Chen W-T, Shih H-M, Hsiung C-N, Wu P-E, and Shen CY (2018). SUMOylation of XRCC1 activated by poly (ADP-ribosyl)ation regulates DNA repair. Hum. Mol. Genet 27, 2306–2317. [DOI] [PubMed] [Google Scholar]
- Huang F, Goyal N, Sullivan K, Hanamshet K, Patel M, Mazina OM, Wang CX, An WF, Spoonamore J, Metkar S, et al. (2016). Targeting BRCA1- and BRCA2-deficient cells with RAD52 small molecule inhibitors. Nucleic Acids Res. 44, 4189–4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungmichel S, Rosenthal F, Altmeyer M, Lukas J, Hottiger MO, and Nielsen ML (2013). Proteome-wide identification of poly(ADP-Ribosyl)ation targets in different genotoxic stress responses. Mol. Cell 52, 272–285. [DOI] [PubMed] [Google Scholar]
- Kagawa W, Kurumizaka H, Ishitani R, Fukai S, Nureki O, Shibata T, and Yokoyama S (2002). Crystal structure of the homologous-pairing domain from the human Rad52 recombinase in the undecameric form. Mol. Cell 10, 359–371. [DOI] [PubMed] [Google Scholar]
- Kan Y, Batada NN, and Hendrickson EA (2017). Human somatic cells deficient for RAD52 are impaired for viral integration and compromised for most aspects of homology-directed repair. DNA Repair (Amst.) 55, 64–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitao H, and Yuan Z-M (2002). Regulation of ionizing radiation-induced Rad52 nuclear foci formation by c-Abl-mediated phosphorylation. J. Biol. Chem 277, 48944–48948. [DOI] [PubMed] [Google Scholar]
- Kohzaki M, Nishihara K, Hirota K, Sonoda E, Yoshimura M, Ekino S, Butler JE, Watanabe M, Halazonetis TD, and Takeda S (2010). DNA polymerases υ and θ are required for efficient immunoglobulin V gene diversification in chicken. J. Cell Biol 189, 1117–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li GC, Ouyang H, Li X, Nagasawa H, Little JB, Chen DJ, Ling CC, Fuks Z, and Cordon-Cardo C (1998). Ku70: a candidate tumor suppressor gene for murine T cell lymphoma. Mol. Cell 2, 1–8. [DOI] [PubMed] [Google Scholar]
- Li M, Lu L-Y, Yang C-Y, Wang S, and Yu X (2013). The FHA and BRCT domains recognize ADP-ribosylation during DNA damage response. Genes Dev. 27, 1752–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- London RE (2015). The structural basis of XRCC1-mediated DNA repair. DNA Repair (Amst.) 30, 90–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundin C, North M, Erixon K, Walters K, Jenssen D, Goldman ASH, and Helleday T (2005). Methyl methanesulfonate (MMS) produces heat-labile DNA damage but no detectable in vivo DNA double-strand breaks. Nucleic Acids Res. 33, 3799–3811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malacaria E, Pugliese GM, Honda M, Marabitti V, Aiello FA, Spies M, Franchitto A, and Pichierri P (2019). Rad52 prevents excessive replication fork reversal and protects from nascent strand degradation. Nat. Commun 10, 1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIlwraith MJ, Van Dyck E, Masson J-Y, Stasiak AZ, Stasiak A, and West SC (2000). Reconstitution of the strand invasion step of double-strand break repair using human Rad51 Rad52 and RPA proteins. J. Mol. Biol 304, 151–164. [DOI] [PubMed] [Google Scholar]
- Mills KD, Ferguson DO, Essers J, Eckersdorff M, Kanaar R, and Alt FW (2004). Rad54 and DNA Ligase IV cooperate to maintain mammalian chromatid stability. Genes Dev. 18, 1283–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison C, Sonoda E, Takao N, Shinohara A, Yamamoto K, and Takeda S (2000). The controlling role of ATM in homologous recombinational repair of DNA damage. EMBO J. 19, 463–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH, Ji J, Takeda S, and Pommier Y (2012). Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 72, 5588–5599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochs F, Somyajit K, Altmeyer M, Rask MB, Lukas J, and Lukas C (2016). 53BP1 fosters fidelity of homology-directed DNA repair. Nat. Struct. Mol. Biol 23, 714–721. [DOI] [PubMed] [Google Scholar]
- Ogorek B, and Bryant PE (1985a). Repair of DNA single-strand breaks in X-irradiated yeast. II. Kinetics of repair as measured by the DNA-unwinding method. Mutat. Res 146, 63–70. [DOI] [PubMed] [Google Scholar]
- Ogorek B, and Bryant PE (1985b). Repair of DNA single-strand breaks in X-irradiated yeast. I. Use of the DNA-unwinding method to measure DNA strand breaks. Mutat. Res 146, 55–61. [DOI] [PubMed] [Google Scholar]
- Panzeter PL, Realini CA, and Althaus FR (1992). Noncovalent interactions of poly(adenosine diphosphate ribose) with histones. Biochemistry 31, 1379–1385. [DOI] [PubMed] [Google Scholar]
- Patel AG, Flatten KS, Schneider PA, Dai NT, McDonald JS, Poirier GG, and Kaufmann SH (2012). Enhanced killing of cancer cells by poly(-ADP-ribose) polymerase inhibitors and topoisomerase I inhibitors reflects poisoning of both enzymes. J. Biol. Chem 287, 4198–4210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier Y, O’Connor MJ, and de Bono J (2016a). Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci. Transl. Med 8, 362ps317. [DOI] [PubMed] [Google Scholar]
- Pommier Y, Sun Y, Huang SN, and Nitiss JL (2016b). Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat. Rev. Mol. Cell Biol 17, 703–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray Chaudhuri A, and Nussenzweig A (2017). The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol 18, 610–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray Chaudhuri A, Hashimoto Y, Herrador R, Neelsen KJ, Fachinetti D, Bermejo R, Cocito A, Costanzo V, and Lopes M (2012). Topoisomerase I poisoning results in PARP-mediated replication fork reversal. Nat. Struct. Mol. Biol 19, 417–423. [DOI] [PubMed] [Google Scholar]
- Rijkers T, Van Den Ouweland J, Morolli B, Rolink AG, Baarends WM, Van Sloun PP, Lohman PH, and Pastink A (1998). Targeted inactivation of mouse RAD52 reduces homologous recombination but not resistance to ionizing radiation. Mol. Cell. Biol 18, 6423–6429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saotome M, Saito K, Yasuda T, Ohtomo H, Sugiyama S, Nishimura Y, Kurumizaka H, and Kagawa W (2018). Structural basis of homology-directed DNA repair mediated by RAD52. iScience 3, 50–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schipler A, and Iliakis G (2013). DNA double-strand-break complexity levels and their possible contributions to the probability for error-prone processing and repair pathway choice. Nucleic Acids Res. 41, 7589–7605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz N, Lopez E, Saleh-Gohari N, and Helleday T (2003). Poly(ADP-ribose) polymerase (PARP-1) has a controlling role in homologous recombination. Nucleic Acids Res. 31, 4959–4964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scully R, Panday A, Elango R, and Willis NA (2019). DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol 20, 698–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simsek D, Furda A, Gao Y, Artus J, Brunet E, Hadjantonakis AK, Van Houten B, Shuman S, McKinnon PJ, and Jasin M (2011). Crucial role for DNA ligase III in mitochondria but not in Xrcc1-dependent repair. Nature 471, 245–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotiriou SK, Kamileri I, Lugli N, Evangelou K, Da-Ré C, Huber F, Padayachy L, Tardy S, Nicati NL, Barriot S, et al. (2016). Mammalian RAD52 functions in break-induced replication repair of collapsed DNA replication forks. Mol. Cell 64, 1127–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan-Reed K, Bolton-Gillespie E, Dasgupta Y, Langer S, Siciliano M, Nieborowska-Skorska M, Hanamshet K, Belyaeva EA, Bernhardy AJ, Lee J, et al. (2018). Simultaneous targeting of PARP1 and RAD52 triggers dual synthetic lethality in BRCA-deficient tumor cells. Cell Rep. 23, 3127–3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland BM, Bennett PV, Sidorkina O, and Laval J (2000). Clustered DNA damages induced in isolated DNA and in human cells by low doses of ionizing radiation. Proc. Natl. Acad. Sci. USA 97, 103–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takata M, Sasaki MS, Sonoda E, Morrison C, Hashimoto M, Utsumi H, Yamaguchi-Iwai Y, Shinohara A, and Takeda S (1998). Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J. 17, 5497–5508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dyck E, Stasiak AZ, Stasiak A, and West SC (1999). Binding of double-strand breaks in DNA by human Rad52 protein. Nature 398, 728–731. [DOI] [PubMed] [Google Scholar]
- Verma P, and Greenberg RA (2016). Noncanonical views of homology-directed DNA repair. Genes Dev. 30, 1138–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZQ, Auer B, Berghammer H, Haidacher D, Schweiger M, and Wagner EF (1995). Mice lacking ADPRT and poly(ADP-ribosyl)ation develop normally but are susceptible to skin disease. Genes Dev. 9, 509–520. [DOI] [PubMed] [Google Scholar]
- Wang H, Zhang X, Wang P, Yu X, Essers J, Chen D, Kanaar R, Takeda S, and Wang Y (2010). Characteristics of DNA-binding proteins determine the biological sensitivity to high-linear energy transfer radiation. Nucleic Acids Res. 38, 3245–3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei L, Nakajima S, Hsieh CL, Kanno S, Masutani M, Levine AS, Yasui A, and Lan L (2013). Damage response of XRCC1 at sites of DNA single strand breaks is regulated by phosphorylation and ubiquitylation after degradation of poly(ADP-ribose). J. Cell Sci 126, 4414–4423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehouse CJ, Taylor RM, Thistlethwaite A, Zhang H, Karimi-Busheri F, Lasko DD, Weinfeld M, and Caldecott KW (2001). XRCC1 stimulates human polynucleotide kinase activity at damaged DNA termini and accelerates DNA single-strand break repair. Cell 104, 107–117. [DOI] [PubMed] [Google Scholar]
- Wood RD, and Doublié S (2016). DNA polymerase q (POLQ), double-strand break repair, and cancer. DNA Repair (Amst.) 44, 22–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi-Iwai Y, Sonoda E, Buerstedde J-M, Bezzubova O, Morrison C, Takata M, Shinohara A, and Takeda S (1998). Homologous recombination, but not DNA repair, is reduced in vertebrate cells deficient in RAD52. Mol. Cell. Biol 18, 6430–6435. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The published article includes data generated in this study, no unique software or code were generated for this study.