

Abstract
Summary
We analyze the transcriptome of baseline and on-therapy tumor biopsies from 101 patients with advanced melanoma treated with nivolumab (anti-PD-1) alone or combined with ipilimumab (anti-CTLA-4). We find that T cell infiltration and interferon-γ (IFNγ) signaling signatures correspond most highly with clinical response to therapy, with a reciprocal decrease in cell cycle and WNT signaling pathways in responding biopsies. We model the interaction in 58 human cell lines, where IFNγ in vitro exposure leads to a conserved transcriptome response unless cells have IFNγ receptor alterations. This conserved IFNγ transcriptome response in melanoma cells serves to amplify the antitumor immune response. Therefore, the magnitude of the antitumor T cell response and the corresponding downstream IFNγ signaling are the main drivers of clinical response or resistance to immune checkpoint blockade therapy.
eTOC Blurb
Analyzing the transcriptome of biopsies of patients during immune checkpoint blockade therapy, Grasso et al. show that the increase of T cell infiltration and the downstream IFNγ signaling drive clinical responses.
Graphical Abstract
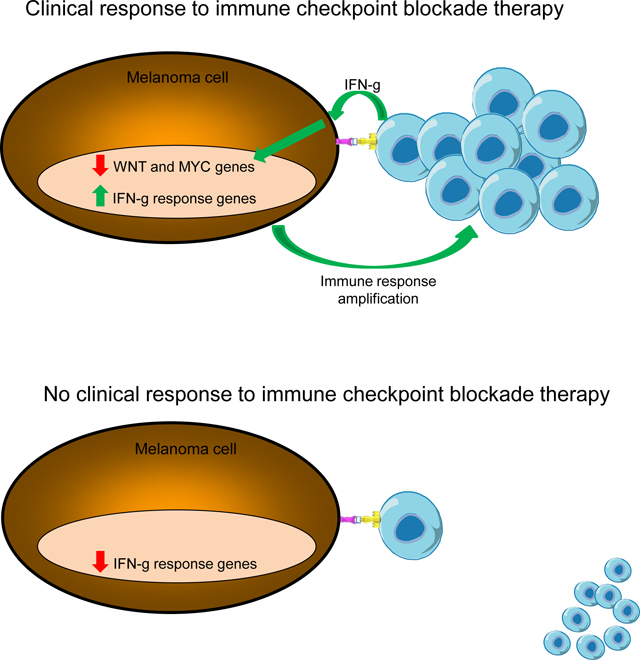
Introduction
Immune checkpoint blockade (ICB) therapy with antibodies targeting the cytotoxic T-lymphocyte–associated protein 4 (CTLA-4) or the programmed cell death-1 receptor (PD-1) blocks two main negative regulators of antitumor immune responses (Curran et al., 2010; Sharma and Allison, 2015a; Wei et al., 2017) and induces durable responses in a subset of patients with melanoma and other cancers (Ribas and Wolchok, 2018; Sharma and Allison, 2015b). Pathological studies performed in tumor biopsies from treated patients support that clinical responses induced by the use of ICB are mediated by tumor-infiltrating T cells that have been re-activated by inhibiting these immune checkpoints (Sharma et al., 2019; Tumeh et al., 2014). Transcriptomic and genomic sequencing of baseline and on-therapy biopsies from patients treated with ICB allow for a comprehensive analysis of mechanisms underlying tumor response and resistance. As the mechanism of action is based on the interaction between immune effector cells with their cancer cell targets, these studies have to focus not only on the genetic alterations and gene expression profiles of cancer cells (Hugo et al., 2016; Liu et al., 2019; Riaz et al., 2017; Rodig et al., 2018), but they also need to analyze the composition of the immune infiltrate and its expression of immune activating gene programs (Auslander et al., 2018; Ayers et al., 2017; Cabrita et al., 2020; Chen et al., 2016; Cristescu et al., 2018; Fehrenbacher et al., 2016; Gide et al., 2019; Helmink et al., 2020; Jerby-Arnon et al., 2018; Jiang et al., 2018; Liu et al., 2019; Petitprez et al., 2020; Rodig et al., 2018; Roh et al., 2017; Sade-Feldman et al., 2018). A major focus has been on the study of biopsies from patients with advanced melanoma treated with anti-PD-1 antibodies administered alone, in sequence or in combination with anti-CTLA-4 antibodies. A study combining immunohistochemistry analyses with RNAseq in biopsies from patients treated sequentially with the anti-CTLA-4 antibody ipilimumab before or after the anti-PD-1 antibody nivolumab concluded that primary response to anti-CTLA-4 required high levels of major histocompatibility complex (MHC) class I expression by cancer cells at baseline, while response to anti-PD-1 was more associated with a preexisting interferon-gamma transcriptome signature (Rodig et al., 2018). Another study combining immunohistochemistry analyses with RNAseq in tumor biopsies from patients treated with anti-PD-1 monotherapy or combined with anti-CTLA-4 confirmed the association of response to PD-1 blockade therapy with baseline evidence of activated T cells using morphological and transcriptome signatures, in particular of an effector memory T cell phenotype (Gide et al., 2019). A third study with a large cohort of baseline biopsies from patients treated with anti-PD-1 therapy, with or without prior anti-CTLA-4 therapy, revealed that response to PD-1 blockade was associated with increased MHC class I and II expression (Liu et al., 2019). In this study, whole exome sequencing revealed occasional genetic alterations in antigen presentation machinery genes in biopsies of patients who did not respond to therapy (Liu et al., 2019).
CheckMate 038 is a prospective, multicenter, international, multi-cohort clinical trial designed to collect tumor biopsies from patients with metastatic melanoma treated with the anti-PD-1 antibody nivolumab as front-line therapy or after progressing on therapy with the anti-CTLA-4 antibody ipilimumab, or receiving the combination of both antibodies. Biopsies of 68 patients receiving nivolumab monotherapy in part 1 of this study have been previously reported (Riaz et al., 2017). These samples were analyzed by whole-exome, transcriptome, and TCR sequencing. Data revealed increases in distinct immune cell subsets and upregulation of immune activation gene programs that was more pronounced in patients with a clinical response to therapy (Riaz et al., 2017). In the current study, we provide information on the transcriptome analysis of tumor biopsies from the complete set of 101 patients treated with single agent nivolumab or with the combination of nivolumab and ipilimumab, which is correlated with the in vitro analysis of how a panel of melanoma cell lines change gene expression upon exposure to interferon-gamma.
Results
Patient characteristics and response to ICB therapy
We analyzed tumor biopsies obtained from patients treated with nivolumab or nivolumab plus ipilimumab within the CheckMate 038 study (NCT01621490), which required baseline and on-therapy biopsies from all patients. The clinical trial had several parts (Figure S1). Part 1 included two cohorts that received nivolumab monotherapy: patients whose melanomas had previously progressed on anti-CTLA-4 monotherapy, and those who were naïve to prior anti-CTLA-4. In Part 2, patients who were anti-CTLA-4-naïve received combination therapy with nivolumab and ipilimumab. Part 3 was also restricted to patients who were anti-CTLA-4-naïve, and randomized patients 1:2 to receive either nivolumab monotherapy or combination therapy with ipilimumab. Part 4 was similar to Part 3 in being restricted to patients naïve to anti-CTLA-4, but included patients with active brain metastases that received either nivolumab monotherapy or combination therapy with ipilimumab. Results of analysis of biopsies of patients from Part 1 have been reported previously by Riaz et al. (Riaz et al., 2017), while analyses from Parts 2–4 have not been previously reported. Biopsies were planned at baseline and during treatment (week 4 for Part 1, and week 2 or 4 for Parts 2–4) and were processed centrally to obtain RNA for sequencing analyses.
Among the 170 patients enrolled in the clinical trial (Figure 1A), 106 patients received nivolumab alone and 62 received nivolumab and ipilimumab combined therapy. RNA was isolated from 101 baseline and 99 on-therapy biopsies (Figure 1A). Several biopsies were not included in this analysis for different reasons, including specimens not meeting quality standards, patients who received a treatment that was not assigned, not having adequate tumor response assessment, and a diagnosis of primary choroidal melanoma (8 patients) due to the distinct biology of this uncommon melanoma subtype (Figure 1A). At the end, there were 84 baseline and 85 on-therapy biopsies (68 of which were paired), that contributed to transcriptome analyses for RNAseq (Figure 1A). Table 1 shows the clinical characteristics of the analysis cohort, including 101 total patients: median age was 56 years (range 22–89). The majority (71%) of patients had cutaneous melanoma, with 11% mucosal and 4% acral melanoma. The majority of patients had stage IV disease (93%), and the adverse prognosis factor of elevated lactate dehydrogenase (LDH) was present in 28% of patients.
Figure 1.

Outline of sample collection, patient treatments and response to therapy. A) Consort diagram describing data generation and selection of final transcriptome cohort. B) Alluvial plot showing the number of patients of each treatment subtype that resulted in each response. The numbers in the treatment subtype box represent the number of patients of that treatment type that had that response (Red = PD, Green = SD, Blue = CRPR). See also Figures S1 and S2.
Table 1.
Baseline patient characteristics
| n (%) N = 101 |
|
|---|---|
| Age (years) | |
| Median | 56 |
| Range | 22–89 |
| Sex, n (%) | |
| Male | 58 (57.4) |
| Female | 43 (42.6) |
| ECOG performance status, n (%) | |
| 0 | 70 (69.3) |
| 1 | 29 (28.7) |
| Not Reported | 2 (2.0) |
| Stage at study entry, n (%) | |
| III | 8 (7.9) |
| IV | 93 (92.1) |
| Prior anti-CTLA-4 therapy, n (%) | |
| Yes | 30 (29.7) |
| No | 71 (70.3) |
| BRAF mutation status, n (%) | |
| Positive | 33 (32.7) |
| Negative | 61 (60.4) |
| Not Reported | 7 (6.9) |
| PD-L1 status at baseline, n (%) | |
| PD-L1 negative (TPSbaseline = 0) | 30 (29.7) |
| PD-L1 positive (TPSbaseline > 0) | 48 (47.5) |
| N/A (TPSbaseline = NA) | 23 (22.8) |
| Metastatic staging, n (%) | |
| M0 | 2 (2.0) |
| M1A | 17 (16.8) |
| M1B | 15 (14.9) |
| M1C | 53 (52.5) |
| Not Reported (Stage III) / Unknown | 14 (13.9) |
| Brain metastases, n (%) | |
| Yes (*) | 13 (12.9) |
| No | 80 (79.2) |
| Not Reported (Stage III) | 8 (7.9) |
| Lactate dehydrogenase, n (%) | |
| Normal (<= ULN) | 72 (71.3) |
| Elevated (> ULN, <= 2*ULN) | 24 (23.8) |
| Highly elevated (> 2*ULN) | 4 (4.0) |
| Not Reported | 1 (1.0) |
| Melanoma primary site, n (%) | |
| Acral | 4 (4.0) |
| Cutaneous | 71 (70.3) |
| Mucosal | 11 (10.9) |
| Other | 15 (14.9) |
ECOG, Eastern Cooperative Oncology Group; M1a, metastases to skin, subcutaneous tissues, or distant lymph nodes; M1b, metastases to lung; M1c, metastases to all other visceral sites or distant metastases at any site combined with an elevated serum concentration of lactate dehydrogenase; PD-L1, programmed death ligand 1.
In addition to all patients enrolled in Part 4, a number of patients in Parts 1–3 also had reported brain metastases.
CheckMate 038 was not designed to assess comparisons between study groups as these were mostly non-randomized cohorts (Figure S1). Therefore, the account of tumor responses and time-to-event outcomes is descriptive and used for the interpretation of the tumor biopsy analyses. Overall, there were more patients with an objective response (complete response or partial response, CRPR) in the group receiving the combination of nivolumab and ipilimumab than among patients treated with nivolumab monotherapy, whether or not naïve to anti-CTLA-4 therapy (Figure 1B). Analyses of overall survival (OS) and progression-free survival (PFS) suggested that survival outcomes were similar among patients receiving nivolumab monotherapy, whether or not they had received prior anti-CTLA-4; OS and PFS among patients receiving combination nivolumab plus ipilimumab therapy appeared to exceed nivolumab monotherapy outcomes (Figure S2A). When analyzing OS and PFS according to tumor response assessment, patients with CRPR unsurprisingly had prolonged survival compared to those with stable disease (SD) or progressive disease (PD) (Figure S2B). Based on these observations, for the RNAseq analyses we compared patients receiving combination therapy to those receiving nivolumab monotherapy (merging the groups that had or had not previously received ipilimumab), and clinical response was defined as CRPR compared to SD or PD.
The dominant signal associated with response or resistance to ICB therapy in patient biopsies is mediated by T cells
Consistently, it has been shown that high levels of CD8 T cell infiltration of tumors at baseline and on-treatment are predictive of clinical response to ICB in patients with advanced melanoma (Chen et al., 2016; Gide et al., 2019; Tumeh et al., 2014). We used the method denominated microenvironment cell populations-counter (MCP-counter) (Becht et al., 2016) for RNAseq data deconvolution to define immune cell types. We applied an optimal pooled t-test, since only a subset of the samples were paired. Using these approaches, we documented that biopsies of patients with a response to therapy had higher baseline levels of T cells than those with PD (Figure 2A). They also had significantly higher baseline levels of B lineage cells, myeloid dendritic cells, and NK cells (Figure 2A). The change from baseline to on-therapy was greater in biopsies of patients with a response to therapy, and there were increases in T cell infiltrates in biopsies of patients whether or not they had an objective clinical response to therapy, and regardless of receiving combination therapy or nivolumab alone (Figure 2A and 2B).
Figure 2.

Immune cell infiltration in patient samples from clinical study CheckMate 038. A) Box plot the MCP-Counter T cell score according to response to therapy combining all treatment groups using an optimal pooled t-test since data is paired and unpaired. B) Principal component analysis showing all RNAseq samples, paired and unpaired, plotted on the first two components. The size of the data point is proportional to the T cell score. Open circles correspond to pre-treatment samples, while closed circles correspond to on-treatment samples. The data points are colored by response (Red = PD, Green = SD, and Blue = CRPR). The vector for the T cell score is plotted as well. Outlier samples are labeled by case. The twelve paired outlier samples were either due to quality control issues that had not been detected, or may be due to an actual different biology. For example, cases 48, 20038, and 30022 all had interferon-gamma signaling either going down or not changing on treatment, while cases 30004 and 20001 have G2M cell cycle genes that either increase or did not respond. C) Box plots showing the average expression of the genes correlated with the T cells broken down by response and pre- and on-treatment using an optimal pooled t-test since data is paired and unpaired. D) Box plots of MCP-Counter immune cell deconvolution types according to response to therapy. Mixed t-test for paired and unpaired samples using an optimal pooled t-test since data is paired and unpaired (*p value < 0.05, **p value < 0.01, ***p value < 0.001, ****p value < 0.0001). See also Figures S3, S4 and S5.
With the goal to define the main drivers explaining the overall biopsy data, we then analyzed the whole transcriptome RNAseq dataset using principal component analysis. Analysis of the first two principal components showed that the signature for T cell score derived from MCP-counter RNAseq data deconvolution was the main factor organizing the data, and it segregated with response to therapy (with the exception of 19 outlier samples, 12 of which were matched from the same patients with baseline and on-therapy biopsies, Figure 2C). Of note, in prior work we had previously validated the MCP-counter T cell score with pathological analysis of immune infiltrates in corresponding patient biopsies (Grasso et al., 2018). In the Chekcmate-038 biopsy dataset, the best organization of the samples was according to the degree of T cell infiltration regardless whether this was observed in baseline or on-therapy biopsies, with or without prior treatment with ipilimumab, single or combination ICB therapy, as well as clinical response or no response to therapy, with the paired CRPR samples showing the most consistent alignment along the T cell score vector (Figure 2C and D). Figure S3 shows the degree to which the T cell score dominates the data, with the large number of genes correlated (n = 2,047) and anticorrelated (n = 1,087) with this score based on a Pearson correlation cut-off of ± 0.3. This supplemental figure also includes information about the effect of the melanoma subtype (cutaneous, mucosal or acral) on the tumor transcriptome. The coordinated alignment of the correlated and anti-correlated genes on treatment is reflected in the significant up-regulation of the correlated genes and the significant down-regulation from baseline to on-treatment in CRPR biopsies (optimal pooled t-test p value < 1e-4 and p value < 1e-4, respectively, Figure 2C). The correlated genes accounted not only for systematic changes in the tumor cell gene expression, but also in the tumor microenvironment. Except for neutrophils, the MCP-counter immune cell signatures were highly correlated with the T cell signature (R^2 ≥ 0.6, Figure S4A and S4B). Use of t-SNE embedding to cluster genes with their closest MCP-Counter immune cell signature resulted in only two main clusters: genes that correlated with the T cell score and genes that were anti-correlated (Figure S4C). These data indicate a truly coordinated immune response, including large numbers of genes, which was dominated by changes in T cells. We acknowledge that this conclusion is limited by being derived from bulk RNAseq analyses as opposed to single cell RNAseq. Based on these analyses, we conclude that ICB therapy has the potential to increase T cell infiltrates regardless of whether there are clinical responses to therapy, and that a greater degree of T cell infiltrate increases from baseline to on-therapy biopsies is associated with improved response to therapy.
Interferon-gamma response genes play an integral role in response and resistance to therapy as mediators of the T cell program
We examined the expression of effector cytokines and toxic granules induced by TCR engagement with cognate antigen, including the cytotoxic molecules perforin and granzyme B, TNF-alpha family members FAS, TRAIL (TNFSF10) and TNF-alpha, and interferon-gamma, applying an optimal pooled t-test. The expression of perforin, granzyme B, TRAIL and TNF-alpha followed the pattern of expression of interferon-gamma, and was higher in biopsies of patients with a clinical response to therapy (Figure S5). As exposure to most of these molecules results in cytotoxic death of cancer cells, we reasoned that the cancer cell expression of interferon-response genes may be relevant to attracting other immune cells into the tumor microenvironment and amplifying the antitumor immune response once it is initiated.
Melanoma cells have a uniform gene set response to interferon-gamma provided that they can signal through the interferon-gamma receptor
With the goal of addressing the question if differential responses to ICB may be due to a different inherent ability of melanoma cells from different patients to respond to interferon-gamma, we set out to examine the transcriptome changes in a panel of ex vivo cultured melanoma cells exposed to interferon-gamma. For this study, we used 57 previously established and described cell lines from cultures of human melanoma biopsies or surgical resections (Atefi et al., 2014; Neubert et al., 2017; Shin et al., 2017; Tsoi et al., 2018) and the primary melanocyte line, HeMa, as a non-malignant pigmented cell control. This cell line panel included three variants of corresponding parental melanoma cell lines that had been rendered resistant to BRAF inhibitors by continuous in vitro drug exposure, denoted as AR for acquired resistance (Atefi et al., 2014; Nazarian et al., 2010; Tsoi et al., 2018); three cell lines that had developed Janus kinase 1 (JAK1) or JAK2 loss of function mutations in patients; as well as variants of parental cell lines that had JAK1 (N=5), JAK2 (N=5) or beta-2 microglobulin (B2M) (N=2) loss of function generated in vitro through CRISPR/Cas9 gene editing, as these are genetic events known to be associated with resistance to anti-PD-1 therapy (Garcia-Diaz et al., 2017; Gettinger et al., 2017; Sade-Feldman et al., 2017; Shin et al., 2017; Sucker et al., 2017; Torrejon et al., 2020; Zaretsky et al., 2016). These cell lines were exposed to 5 ng/ml of interferon-gamma for 6 hours to assess immediate whole transcriptome changes by RNAseq analysis.
We found that a cell line’s gene expression profile was most correlated with itself at baseline and upon exposure to interferon-gamma, indicating that cell line identity is a major contributor to gene expression (Figure S6). In addition, we observed that the baseline samples were not correlated with each other, nor were the interferon-gamma exposed samples correlated with each other (Figure S6). However, for the 46 cell lines that responded to interferon-gamma, the changes between on-treatment and baseline for interferon-gamma were highly correlated as a result of consistent large changes in interferon-gamma response genes (Figure S7). Figure S8A shows the high-level of concordance among expression changes of known interferon-gamma response genes, making it possible to identify outlier gene expression changes, such as the two samples whose JAK2 expression decreased significantly compared to the rest of the samples. Analysis of all increased and decreased gene expression showed that there were two broad groups of cell lines: the great majority (n = 46) that could signal through the interferon-gamma receptor and had large changes in the expression of the same set of interferon-gamma response genes; and a distinct set of 12 cell lines that had JAK1 or JAK2 natural or induced loss of function mutations and were largely unable to signal through the interferon-gamma receptor (Figure 3 and Figure S8B). Of note, there were no major differences in gene expression profiles induced by interferon-gamma exposure in cell lines with acquired resistance to BRAF inhibitors or in B2M knockout cell lines. Therefore, this large panel of melanoma cell lines, each of which had a unique gene expression signature, responded consistently to interferon-gamma exposure.
Figure 3.

Interferon-gamma-induced changes in gene expression in 58 human cell lines. A) Heatmap of changes in key interferon-gamma response genes after 6 hours of treatment at 5 ng/ml expressed as fold changes pre-treatment to post-treatment. Genes are organized by class: anti-proliferative (Black), antigen presentation (White), chemoattractants (Cyan), cytotoxic effectors (Orange), feedback/signaling (Purple). B) The pre- and post-treatment gene expression levels of IRF1, the transcription factor executing the down-stream interferon gamma program, shown as arrows (Red = up, Blue = down). C) The pre- and post-treatment gene expression levels of CD274, the gene that codes for PD-L1, shown as arrows (Red = up, Blue = down). See also Figures S6, S7, S8, S9 and S10.
To identify a set of genes up- and down-regulated in response to interferon-gamma in typical melanoma cell lines, we used all the samples that were not experimentally modified (excluding cases with JAK1, JAK2, and B2M loss generated by CRISPR-CAS9, as well as the AR samples but including naturally occurring JAK and B2M loss cases). We performed a paired t-test between cell lines before and after treatment with interferon gamma and observed a significant decrease in the expression of 1,176 genes and increase in the expression of 549 genes after applying an FDR cut-off of 0.01. These changes clustered with the changes in the primary melanocyte line HeMa, included as a normal control, indicating that normal interferon-gamma signaling was intact in all of these cases (Figure S8B). This transcriptome signature includes previously reported interferon-gamma response genes, melanoma-specific responses, as well as a deeper look into the genes whose expression decreases in response to interferon-gamma (Figure S9).
Interferon-gamma triggers melanoma cells to increase expression of interferon pathway genes, antigen presenting machinery and T cell attracting chemokines
We next focused the analysis on changes in known genes that are related to interferon-gamma exposure, including interference with cell proliferation (the first functional effect that led to the description of interferons), upregulation of antigen presentation machinery, enhancement of immune cell-attracting chemokines, as well as genes involved in positive and negative downstream interferon signaling pathways (Bach et al., 1997). Figure 3A shows the response as fold changes, while Figure S10 shows it in terms of baseline and on-treatment gene expression. The overall change in transcripts involved in cell proliferation was low after 6 hours of in vitro interferon-gamma exposure (Figure 3A). There was evidence of increased expression of multiple HLA class I and II genes, with the most consistent increase being in HLA-E. The two major clusters in the data were defined by whether or not HLA class II genes are expressed at baseline or not. There was a very strong and consistent increase in the expression of additional genes in the antigen presentation pathway, in particular NLRC5 (also known as HLA class I transactivator) and CIITA (HLA class II transactivator), TAP transporters and proteasome subunits. Another set of genes with large increases were those involved in the interferon signaling pathway itself, representing an amplification of the interferon-gamma signal, including JAK2 (but not JAK1), several signal transductors and activators of transcription (STATs) and interferon response factors (IRFs), in particular the transcription factor IRF1 which serves as a well-defined positive control gene of interferon-gamma response (Garcia-Diaz et al., 2017), going up in all the samples with JAKs intact (Figure 3B). With this gene group, there was also an increase in negative regulators of the pathway including SOCS genes, as well as PD-L1 (CD274), which are also well characterized interferon-gamma immediate response genes (Garcia-Diaz et al., 2017; Shin et al., 2017) (Figure 3C). Chemokines, in particular CXCL9, CXCL10 and CXCL11, were increased more consistently than immune effector molecules and physiologically serve to attract more T cells in response to interferon-gamma exposure. These increases in gene expression were very inconsistent or non-existent in the 12 cell lines that had JAK1 or JAK2 natural or induced loss-of-function mutations. Overall, melanoma cell lines have a near uniform transcriptome response to interferon-gamma exposure, with increases in transcripts for antigen presentation, interferon pathway and chemokine genes, that is lost if there are loss-of-function mutations in interferon-gamma receptor pathway signaling at the level of JAK1 or JAK2.
Analysis of interferon-gamma response genes in patient biopsies reveals increased HLA expression as the major difference between response and lack of response to ICB therapy
We analyzed the Checkmate-038 patient biopsies for interferon-gamma response genes before and during ICB therapy. For this analysis, we acknowledge that cells other than melanoma cells may be responsible for the expression of interferon-gamma response genes when analyzing bulk RNAseq from biopsies as opposed to the experiment using human cell lines or single cell RNAseq from fresh tumor biopsies. Biopsies of patients with PD and SD included samples with and without expression of interferon response genes at baseline (Figure 4). There was evidence of an increase in the frequency and intensity of gene expression in on-therapy biopsies, in particular for chemokines and immune effector molecules (Figure 4). Biopsies of patients with CRPR had higher baseline expression of interferon response genes, which increased substantially in the on-therapy biopsies (Figure 4). The largest difference between the PD and SD groups compared to the CRPR group was in the marked increase in antigen presentation pathway genes, in particular in HLA class I and class II genes, B2M, TAP1, TAP2, NLRC5 and CIITA. After normalizing the data by leukocyte common antigen (LCA, CD45 gene) expression to account for changes in immune cell infiltrates, the HLA class I gene expression did not increase. This does not mean that it did not increase in the tumor, but rather that the predicted increase in HLA class I expression due to interferon-gamma signaling was not large enough to be detected against the background of other changes in the tumor microenvironment (Figures S11 and S12). Therefore, the main difference between interferon-gamma genes in biopsies of patients with response and resistance to ICB was an increase in antigen presentation genes in biopsies obtained during therapy, a conclusion that can be confounded by being derived from bulk RNAseq analyses.
Figure 4.
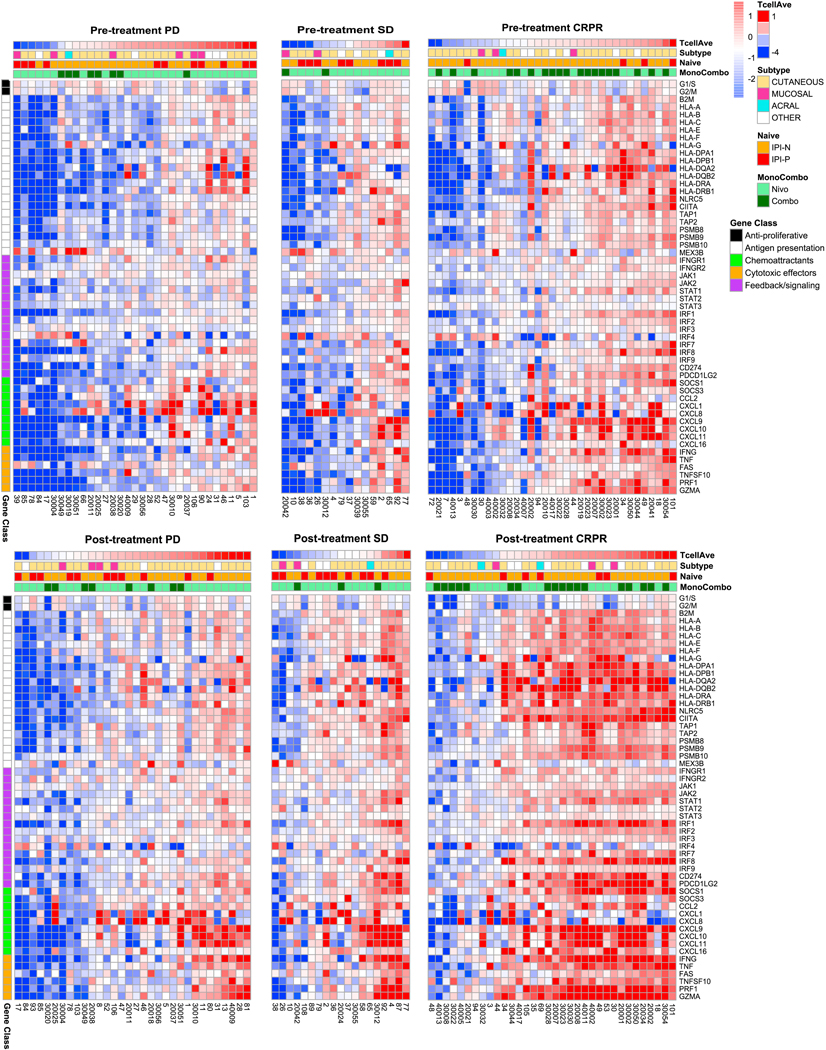
Interferon-gamma-induced changes in gene expression in specimens in clinical study CheckMate 038. Heatmap of key interferon-gamma response genes in biopsies obtained at baseline and on-treatment with immune blockade therapy, separated by response to therapy. Samples are ordered by T cell score and annotated by ipilimumab naïve status, monotherapy versus combination therapy and melanoma subtype. Genes are organized by class: anti-proliferative (Black), antigen presentation (White), chemoattractants (Cyan), cytotoxic effectors (Orange), feedback/signaling (Purple). See also Figures S11 and S12.
CRPR cases were associated with consistent changes in hallmark pathways, while PD cases involve different pathways for each sample
We found a large set of genes that were significantly different between on-treatment CRPR and on-treatment PD, likely driven in part by the large number of consistent changes in the on-treatment CRPR relative to the pre-treatment CRPR (1,935 up and 3,503 down after applying an optimal pooled t-test with an FDR cut-off of 0.05, Figure S13). The SD and PD samples did not change consistently from pre-treatment to on-treatment, except for four genes, ADI1, ARHGEF9, POU6F2, RGPD4, which were significantly different on-treatment relative to pre-treatment SD cases (after applying an optimal pooled t-test with an FDR cut-off of 0.05). Only one gene in baseline samples was associated with response, as expression of GPR31 was significantly different between biopsies of patients with CRPR or PD (p = 8.4e-7, after applying an optimal pooled t-test with an FDR cut-off of 0.05).
To understand the major pathways driving response or lack of response, we applied Gene Set Enrichment Analysis (GSEA) using the Hallmark Gene Signatures to identify enrichment of gene sets changing on-treatment in CRPR (Figure 5A). We identified a set of gene signatures that were also present when we applied GSEA to genes significantly altered in cell lines after interferon-gamma treatment (Figure 5B). When analyzed individually for the two top-scoring pathways according to response to therapy, most biopsies with CRPR had increases in interferon-gamma genes and decreases in G2M checkpoint genes, while biopsies from patients with PD had increases or decreases of these gene sets with no apparent directionality (Figure S14). This implies that interferon-gamma response, presumably resulting from higher tumor antigen-specific T cell infiltration, is the major driver of clinical response in the CRPR samples. Despite the caveat of the mixed cell content of the tumor microenvironment contributing to the bulk RNAseq transcriptome analysis, the CRPR and SD cases had high increases in interferon-gamma response gene sets that were consistent with the interferon-gamma 6 hour in vitro human cell line data, while there were PD cases for which all the interferon-gamma genes were downregulated (Supplemental Figures 14 and 15). Similarly, for the HALLMARK_G2M_CHECKPOINT gene set, most of the CRPR and SD cases were decreased, consistent with the interferon-gamma 6-hour in vitro exposure data (Figure S14 and S15). However, there were biopsies from multiple patients with PD and one with SD for which all the cell cycle genes increased. Other individual Hallmark Gene Sets similarly revealed fold changes consistent with interferon-gamma treatment for CRPR cases, while changes for the PD cases were the opposite of what would be expected based on interferon-gamma biology. This is true both for additional immune gene sets (Figures S16 and S17) and for additional cell cycle gene sets for E2F targets and mitotic spindle (Figure S18). The main observation was that the gene expression profile of biopsies from patients with clinical response is well-defined and consistent with exposure to interferon-gamma, while there was a large diversity of gene expression changes in SD and PD cases.
Figure 5.

Global changes in gene expression in clinical study CheckMate 038 consistent with interferon-gamma induced changes in biopsies from patients with response but not without response to therapy. A and B) Gene Set Enrichment Analysis (GSEA) using Hallmark Gene Sets for significantly up- or down-regulated genes for CRPR post-treatment relative to pre-treatment for clinical data set CheckMate 038 (A) and significantly up- or down-regulated genes for interferon gamma 6-hour treatment for the unmodified human cell lines (B). At most 2,000 of most significant genes by q-value submitted to GSEA for each class (maximum set by GSEA). See also Figures S13, S14, S15, S16, S17 and S18.
Changes in WNT, MYC and T cell exclusion programs as tumor-intrinsic responses to interferon-gamma
Several publications have recently shown that high WNT signaling contributes to immune exclusion (Grasso et al., 2018; Luke et al., 2019; Nsengimana et al., 2018; Spranger et al., 2015; Spranger and Gajewski, 2018). We used a 9-gene RNAseq based WNT score to assess the level of WNT signaling in each biopsy (Nsengimana et al., 2018). The WNT score is the geometric average of APC, APC2, CTNNB1, MYC, SOX11, SOX2, TCF12, TCF7, VEGFA. Analysis of WNT gene score revealed that biopsies of patients with CRPR exhibited a consistent significant decrease in WNT score in the on-therapy biopsies, while there was no change in the score in biopsies of patients with SD or PD (after applying an optimal pooled t-test) (Figure 6A). To explore if this effect was correlated with the degree of change in T cell infiltration, we plotted the WNT score with T cell infiltration by RNAseq deconvolution with baseline and on-therapy biopsies tracked with arrows. Again, we acknowledge that this analysis is limited by the bulk RNAseq that includes cellular changes in the tumor microenvironment in responding biopsies, which may make it harder to interpret the results. Despite this caveat, we noted that biopsies of patients with PD or SD had a rather random distribution in this WNT/T cell space, while the majority of biopsies from patients with CRPR had a trend of going from a high WNT/low T cell to a low WNT/high T cell when comparing the baseline and on-therapy biopsies (Figure 6B). A key advantage of the data derived from the cell lines before and after exposure to interferon-gamma in vitro was that the results were not confounded by tumor purity. We observed the same significant downward trend in the WNT gene score (p value < 7.8e-06 after applying a paired t-test) in the interferon-gamma responsive melanoma cell lines (Figure 6C), as six of the genes comprising the 9-gene WNT score were significantly decreased in response to interferon-gamma exposure. MYC, a WNT target gene used to monitor changes in WNT signaling, was similarly down-regulated in response to interferon-gamma exposure across most samples (Figure 6D and Figure S19). Upstream WNT signaling genes also changed consistently in response to interferon-gamma exposure, including DKK1 and the frizzled gene family (Figure S20A). In fact, up-regulation of FZD5 was one of the highest increases across all samples (Figure S20B).
Figure 6.

Immune exclusion signatures and WNT signaling in biopsies of patients receiving ICB therapy. A) Box plot of the 9-gene Wnt Score (the geometric average of APC, APC2, CTNNB1, MYC, SOX11, SOX2, TCF12, TCF7, VEGFA), by RNAseq according to response to therapy in patient biopsies using an optimal pooled t-test since data is paired and unpaired (*p value < 0.05, **p value < 0.01, ***p value < 0.001). B) Plot of Wnt score versus T cell score with arrows connecting pre-treatment and on-treatment and one figure for each response PD (Red), SD (Green), and CRPR (Blue). C to E) Pre- and post-treatment gene expression levels of the Wnt Score (C), MYC (D), and WNT5A (E), the Wnt ligand initiating the down-stream Wnt signaling programming (Red = up, Blue = down). F to J) Box plots of immune exclusion genes/signatures by RNAseq according to response to therapy: Jerby-Arnon, et al. associated immune exclusion gene sets 1 up in immune excluded tissues (F) and 2, down in immune excluded samples (G), CDK4 (H), MYC (I) and PAK4 (J) using an optimal pooled t-test since data is paired and unpaired (*p value < 0.05, **p value < 0.01, ***p value < 0.001). K) Heatmap showing the level of correlation between the immune exclusion genes and signatures. See also Figures S19, S20 and S21.
Expression of genes that drive immune exclusion are selectively decreased in biopsies during response to therapy
Recently, Jerby-Arnon et al. (Jerby-Arnon et al., 2018) developed an immune exclusion signature by single cell RNA sequencing of melanoma cells from biopsies with low levels of immune infiltration. This work identified a set of transcripts originating from tumor cells that were positively or negatively associated with immune exclusion by that tumor. Figure S21 shows a heatmap of these two sets of genes for the Checkmate-038 dataset. CRPR cases showed a significant decrease in the immune excluded-high genes (p value < 1.4e-4, after applying an optimal pooled t-test, Figure 6F and 6G). These genes did not change significantly in response to interferon-gamma in our in vitro testing in melanoma cell lines (the immune excluded-down genes do go up significantly, p value < 0.006, after applying a paired t-test). We similarly observed significant down-regulation in CDK4 restricted to CRPR cases (after applying an optimal pooled t-test, Figure 6H), which was reported as the driver of the immune exclusion signature (Jerby-Arnon et al., 2018). Based on our melanoma cell line cohort, interferon-gamma does not appear to significantly alter CDK4 expression. On the other hand, MYC genes followed the same pattern (after applying an optimal pooled t-test, Figure 6I), consistent with the WNT gene score (Figure 6A), while also going down significantly in response to interferon-gamma. We previously reported, using a separate set of biopsies, that PAK4 was over-expressed in biopsies of patients with low T cell infiltration and lack of response to PD-1 blockade therapy, and that in experimental mouse models the T cell exclusion and anti-PD-1 resistance could be reversed by PAK4 inhibition (Abril-Rodríguez et al., 2020). In the Checkmate 038 biopsy dataset, PAK4 expression did not differ significantly between baseline and on-treatment for patients with PD or SD, but expression was significantly (p value < 2.7e-4) downregulated in on-treatment biopsies of patients with CRPR (after applying an optimal pooled t-test, Figure 6J). This change was not explained by interferon-gamma signaling, since in our melanoma cell line cohort, PAK4 did not change significantly in response to interferon-gamma. Interestingly, all of these biomarkers of immune exclusion were highly correlated, except for the T cell exclusion-up signature, which, as expected, was anti-correlated (Pearson correlation, Figure 6K). Together, these data indicate that ICB therapy results in decreased expression of immune exclusion gene programs in biopsies of patients who experience clinical response to therapy. While interferon-gamma from T cells directly decreases WNT signaling and MYC, decreases in the Jerby-Arnon immune exclusion down signature, CDK4 or PAK4 expression are likely downstream effects in clinically responding tumors.
Discussion
Results from the current study provide a possible scenario by which response to ICB therapy, either PD-1 blockade alone or in combination with CTLA-4 blockade, can be explained by the development of a strong T cell response that can overcome immune cell-intrinsic, tumor-intrinsic and tumor microenvironment limitations to a clinically-effective antitumor immune response (Kalbasi and Ribas, 2020). Key contributing factors are: i) the pre-existing level of T cell infiltration of the tumor (Herbst et al., 2014; Taube et al., 2014; Tumeh et al., 2014), which reflects both the immunogenicity of the cancer cells and the ability of the host immune system to have made a serious attempt to attack them specifically; ii) the baseline expression of immune suppressive gene programs by cancer cells that are detrimental to the initiation of an antitumor immune response, such as WNT signaling (Grasso et al., 2018; Spranger et al., 2015; Spranger and Gajewski, 2018); and iii) the strength of the antitumor immune response that results from the release of the immune checkpoint PD-1, alone or together with the checkpoint CTLA-4, driving the expression of interferon-gamma response genes in the tumor microenvironment (Ayers et al., 2017; Cristescu et al., 2018). The release of interferon-gamma from T cells recognizing cognate antigen on cancer cells serves to amplify the nascent antitumor immune response, which our and prior data (Neubert et al., 2017) suggest is mediated by the conserved ability of the great majority of melanoma cells to signal through the interferon-gamma receptor. Amplification of the immune response is a result of an increase in antigen-presentation machinery, positive feed-back resulting in increased interferon-gamma pathway signaling, production of chemokines that attract other immune cells favorably altering the tumor microenvironment and inhibition of immune exclusion cancer signatures.
The strength of the antitumor immune response is dependent on the interplay of these key contributing factors, which could be individually modulated with additional interventions as there does not seem to be a dominant process that would always inhibit mounting the antitumor response in the majority of cases without clinical response to ICB therapy. In preclinical modeling (Curran et al., 2010; Wei et al., 2019; Wei et al., 2017) and in clinical series (Larkin et al., 2019), the combination of anti-PD-1 and anti-CTLA-4 is arguably a stronger immune stimulation than either therapy alone, thereby shifting the balance in favor of an antitumor immune response in a greater number of cases. Another way to shift this balance would be to induce a physiological process of intratumoral interferon production by triggering pattern recognition receptors, such as the use of intratumoral administration of oncolytic viruses or Toll-like receptor agonists, two approaches already successfully reported to improve response rates of PD-1 blockade therapy (Ribas et al., 2018a; Ribas et al., 2018b; Vanpouille-Box et al., 2019). Additional combinatorial approaches to enhance responses to ICB, not yet demonstrated to be clinically active in patients, include the triggering of the STING pathway (Li and Chen, 2018), inhibition of immune suppressive factors such as WNT signaling or the adenosine pathway (Abril-Rodríguez et al., 2020; Grasso et al., 2018; Smyth et al., 2016; Spranger and Gajewski, 2018), or the release of other immune checkpoints, such as LAG-3, TIM-3 or TIGIT, and others in T cells (Smyth et al., 2016). These observations provide hope that the benefit of cancer immunotherapy can be expanded to more patients and indications by using a combination therapy that reaches the level where antitumor T cells are potent enough to overcome the different nodes that restrict immune responses to cancer (Smyth et al., 2016).
It was possible that cancer cells may have different inherent abilities to respond to interferon-gamma, and this may lead to some patients responding or not to ICB because some cancers may not induce the full set of interferon-gamma response genes that are needed to mount a productive antitumor immune response. However, our data using a panel of melanoma cell lines suggests that this is very unlikely, at least in patients with metastatic melanoma. The data on interferon-gamma response in these cell lines was dichotomous, clearly separating the cell lines with or without the ability to signal from the interferon-gamma receptor. The cell lines unable to signal had natural or modelled homozygous loss-of-function mutations in JAK1 or JAK2. These mutations can develop sporadically in patients with melanoma and other cancers before receiving ICB immunotherapy, but they are infrequent, likely less than 1% of the cases (Liu et al., 2019; Shin et al., 2017). Upon selective immune pressure they may become more frequent and lead to acquired resistance, but these seem to also be rather infrequent cases (Sucker et al., 2017; Zaretsky et al., 2016). Given the low baseline frequency of loss-of-function mutations in the interferon-gamma signaling pathway, signaling through the interferon-gamma receptor inducing the full set of interferon-gamma response genes may be a positively selected feature of melanoma cells, which is rather counter-intuitive. At baseline, it may be favorable to the cancer cells to maintain this signaling and express immune-suppressive molecules such as PD-L1, indoleamine 2,3-dioxygenase (IDO), colony stimulating factor-1 (CSF-1), vascular endothelial growth factor (VEGF), or IL-6, for example, to stop the antitumor immune response, but this is at the expense of making the cancer cells more vulnerable to a future immune response.
A common feature of biopsies from patients who respond to ICB is the expression of immune activation genes with mostly overlapping signatures (Ayers et al., 2017; Cristescu et al., 2018; Fehrenbacher et al., 2016; Jerby-Arnon et al., 2018; Liu et al., 2019; Rodig et al., 2018; Roh et al., 2017; Rooney et al., 2015; Sade-Feldman et al., 2018). Our studies suggest that the common denominator of these immune activation transcriptomes is the expression of interferon-gamma response genes initiated by the activation of tumor antigen-specific T cells, which increases upon ICB. Key among these gene sets is the expression of antigen presenting machinery, most notably MHC class I and class II, among biopsies of patients that go on to respond to ICB therapy (Liu et al., 2019; Rodig et al., 2018). This was the most relevant feature separating patients who responded or not to therapy in our series, in particular the increase in MHC genes in on-therapy biopsies. However, a significant component of the increase in expression of MHC genes had to be from the infiltration with hematopoietic lineage cells into responding tumors as assessed by LCA correction. Therefore, future analyses would need to use techniques allowing determination of cancer cell-intrinsic antigen processing and presentation pathway.
The baseline expression of several T cell immune exclusion signatures has been associated with lack of response to ICB in different series, most of which also have overlapping gene sets (Auslander et al., 2018; Jerby-Arnon et al., 2018; Jiang et al., 2018; Liu et al., 2019). In our series, baseline and on-therapy biopsies of patients without a response to ICB therapy had a higher expression of the Jerby-Arnon signatures of T cell exclusion (Jerby-Arnon et al., 2018), with the relevant feature that responding biopsies displayed decreases in expression of WNT and MYC genes on-therapy, while non-responding biopsies continued to express the immune exclusion genes. Overall, our data together with transcriptome analyses of other biopsy series (Auslander et al., 2018; Ayers et al., 2017; Cabrita et al., 2020; Chen et al., 2016; Cristescu et al., 2018; Fehrenbacher et al., 2016; Gide et al., 2019; Helmink et al., 2020; Hugo et al., 2016; Jerby-Arnon et al., 2018; Jiang et al., 2018; Liu et al., 2019; Petitprez et al., 2020; Riaz et al., 2017; Rodig et al., 2018; Roh et al., 2017; Sade-Feldman et al., 2018), implies the presence of feedback loops between the tumor and CD8 T cells mediated by interferon-gamma that modulates immune exclusion secondary to WNT signaling (Luke et al., 2019).
In conclusion, T cell infiltration and expression of interferon gamma-regulated genes increase in biopsies of patients receiving ICB therapy regardless of clinical response, whereas the degree of HLA upregulation on-therapy and the decrease in expression of genes associated with immune exclusion are features of clinically responding biopsies. The difference between responsive and non-responsive melanomas does not seem to be due to an inherent ability of some cancer cells to respond differently to interferon-gamma, as the quality of the interferon-gamma response program was similar in the great majority of melanoma cell lines tested. As we did not have single cells for RNA sequencing analyses, we were not able to directly determine if the same was true in the tumor biopsies. In most cases, ICB was able to change the transcriptome of melanoma biopsies, but it did so to different degrees that correlate with favorable changes in the immune cell infiltrate. The ability to provide a more powerful anti-melanoma immune response with combination ICB explains a higher rate of responses. This information also provides hope that additional combinatorial strategies may dial up the antitumor immune response to become clinically meaningful in more patients, in particular when combining immune checkpoint blockade therapy with treatments that increase interferon signaling inside tumors to jump-start an antitumor immune response when it is not already pre-existing, such as the intratumoral administration of oncolytic viruses or nucleotide sequences that trigger pattern recognition receptors to produce interferons (Ribas et al., 2018a; Ribas et al., 2018b; Torrejon et al., 2020; Vanpouille-Box et al., 2019).
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Antoni Ribas (aribas@mednet.ucla.edu).
Materials Availability
Melanoma cell lines are available from the Lead Contact with a completed Materials Transfer Agreement following institutional policies.
Data and Code Availability
RNA-seq data from patients who consented to deposition has been deposited in the European Genome-phenome Archive, with accession numbers CM38_DNA - EGAS00001004548 and CM38_RNA - EGAS00001004545. The RNA-seq data for human melanoma cell lines with and without interferon-gamma exposure for 6-hours is deposited in the Gene Expression Omnibus (GEO), with accession number GSE154996. Both are listed in the Key Resources Table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological Samples | ||
| Patient tumor biopsies collected from advanced melanoma patients in clinical study CheckMate 038 | This article | https://clinicaltrials.gov/ct2/show/NCT01621490 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant Human IFN-gamma | BD Biosciences | Cat No. 554617 |
| Critical Commercial Assays | ||
| RNA extraction with miRNeasy Mini Kit | Qiagen | Cat No. 217004 |
| Illumina Truseq Stranded mRNA kit | Illumina |
https://www.illumina.com/products/by-type/sequencingkits/library-prep-kits/truseq-stranded-mrna.html
Cat No. 20020595 |
| Deposited Data | ||
| Checkmate-038 biopsies RNAseq raw and analyzed data | This paper | European Genome-phenome Archive CM38_DNA - EGAS00001004548 CM38_RNA - EGAS00001004545 |
| M series human melanoma cell line RNAseq raw and analyzed data, with and without interferon-gamma exposure | This paper | Gene Expression Omnibus (GEO) GSE154996 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE154996 |
| Experimental Models: Cell Lines | ||
| Human: M-series patient derived cell lines | This paper; Drs. James Economou, Antoni Ribas, and Roger Lo Labs | Søndergaard et al., 2010 |
| Human: Primary Epidermal Melanocytes; Normal, Adult (HEMa) | ATCC | PCS-200–013 |
| Software and Algorithms | ||
| HISAT2 (v.2.0.5) | (Kim et al., 2015) | https://ccb.jhu.edu/software/hisat2/index.shtml |
| HTSeq (0.6.1) | (Anders et al., 2015) | https://ccb.jhu.edu/software/hisat2/index.shtml |
| R (v3.2) | N/A | https://www.r-project.org/ |
| MCPcounter R Package | N/A | https://github.com/ebecht/MCPcounter |
EXPERIMETNAL MODEL AND SUBJECT DETAILS
Clinical Trial and Biopsy Collections
Study CheckMate 038 (NCT01621490) was a multi-arm, multi-institutional, prospective study to investigate the effects of nivolumab (3 mg/kg every 2 weeks) single agent, or the combination of nivolumab (1 mg/kg every 3 weeks) plus ipilimumab (3 mg/kg every 3 weeks) given for four doses and followed by nivolumab (3 mg/kg every 2 weeks) single agent. The protocol and its amendments were approved by the relevant institutional review boards, and the study was conducted in accordance with the Declaration of Helsinki and the International Conference on Harmonization Guidelines for Good Clinical Practice. All patients signed written informed consent prior to having any study procedures performed. Patients were treated until progression or for a maximum of 2 years, or were stopped due to toxicities. Radiographic assessment of response was performed approximately every 8 weeks until progression. Progression was confirmed with a repeat CT scan at least four weeks later. Tumor response for patients was defined by RECIST v1.1. Response to therapy indicates best overall response unless otherwise indicated. All patients underwent a baseline biopsy before commencing therapy (1 to 7 days before the first dose of therapy) and a repeat biopsy, on cycle 1, day 29 (between days 23–29).
Human Melanoma Cell Lines
Human melanoma cell lines were established from patient’s biopsies under UCLA IRB approval # 11–003254 as previously described (Atefi et al., 2014; Nazarian et al., 2010; Sondergaard et al., 2010; Tsoi et al., 2018).
METHODS DETAILS
RNA Sequencing
Tumor tissue was divided for formalin-fixed paraffin-embedding (FFPE) for IHC analysis, and storage with RNAlater (Ambion) for subsequent RNA extraction using Qiagen kits. Of 170 patients, 101 had enough RNA for RNAseq. RNAseq library was prepared using Illumina Truseq Stranded mRNA kit. Sequencing was done on an Illumina Hiseq sequencer using paired end sequencing of 50 bp for each mate pair. RNAseq reads were mapped using HISAT2 version 2.0.4 (Kim et al., 2019) and aligned to the hg19 genome using default parameters. Reads were quantified by HTSeq version 0.6.1 (Anders et al., 2015) with the intersection-non-empty mode and counting ambiguous reads if fully overlapping. Raw counts were then normalised to fragments per kilobase of exon per million fragments mapped (FPKM) expression values. Heatmaps of log2 FPKM expression data and z-scores were generated using the pheatmap R package. Progression free and overall survival plots were generated using the survival and ggsurv R packages. For melanoma cell line RNAseq analysis with and without interferon-gamma exposure, RNAseq reads were mapped using HISAT2 version 2.0.4 (Kim et al., 2019) and aligned to the hg19 genome using default parameters. Reads were quantified by HTSeq version 0.6.1 (Anders et al., 2015) with the intersection-non-empty mode and counting ambiguous reads if fully overlapping. Raw counts were then normalized to fragments per FPKM expression values.
RNA Immune Deconvolution Using MCP-Counter
RNA sequencing (RNA-seq)–based cell deconvolution of tissue-infiltrating immune and stromal populations using MCP-counter (Becht et al., 2016),based on 111 genes, including CD8A and ICOS, two genes in the T cell average measurement, was applied to all the samples to assess tumor infiltration of T cells, cytotoxic lymphocytes, CD8 T cells, myeloid dendritic cells, monocytic lineages, B lineage cells, NK cells, endothelial cells and fibroblast cells. A key advantage of MCP-Counter is that it accounts for the tumor cell fraction. As a result, T cell average from MCP-Counter is correlated with tumor-infiltrating lymphocyte (TIL) score, a pathology-based measure of T cell infiltration, based on 429 pathology slides available for TCGA samples (Grasso et al., 2018). This measurement is highly correlated with CD8A expression by itself (R2 = 0.73) and the “CYT” score (the average of GZMA and PRF1 expression, a measure of immune cytolytic activity based on two genes not present in the T cell average) (Rooney et al., 2015). We use the measurement with more genes here for robustness and for consistency with recent analyses, in addition, the including of stromal populations increases the overall accuracy.
Interferon-gamma in vitro exposure and transcriptome analysis
Melanoma cell lines were seeded on 10 cm tissue culture plates in RPMI media with 10% FBS and 1% PSA. After allowing 2–3 passages during exponential growth phase at 70% confluence cells were exposed to interferon-gamma at concentration 5 ng/ml (Recombinant Human IFN-gamma, 0.1–2.0×108 units/mg, BD Biosciences, Cat No. 554617). Culture media without interferon-gamma was added simultaneously to the controls. Cells were collected, cell pellets were frozen immediately and stored at −80°C. RNA was extracted with miRNeasy Mini Kit (Qiagen, Cat No. 217004). RNA sequencing was performed on Illumina HiSeq 3000, with a 1×50 run and with data quality check done on Illumina SAV and demultiplexing performed with Illumina Bcl2fastq2 v 2.17 program.
QUANTIFICATION AND STATISTICA ANALYSIS
Statistics and Survival Analysis
Statistical analyses were performed in R. Paired t-tests were used on the melanoma cell lines before and after interferon gamma treatment. The patient data is a combination of paired and unpaired observations (partially paired data), so we used the “optimal pooled t-test” described by Guo and Yuan in their comparative review of such methods (Guo and Yuan, 2017). An FDR was applied when we used either t-test to all the genes in a cohort in order to yield a list of significantly altered genes. An FDR was not applied when we looked at individual genes that we selected on the basis of biological interest, which is standard practice.
Supplementary Material
Highlights.
-
-
T cell-induced IFNγ correlates the best to immune checkpoint blockade (ICB) therapy
-
-
Immune signatures increase in on-therapy patients regardless response to therapy
-
-
ICB therapy decreases MYC and WNT signalings in patients with clinical response
-
-
IFNγ exposure of melanoma cells leads to a conserved transcriptome signature
Acknowledgements
C.S.G. was funded by the Parker Institute for Cancer Immunotherapy. J.T. was supported by the NIH Ruth L. Kirschstein Institutional National Research Service Award #T32-CA009120. G.A-R. was supported by the Isabel & Harvey Kibel Fellowship award and the Alan Ghitis Fellowship Award for Melanoma Research. D.Y.T was supported by a Young Investigator Award from ASCO, a grant from the Spanish Society of Medical Oncology for Translational Research in Reference Centers and the V Foundation-Gil Nickel Family Endowed Fellowship in Melanoma Research. K.C. was supported by the NIH Ruth L. Kirschstein Institutional National Research Service Award #T32-CA009120 and a Cancer Research Institute Irvington Postdoctoral Fellowship.J.D.W. was funded by the Parker Institute for Cancer Immunotherapy, and NIH grant P30 CA008748. V.A. was funded by US National Institutes of Health grants CA121113, the V Foundation, Swim Across America, the Allegheny Health Network – Johns Hopkins Research Fund, and the LUNGevity Foundation. D.M.P. and S.L.T. were funded by NCI R01 CA142779, the Bloomberg-Kimmel Institute for Cancer Immunotherapy, and a Cancer Immunology Translational Cancer Research Grant (SU2C-AACR-DT1012) from Cancer Research Institute–Stand Up 2 Cancer (A.R., D.M.P., S.L.T.). Stand Up 2 Cancer is a program of the Entertainment Industry Foundation administered by the American Association for Cancer Research. V.E.V. was funded in part by US National Institutes of Health grants CA121113, CA006973, CA233259, the Commonwealth Foundation, the Bloomberg-Kimmel Institute for Cancer Immunotherapy, the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation, the V Foundation, the Allegheny Health Network – Johns Hopkins Research Fund, and the Mark Foundation for Cancer Research. A.K. was supported by UCLA CTSI KL2 Award, Sarcoma Alliance for Research Through Collaboration Career Enhancement Program, Tower Cancer Research Foundation Young Investigator Award, and Radiological Society for North America Research Scholar Grant. A.R. was funded by the Parker Institute for Cancer Immunotherapy, NIH grants R35 CA197633, P01 CA244118 and P30 CA016042, the Ressler Family Fund, Ken and Donna Schultz Fund, and Cancer Immunology Translational Cancer Research Grant (SU2C-AACR-DT1012) from Cancer Research Institute–Stand Up 2 Cancer. Stand Up 2 Cancer is a program of the Entertainment Industry Foundation administered by the American Association for Cancer Research.
Declaration of Interest
C.S.G. reports a pending patent on the use of PAK4 inhibitors. J.T. is currently an employee of Tango Therapeutics. M.O. reports no conflicts of interest. G.A.R. reports a pending patent on the use of PAK4 inhibitors. P.R-M. is an employee and stockholder for Bristol-Myers Squibb. M.W.-R. is an employee and stockholder for Bristol-Myers Squibb. A.C. reports no conflicts of interest. E.M. reports no conflicts of interest. D.Y.T. reports no conflicts of interest. D.S.S. received honoraria from Genentech (speaker’s bureau) and NovMetaPharma (consultant). P.T. reports no conflicts of interest. Y.J.K. reports no conflicts of interest. C.P.S. has received payment for licensing a patent on non-viral T cell gene editing to Arsenal. K.M.C. is a shareholder in Geneoscopy LLC. A.V-C. reports no conflicts of interest. M.Q. reports no conflicts of interest. C.M. reports no conflicts of interest. J.J.L. discloses Data and Safety Monitoring Board: TTC Oncology; Scientific Advisory Board: 7 Hills, Actym, Alphamab Oncology, Kanaph, Mavu (now part of AbbVie), Onc.AI, Pyxis, Springbank, Tempest, Consultancy: Abbvie, Akrevia, Algios, Array, Astellas, Bayer, Bristol-Myers Squibb, Eisai, EMD Serono, Ideaya, Incyte, Janssen, Merck, Mersana, Novartis, PTx, RefleXion, Silicon, Tesaro, Vividion; Research Support: (all to institution for clinical trials unless noted) AbbVie, Agios (IIT), Array (IIT), Astellas, Bristol-Myers Squibb, CheckMate (SRA), Compugen, Corvus, EMD Serono, Evelo (SRA), Five Prime, FLX Bio, Genentech, Immatics, Immunocore, Incyte, Leap, MedImmune, Macrogenics, Necktar, Novartis, Palleon (SRA), Merck, Springbank, Tesaro, Tizona, Xencor; Travel: Akrevia, Bayer, Bristol-Myers Squibb, EMD Serono, Incyte, Janssen, Merck, Mersana, Novartis, Pyxis, RefleXion; Patents: (both provisional) Serial #15/612,657 (Cancer Immunotherapy), PCT/US18/36052 (Microbiome Biomarkers for Anti-PD-1/PD-L1 Responsiveness: Diagnostic, Prognostic and Therapeutic Uses Thereof). J.D.W. is consultant for: Adaptive Biotech; Amgen; Apricity; Ascentage Pharma; Astellas; AstraZeneca; Bayer; Beigene; Bristol Myers Squibb; Celgene; Eli Lilly; F Star; Imvaq; Kyowa Hakko Kirin; Linneaus; AstraZeneca; Merck; Neon Therapuetics; Polynoma; Psioxus; Recepta; Takara Bio; Trieza; Truvax; Serametrix; Surface Oncology; Syndax; Syntalogic. J.D.W receives research support from: Bristol Myers Squibb; AstraZeneca; Sephora. J.D.W. has Equity in: Tizona Pharmaceuticals; Adaptive Biotechnologies; Imvaq; Beigene; Linneaus. DBJ serves on advisory boards for Array Biopharma, BMS, Jansen, Merck, and Novartis, receives research funding from BMS and Incyte, and has a patent pending on using MHC-II as a biomarker for immunotherapy response. B.C. is a member of the Speaker’s Bureau for Regeneron and Sanofi. F.S.H. reports funding from Bristol-Myers Squibb to institution, during the conduct of the study; grants, personal fees and other from Bristol-Myers Squibb, personal fees from Merck, personal fees from EMD Serono, grants, personal fees and other from Novartis, personal fees from Takeda, personal fees from Surface, personal fees from Genentech/Roche, personal fees from Compass Therapeutics, personal fees from Apricity, personal fees from Bayer, personal fees from Aduro, personal fees from Partners Therapeutics, personal fees from Sanofi, personal fees from Pfizer, personal fees from Pionyr, personal fees from 7 Hills Pharma, personal fees from Verastem, personal fees from Torque, personal fees from Rheos, personal fees from Kairos, personal fees from Bicara, from Psioxus Therapeutics, personal fees from Amgen, other from Pieris Pharmaceutical, from Boston Pharmaceuticals, from Zumutor, outside the submitted work; In addition, Dr. Hodi has a patent Methods for Treating MICA-Related Disorders (#20100111973) with royalties paid, a patent Tumor antigens and uses thereof (#7250291) issued, a patent Angiopoiten-2 Biomarkers Predictive of Anti-immune checkpoint response (#20170248603) pending, a patent Compositions and Methods for Identification, Assessment, Prevention, and Treatment of Melanoma using PD-L1 Isoforms (#20160340407) pending, a patent Therapeutic peptides (#20160046716) pending, a patent Therapeutic Peptides (#20140004112) pending, a patent Therapeutic Peptides (#20170022275) pending, a patent Therapeutic Peptides (#20170008962) pending, a patent THERAPEUTIC PEPTIDES Therapeutic Peptides patent number: 9402905 issued, a patent METHODS OF USING PEMBROLIZUMAB AND TREBANANIB pending, a patent Vaccine compositions and methods for restoring NKG2D pathway function against cancers Patent number: 10279021 issued, a patent antibodies that bind to MHC class I polypeptide-related sequence A Patent number: 10106611 issued, and a patent ANTI-GALECTIN ANTIBODY BIOMARKERS PREDICTIVE OF ANTI-IMMUNE CHECKPOINT AND ANTI-ANGIOGENESIS RESPONSES Publication number: 20170343552 pending. S.B. reports advisory board participation (with honorarium) from Genentech, EMD-Serono, BMS and Sanofi-Genzyme; and research funding to his institution (University of Washington) from BMS, Oncosec, EMD-Serono, Merck, NantKwest, Novartis, Exicure, Incyte and Immune Design. W.S. discloses consulting fees from BMS, Merck, Novartis, Regeneron and research grant support from BMS, Merck, Novartis. W.J.U. serves on a compensated data and safety monitoring committee for Astra Zeneca, and reports institutional grants from BMS and Merck. C.L.S. discloses research funding to his University from Merck, Celldex, and GlaxoSmithKline; research support in kind to his University from Theraclion and 3M; Scientific Advisory Board role with Immatics (prior), and Curvac (planned); PI role for Polynoma with compensation to his University; and patent royalties as co-inventor of peptides for use in cancer vaccines (patents held by the UVA Licensing and Ventures Group). A.D. reports receiving honoraria from Nektar, Idera, Novartis and Array BioPharma. J.B.A.G.H. Advisory roles: Aimm, Amgen, AZ, Bayer, BMS, Celsius, Gadeta, GSK, Immunocore, MSD, Merck Serono, Neon, Neogene, Novartis, Pfizer, Roche, Sanofi, Seattle Genetics, Vaximm. Grant support: Neon, BMS, MSD, Novartis. Stock options: Neogene Therapeutics. S.M.A. participated in advisory boards with Amgen, BMS, Merck-Serono, MSD, Pierre-Fabre, Sanofi-Aventis and in educational events organized by BMS, MSD, Pierre-Fabre, Roche, Sanofi-Aventis. V.A. receives research funding from Bristol-Myers Squibb. D.M.P. and S.L.T. report stock and other ownership interests from Aduro Biotech, Compugen, DNAtrix, Dragonfly Therapeutics, Ervaxx, Five Prime Therapeutics, FLX Bio, Jounce Therapeutics, Potenza Therapeutics, Tizona Therapeutics, and WindMIL; consulting or advisory roles with AbbVie, Amgen, Bayer, Compugen, DNAtrix, Dragonfly Therapeutics, Dynavax, Ervaxx, Five Prime Therapeutics, FLX Bio, Immunocore, lmmunomic Therapeutics, Janssen Oncology, Medlmmune, Merck, Tizona Therapeutics, and Wind MIL; research funding from Bristol-Myers Squibb, Compugen, and Potenza Therapeutics; patents, royalties, and other intellectual property from Aduro Biotech, Bristol-Myers Squibb, and lmmunonomic Therapeutics; and travel, accommodations, and expenses from Bristol-Myers Squibb and Five Prime Therapeutics. V.E.V. is a founder of Delfi Diagnostics and Personal Genome Diagnostics, serves on the Board of Directors and as a consultant for both organizations, and owns Delfi Diagnostics and Personal Genome Diagnostics stock, which are subject to certain restrictions under university policy. Additionally, Johns Hopkins University owns equity in Delfi Diagnostics and Personal Genome Diagnostics. V.E.V. is an advisor to Bristol-Myers Squibb, Genentech, Merck, and Takeda Pharmaceuticals. Within the last five years, V.E.V. has been an advisor to Daiichi Sankyo, Janssen Diagnostics, and Ignyta. These arrangements have been reviewed and approved by the Johns Hopkins University in accordance with its conflict of interest policies. D.E.S. reports no conflicts of interest. A.K. reports no conflicts of interest. A.R. has received honoraria from consulting with Amgen, Bristol-Myers Squibb, Chugai, Genentech, Merck, Novartis, Roche and Sanofi, is or has been a member of the scientific advisory board and holds stock in Advaxis, Apricity, Arcus Biosciences, Bioncotech Therapeutics, Compugen, CytomX, Five Prime, FLX-Bio, ImaginAb, Isoplexis, Kite-Gilead, Lutris Pharma, Merus, PACT Pharma, Rgenix and Tango Therapeutics, has a reports a pending patent on the use of PAK4 inhibitors, has received research funding from Agilent and from Bristol-Myers Squibb through Stand Up to Cancer (SU2C), and has received payment for licensing a patent on non-viral T cell gene editing to Arsenal.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abril-Rodríguez G, Torrejon DY, Liu W, Zaretsky JM, Nowicki TS, Tsoi J, Puig-Saus C, Baselga Carretero I, Medina E, Quist MJ, et al. (2020). PAK4 inhibition improves PD-1 blockade immunotherapy. Nature Cancer 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atefi M, Avramis E, Lassen A, Wong DJ, Robert L, Foulad D, Cerniglia M, Titz B, Chodon T, Graeber TG, et al. (2014). Effects of MAPK and PI3K Pathways on PD-L1 Expression in Melanoma. Clin Cancer Res 20, 3446–3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auslander N, Zhang G, Lee JS, Frederick DT, Miao B, Moll T, Tian T, Wei Z, Madan S, Sullivan RJ, et al. (2018). Robust prediction of response to immune checkpoint blockade therapy in metastatic melanoma. Nat Med 24, 1545–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, Albright A, Cheng JD, Kang SP, Shankaran V, et al. (2017). IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest 127, 2930–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach EA, Aguet M, and Schreiber RD (1997). The IFN gamma receptor: a paradigm for cytokine receptor signaling. Annu Rev Immunol 15, 563–591. [DOI] [PubMed] [Google Scholar]
- Becht E, Giraldo NA, Lacroix L, Buttard B, Elarouci N, Petitprez F, Selves J, Laurent-Puig P, Sautes-Fridman C, Fridman WH, et al. (2016). Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol 17, 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrita R, Lauss M, Sanna A, Donia M, Skaarup Larsen M, Mitra S, Johansson I, Phung B, Harbst K, Vallon-Christersson J, et al. (2020). Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 577, 561–565. [DOI] [PubMed] [Google Scholar]
- Chen PL, Roh W, Reuben A, Cooper ZA, Spencer CN, Prieto PA, Miller JP, Bassett RL, Gopalakrishnan V, Wani K, et al. (2016). Analysis of Immune Signatures in Longitudinal Tumor Samples Yields Insight into Biomarkers of Response and Mechanisms of Resistance to Immune Checkpoint Blockade. Cancer Discov 6, 827–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristescu R, Mogg R, Ayers M, Albright A, Murphy E, Yearley J, Sher X, Liu XQ, Lu H, Nebozhyn M, et al. (2018). Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran MA, Montalvo W, Yagita H, and Allison JP (2010). PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A 107, 4275–4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehrenbacher L, Spira A, Ballinger M, Kowanetz M, Vansteenkiste J, Mazieres J, Park K, Smith D, Artal-Cortes A, Lewanski C, et al. (2016). Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): a multicentre, open-label, phase 2 randomised controlled trial. Lancet 387, 1837–1846. [DOI] [PubMed] [Google Scholar]
- Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, Zaretsky JM, Sun L, Hugo W, Wang X, et al. (2017). Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell reports 19, 1189–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gettinger S, Choi J, Hastings K, Truini A, Datar I, Sowell R, Wurtz A, Dong W, Cai G, Melnick MA, et al. (2017). Impaired HLA Class I Antigen Processing and Presentation as a Mechanism of Acquired Resistance to Immune Checkpoint Inhibitors in Lung Cancer. Cancer Discov. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gide TN, Quek C, Menzies AM, Tasker AT, Shang P, Holst J, Madore J, Lim SY, Velickovic R, Wongchenko M, et al. (2019). Distinct Immune Cell Populations Define Response to Anti-PD-1 Monotherapy and Anti-PD-1/Anti-CTLA-4 Combined Therapy. Cancer Cell 35, 238–255 e236. [DOI] [PubMed] [Google Scholar]
- Grasso CS, Giannakis M, Wells DK, Hamada T, Mu XJ, Quist M, Nowak JA, Nishihara R, Qian ZR, Inamura K, et al. (2018). Genetic Mechanisms of Immune Evasion in Colorectal Cancer. Cancer Discov 8, 730–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo B, and Yuan Y (2017). A comparative review of methods for comparing means using partially paired data. Stat Methods Med Res 26, 1323–1340. [DOI] [PubMed] [Google Scholar]
- Helmink BA, Reddy SM, Gao J, Zhang S, Basar R, Thakur R, Yizhak K, Sade-Feldman M, Blando J, Han G, et al. (2020). B cells and tertiary lymphoid structures promote immunotherapy response. Nature 577, 549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, Sosman JA, McDermott DF, Powderly JD, Gettinger SN, et al. (2014). Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 515, 563–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, Berent-Maoz B, Pang J, Chmielowski B, Cherry G, et al. (2016). Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 165, 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerby-Arnon L, Shah P, Cuoco MS, Rodman C, Su MJ, Melms JC, Leeson R, Kanodia A, Mei S, Lin JR, et al. (2018). A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell 175, 984–997 e924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang P, Gu S, Pan D, Fu J, Sahu A, Hu X, Li Z, Traugh N, Bu X, Li B, et al. (2018). Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med 24, 1550–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalbasi A, and Ribas A (2020). Tumour-intrinsic resistance to immune checkpoint blockade. Nat Rev Immunol 20, 25–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Paggi JM, Park C, Bennett C, and Salzberg SL (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol 37, 907–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Lao CD, Cowey CL, Schadendorf D, Wagstaff J, Dummer R, et al. (2019). Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N Engl J Med 381, 1535–1546. [DOI] [PubMed] [Google Scholar]
- Li T, and Chen ZJ (2018). The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J Exp Med 215, 1287–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Schilling B, Liu D, Sucker A, Livingstone E, Jerby-Amon L, Zimmer L, Gutzmer R, Satzger I, Loquai C, et al. (2019). Integrative molecular and clinical modeling of clinical outcomes to PD1 blockade in patients with metastatic melanoma. Nat Med 25, 1916–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luke JJ, Bao R, Sweis RF, Spranger S, and Gajewski TF (2019). WNT/beta-catenin Pathway Activation Correlates with Immune Exclusion across Human Cancers. Clin Cancer Res 25, 3074–3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, Chen Z, Lee MK, Attar N, Sazegar H, et al. (2010). Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 468, 973–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neubert NJ, Tille L, Barras D, Soneson C, Baumgaertner P, Rimoldi D, Gfeller D, Delorenzi M, Fuertes Marraco SA, and Speiser DE (2017). Broad and Conserved Immune Regulation by Genetically Heterogeneous Melanoma Cells. Cancer Res 77, 1623–1636. [DOI] [PubMed] [Google Scholar]
- Nsengimana J, Laye J, Filia A, O’Shea S, Muralidhar S, Pozniak J, Droop A, Chan M, Walker C, Parkinson L, et al. (2018). beta-Catenin-mediated immune evasion pathway frequently operates in primary cutaneous melanomas. J Clin Invest 128, 2048–2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petitprez F, de Reynies A, Keung EZ, Chen TW, Sun CM, Calderaro J, Jeng YM, Hsiao LP, Lacroix L, Bougouin A, et al. (2020). B cells are associated with survival and immunotherapy response in sarcoma. Nature 577, 556–560. [DOI] [PubMed] [Google Scholar]
- Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, Hodi FS, Martin-Algarra S, Mandal R, Sharfman WH, et al. (2017). Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 171, 934–949 e916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas A, Dummer R, Puzanov I, VanderWalde A, Andtbacka RHI, Michielin O, Olszanski AJ, Malvehy J, Cebon J, Fernandez E, et al. (2018a). Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell 174, 1031–1032. [DOI] [PubMed] [Google Scholar]
- Ribas A, Medina T, Kummar S, Amin A, Kalbasi A, Drabick JJ, Barve M, Daniels GA, Wong DJ, Schmidt EV, et al. (2018b). SD-101 in Combination with Pembrolizumab in Advanced Melanoma: Results of a Phase Ib, Multicenter Study. Cancer Discov 8, 1250–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas A, and Wolchok JD (2018). Cancer immunotherapy using checkpoint blockade. Science 359, 1350–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodig SJ, Gusenleitner D, Jackson DG, Gjini E, Giobbie-Hurder A, Jin C, Chang H, Lovitch SB, Horak C, Weber JS, et al. (2018). MHC proteins confer differential sensitivity to CTLA-4 and PD-1 blockade in untreated metastatic melanoma. Sci Transl Med 10. [DOI] [PubMed] [Google Scholar]
- Roh W, Chen PL, Reuben A, Spencer CN, Prieto PA, Miller JP, Gopalakrishnan V, Wang F, Cooper ZA, Reddy SM, et al. (2017). Integrated molecular analysis of tumor biopsies on sequential CTLA-4 and PD-1 blockade reveals markers of response and resistance. Sci Transl Med 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney MS, Shukla SA, Wu CJ, Getz G, and Hacohen N (2015). Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 160, 48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sade-Feldman M, Jiao YJ, Chen JH, Rooney MS, Barzily-Rokni M, Eliane JP, Bjorgaard SL, Hammond MR, Vitzthum H, Blackmon SM, et al. (2017). Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat Commun 8, 1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, Lieb DJ, Chen JH, Frederick DT, Barzily-Rokni M, et al. (2018). Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell 175, 998–1013 e1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Subudhi SK, Blando J, Vence L, Wargo J, Allison JP, Ribas A, and Sharma P (2019). Anti-CTLA-4 Immunotherapy Does Not Deplete FOXP3(+) Regulatory T Cells (Tregs) in Human Cancers-Response. Clin Cancer Res 25, 3469–3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P, and Allison JP (2015a). The future of immune checkpoint therapy. Science 348, 56–61. [DOI] [PubMed] [Google Scholar]
- Sharma P, and Allison JP (2015b). Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell 161, 205–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A, Grasso CS, Hugo W, Sandoval S, Torrejon DY, et al. (2017). Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov 7, 188–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth MJ, Ngiow SF, Ribas A, and Teng MW (2016). Combination cancer immunotherapies tailored to the tumour microenvironment. Nat Rev Clin Oncol 13, 143–158. [DOI] [PubMed] [Google Scholar]
- Sondergaard JN, Nazarian R, Wang Q, Guo D, Hsueh T, Mok S, Sazegar H, Macconaill LE, Barretina JG, Kehoe SM, et al. (2010). Differential sensitivity of melanoma cell lines with BRAFV600E mutation to the specific raf inhibitor PLX4032. J Transl Med 8, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spranger S, Bao R, and Gajewski TF (2015). Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 523, 231–235. [DOI] [PubMed] [Google Scholar]
- Spranger S, and Gajewski TF (2018). Impact of oncogenic pathways on evasion of antitumour immune responses. Nat Rev Cancer 18, 139–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sucker A, Zhao F, Pieper N, Heeke C, Maltaner R, Stadtler N, Real B, Bielefeld N, Howe S, Weide B, et al. (2017). Acquired IFNgamma resistance impairs anti-tumor immunity and gives rise to T-cell-resistant melanoma lesions. Nat Commun 8, 15440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taube JM, Klein AP, Brahmer JR, Xu H, Pan X, Kim JH, Chen L, Pardoll DM, Topalian SL, and Anders RA (2014). Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrejon DY, Abril-Rodriguez G, Champhekar AS, Tsoi J, Campbell KM, Kalbasi A, Parisi G, Zarestsky JM, Garcia-Diaz A, Puig-Saus C, et al. (2020). Overcoming genetically-based resistance mechanisms to PD-1 blockade. Cancer Discov (epub), May 28: CD-19-1409. doi: 1410.1158/2159-8290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsoi J, Robert L, Paraiso K, Galvan C, Sheu K, Lay J, Wong DJL, Atefi M, Shirazi R, Wang X, et al. (2018). Multi-stage differentiation defines melanoma subtypes with differential vulnerability to drug-induced iron-dependent oxidative stress. Cancer Cell (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. (2014). PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanpouille-Box C, Hoffmann JA, and Galluzzi L (2019). Pharmacological modulation of nucleic acid sensors - therapeutic potential and persisting obstacles. Nature reviews Drug discovery 18, 845–867. [DOI] [PubMed] [Google Scholar]
- Wei SC, Anang NAS, Sharma R, Andrews MC, Reuben A, Levine JH, Cogdill AP, Mancuso JJ, Wargo JA, Pe’er D, et al. (2019). Combination anti-CTLA-4 plus anti-PD-1 checkpoint blockade utilizes cellular mechanisms partially distinct from monotherapies. Proc Natl Acad Sci U S A 116, 22699–22709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei SC, Levine JH, Cogdill AP, Zhao Y, Anang NAS, Andrews MC, Sharma P, Wang J, Wargo JA, Pe’er D, et al. (2017). Distinct Cellular Mechanisms Underlie Anti-CTLA-4 and Anti-PD-1 Checkpoint Blockade. Cell 170, 1120–1133 e1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L, et al. (2016). Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N Engl J Med 375, 819–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data from patients who consented to deposition has been deposited in the European Genome-phenome Archive, with accession numbers CM38_DNA - EGAS00001004548 and CM38_RNA - EGAS00001004545. The RNA-seq data for human melanoma cell lines with and without interferon-gamma exposure for 6-hours is deposited in the Gene Expression Omnibus (GEO), with accession number GSE154996. Both are listed in the Key Resources Table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological Samples | ||
| Patient tumor biopsies collected from advanced melanoma patients in clinical study CheckMate 038 | This article | https://clinicaltrials.gov/ct2/show/NCT01621490 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant Human IFN-gamma | BD Biosciences | Cat No. 554617 |
| Critical Commercial Assays | ||
| RNA extraction with miRNeasy Mini Kit | Qiagen | Cat No. 217004 |
| Illumina Truseq Stranded mRNA kit | Illumina |
https://www.illumina.com/products/by-type/sequencingkits/library-prep-kits/truseq-stranded-mrna.html
Cat No. 20020595 |
| Deposited Data | ||
| Checkmate-038 biopsies RNAseq raw and analyzed data | This paper | European Genome-phenome Archive CM38_DNA - EGAS00001004548 CM38_RNA - EGAS00001004545 |
| M series human melanoma cell line RNAseq raw and analyzed data, with and without interferon-gamma exposure | This paper | Gene Expression Omnibus (GEO) GSE154996 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE154996 |
| Experimental Models: Cell Lines | ||
| Human: M-series patient derived cell lines | This paper; Drs. James Economou, Antoni Ribas, and Roger Lo Labs | Søndergaard et al., 2010 |
| Human: Primary Epidermal Melanocytes; Normal, Adult (HEMa) | ATCC | PCS-200–013 |
| Software and Algorithms | ||
| HISAT2 (v.2.0.5) | (Kim et al., 2015) | https://ccb.jhu.edu/software/hisat2/index.shtml |
| HTSeq (0.6.1) | (Anders et al., 2015) | https://ccb.jhu.edu/software/hisat2/index.shtml |
| R (v3.2) | N/A | https://www.r-project.org/ |
| MCPcounter R Package | N/A | https://github.com/ebecht/MCPcounter |