

Abstract
Background
The purpose of this study was to explore the genomic landscape of head and neck squamous cell carcinoma (HNSCC) in circulation (circulating tumor DNA [ctDNA]) and tumor (tumor tissue DNA [tDNA]) and understand the implications of ctDNA sequencing for prognosis and precision oncology treatments.
Materials and Methods
This is a retrospective review of 75 patients with HNSCC for both tDNA and ctDNA. Results were analyzed for concordance between tDNA and ctDNA and for their individual and combined association with demographics, survival, and presence and extent of disease at last visit (DLV).
Results
The five most frequently altered genes were TP53, CDKN2A, TERT, BRCA2, and NOTCH1. Twenty percent of patients had NOTCH1 alterations in tDNA, with none found in ctDNA. Concordance among altered genes was 13.0%, and 65.3% of patients had actionable ctDNA alterations. ctDNA alterations were significantly associated with decreased overall survival (OS) and presence and extent of DLV. In DNA repair genes, alterations in ctDNA alone and combined with tDNA were significantly associated with decreased OS and presence of DLV. Similar significant associations were found in TP53 for ctDNA alone and combined with tDNA. DNA repair gene alterations in ctDNA and unique ctDNA alterations within partially concordant genes were significantly associated with decreased OS in multivariate analysis.
Conclusion
This study illustrates the circulating and tumor genomic profile in the largest HNSCC cohort to date, underscoring the potential utility of ctDNA in prognostication and precision oncology treatment. For the first time, the presence of ctDNA alterations and specific ctDNA sequencing results were shown to be significantly associated with poor prognosis in HNSCC.
Implications for Practice
The use of precision genomic targeted therapies in head and neck squamous cell carcinoma (HNSCC) lags behind many other cancers, and poor survival in advanced stages indicates the urgent need for improved treatment options. This exploratory analysis of circulating tumor DNA (ctDNA) and tumor tissue DNA (tDNA) sequencing in the largest cohort to date of patients with HNSCC provides a novel depiction of the ctDNA genome, with two thirds of patients having actionable ctDNA alterations. This study reports for the first time the prognostic value of ctDNA sequencing, with the presence of ctDNA alterations, specific ctDNA alterations in DNA repair genes and TP53, and unique ctDNA alterations within partially concordant genes predicting poor survival.
Keywords: Circulating tumor DNA, Liquid biopsy, Cell‐free DNA, Head and neck squamous cell carcinoma, Head and neck neoplasms, Genomic testing, Next‐generation sequencing, Prognosis, Overall survival, Precision medicine
Short abstract
This article reports on the genomic landscape of circulating tumor DNA in head and neck squamous cell carcinoma and analyzes its prognostic significance and potential contribution to precision oncology treatment strategies alone and in combination with tumor DNA analysis.
Introduction
Next‐generation sequencing (NGS) of tumor tissue DNA (tDNA) emerged a decade ago as the standard of care of genetic tumor analysis in head and neck squamous cell cancer (HNSCC) being used to analyze tumor genomics and direct precision oncology treatment strategies [1, 2, 3]. Performing tDNA sequencing to determine tumor genomics comes with significant challenges and limitations. Conventional biopsies of a dynamic tumor fail to detect tumor heterogeneity, may miss alterations amenable to precision oncology treatments, and may be of insufficient quantity to complete sequencing [4]. Furthermore, sequelae of adjuvant radiation therapy also create significant difficulty in obtaining tissue evaluable for residual disease or recurrence.
Performing NGS on circulating tumor DNA (ctDNA) in blood, often referred to as a “liquid biopsy,” recently emerged as an alternative source of tumor DNA with the ability to avoid many of the constraints of tDNA sequencing. Obtaining a simple blood draw in clinic avoids the delays and risks associated with repeated biopsies or surgical intervention, detects tumor heterogeneity, and can characterize occult metastases throughout the body that shed ctDNA into circulation [5]. Finally, studies have shown that levels of ctDNA may also be used to monitor tumor burden and that ctDNA burden predicts survival [5, 6].
Exploration of ctDNA sequencing is underway in many types of cancer to determine its potential prognostic and therapeutic applications [7, 8, 9, 10, 11, 12]. A recent analysis of ctDNA sequencing in gastrointestinal, brain, breast, lung, and head and neck tumors found that patients with head and neck cancer had the highest number of patients with ctDNA alterations detected (88%) and patients with three or more ctDNA alterations (48%) [5]. These results speak to the potential utility of ctDNA sequencing in HNSCC. The only U.S. Food and Drug Administration (FDA)–approved targeted drug therapy in HNSCC is the anti‐EGFR monoclonal antibody cetuximab, but there are many off‐label precision oncology therapies and new therapies currently in development that could benefit from increased knowledge of the tumor genome.
Whereas the genomic signature of HNSCC tumor tissue has been characterized through extensive research, the genomic landscape of ctDNA in HNSCC and its significance in clinical practice remains to be examined [1, 2, 3]. Our primary aim is to characterize the genomic landscape of ctDNA in HNSCC and analyze its prognostic significance and potential contribution to precision oncology treatment strategies alone and in combination with tDNA sequencing. We also aim to analyze concordance between tDNA and ctDNA sequencing in HNSCC.
Materials and Methods
This study is a single‐institution retrospective review of adult patients diagnosed with HNSCC and treated at the Wake Forest Baptist Health Hospital from May 2014 to July 2019. The Wake Forest School of Medicine Institutional Review Board granted approval for this study. To be included in this study, patients with HNSCC must have been tested for both tDNA and ctDNA. Exclusion criteria included other types of cancer within the head and neck such as cutaneous squamous cell carcinoma or salivary gland cancers, incomplete documentation, diagnosis with another synchronous tumor, and inadequate or incomplete sequencing data or quality of tDNA. From electronic medical records, we collected demographics, human papillomavirus (HPV) status (defined by polymerase chain reaction or/and by surrogate p16 marker), smoking status, alcohol consumption, cancer stage at diagnosis by American Joint Committee on Cancer version 8 criteria (stage I–III vs. stage IVA–C), tumor progression status (disease free, primary or locoregional tumor, metastatic tumor, and recurrent or progressive tumor) at key time points (tDNA collection, ctDNA collection, and last visit), and treatment received before and after collection of tDNA and ctDNA. Outcome measures collected include overall survival (OS), survival at 1 or 2 years measured from the date of ctDNA collection, and extent of disease (tumor) status at last visit (DLV) as defined above. Patients with follow‐up shorter than 6 months from the date of NGS testing were excluded from the outcome analysis.
NGS of tDNA was performed using the FoundationOne platform (Foundation Medicine, Cambridge, MA), which is capable of detecting substitutions, insertion and deletion alterations (indels), and copy number alterations (CNAs) in 323 genes as well as select gene rearrangements. Sequencing of ctDNA was performed using the Guardant360 platform (Guardant Health, Redwood City, CA), which assesses for single nucleotide variants in 73 genes, indels and fusion alterations, and CNAs in select genes. For all analyses, variants of unknown significance (VUS) were included, and synonymous alterations were excluded. CNAs were excluded only from analysis of concordance.
tDNA was isolated from the most recent pathologic specimen available either from surgery or core biopsy using the instructions provided by Foundation Medicine. The portion of one block with the greatest percentage of tumor nuclei (>20%) was sectioned, and at least 10 unstained slides were submitted for this analysis. Blood plasma samples were drawn and sent to Guardant Health within 24 hours for ctDNA isolation and sequencing to be performed.
Concordance analysis was performed for genes sequenced by both FoundationOne and Guardant360 platforms (70 genes). Concordance was calculated per patient at the gene level, and results were classified in five categories depending on detection of alterations in ctDNA and tDNA. Full concordance is defined as detection of matching, identical alterations in ctDNA and tDNA per gene, per patient. Full concordance was calculated with two methods: first including wild‐type genes, in which detecting a wild‐type gene in ctDNA and tDNA would be considered concordant, and second in which only altered genes were considered in each patient with wild‐type genes excluded. Partial concordance is defined as detection of identical alterations in ctDNA and tDNA and additional alterations in ctDNA and/or tDNA within a gene. Discordance is defined as detection of different alterations by ctDNA and tDNA in a gene. Concordance may also be calculated as an overall value per patient, in which the number of concordant genes is divided by the total number of altered genes.
Several sets of genes, including 15 genes with known clinical relevance and a high alteration rate in our analysis, were selected for analysis of sensitivity, specificity, positive predictive value, negative predictive value, diagnostic accuracy, and Youden's J index. Additional analyses of four highly altered genes involved in DNA repair—APC, ATM, BRCA1, and BRCA2—were also performed in ctDNA, tDNA, and together.
Statistical Analysis
Descriptive statistics of means and standard deviations were presented for continuous variables, as well as counts and percentages for categorical variables. For comparisons among categorical variables, we used Fisher's exact tests when comparing two binary variables and chi‐square tests when comparing groups with more than two categories. For comparisons between continuous measures we used correlation analyses. For analyses comparing mean values between two groups we used two‐sample t tests, and when there were three or more groups we used a general linear model approach to compare means across groups, followed by post hoc pairwise comparisons of relevant groups using two‐sample t tests. For comparing survival curves, we generated Kaplan‐Meier curves and compared groups using log‐rank tests. For some survival models, we compared groups after accounting for a stratification variable such as staging at diagnosis or HPV and/or p16 testing status. Next, we used Cox proportional hazards regression models to examine the relationship of OS to a number of potential risk factors and predictors in the same model. A stepwise selection approach was then used to identify the covariates/predictors to include in the Cox proportional hazards regression model. The first step considered all potential predictors, and one variable was entered (or removed) at a time based on its level of significance in the model (p < .05 for entry or removal from the model). Age, gender, smoking history, alcohol consumption, and stage at diagnosis were included in the final models independently of statistical significance in order to ensure that they were accounted for in the multivariate model. Hazard ratios and corresponding 95% confidence intervals were estimated from these proportional hazards regression models. In all analyses we used two‐sided tests with an alpha level of .05 to determine significance. SAS version 9.4 (SAS Institute, Cary, NC) was used to perform all analyses.
Results
Seventy‐five total patients met criteria for enrollment. Demographics and disease characteristics are listed in Table 1 and supplemental online Figure 1. The median time between collection of tDNA and ctDNA was 54 days with a mean of 185 days and a range from 4 to 1,495 days. HPV and p16 testing results were negative for HPV and p16 in 44.0% of patients, positive for HPV or p16 in 26.7%, and not performed in 29.3%.
Table 1.
Demographics and disease characteristics
| Characteristic | n (%) |
|---|---|
| Age at ctDNA Collection | |
| Median, range | 60 (34–85) |
| Gender | |
| Male | 52 (69.3) |
| Female | 23 (30.7) |
| Race | |
| White | 66 (88.0) |
| Black | 7 (9.3) |
| Asian | 2 (2.7) |
| Smoking history | |
| Never | 18 (24.0) |
| Former | 27 (36.0) |
| Active | 30 (40.0) |
| Alcohol use | |
| Current | 30 (40.0) |
| Former | 18 (24.0) |
| None | 27 (36.0) |
| HPV and p16 testing | |
| Negative | 33 (44.0) |
| Positive | 20 (26.7) |
| Not tested | 22 (29.3) |
| tDNA tissue source | |
| Primary tumor tissue | 50 (66.7) |
| Metastasis | 8 (10.7) |
| Recurrence | 17 (22.7) |
| Tumor location | |
| Sinonasal | 3 (4.0) |
| Nasopharynx | 1 (1.3) |
| Oropharynx | 22 (29.3) |
| Oral cavity | 28 (37.3) |
| Hypopharynx | 7 (9.3) |
| Larynx | 14 (18.7) |
| Initial tumor stage | |
| Stage I–III | 28 (37.3) |
| Stage IVA–C | 47 (62.7) |
| Tumor progression at key timepoints | |
| tDNA collection | |
| Primary/locoregional | 49 (65.3) |
| Recurrent/progressive | 16 (21.3) |
| Metastatic | 10 (13.3) |
| ctDNA collection | |
| No evidence of disease | 12 (16.0) |
| Primary/locoregional | 21 (28.0) |
| Recurrent/progressive | 20 (26.7) |
| Metastatic | 22 (29.3) |
| Last visit | |
| No evidence of disease | 24 (32.0) |
| Recurrent/progressive | 26 (34.7) |
| Metastatic | 25 (33.3) |
Abbreviations: ctDNA, circulating tumor DNA; HPV, human papillomavirus; tDNA, tumor tissue DNA.
Sequencing Results
All patients had tDNA alterations, and all had tDNA alterations detected in genes that were not sequenced in the ctDNA analysis. ctDNA alterations were detected in 76% of patients (Figs. 1, 2, 3). Genes with similar alteration rates in tDNA and ctDNA (total ctDNA:tDNA alteration ratio of 0.5–1.5) included TP53; DNA repair genes such as APC, ATM, BRCA1, and BRCA2; and known drivers of metastasis such as EGFR, KIT, MET, BRAF, FGFR2, and FGFR3. The top three genes with the greatest disparity of alteration number (tDNA alterations minus ctDNA alterations) were NOTCH1 (20), TERT (15), and CDKN2A (11), all of which were among the top five most altered genes. TP53 was the most altered gene, with 127 total alterations spread among 73.3% of patients, yielding an average of 1.67 ± 1.48 TP53 alterations per patient. NOTCH1 was the fourth most altered gene with 20 total alterations found in 20% of patients. All NOTCH1 alterations were found in tDNA and none in ctDNA. In FBXW7, a member of the NOTCH1 pathway, seven tDNA alterations and one ctDNA alteration were detected. Other genes with alterations found only in tDNA include RB1, JAK2, JAK3, RET, IDH1, VHL, HER2, and PDGFRA.
Figure 1.
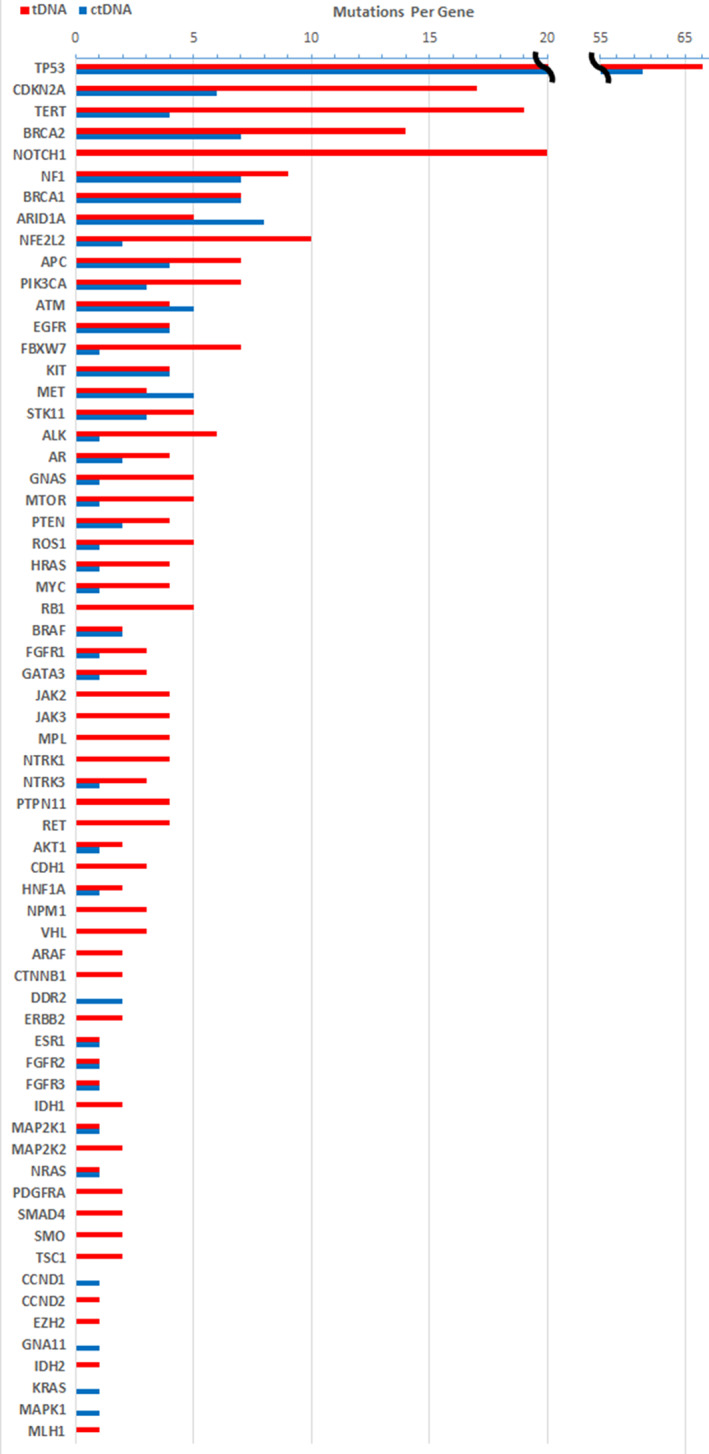
Histogram of alterations per gene.Abbreviations: ctDNA, circulating tumor DNA; tDNA, tumor tissue DNA.
Figure 2.
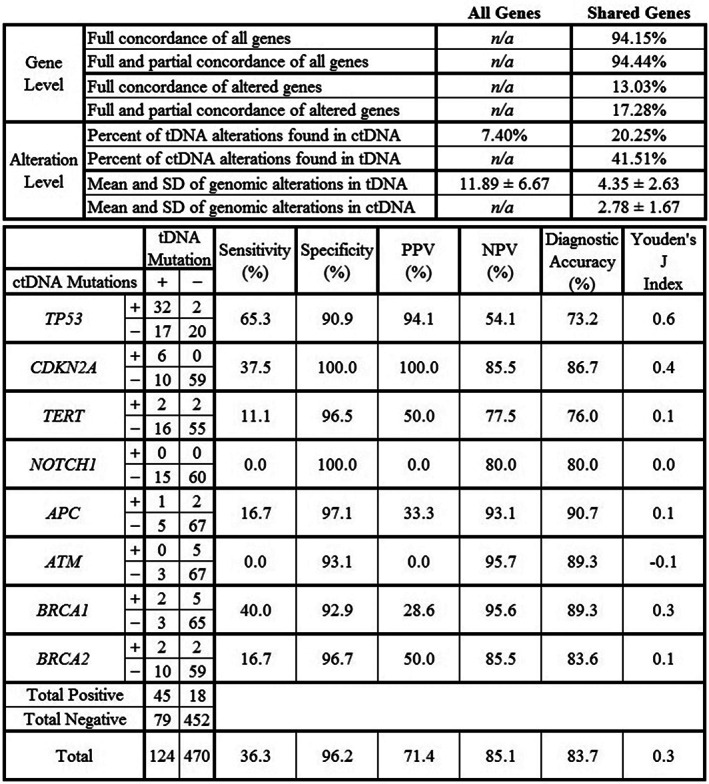
Concordance and alteration analysis with diagnostic accuracy of eight genes. “All Genes” includes 323 genes sequenced in tDNA using the FoundationOne Platform; “Shared Genes” includes 70 common genes sequenced in both tDNA and ctDNA. Genes selected for diagnostic accuracy analysis were the top five altered genes (TP53, CDKN2A, TERT, NOTCH1, BRCA2) and four DNA repair genes (APC, ATM, BRCA1, BRCA2). The Materials and Methods section defines concordance, and supplemental online Figure 2 shows analysis of the top 15 genes. Figure formatting adapted from Chae et al. [10].Abbreviations: ctDNA, circulating tumor DNA; PPV, positive predictive value; n/a, non‐applicable; NPV, negative predictive value; tDNA, tumor tissue DNA.
Figure 3.

Oncoprint of concordance for 15 representative genes. Four patients with no alterations in the top 15 genes were excluded from this figure. Figure formatting adapted from Chae et al. [10].Abbreviations: ctDNA, circulating tumor DNA; tDNA, tumor tissue DNA.
ctDNA sequencing of the top 15 genes had an overall sensitivity of 33.8% and specificity of 95.9% when using tDNA results as the benchmark (supplemental online Fig. 2). The same analysis of the top five altered genes showed a sensitivity of 38.2% and specificity of 97.7%.
Alterations in DNA repair genes (APC, ATM, BRCA1, and/or BRCA2) were present in 38.8% of patients, with a total of 32 tDNA alterations and 23 ctDNA alterations. DNA repair gene alterations were present in 25.3% and 25.3% of patients in tDNA and ctDNA, respectively, in a nonexclusive manner. The ctDNA results of these four DNA repair genes had a sensitivity of 19.2% and specificity of 94.9%.
Patients with positive HPV and/or p16 testing had significantly fewer total TP53 alterations in tDNA (0.29 vs. 1.0 per patient, p = .0003), fewer TP53 alterations in ctDNA (0.30 vs. 0.97 per patient, p = .042), and fewer DNA repair gene alterations in tDNA (0.059 vs. 0.50 per patient, p = .041) compared with patients with negative HPV and/or p16 testing (supplemental online Fig. 3).
Sequencing Results and Tumor Progression Status at ctDNA Collection
Advanced tumor progression status at time of ctDNA collection was associated with presence of ctDNA alterations (p = .0013), an increased number of ctDNA alterations (p = .0036), and presence of TP53 alterations in ctDNA (p = .015) (supplemental online Fig. 3). At time of ctDNA collection, patients with recurrent and metastatic disease had an average of 2.35 and 2.45 ctDNA alterations, respectively, compared with 0.36 alterations in patients with no evidence of disease (p = .0036). Eighty‐eight percent of patients with recurrent disease and 86% of patients with metastatic disease had ctDNA alterations present at time of ctDNA collection compared with 27% of patients with no evidence of disease (p = .0013). Finally, at the time of ctDNA collection, only 9.1% of patients with no evidence of disease had ctDNA alterations in TP53 compared with 64.7% of patients with recurrent disease and 63.6% with metastatic disease (p = .015).
Concordance Analysis
Among genes common to both assays, average concordance per patient was 14.6% ± 21.0% among altered genes (Figs. 2 and 3). TP53 was the most concordant gene in our analysis, with 28.0% of patients concordant, 14.7% partially concordant, 2.3% discordant, 22.7% with only tDNA alterations, and 2.7% with only ctDNA alterations. Average concordance among the top 15 representative genes was 18.8% ± 28.4%, and 46.7% of patients had no concordance among these 15 genes. There was no statistically significant relationship between concordance and ctDNA percent, tDNA variant allele frequency, or time between tDNA and ctDNA collection.
Actionability of ctDNA Alterations
Overall, 65.3% of patients had actionable alterations detected in ctDNA, defined as alterations with FDA‐approved therapies, off‐label therapies, and/or active clinical trial options. On the alteration level in a nonexclusive manner, 45.3% of ctDNA alterations had FDA‐approved off‐label therapies (such as PARP inhibitors for BRCA1 or BRCA2 alterations, copanlisib or duvelisib for PIK3CA alterations, etc.), 65.0% had active clinical trial options (opened for multiple tumors including HNSCC or specifically for HNSCC), and 34.3% were VUS. On a nonexclusive per‐patient basis, 13.3% of patients had ctDNA alterations detected with FDA‐approved off‐label therapies, 62.7% had ctDNA alterations with clinical trial treatment options, and 33.3% of patients did not have altered ctDNA targets for treatment or clinical trial options, including 10.7% of patients who had only VUS alterations.
Clinical Outcomes
Eight patients were excluded from analysis of outcomes because of ctDNA sampling less than 6 months before the time of this analysis. For the 67 remaining patients, median OS was 13 months, and median follow‐up was 8 months. At last visit, 32.0% of patients had no evidence of disease, 34.7% had recurrent or progressive disease, and 33.3% had metastatic disease (Table 1). Positive HPV and/or p16 testing of tumor tissue neared association with increased OS (p = .073; hazard ratio [HR], 0.3) and achieved association with increased OS at 1 year after ctDNA sampling (p = .010; Table 2). Increased tDNA tumor mutational burden was associated with decreased OS at 2 years after ctDNA sampling (p = .0015). NOTCH1 alterations were not associated with prognosis. No other demographic variables associated significantly with survival or with genomic alterations.
Table 2.
Analysis of prognostic variables
| Variable | Kaplan‐Meier overall survival curve: p value | Overall survival: univariate analysis | Overall survival: stepwise analysis | 1‐year overall survival: p value | 2‐year overall survival: p value | Presence of disease at last visit: p value | Extent of disease at last visit: p value | ||
|---|---|---|---|---|---|---|---|---|---|
| p value | HR (95% CI) | p value | HR (95% CI) | ||||||
| Presence vs. absence of variable | |||||||||
| ctDNA alterations | .030 | .042 | 3.5 (1.0–11.5) | .093 | .018 | .030 | .039 | ||
| Fully concordant genes | .24 | .24 | 1.6 (0.74–3.3) | .56 | .096 | .17 | .36 | ||
| Partially concordant genes | .25 | .30 | 1.7 (0.68–4.2) | .25 | .65 | .26 | .55 | ||
| Partially concordant genes with additional ctDNA mutations | .034 | .041 | 2.6 (1.0–6.4) | .0019 | 6.4 (2.0–20.6) | .097 | 1.00 | .42 | .22 |
| Fully concordant or partially concordant genes | .20 | .20 | 1.7 (0.76–3.6) | .39 | .40 | .061 | .14 | ||
| tDNA DNA repair alterations | .17 | .18 | 1.7 (0.78–3.9) | .072 | .64 | .12 | .26 | ||
| ctDNA DNA repair alterations | .0028 | .0044 | 3.0 (1.4–6.5) | .0054 | 4.1 (1.5–10.9) | .049 | .070 | .027 | .043 |
| DNA repair alterations | .0037 | .0055 | 3.0 (1.4–6.4) | .0065 | .088 | .025 | .054 | ||
| tDNA TP53 alterations | .021 | .031 | 3.7 (1.1–12.4) | .0018 | .079 | .064 | .17 | ||
| ctDNA TP53 alterations | .045 | .051 | 2.2 (0.99–4.9) | .042 | .38 | .0029 | .014 | ||
| TP53 alterations | .021 | .031 | 3.7 (1.1–12.4) | .0018 | .079 | .064 | .17 | ||
| NOTCH1 alterations | .75 | .75 | 0.86 (0.35–2.1) | .74 | .99 | .99 | .90 | ||
| NOTCH1 or FBXW7 alterations | .77 | .77 | 0.89 (0.39–2.0) | .99 | .65 | .76 | .47 | ||
| + HPV or + p16 testing | .058 | .073 | 0.3 (0.08–1.1) | .010 | .11 | .080 | .10 | ||
| Continuous variables | |||||||||
| Number of tDNA alterations | n/a | .11 | 0.94 (0.88–1.0) | .081 | .0003 | .20 | .59 | ||
| Number of tDNA alterations within ctDNA genes sequenced | n/a | .61 | 0.96 (0.84–1.1) | .80 | .14 | .78 | .93 | ||
| Number of TP53 alterations | n/a | .020 | 1.4 (1.0–1.8) | .0053 | .35 | .049 | .12 | ||
| Tumor mutational burden score | n/a | .093 | 0.93 (0.9–1.0) | .099 | .0015 | .11 | .33 | ||
Analyses with p < .05 are bolded in italics; findings with .05 < p < .10 are in italics. DNA repair genes are APC, ATM, BRCA1, and BRCA2. The Materials and Methods section defines partial and full concordance and statistical methods.
Abbreviations: CI, confidence interval; ctDNA, circulating tumor DNA; HPV, human papillomavirus; HR, hazard ratio; n/a, non‐applicable; tDNA, tumor tissue DNA.
Prognostic Value of the Presence of ctDNA Alterations
ctDNA alterations were detected in 73.1% of patients included in the outcome analysis. Presence of ctDNA alterations was associated with decreased OS (p = .042; HR, 3.5) and both the presence (p = .030) and extent (p = .039) of DLV (Table 2; Fig. 4). At 2 years after ctDNA collection, 57.1% of patients without alterations and 10.7% of patients with alterations were still living (p = .018). Of patients with metastatic disease at last visit, 91.7% had ctDNA alterations, and 8.3% did not (p = .039). When stratified by tumor staging at diagnosis, presence of ctDNA alterations remained associated with OS (p = .043; HR, 3.5) (supplemental online Fig. 4).
Figure 4.
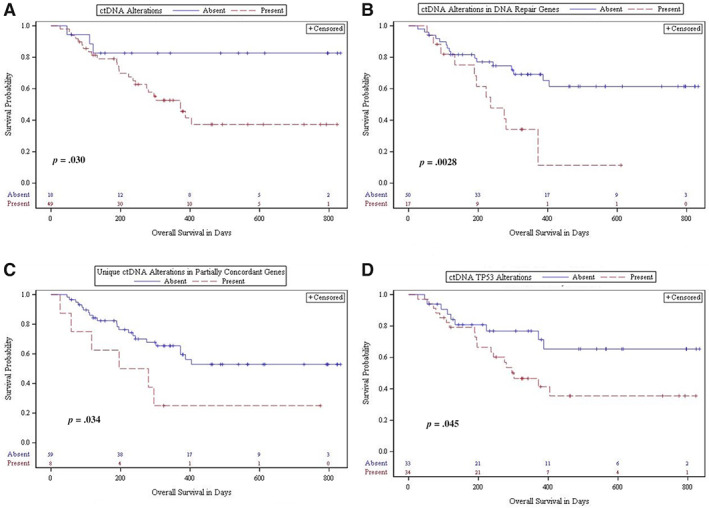
Analyses of overall survival. Kaplan‐Meier analyses of overall survival for presence versus absence of variables. (A): ctDNA alterations (p = .030). (B): ctDNA alterations in DNA repair genes (APC, ATM, BRCA1, BRCA2) (p = .0028). (C): Unique ctDNA alterations within partially concordant genes (p = .034). (D): ctDNA TP53 alterations (p = .045). The Materials and Methods section defines partial concordance.Abbreviations: ctDNA, circulating tumor DNA; tDNA, tumor tissue DNA.
Prognostic Value of Concordance
Analyses of the impact of full concordance per patient yielded no results near or achieving significance as a prognosticator (Table 2). Partially concordant alterations were present in 14.9% of patients in the outcome analysis (n = 10). Percentage of partially concordant genes achieved significance when analyzed as a continuous variable (p = .0012; HR, 1.054). Presence of unique ctDNA alterations within partially concordant genes (n = 8) was markedly associated with decreased OS in univariate analysis (p = .041; HR, 2.6) and stepwise multivariate analysis when controlled for age, gender, smoking history, alcohol history, and stage at diagnosis (p = .0019; HR, 6.4; Table 2; Fig. 4).
Prognostic Value of Alterations in DNA Repair Genes
Presence of DNA repair gene alterations (APC, ATM, BRCA1, and/or BRCA2) in ctDNA was markedly associated with decreased OS in univariate analysis (p = .0044; HR, 3.0), when stratified by tumor staging at diagnosis (p = .0040; HR, 3.0) or by HPV and/or p16 status (p = .0246), and in multivariate analysis when controlled for age, gender, smoking history, alcohol history, and stage at diagnosis (p = .0054; HR, 4.1) (Table 2; Fig. 4; supplemental online Fig. 4). Presence of DNA repair gene alterations in ctDNA was also associated with decreased OS at 1 year after ctDNA sampling (p = .049), and presence (p = .027) and extent (p = .043) of DLV (Table 2). Presence of DNA repair alterations in tDNA was not associated with prognosis. Presence of DNA repair genes alterations in ctDNA and/or tDNA was associated with decreased OS (p = .0055; HR, 3.0), maintained association when stratified for tumor stage at diagnosis (p = .0048; HR, 3.0) or HPV and/or p16 status (p = .028; HR, 3.4), and was associated with presence of DLV (p = .025). At last visit, evidence of disease was present in 88.5% of patients with and 61.0% of patients without DNA repair alterations (p = .025).
Prognostic Value of TP53 Alterations in tDNA and ctDNA
TP53 alterations were detected in tDNA in 74.6% and ctDNA in 50.7% of patients included in the outcome analysis. All patients with TP53 alterations had TP53 alterations in tDNA. Presence of TP53 alterations in ctDNA trended toward association with decreased OS (p = .051; HR, 2.2) and was significantly associated with OS at 1 year (p = .042) and presence (p = .0029) and extent (p = .014) of DLV (Table 2; Fig. 4). Presence of TP53 alterations in tDNA was associated with decreased OS (p = .031; HR, 3.7) and decreased OS at 1 year (p = .0018). Number of total TP53 alterations was also associated with decreased OS (p = .020; HR, 1.4), decreased OS at 1 year (p = .0053), and presence of DLV (p = .049). Concordance of TP53 was not associated with prognosis. When stratified by tumor staging at diagnosis, presence of tDNA alterations in TP53 and presence of either tDNA or ctDNA alterations in TP53 maintained significant association with OS (supplemental online Fig. 4).
Discussion
This study is a single‐institution retrospective analysis examining the genomic landscape of HNSCC in ctDNA in a dedicated cohort of patients with HNSCC with both tDNA and ctDNA sequencing results. To our knowledge, this study is the first to determine the concordance, evaluate the prognostic significance, and explore the therapeutic implications of ctDNA sequencing alone and in combination with tDNA sequencing in HNSCC.
Our patient population is validated by several results consistent with literature. The presence and number of TP53 alterations were associated with decreased OS, positive HPV or p16 results were associated with increased OS, and decreased TP53 and DNA repair gene alterations and the top five most altered genes in our analysis were TP53, CDKN2A, TERT, BRCA2, and NOTCH1 [1, 13, 14, 15, 16]. Our findings in HNSCC of ctDNA alterations in 76% of patients and three or more ctDNA alterations in 44% of patients are also similar to a recent study of 28 patients with head and neck cancer that found 88% and 48%, respectively [5]. Furthermore, advanced tumor status at the time of ctDNA collection (recurrent tumor or presence of metastasis) was associated with presence of ctDNA alterations, increased number of ctDNA alterations, and presence of TP53 alterations in ctDNA. Although expected, these results were not previously published in HNSCC studies [5].
The comparison of ctDNA sequencing to tDNA sequence is imperfect but has been performed in many cancers to validate the advent of ctDNA sequencing against the current standard of tDNA sequencing. Recent studies in breast, urothelial, lung, and solid cancers have analyzed ctDNA sequencing on the Guardant360 platform against tDNA sequencing on the FoundationOne platform to determine concordance [7, 8, 9, 10, 11]. In our analysis of HNSCC, we are the first to report a concordance of 13.0% when considering only altered genes, which bears similarity to results of 15.1% in breast cancer, 16.4% in urothelial cancer, and 17.1% in a study of lung and other solid tumors [8, 9, 10, 11].
This low level of concordance across many cancers is likely multifactorial and due in part to the limitations and sensitivities of the two platforms. The analysis of concordance is restricted to the 70 genes sequenced by both platforms based upon precedent from past literature [7, 8, 9, 10, 11]. tDNA sequencing through FoundationOne performs complete exon sequencing in these genes, but Guardant360 performs critical exon sequencing in 36 of these genes and complete exon sequencing in the remaining genes [17, 18]. Therefore, within the noncritical exons of 34 of the 70 overlapping genes, there may be alterations in both tDNA and ctDNA that can be detected in tDNA sequencing but not ctDNA sequencing.
Furthermore, there is a significant difference in the sensitivities of the two platforms. Each is highly specific, but an allelic fraction of 0.05%–0.20% in ctDNA corresponds with Guardant360 sensitivities of 64%, 68%, and 83% for single nucleotide variants, indels, and fusions, respectively [18]. Sensitivity increases dramatically to >95% for an allelic fraction >0.20%, but abundance of DNA released by nonmalignant cells into a large volume of blood plasma may dilute the fraction of altered alleles to below the threshold of detection for Guardant360 [18]. FoundationOne sequencing of tDNA quotes a higher allelic fraction of 2.0% for its limit of detection, but the fact that tDNA is isolated directly from tumor tissue with much less dilution likely makes it easier to achieve this threshold [10, 17]. These assertions are supported by our results that ctDNA was 33.8% sensitive among the top 15 genes and that the average number of alterations per patient among the 70 overlapping genes was greater in tDNA (4.35) than ctDNA (2.78) (Fig. 2).
In our analysis, timing between DNA sampling was not significantly associated with concordance but may still contribute to a lack of concordance because additional alterations may have accumulated in ctDNA during the mean of 185 days between tDNA and ctDNA sampling. Finally, sampling bias in tDNA isolation may contribute as well. tDNA was isolated from tumor sections with the largest visible tumor burden and highest grade in order to determine the greatest number of alterations, but sequencing tDNA from a single portion of a dynamic tumor likely fails to capture tumor heterogeneity [5]. Alternatively, all portions of a tumor have the potential to release DNA for ctDNA detection [5].
Our ctDNA sequencing results have several intriguing findings not yet demonstrated in literature. NOTCH1 is the third most altered gene in HNSCC, with tDNA alterations seen in 15%–19% of patients and a hypothesized role as a tumor suppressor [2]. In our analysis, NOTCH1 was the fourth most altered gene with alterations in 20% of patients, but all alterations were in tDNA. Previous studies demonstrated a NOTCH1 alteration rate of 5.8% and 23.0% in the ctDNA of various solid cancers, but a lack of NOTCH1 ctDNA alterations in HNSCC has not yet been reported [19, 20]. Further analysis of these patients found no difference in survival, staging, disease state, smoking, or HPV status, which indicates a lack of prognostic benefit to the detection of NOTCH1 alterations in HNSCC. It could be hypothesized that the lack of NOTCH1 alterations detected in the ctDNA of our patients with HNSCC is due to a lower level of altered genetic DNA released by the malignant squamous cells of the head and neck tumors, but further studies are needed to confirm our finding and investigate the possible mechanism.
Genes with alterations found only in tDNA, such as RB1, JAK2, JAK3, RET, IDH1, VHL, HER2, and PDGFRA, may not benefit from ctDNA sequencing. Alternatively, a number of genes, including TP53, EGFR, KIT, BRAF, FGFR2, and FGFR3, showed similar numbers of alterations in tDNA and ctDNA, and several genes, such as ARID1A, ATM, and MET, showed more alterations in ctDNA than in tDNA. Many of these genes have targetable alterations; therefore, ctDNA sequencing of these genes in HNSCC may be of significant utility in precision oncology treatment strategies.
This study is the first to analyze the prognostic utility of ctDNA sequencing in HNSCC. We found that decreased OS and extent of DLV were independently associated with the presence of ctDNA alterations and ctDNA TP53 alterations. We specifically examined DNA repair genes as potential targetable alterations for PARP inhibitors, which are currently in clinical trials for HNSCC, and found similar alteration rates of APC, ATM, BRCA1, and BRCA2 in tDNA and ctDNA. The presence of DNA repair alterations in ctDNA had the second strongest association with decreased OS in our analysis after controlling for age, gender, smoking history, alcohol history, and stage at diagnosis and was also associated with the extent of DLV. These results support the use of ctDNA testing of DNA repair genes for prognostic and therapeutic purposes in HNSCC. Our retrospective study lacked the opportunity to evaluate diagnostic and prognostic value of a longitudinal monitoring of ctDNA. In the field of colorectal cancer, for example, emerging literature has demonstrated that ctDNA can act as a real‐time monitor of the disease because of the short half‐life of ctDNA and can be further used as a prognostic biomarker for metastatic colorectal cancer [21].
Statistical power restricted the analysis of outcomes on a per‐gene basis to the most highly altered genes such as TP53. Presence of TP53 alterations in ctDNA was associated with OS and had the strongest association of all variables with the presence and extent of DLV. Analyses of CDKN2A, TERT, and NOTCH1 showed no prognostic utility. Although these results are not unexpected, they have not yet been presented in HNSCC.
A partially concordant gene is defined as having at least one identical alteration in ctDNA and tDNA (e.g., TP53 H178 frameshift) and additional unique alterations in ctDNA and/or tDNA (e.g., TP53 H214R and Y236C in ctDNA). The presence of partially concordant genes (n = 10) was insignificant, but patients with unique ctDNA alterations within partially concordant genes (n = 8, seven of which were partially concordant for TP53) showed decreased OS in multivariate analysis after controlling for age, gender, smoking history, alcohol history, and stage at diagnosis. This result demonstrates the prognostic utility of performing ctDNA sequencing after tDNA sequencing, because ctDNA was sampled on average 185 days after tDNA sampling in these patients. We hypothesize that the development of unique ctDNA alterations within a partially concordant gene between the time of tDNA and ctDNA sampling may be a distinct marker of disease progression that contributes to decreased OS.
A recent analysis found that 76% of patients with head and neck cancer had actionable ctDNA alterations, which is greater than in gastrointestinal, brain, lung, or breast cancer and similar to our finding of 65% in HNSCC [5]. In a nonexclusive manner, 13% of our patients had ctDNA alterations with available off‐label targeted therapies, and 63% had ctDNA alterations with clinical trial options. Despite the wealth of targetable alterations, use of precision oncology treatments is limited in HNSCC. Application of off‐label targeted therapy guided by genomic results is increasing, with more clinical trials underway in patients with HNSCC, underscoring the need to determine actionable alterations. ctDNA sequencing is poised as a promising method to overcome this restriction that needs investigation. Fifty‐three percent of patients had genes with ctDNA alterations that were wild type in tDNA, indicating an additive therapeutic utility of ctDNA sequencing to the current standard of tDNA sequencing. Accordingly, our analysis provides strong evidence that ctDNA sequencing increases the chance to identify targets for precision medicine treatments in HNSCC.
A number of limitations are present in our analysis. The evaluation of concordance between platforms is an imperfect analysis because of a difference in the detection threshold for ctDNA and tDNA sequencing, the potential impact of tumor heterogeneity on the sensitivity of tDNA sequencing, and the effect of tumor burden upon the sensitivity of ctDNA sequencing. In the context of a heterogenous population with regard to initial tumor staging, various timing between ctDNA and tDNA collection, tumor progression status at the time of tissue collection, and treatments received, the modest size of our cohort limits our power to draw conclusions about genes that are not heavily altered and compare across disease stages. Additional limitations include the single‐institution nature of this analysis, the lack of long‐term longitudinal follow‐up, and the retrospective nature of this study, which predisposes it to several types of bias. Nevertheless, the finding of genomic alterations being predictive of survival in the context of such a heterogenous population of patients with HNSCC raises the possibility that DNA alterations detected in blood and tumor could potentially circumvent canonical predictors of response such as tumor staging.
Conclusion
This exploratory analysis of the largest cohort to date of patients with HNSCC with ctDNA and tDNA sequencing provides a novel depiction of the ctDNA genome in HNSCC. We demonstrated the prognostic impact of ctDNA sequencing results with and without prior tDNA sequencing and provided indication for the utility of ctDNA sequencing in advancing precision medicine in HNSCC. For the first time, presence of ctDNA alterations, ctDNA alterations in TP53 and DNA repair genes, and unique ctDNA alterations within partially concordant genes were shown to be significantly associated with poor prognosis in HNSCC. We expect our results to provide an impetus for future research to fully validate this new tool in the setting of HNSCC.
Author Contributions
Conception/design: Harper L. Wilson, Ralph B. D'Agostino Jr., Robin Petro, Sara Commander, Umit Topaloglu, Wei Zhang, Mercedes Porosnicu
Provision of study material or patients: Harper L. Wilson, Ralph B. D'Agostino Jr., Robin Petro, Sara Commander, Umit Topaloglu, Wei Zhang, Mercedes Porosnicu
Collection and/or assembly of data: Harper L. Wilson, Ralph B. D'Agostino Jr., Robin Petro, Sara Commander, Umit Topaloglu, Wei Zhang, Mercedes Porosnicu
Data analysis and interpretation: Harper L. Wilson, Ralph B. D'Agostino Jr., Robin Petro, Sara Commander, Umit Topaloglu, Wei Zhang, Mercedes Porosnicu
Manuscript writing: Harper L. Wilson, Ralph B. D'Agostino Jr., Umit Topaloglu, Wei Zhang, Mercedes Porosnicu
Final approval of manuscript: Harper L. Wilson, Ralph B. D'Agostino Jr., Nuwan Meegalla, Robin Petro, Sara Commander, Umit Topaloglu, Wei Zhang, Mercedes Porosnicu
Disclosures
The authors indicated no financial relationships.
Supporting information
See http://www.TheOncologist.com for supplemental material available online.
Appendix S1: Supporting information
Acknowledgments
The authors thank Dr. Liang Liu, Ph.D., Department of Cancer Biology and Center of Cancer Genomics and Precision Oncology, Wake Forest Baptist Comprehensive Cancer Center, for his thoughtful review and discussions concerning the analysis of next‐generation sequencing data. Biostatistical and bioinformatics services were supported by the Comprehensive Cancer Center of Wake Forest University National Cancer Institute Cancer Center Support Grant P30CA012197.
Disclosures of potential conflicts of interest may be found at the end of this article.
References
- 1. Stransky N, Egloff AM, Tward AD et al. The alterational landscape of head and neck squamous cell carcinoma. Science 2011;333:1157–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Agrawal N, Frederick MJ, Pickering CR et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating alterations in NOTCH1. Science 2011;333:1154–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hoesli RC, Ludwig ML, Michmerhuizen NL et al. Genomic sequencing and precision medicine in head and neck cancers. Eur J Surg Oncol 2017;43:884–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bedard PL, Hansen AR, Ratain MJ et al. Tumor heterogeneity in the clinic. Nature 2013;501:355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schwaederle M, Chattopadhyay R, Kato S et al. Genomic alterations in circulating tumor DNA from diverse cancer patients identified by next‐generation sequencing. Cancer Res 2017;77:5419–5427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bettegowda C, Sausen M, Leary RJ et al. Detection of circulating tumor DNA in early‐ and late‐stage human malignancies. Sci Transl Med 2014;6:224ra24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Leighl NB, Page RD, Raymond VM et al. Clinical utility of comprehensive cell‐free DNA analysis to identify genomic biomarkers in patients with newly diagnosed metastatic non‐small cell lung cancer. Clin Cancer Res 2019;25:4691–4700. [DOI] [PubMed] [Google Scholar]
- 8. Chae YK, Davis AA, Jain S et al. Concordance of genomic alterations by next‐generation sequencing in tumor tissue versus circulating tumor DNA in breast cancer. Mol Cancer Ther 2017;16:1412–1420. [DOI] [PubMed] [Google Scholar]
- 9. Barata PC, Koshkin VS, Funchain P et al. Next‐generation sequencing (NGS) of cell‐free circulating tumor DNA and tumor tissue in patients with advanced urothelial cancer: A pilot assessment of concordance. Ann Oncol 2017;28:2458–2463. [DOI] [PubMed] [Google Scholar]
- 10. Chae YK, Davis AA, Carneiro BA et al. Concordance between genomic alterations assessed by next‐generation sequencing in tumor tissue or circulating cell‐free DNA. Oncotarget 2016;7:65364–65373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zill OA, Greene C, Sebisanovic D et al. Cell‐free DNA next‐generation sequencing in pancreatobiliary carcinomas. Cancer Discovery 2015;5:1040–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yang M, Topaloglu U, Petty WJ et al. Circulating mutational portrait of cancer: Manifestation of aggressive clonal events in both early and late stages. J Hematol Oncol 2017;10:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nichols AC, Yoo J, Palma DA et al. Frequent alterations in TP53 and CDKN2A found by next‐generation sequencing of head and neck cancer cell lines. Arch Otolaryngol Head Neck Surg 2012;138:732–739. [DOI] [PubMed] [Google Scholar]
- 14. Dubot C et al. Comprehensive genomic profiling of head and neck squamous cell carcinoma reveals FGFR1 amplifications and tumour genomic alterations burden as prognostic biomarkers of survival. Eur J Cancer 2018;91:47e55. [DOI] [PubMed] [Google Scholar]
- 15. Chung CH, Guthrie VB, Masica DL. Genomic alterations in head and neck squamous cell carcinoma determined by cancer gene‐targeted sequencing. Ann Oncol 2015;26:1216–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Seiwert TY, Zuo Z, Keck MK. Integrative and comparative genomic analysis of HPV‐positive and HPV‐negative head and neck squamous cell carcinomas. Clin Cancer Res 2015;21:632–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. FoundationOne CDx Technical Information Sheet. Cambridge, MA: Foundation Medicine, Inc; Available at https://assets.ctfassets.net/vhribv12lmne/6Rt6csmCPuaguuqmgi2iY8/26c3601c30e0d64c4f7e908543b8d62a/P170019.S015.Label.Technical_Info.FINAL.pdf. Accessed May 25, 2020. [Google Scholar]
- 18. Guardant360 Assay Specification Sheet. Redwood City, CA: Guardant Health; Available at https://www.therapyselect.de/sites/default/files/downloads/guardant360/guardant360_specification‐sheet_en.pdf. Accessed May 25, 2020. [Google Scholar]
- 19. Schwaederle M, Husain H, Fanta PT et al. Detection rate of actionable alterations in diverse cancers using a biopsy‐free (blood) circulating tumor cell DNA assay. Oncotarget 2016;7:9707–9718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kuziora M, Si H, Higgs B et al. Somatic alterations in BRCA2, NFE2L2, ARID1A, and NOTCH1 sensitize to anti‐PDL1 therapy in multiple tumor types. Ann Oncol 2018;29(suppl 10):X1. [Google Scholar]
- 21. Feifei B, Wang Q et al. Circulating tumor DNA in colorectal cancer: Opportunities and challenges. Am J Transl Res 2020;12:1044–1055. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
See http://www.TheOncologist.com for supplemental material available online.
Appendix S1: Supporting information