

Abstract
The NOTCH1 gene encodes a transmembrane receptor protein with activating mutations observed in many T‐cell acute lymphoblastic leukemias (T‐ALLs) and lymphomas, as well as in other tumor types, which has led to interest in inhibiting NOTCH1 signaling as a therapeutic target in cancer. Several classes of Notch inhibitors have been developed, including monoclonal antibodies against NOTCH receptors or ligands, decoys, blocking peptides, and γ‐secretase inhibitors (GSIs). GSIs block a critical proteolytic step in NOTCH activation and are the most widely studied. Current treatments with GSIs have not successfully passed clinical trials because of side effects that limit the maximum tolerable dose. Multiple γ‐secretase–cleavage substrates may be involved in carcinogenesis, indicating that there may be other targets for GSIs. Resistance mechanisms may include PTEN inactivation, mutations involving FBXW7, or constitutive MYC expression conferring independence from NOTCH1 inactivation. Recent studies have suggested that selective targeting γ‐secretase may offer an improved efficacy and toxicity profile over the effects caused by broad‐spectrum GSIs. Understanding the mechanism of GSI‐induced cell death and the ability to accurately identify patients based on the activity of the pathway will improve the response to GSI and support further investigation of such compounds for the rational design of anti‐NOTCH1 therapies for the treatment of T‐ALL.
Implications for Practice
γ‐secretase has been proposed as a therapeutic target in numerous human conditions, including cancer. A better understanding of the structure and function of the γ‐secretase inhibitor (GSI) would help to develop safe and effective γ‐secretase–based therapies. The ability to accurately identify patients based on the activity of the pathway could improve the response to GSI therapy for the treatment of cancer. Toward these ends, this study focused on γ‐secretase inhibitors as a potential therapeutic target for the design of anti‐NOTCH1 therapies for the treatment of T‐cell acute lymphoblastic leukemias and lymphomas.
Keywords: Broad‐spectrum γ‐secretase inhibitors, PF‐03084014 treatment, MYC gene dosage, T‐cell lymphoblastic cell lines, New resistance factor, Selective γ‐secretase inhibitors
Short abstract
Understanding the mechanism of γ‐secretase inhibitor (GSI)–induced cell death and the ability to accurately identify patients based on the activity of the pathway could improve the response to GSI therapy for the treatment of cancer. This article focuses on γ‐secretase inhibitors as a potential therapeutic target to treat T‐cell acute lymphoblastic leukemias and lymphomas.
Notch Signaling Pathway
The Notch signaling pathway is an evolutionarily conserved pathway whose deregulation is implicated in multiple pathologies, including malignant transformation. The first observation of the oncogenic potential of the Notch signaling pathway was in 1991 in specific forms of leukemia where the NOTCH1 gene was involved in the t(7;9)(q34;q34.3) chromosomal translocation in T‐cell acute lymphoblastic leukemia (T‐ALL) [1], resulting in a fusion protein that leads to inappropriate activation of Notch signaling. This signaling stimulates proliferation, restricts differentiation, and prevents apoptosis in cancer cells [2]. Beyond the chromosomal translocation, present in about 1% of patients with T‐ALL, activating missense mutations in the NOTCH1 gene are found in over 60% of T‐cell malignancies. Thus, strategies to therapeutically modulate Notch signaling are of interest, and current approaches include inhibition of the ligand‐receptor interaction or interference with the proteolytic processing of the receptor [3, 4] (Fig. 1A).
Figure 1.
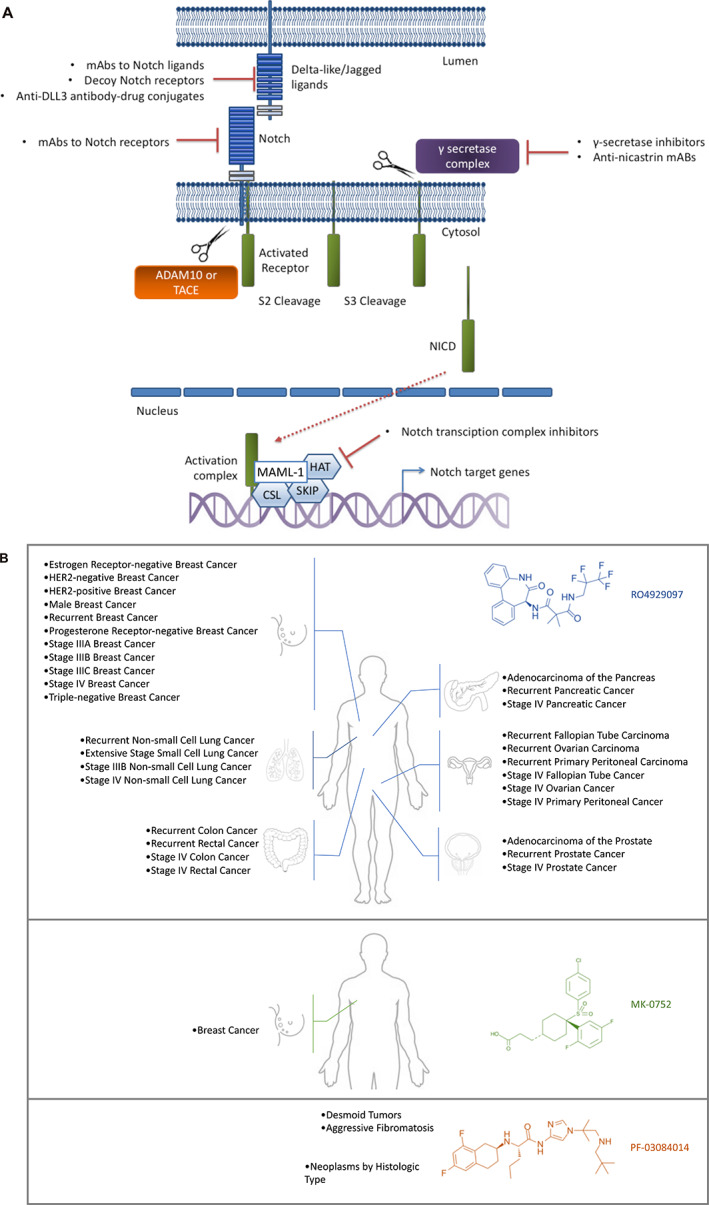
Notch signaling pathway and clinical trials with γ‐secretase inhibitors. (A): Schematic representation of Notch signaling pathway. Signaling is initiated by the interaction of Notch with Delta‐like ligands or Jagged ligands on the surface of instructing cells. Then two sequential proteolytic cleavages occur, the first mediated by an ADAM family protease (S2 cleavage) and the next by a γ‐secretase complex (S3 cleavage), resulting in the release of NICDs. NICDs are translocated to the nucleus and bind with transcriptional regulators to activate the expression of Notch downstream targets. The downstream proteins promote cell proliferation, inhibit cell apoptosis, and maintain cancer stem‐like phenotypes. (B): Clinical trials. A summary of the clinical trials with results employing γ‐secretase inhibitors in the treatment of cancer (https://clinicaltrials.gov). Abbreviations: ADAM, a disintegrin and metalloproteinase; CSL, C promoter‐binding factor; DLL3, Delta‐like ligand 3; HAT, histone acetyltransferase; mAb, monoclonal antibody; MAML‐1, Mastermind‐like 1; NICD, Notch intracellular domain; SKIP, ski‐interacting protein; TACE, TNF‐α–converting enzyme.
In brief, Notch signaling is a cell‐cell communication system between a Notch receptor (NOTCH1, 2, 3 or 4) and its ligands Jagged 1–2 (JAG1 and JAG2) or Delta‐like ligand (DLL1, DLL3, and DLL4). NOTCH receptors are transmembrane proteins with large extracellular domains that consist primarily of epidermal growth factor–like repeats, transmembrane regions, and intracellular regions. Binding of Notch ligands promotes two proteolytic cleavage events in the Notch receptor; the first one is catalyzed by ADAM family metalloproteases at the cell surface, whereas the second is mediated by the multiprotein complex γ‐secretase in the transmembrane domain inserted in the membrane. These cleavage events trigger notch translocation to the nucleus, where Notch cooperates with the DNA‐binding protein C promotor‐binding factor and its coactivator Mastermind to stimulate transcription of downstream target genes such as HES1 or MYC [5]. γ‐secretase is a multiprotein complex that includes presenilin (PSEN1 or 2), nicastrin, presenilin enhancer protein 2, and anterior pharynx‐defective 1. The bipartite protein PSEN1 or PSEN2 provides the catalytic subunit. γ‐secretase inhibitors (GSIs) lock γ‐secretase in a closed conformation, rendering it unable to cleave substrates such as NOTCH and other additional proteins; inhibit the proteasome; and can elicit endoplasmic reticulum stress [6, 7, 8, 9, 10, 11]. Therefore, this class of drugs inhibits the cleavage of the Notch receptor intracellular domain, which is necessary for transactivation of Notch targets, suggesting a promising clinical application in cancer therapeutics (Fig. 1A).
Broad‐Spectrum γ‐Secretase Inhibitors
Inhibitors for γ‐secretase have been investigated for their potential to catalyze proteolysis of the transmembrane region of the amyloid β–protein precursor (APP) to generate the amyloid β protein, thereby blocking the generation of the amyloid β peptide associated with Alzheimer's disease [12].
Considering that these compounds prevent Notch receptor activation, a wide range of GSIs have been tested in animals and humans for antitumor effects. Inhibitors of γ‐secretase prevent the second Notch cleavage, effectively blocking Notch signaling, and can induce G0/G1 arrest, decrease cell viability, and cause apoptosis of T‐ALL cell lines carrying gain‐of‐function NOTCH mutations (~60% in human T‐ALL). This treatment strategy was supported by in vitro and in vivo studies, although the results of clinical trials have shown only limited activity to date [13]. in vitro data vary, depending on the compound and the cell type. For example, MK‐0752 has been shown to inhibit γ‐secretase–mediated cleavage of Notch with a half‐maximal inhibitory concentration (IC50) of 55 nM [14]; the IC50 of PF‐3084014 is 13.3 nmol/L in HPB‐ALL cells, whereas in HeLa cells the IC50 was determined to be 6.2 nmol/L [15].
One problem in the clinical development of GSIs has been the occurrence of adverse effects based on Common Terminology Criteria for Adverse Events, limiting the maximum tolerated dose. In fact, the first attempts to treat patients with cancer and activating NOTCH1 mutations were reported in 2006, although treatment had to be interrupted because of undesired side effects (gastrointestinal, infections, and skin cancer related) [16]. Forty‐five trials focused on the study of patients with cancer can be identified in the ClinicalTrials.gov database (https://clinicaltrials.gov); this includes 287 tumor types. Twenty‐six of these 45 studies use this type of treatment as monotherapy; the remaining 19 involve the use of these drugs in a combination with other therapy. Seventeen are already terminated (seven with results), 16 are completed (eight with results), 6 have been withdrawn, and 1 is not available. All studies that have results posted involve three drugs (RO4929097, MK‐0752, and PF‐03084014) (Fig. 1B). Regarding the remaining studies (5/45), one is not yet recruiting, two are active (not recruiting), and two are recruiting.
The limited response to GSIs may be attributed to different causes. Firstly, there are multiple γ‐secretase–cleavage substrates in addition to APP or NOTCH1 (ERBBP4, E‐cadherin, ephrinB2, CD44, and others) that may be involved in carcinogenesis, indicating the need to systematically identify other targets as potential biomarkers for sensitivity in clinical trials [17]. In addition, GSIs used in different clinical trials are not pharmacologically equivalent [18], limiting the generalizability of the results. Another point to be considered is the toxicity induced by these drugs, which can be mitigated by glucocorticoid treatment [19] or via intermittent scheduling [20]. The synergistic interactions of some combination treatments suggest that lower doses of individual agents may be used, thus having the potential to limit tissue toxicity at the same therapeutic efficacy [21]. There are multiple published preclinical studies, using GSIs alone and in combination with glucocorticoids, bortezomib, or rapamycin, with encouraging results [14, 22, 23]. NOTCH1 is also central to the control of leukemia cell metabolism; inactivation of glutaminolysis in combination with NOTCH inhibition with GSIs has also proven efficacious in preclinical animal models [24].
Mechanisms of resistance to NOTCH inhibition have been studied. One important point that should be considered in development is that GSIs are more effective against tumors with upregulated Notch signaling [8]. Clinical trials should enroll patients with tumors displaying Notch activation (via mutation and/or overexpression), to avoid resistance because of lack of the therapeutic target. Acquired PTEN‐inactivating events [25] in NOTCH1‐dependent T‐cell lymphoblastic neoplasms could result in strong activation of PI3K‐AKT signaling, increased glycolysis and glutaminolysis, and consequently γ‐secretase inhibitor resistance. In addition, cell lines with mutations involving FBXW7, which encodes a negative regulator of NOTCH1 and MYC [26, 27], display resistance to GSIs. Last, as we describe below, our laboratory studies have shown that high levels of MYC expression confer resistance and could serve as a biomarker to identify patients not expected to experience response to to GSI treatment.
MYC Gene Dosage as a New Biomarker For γ‐Secretase Inhibitors
Activation of NOTCH receptor signaling sustains a broad transcriptional program of cell growth and proliferation in which the MYC oncogene seems to play a major role [28, 29, 30]. MYC has been shown to regulate mitochondrial biogenesis in T‐ALL cells [31] by directly targeting mitochondrial genes [32]. It has been shown that the mitochondria‐dependent process is regulated by NOTCH through MYC [33] and that MYC regulates the expression of anabolic genes and pathways downstream of NOTCH1 [25]. This NOTCH1‐MYC transcriptional regulatory loop places the control of cell growth anabolic pathways at the core of the mechanisms mediating T‐cell transformation by oncogenic NOTCH1 [34], so MYC could be a good biomarker for NOTCH activation. However, MYC overexpression can occur by mechanisms other than activation of the NOTCH signaling pathway, such as upon retroviral promoter insertion, chromosomal translocation/amplification, and activation of super‐enhancers within the MYC gene [35].
We hypothesized that constitutive MYC expression with independence of NOTCH1 activation could be a strong biomarker for resistance to GSI treatments. To develop this idea, we carried out a proof of concept in our laboratory. We used the γ‐secretase inhibitor PF‐03084014 to treat three NOTCH1‐dependent T‐cell lymphoblastic cell lines (HPB‐ALL, MOLT‐4, and SUP‐T1), characterized by the absence of FBXW7 mutations (polymerase chain reaction and Sanger sequencing oligonucleotides; supplemental online data). The small‐molecule GSI classified as a tetralin imidazole, PF‐03084014 (Pfizer Inc., Groton, CT), has been clinically evaluated to treat advanced breast cancer and other solid tumors [15]. PF‐03084014 is considered a selective inhibitor of NOTCH1 signaling that specifically inhibits γ‐secretase, triggering cell cycle arrest and apoptosis in multiple tumor types including T‐ALL and T‐cell lymphoblastic lymphoma (T‐LBL) [15, 19, 36, 37]. When evaluated for NOTCH activity, PF‐03084014 significantly decreased NOTCH intracellular domain (NICD) levels and mRNA expression of the NOTCH target genes HES1 and MYC in the cell line HPB‐ALL, and it is able to inhibit cell growth or survival in several NOTCH‐dependent cell lines via cell cycle arrest and induction of apoptosis [15]. Notably, the T‐cell lines displayed significant differences in GSI sensitivity, judging by the levels of cell proliferation and increased cell death (see below). In trying to understand this differential sensitivity, we noted that the abovementioned cell lines exhibited significant differences in MYC gene dosage in such a way that the higher MYC gene dosage, the higher level of resistance was observed. Taking into account that GSI‐persistent cells showed upregulation of MYC expression [38], changes in the copy number of MYC could be contributing to modulate the response to GSI treatment. These support the notion that the antileukemic effects of NOTCH1 inhibition can be bypassed by different, sometimes convergent, genetic and epigenetic mechanisms capable of supporting NOTCH‐independent leukemia cell growth [28]. High resolution microarray‐based comparative genomic hybridization analyses revealed that the HPB‐ALL cell line is pseudodiploid without significant copy number variations affecting chromosome 8, where MYC resides; MOLT‐4 cells exhibits mosaic duplication of chromosome 8, which is compatible with a partial trisomy; and SUP‐T1 cells combine a partial tetrasomy at 8q24.13q24.21 region that includes the MYC locus (Fig. 2A). To confirm the number of MYC copies, we performed fluorescence in situ hybridization using the DAKO Y5410 probe, a break‐apart probe that allows detection of two different colored signals over the MYC DNA sequence: one proximal (green) and another distal (red). Results indicate the existence of mosaicism with different cell populations exhibiting a different number of MYC copies (Fig. 2B). Of note, we always detected colocalization of green and red signals in all cells of the three cell lines, indicating that there is no evidence of translocation that could be activating MYC expression.
Figure 2.
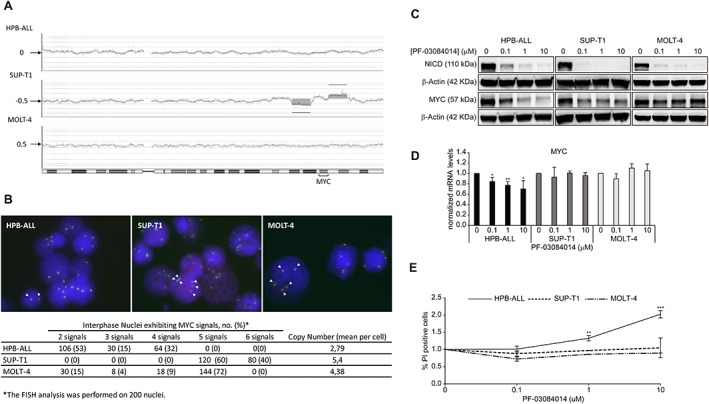
MYC gene dosage and γ‐secretase inhibitors. (A): Microarray‐based comparative genomic hybridization analysis (aCGH) of chromosome 8 to screen for copy number alterations of MYC in HPB‐ALL, SUP‐T1, and MOLT‐4 cell lines. The aCGH plots of chromosome 8 show alterations outside the thresholds of +0.5 for gain and −0.5 for loss, as well as borderline alterations at the +0.5 threshold. (B): Representative FISH signal patterns using the Myc break‐apart and DAKO Y5410 probe in T‐cell acute lymphoblastic leukemia (T‐ALL) cell lines. Interphase FISH analyses in at least 200 nuclei were evaluated. The colocalization of red and green signals excludes the presence of a translocation affecting the MYC. In the HPB‐ALL image, there are >50% of nuclei with two signals both for MYC. This case has a normal disomic status. In the other two images (SUP‐T1 and MOLT‐4) there are >50% of nuclei with five signals for MYC. These cases are representative of a gain for chromosome 8. (C–E): Effects of treatment of HPB‐ALL, SUP‐T1, and MOLT‐4 cells lines with γ‐secretase inhibitor (GSI) PF‐03084014 (0, 0.1, 1, and 10 μM) for 72 hours. (C): GSI PF‐03084014 treatment in T‐ALL cells inhibits Notch and induces apoptosis. Western blotting for detection of 120‐kDa NICD and MYC. (D): MYC mRNA expression is downregulated by PF‐03084014 in HPB‐ALL cell line. Real‐time polymerase chain reaction was performed for mRNAs expression levels of MYC (n = 3). The expression of specific mRNA is relative to housekeeping genes and was normalized to that the same ratio in unstimulated cells. (E): Flow cytometry analysis of PF‐03084014–treated cells (0–10 μM) stained with PI. Cell death was quantified by staining with propidium iodide using flow cytometry; treatment with PF‐03084014 dramatically increased the number of HPB‐ALL PI‐positive cells (n = 3). Each line shows the normalized data of stained cells percentage. Data are expressed as means ± SD. Significant differences using Student's t test (*p < .05, **p < .01, ***p < .001). Data normalized to vehicle treated cells (0 μM). Abbreviations: FISH, fluorescence in situ hybridization; NICD, Notch intracellular domain; PI, propidium iodide.
Our results showed that PF‐03084014 treatment, using the time and concentrations previously reported [15], decreased levels of intracellular domain of NOTCH protein in all cell lines (Fig. 2C). The level of expression of MYC underwent a significant decrease at the protein and transcriptional levels in the pseudodiploid line HPB‐ALL (Fig. 2C and D), indicating that the expression of MYC in this cell line must be essentially sustained by NOTCH1 activation. The expression levels of MYC remained relatively high in SUP‐T1 and MOLT‐4 (Fig. 2C), independent of the GSI dose treatments, probably because these cell lines also sustain the expression of MYC by gene dosage amplification in their genomes. We thus determined that PF‐03084014 decreased cell proliferation and increased cell death significantly only in HPB‐ALL cells. These results demonstrated that GSI was associated with antitumor activity that is practically undetectable in SUP‐T1 and MOLT cells (Fig. 2E).
Thus, constitutive MYC expression with independence of NOTCH1 activation may be a biomarker for resistance that can overcome the antiproliferative effects caused by the inhibition of NOTCH1 signaling after treatment with the γ‐secretase inhibitor PF‐03084014. Our findings underline the idea that gene dosage of MYC could be a mechanism of resistance for the GSIs.
Selective Targeting of γ‐Secretase Inhibitors
Recently, findings have been presented as a proof of principle that selective targeting of γ‐secretase may improve upon the activity of broad‐spectrum GSIs in preclinical models by reducing toxicities while showing significant therapeutic efficacy [39]. In a seminal work, the team led by Jan Cools and Bart de Strooper showed that T‐ALL cell lines and primary samples predominantly express a subclass of γ‐secretase that contains PSEN1 at its catalytic center. Therefore, they tested the PSEN1‐selective inhibitor MRK‐560 to treat human T‐ALL cell lines and different patient‐derived xenografts in mice with mutated NOTCH1. Interestingly, this compound showed a clear antitumor activity without adverse impact in the gastrointestinal tract or in T‐cell development [40].
Conclusion
One of the main limitations with medicine today is our limited understanding of the biology of disease. Thus, a deeper understanding of GSI structure and function would help to develop safe and effective γ‐secretase–based therapies. The National Cancer Institute defines precision medicine as “a form of medicine that uses information about person's genes, proteins, and environment to prevent, diagnose, and treat disease (National Cancer Institute, USNIH (2011) http://www.cancer.gov/dictionary/?CdrID=561717).” Personalized medicine uses specific markers in patients’ tumors to make treatment decisions. Genetics‐enabled clinical trials with molecularly defined subpopulations could potentially inform drug efficacy and safety profiling for GSIs, because maximum benefits can be obtained by minimizing adverse effects.
Moreover, γ‐secretase has been proposed as a therapeutic target in numerous human conditions, such us Alzheimer's disease, Kaposi's sarcoma, various cancers [2, 15, 41, 42, 43, 44], immunologic disorders [45], hearing loss [46], etc. Targeting the Notch signaling pathway in cancer via downregulation with GSIs is most promising in the context of a personalized precision medicine strategy. Genomic tests that detect mutations in the NOTCH genes, as well as mRNA in situ hybridization and immunohistochemical analyses that detect overexpression of Notch family receptors, would enhance the benefits of Notch pathway inhibitors, such as blocking monoclonal antibodies (mAbs) and γ‐secretase inhibitors, through successful positive selection of putative responders [47] (Fig. 3). Several biomarkers of resistance have already been pointed out, such as PTEN inactivation and mutations involving FBXW7 and now the level of MYC gene dosage. Integration of big data approaches can provide us with predictive models to construct a optimized Notch targeted strategy. In the same way, further studies will be required to assess the efficacy of selective targeting of GSIs against broad‐spectrum GSIs to treat T‐ALL and other tumors with driver‐activating mutations in the NOTCH1 receptor.
Figure 3.
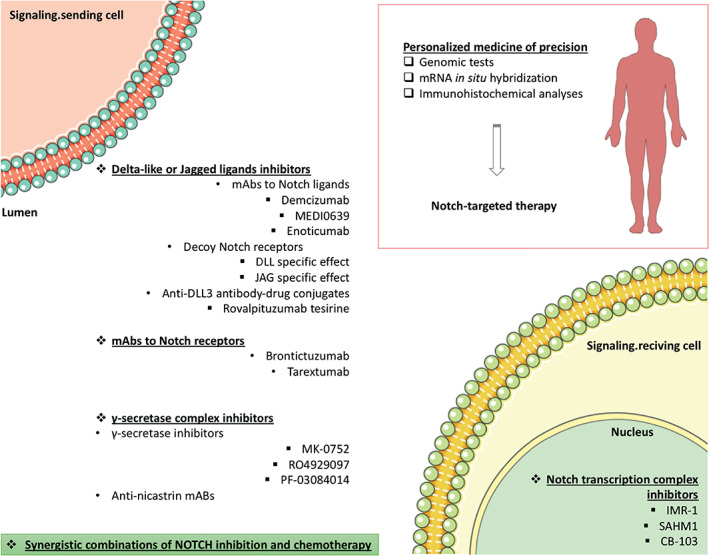
Omics‐based tests, including genomic tests, mRNA in situ hybridization, and immunohistochemical (among others) analyses, should be integrated to construct a Notch‐related knowledge base for the optimization of Notch‐targeted therapy. Synergistic combinations of NOTCH inhibition and chemotherapy should be considered. Templates to build this figure were obtained from SMART Servier Medical Art (Attribution 3.0 Unported, CC BY 3.0). Abbreviations: DLL3, Delta‐like ligand 3; mAb, monoclonal antibody.
Author Contributions
Conception/design: Raúl Córdoba, José Fernández‐Piqueras
Collection and/or assembly of data: Pilar López‐Nieva, Laura González‐Sánchez
Data analysis and interpretation: Pilar López‐Nieva, Laura González‐Sánchez, María Ángeles Cobos‐Fernández
Manuscript writing: Pilar López‐Nieva, Laura González‐Sánchez, José Fernández‐Piqueras
Final approval of manuscript: Pilar López‐Nieva, Laura González‐Sánchez, María Ángeles Cobos‐Fernández, Raúl Córdoba, José Fernández‐Piqueras
Disclosures
The authors indicated no financial relationships.
Supporting information
See http://www.TheOncologist.com for supplemental material available online.
Appendix S1: Supporting Information
Acknowledgments
The authors would like to thank the Cytometry and Cell Culture services of the Centro de Biología Molecular Severo Ochoa (CBMSO) and Isabel Sastre for the technical support. This work was financed by grants from the Spanish Ministry of Science, Innovation and Universities (MCIU)(RTI2018‐093330‐B‐I00; MCIU/FEDER, EU), Ramón Areces Fundation (CIVP19S7917) Spanish Ministry of Economy and Competitiveness (SAF2015‐70561‐R; MINECO/FEDER, EU; BES‐2013‐065740), the Autonomous Community of Madrid, Spain (B2017/BMD‐3778; LINFOMAS‐CM), the Spanish Association Against Cancer (AECC, 2018; PROYE18054PIRI), and the Instituto de Salud Carlos III (ISCIII; ACCI‐CIBERER‐17). Institutional grants from the Fundación Ramón Areces and Banco de Santander to the CBMSO are also acknowledged.
No part of this article may be reproduced, stored, or transmitted in any form or for any means without the prior permission in writing from the copyright holder. For information on purchasing reprints contact commercialreprints@wiley.com. For permission information contact permissions@wiley.com.
Disclosures of potential conflicts of interest may be found at the end of this article.
References
- 1. Ellisen LW, Bird J, West DC et al. TAN‐1, the human homolog of the Drosophila notch gene, is broken by chromosomal translocations in T lymphoblastic neoplasms. Cell 1991;66:649–661. [DOI] [PubMed] [Google Scholar]
- 2. Rizzo P, Osipo C, Foreman K et al. Rational targeting of Notch signaling in cancer. Oncogene 2008;27:5124–5131. [DOI] [PubMed] [Google Scholar]
- 3. Shao H, Huang Q, Liu ZJ. Targeting Notch signaling for cancer therapeutic intervention. Adv Pharmacol San Diego Calif 2012;65:191–234. [DOI] [PubMed] [Google Scholar]
- 4. Groth C, Fortini ME. Therapeutic approaches to modulating Notch signaling: Current challenges and future prospects. Semin Cell Dev Biol 2012;23:465–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Belver L, Ferrando A. The genetics and mechanisms of T cell acute lymphoblastic leukaemia. Nat Rev Cancer 2016;16:494–507. [DOI] [PubMed] [Google Scholar]
- 6. Dovey HF, John V, Anderson JP et al. Functional gamma‐secretase inhibitors reduce beta‐amyloid peptide levels in brain. J Neurochem 2001;76:173–181. [DOI] [PubMed] [Google Scholar]
- 7. Marambaud P, Shioi J, Serban G et al. A presenilin‐1/gamma‐secretase cleavage releases the E‐cadherin intracellular domain and regulates disassembly of adherens junctions. EMBO J 2002;21:1948–1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Olsauskas‐Kuprys R, Zlobin A, Osipo C. Gamma secretase inhibitors of Notch signaling. OncoTargets Ther 2013;6:943–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rosati E, Sabatini R, De Falco F et al. γ‐Secretase inhibitor I induces apoptosis in chronic lymphocytic leukemia cells by proteasome inhibition, endoplasmic reticulum stress increase and notch down‐regulation. Int J Cancer 2013;132:1940–1953. [DOI] [PubMed] [Google Scholar]
- 10. De Strooper B, Annaert W, Cupers P et al. A presenilin‐1‐dependent gamma‐secretase‐like protease mediates release of Notch intracellular domain. Nature 1999;398:518–522. [DOI] [PubMed] [Google Scholar]
- 11. Andersson ER, Lendahl U. Therapeutic modulation of Notch signalling–are we there yet? Nat Rev Drug Discov 2014;13:357–378. [DOI] [PubMed] [Google Scholar]
- 12. Selkoe D, Notch Kopan R. and Presenilin: Regulated intramembrane proteolysis links development and degeneration. Annu Rev Neurosci 2003;26:565–597. [DOI] [PubMed] [Google Scholar]
- 13. Sanchez‐Martin M, Ambesi‐Impiombato A, Qin Y et al. Synergistic antileukemic therapies in NOTCH1‐induced T‐ALL. Proc Natl Acad Sci USA 2017;114:2006–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. A phase I clinical trial of the notch inhibitor MK‐0752 in patients with T‐cell acute lymphoblastic leukemia/lymphoma (T‐ALL) and other leukemias. J Clin Oncol 2006;24(suppl 18):6585a. [Google Scholar]
- 15. Wei P, Walls M, Qiu M et al. Evaluation of selective gamma‐secretase inhibitor PF‐03084014 for its antitumor efficacy and gastrointestinal safety to guide optimal clinical trial design. Mol Cancer Ther 2010;9:1618–1628. [DOI] [PubMed] [Google Scholar]
- 16. Staal Frank J.T., Anton W. Langerak. Signaling pathways involved in the development of T‐cell acute lymphoblastic leukemia. Haematologica 2008;93:493–497. [DOI] [PubMed] [Google Scholar]
- 17. Pine SR. Rethinking gamma‐secretase inhibitors for treatment of non‐small‐cell lung cancer: Is notch the target? Clin Cancer Res 2018;24:6136–6141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ran Y, Hossain F, Pannuti A et al. γ‐Secretase inhibitors in cancer clinical trials are pharmacologically and functionally distinct. EMBO Mol Med 2017;9:950–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Samon JB, Castillo‐Martin M, Hadler M et al. Preclinical analysis of the γ‐secretase inhibitor PF‐03084014 in combination with glucocorticoids in T‐cell acute lymphoblastic leukemia. Mol Cancer Ther 2012;11:1565–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yahyanejad S, King H, Iglesias VS et al. NOTCH blockade combined with radiation therapy and temozolomide prolongs survival of orthotopic glioblastoma. Oncotarget 2016;7:41251–41264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sosa Iglesias V, Theys J, Groot AJ et al. Synergistic effects of NOTCH/γ‐secretase inhibition and standard of care treatment modalities in non‐small cell lung cancer cells. Front Oncol 2018;8:460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Plentz R, Park J, Rhim AD et al. Inhibition of γ‐secretase activity inhibits tumor progression in a mouse model of pancreatic ductal adenocarcinoma. Gastroenterology 2009;136:1741–1749.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ferrando A. Can one target T‐cell ALL? Best Pract Res Clin Haematol 2018;31:361–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Herranz D, Ambesi‐Impiombato A, Sudderth J et al. Metabolic reprogramming induces resistance to anti‐NOTCH1 therapies in T cell acute lymphoblastic leukemia. Nat Med 2015;21:1182–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Palomero T, Sulis ML, Cortina M et al. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T‐cell leukemia. Nat Med 2007;13:1203–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. O'Neil J, Grim J, Strack P et al. FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to gamma‐secretase inhibitors. J Exp Med 2007;204:1813–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Herranz D, Ferrando AA. Targeting NOTCH1 in T‐ALL: Starving the dragon. Cell Cycle Georget Tex 2016;15:483–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sanchez‐Martin M, Ferrando A. The NOTCH1‐MYC highway toward T‐cell acute lymphoblastic leukemia. Blood 2017;129:1124–1133. [DOI] [PubMed] [Google Scholar]
- 29. Sharma VM, Calvo JA, Draheim KM et al. Notch1 contributes to mouse T‐cell leukemia by directly inducing the expression of c‐myc. Mol Cell Biol 2006;26:8022–8031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Weng AP, Millholland JM, Yashiro‐Ohtani Y et al. c‐Myc is an important direct target of Notch1 in T‐cell acute lymphoblastic leukemia/lymphoma. Genes Dev 2006;20:2096–2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Margolin AA, Palomero T, Sumazin P et al. ChIP‐on‐chip significance analysis reveals large‐scale binding and regulation by human transcription factor oncogenes. Proc Natl Acad Sci USA 2009;106:244–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kim J, Lee J, Iyer VR. Global identification of Myc target genes reveals its direct role in mitochondrial biogenesis and its E‐box usage in vivo. PloS One 2008;3:e1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tammam J, Ware C, Efferson C et al. Down‐regulation of the Notch pathway mediated by a gamma‐secretase inhibitor induces anti‐tumour effects in mouse models of T‐cell leukaemia. Br J Pharmacol 2009;158:1183–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Palomero T, Ferrando A. Oncogenic NOTCH1 control of MYC and PI3K: Challenges and opportunities for anti‐NOTCH1 therapy in T‐cell acute lymphoblastic leukemias and lymphomas. Clin Cancer Res 2008;14:5314–5317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Meyer N, Penn LZ. Reflecting on 25 years with MYC. Nat Rev Cancer 2008;8:976–990. [DOI] [PubMed] [Google Scholar]
- 36. Zhang CC, Pavlicek A, Zhang Q et al. Biomarker and pharmacologic evaluation of the γ‐secretase inhibitor PF‐03084014 in breast cancer models. Clin Cancer Res 2012;18:5008–5019. [DOI] [PubMed] [Google Scholar]
- 37. Papayannidis C, DeAngelo DJ, Stock W et al. A phase 1 study of the novel gamma‐secretase inhibitor PF‐03084014 in patients with T‐cell acute lymphoblastic leukemia and T‐cell lymphoblastic lymphoma. Blood Cancer J 2015;5:e350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Knoechel B, Roderick JE, Williamson KE et al. An epigenetic mechanism of resistance to targeted therapy in T cell acute lymphoblastic leukemia. Nat Genet 2014;46:364–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Baratta MG. Adjusting the focus on γ‐secretase inhibition. Nat Rev Cancer 2019;19:419. [DOI] [PubMed] [Google Scholar]
- 40. Habets RA, de Bock CE, Serneels L et al. Safe targeting of T cell acute lymphoblastic leukemia by pathology‐specific NOTCH inhibition. Sci Transl Med 2019;11:eaau6246. [DOI] [PubMed] [Google Scholar]
- 41. Li T, Wen H, Brayton C et al. Epidermal growth factor receptor and notch pathways participate in the tumor suppressor function of gamma‐secretase. J Biol Chem 2007;282:32264–32273. [DOI] [PubMed] [Google Scholar]
- 42. Wang M, Wu L, Wang L et al. Down‐regulation of Notch1 by gamma‐secretase inhibition contributes to cell growth inhibition and apoptosis in ovarian cancer cells A2780. Biochem Biophys Res Commun 2010;393:144–149. [DOI] [PubMed] [Google Scholar]
- 43. Rodilla V, Villanueva A, Obrador‐Hevia A et al. Jagged1 is the pathological link between Wnt and Notch pathways in colorectal cancer. Proc Natl Acad Sci USA 2009;106:6315–6320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yabuuchi S, Pai SG, Campbell NR et al. Notch signaling pathway targeted therapy suppresses tumor progression and metastatic spread in pancreatic cancer. Cancer Lett 2013;335:41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Roderick JE, Gonzalez‐Perez G, Kuksin CA et al. Therapeutic targeting of NOTCH signaling ameliorates immune‐mediated bone marrow failure of aplastic anemia. J Exp Med 2013;210:1311–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mizutari K, Fujioka M, Hosoya M et al. Notch inhibition induces cochlear hair cell regeneration and recovery of hearing after acoustic trauma. Neuron 2013;77:58–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Katoh M, Katoh M. Precision medicine for human cancers with Notch signaling dysregulation (review). Int J Mol Med 2020;45:279–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
See http://www.TheOncologist.com for supplemental material available online.
Appendix S1: Supporting Information