

Abstract
Colorectal cancer (CRC) has a high mortality rate and poor prognosis. Despite chemotherapeutic agents such as cisplatin, which has achieved a better prognosis and survival rate against cancer, drug resistance leads to significant challenges. Accumulating evidence suggests that YTHDF1, the N6-methyladenosine (m6A) “reader,” is an important regulator in tumor progresses. Herein, we report that YTHDF1 was significantly upregulated in human colon tumors and cell lines. Overexpression of YTHDF1 decreased the cisplatin sensitivity of colon cancer cells. From the established cisplatin-resistant CRC cell line (LoVo CDDP R), we detected that YTHDF1 was significantly upregulated in cisplatin-resistant CRC cells. Intriguingly, RNA sequencing (RNA-seq) results revealed that glutamine metabolism enzymes were clearly upregulated in LoVo CDDP R cells. Glutamine uptake, that is, glutaminase (GLS) activity, was upregulated in LoVo CDDP R cells. Furthermore, bioinformatics analysis indicated that the 3′ UTR of GLS1 contained a putative binding motif of YTHDF1, and an interaction was further validated by a protein-RNA interaction assay (RNA immunoprecipitation [RIP]). Furthermore, we demonstrated that YTHDF1 promoted protein synthesis of GLS1. Inhibiting GLS1 effectively synergizes with cisplatin to induce colon cancer cell death. Finally, that YTHDF1 mediated cisplatin through the GLS1-glutamine metabolism axis was validated by an in vivo xenograft mouse model. In summary, our study reveals a new mechanism for YTHDF1-promoted cisplatin resistance, contributing to overcoming chemoresistant colon cancers.
Graphical Abstract
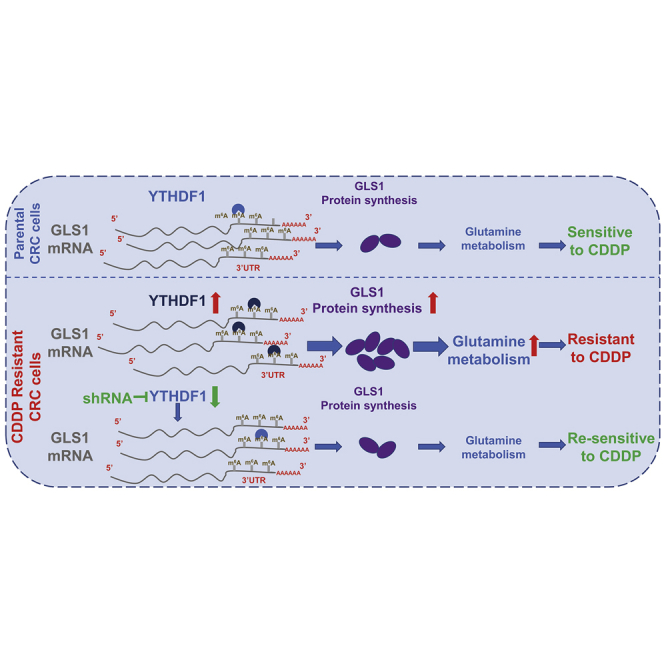
YTHDF1 is positively associated with cisplatin resistance through promoting glutamine metabolism, which is elevated in cisplatin-resistant CRC cells. YTHDF1 binds to the 3′ UTR of glutaminase 1 (GLS1) to promote protein synthesis of GLS1. Silencing the YTHDF1-mediated GLS1 and glutamine metabolism re-sensitizes cisplatin-resistant CRC cells in vitro and in vivo.
Introduction
Colorectal cancer (CRC), one of the leading causes of cancer-related death worldwide, has a high mortality rate and poor prognosis among cancer patients.1 Recently, despite that the mortality rate of colon cancer has declined due to the improvement of therapeutic strategies, the 5-year overall survival rate of CRC patients is still below expectations.2,3 Cisplatin, a platinum-based chemotherapeutic agent, has been clinically proven to combat a variety of malignancies.4 It acts through crosslinking with the purine bases on the DNA to impair the DNA repair processes, subsequently resulting in apoptosis of cancer cells.4,5 Although clinical application of cisplatin has recently achieved a better prognosis and survival rate for cancer patients, significant challenges remain such as drug resistance and considerable side effects,5 presenting remarkable limitations for its wide application. Therefore, investigating the underlying mechanisms for acquired cisplatin resistance will contribute to the development of effective therapeutic approaches for overcoming cisplatin resistance.
Accumulating studies have revealed that the aberrant metabolic reprogramming of tumors, including glucose and glutamine metabolism, is recognized as a new hallmark of cancers.6 In addition to glucose, glutamine is an essential growth-supporting amino acid in diverse types of cancer to refill the precursor molecules or metabolic intermediates for nucleic acid, lipid, and amino acid synthesis.7 CRCs display dysregulated metabolic profiling during tumor progression.8 Recent studies have elucidated that glutamine metabolism was upregulated in CRC, having been associated with tumor malignancy and poor prognosis.9 Oncogene c-MYC was reported to promote glutamine metabolism through inducing the expression of glutamine transporters and glutaminase (GLS).10 Furthermore, glutamine metabolism inhibitors, such as bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide (BPTES) and aminooxyacetic acid (AOA), have been reported to effectively suppress cancer proliferation.11,12 However, the precise mechanisms for the glutamine metabolism-mediated cisplatin sensitivity of colon cancer remain unclear.
N6-methyladenosine (m6A) is a prevalent modification of eukaryotic mRNA,13 leading to regulations of RNA metabolism, such as RNA splicing, decay, stability, and translation.14 m6A RNA methylation is known to be catalyzed by protein complexes consisting of “writer proteins,” which are methyltransferases, and demethylases acting as “erasers.”15 In addition, the YT521-B homology (YTH) domain-containing proteins, including YTHDF1–3, YTHDC1, and YTHDC2, are known as readers for identification of m6A modification.16 Increasing evidence has shown that dysregulated m6A modification is implicated in diverse tumor progressions. A recent study demonstrated that YTHDF1 is regulated by c-Myc, being associated with metastasis and poor overall survival of colon cancer.17 However, the roles of YTHDF1 in cisplatin resistance in colon cancer and the precise mechanisms for YTHDF1-mediated chemosensitivity remain unclear. This study focused on the roles of YTHDF1 in colon cancer cisplatin sensitivity and investigated the molecular mechanisms for YTHDF1-regulated glutamine metabolism. The potential protein-RNA interacting target will be identified. This study reveals a new mechanism for YTHDF1-promoted cisplatin resistance through the GLS1-glutamine metabolism axis, presenting it as a therapeutic target against chemo-resistant colon cancer.
Results
YTHDF1 is upregulated in colon cancer
Recent studies revealed important functions of m6A modification in the progressions of various cancers.13, 14, 15 To elucidate the roles of the RNA methylation reader YTHDF1 in human CRC, we compared the expression levels of YTHDF1 in colon tumor specimens and their adjacent normal tissues from 50 colon cancer patients. As we expected, both qRT-PCR data and analysis from The Cancer Genome Atlas database demonstrated that YTHDF1 was significantly elevated in colon cancers compared with normal tissues (Figure 1A; Figures S1A–S1C). Moreover, western blot results in Figure 1B clearly demonstrate that the protein expressions of YTHDF1 were markedly upregulated in CRC patient specimens. Consistent results were detected showing that the YTHDF1 protein expression was significantly upregulated in colon tumors by immunohistochemistry (IHC) staining (Figure 1C). Among the cancerous tissues, most (45 cases, 90%) stained strongly, 4 cases (8%) stained at a medium level, and only 1 case (2%) showed weak staining for YTHDF1. In contrast, only 2 (4%) of the adjacent normal colon tissues showed medium staining, and the rest (48 cases, 96%) displayed relative low staining for YTHDF1 (Figure 1C). Furthermore, the expressions of YTHDF1 were found to be significantly upregulated in five colon cancer cell lines compared with a normal colon epithelial cell line, CRL-1790 (Figure 1D). Taken together, the above results clearly demonstrated that YTHDF1 is a potential oncogene and is positively associated with colon cancer.
Figure 1.

Increased YTHDF1 expression is associated with CRC
(A) Expression levels of YTHDF1 from 50 colon tumor tissues and their matched normal colon tissues were detected by qRT-PCR. (B) YTHDF1 protein expression levels were detected from eight CRC tumors and their matched normal colon tissues by western blot. β-Actin was a loading control. (C) Representative immunohistochemical staining of YTHDF1 protein expression levels from colon tumor tissues and their matched normal colon tissues. (D) mRNA expression levels of YTHDF1 in normal colon epithelial cells and five CRC cell lines were detected by qRT-PCR. (E and F) DLD-1 (E) and LoVo (F) cells were transfected with control or YTHDF1 overexpression plasmid for 48 h, followed by cisplatin treatments at the indicated concentrations. Cell viability was determined by MTT and clonogenic assays. Columns include mean of three independent experiments; data are presented as mean ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
YTHDF1 is positively associated with cisplatin resistance of colon cancer
Since cisplatin resistance is one of the contributors to chemotherapy failure,5 we thus evaluated the roles of YTHDF1 in cisplatin sensitivity of colon cancer cells. YTHDF1 was overexpressed in DLD-1 and LoVo cells following exposure to elevated concentrations of cisplatin. As expected, results in Figures 1E and 1F demonstrated that colon cancer cells with higher YTHDF1 expression were less sensitive to cisplatin. The 50% inhibitory concentration (IC50) levels of cisplatin for DLD-1 and LoVo cells with YTHDF1 overexpression increased to 102.24 and 56.83 μM, respectively, which were significantly higher than those of control cells (30.53 and 17.84 μM). Furthermore, CRC cells with stable knockdown of YTHDF1 displayed increased cisplatin sensitivity (Figures S2A and S2B). To further validate the functions of YTHDF1 in cisplatin resistance, we established a cisplatin-resistant colon cancer cell line (LoVo CDDP R) via exposing cells to elevated concentrations of cisplatin to select the surviving (resistant) cells. As shown in Figures 2A and 2B, the LoVo CDDP R cells could tolerate higher concentrations of cisplatin treatments compared with LoVo parental cells. A cell viability assay showed that the IC50 value of LoVo CDDP R cells increased to 43.55 μM from that of parental cells (16.32 μM). Consistent results from an annexin V cell apoptosis assay showed that cisplatin treatment at 5 μM led to slightly different inhibition on cell viabilities of LoVo parental and CDDP R cells. However, significant cell death difference was observed with 10 and 20 μM cisplatin treatments (Figure 2C). Moreover, we detected that the protein and mRNA expression levels of YTHDF1 were clearly upregulated in cisplatin-resistant cells (Figures 2D and 2E). To evaluate the direct functions of YTHDF1 in cisplatin resistance, YTHDF1 was stably knocked down in LoVo CDDP R cells (Figure 2F). As expected, silencing YTHDF1 markedly sensitized cisplatin-resistant CRC cells to cisplatin compared with control short hairpin RNA (shRNA)-transfected cells (Figure 2F). Taken together, these results suggest that targeting YTHDF1 could be an effective approach for overcoming cisplatin resistance of colon cancer.
Figure 2.

Positive correlation between YTHDF1 and cisplatin resistance in colon cancer cells
(A) LoVo CDDP R cells were established according to the description in Materials and methods. (B and C) LoVo parental and CDDP R cells were treated with cisplatin at the indicated concentrations for 48 h, and cell viability was measured by an MTT assay, clonogenic assay (B), and annexin V assay (C). (D and E) The protein (D) and mRNA (E) expression levels of EGFR were measured in DLD-1 parental and 5-fluorouracil (5-Fu)-resistant cells. (F) YTHDF1 was stably knocked down in LoVo CDDP R cells. Cell were treated with cisplatin at the indicated concentrations for 48 h, and cell viability was measured by an MTT assay. Columns include mean of three independent experiments; data are presented as mean ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Glutamine metabolism is elevated in cisplatin-resistant cells
We then explored the molecular mechanisms for YTHDF1-promoted cisplatin resistance. Accumulating evidence has indicated that elevated and fast glutamine consumption levels play a critical role in cancer cell progression.6,7 Moreover, blocking glutamine metabolism by depletion of glutamine supplementation or inhibiting specific regulators in glutamine metabolism has been shown to effectively suppress cancer cell growth.8 To evaluate whether glutamine metabolism was associated with cisplatin sensitivity of colon cancer cells, we performed RNA sequencing (RNA-seq) analysis with LoVo parental and CDDP R cells. Intriguingly, a total of 1,251 genes were altered globally in cisplatin-resistant cells, including 813 upregulated genes and 438 downregulated genes (Figure 3A). Among them, we observed that a group of genes that are essential catalytic enzymes in glutamine metabolism were upregulated, indicating that glutamine metabolism is positively associated with cisplatin resistance. Moreover, a pan-metabolomic analysis was performed based on the RNA-seq results. The principal-component analysis (PCA) was applied to identify metabolic alterations between LoVo parental and CDDP R cells. As expected, cisplatin-resistant cells had markedly distinct glutamine metabolic profiles compared to parental cells (Figure 3B). Metabolic pathways involved in glucose metabolism, amino acid metabolism, and nucleotide synthesis were shown to be significantly upregulated in cisplatin-resistant cells (Figure 3B), suggesting that the glutamine-related metabolism was hyper-active, leading to cisplatin resistance of colon cancer. These upregulated glutamine metabolism enzymes from RNA-seq analysis were further validated by qRT-PCR experiments. Consistent results in Figure 3C demonstrated that GLS2, GOT1, GOT2, GPT2, GDH, and GLUD1, which are important glutamine metabolism enzymes or transporters, were significantly upregulated in LoVo CDDP R cells compared with parental cells. Since glutamine is an amidohydrolase enzyme that catalyzes the conversion of glutamate from glutamine, the first irreversible reaction of glutamine metabolism,6 we assessed the protein expressions of GLS1 and GLS2 in LoVo parental and CDDP R cells. Although we did not detect a significantly upregulation of GLS1 mRNA in cisplatin-resistant cells (Figure 3C), western blot results clearly illustrated that the protein level of GLS1 was upregulated (Figure 3D), suggesting that GLS1 was also involved in cisplatin resistance. Consistently, glutamine uptake and GLS activity were apparently higher in cisplatin-resistant colon cancer cells (Figures 3E and 3F). Accumulating evidence has revealed that diverse cancer cells display glutamine dependence for progress, and we thus evaluated whether glutamine deprivation could increase the cytotoxicity of cisplatin on cisplatin-resistant cells. As expected, results showed that compared with parental cells, a low glutamine supply suppressed a higher percentage of cell growth and stimulated cell death of LoVo CDDP R cells (Figures 3G and 3H). Moreover, a clonogenic assay and annexin V assay consistently demonstrated that LoVo CDDP R cells were more susceptible to cisplatin treatments under low glutamine supplementation (Figures 3I and 3J). Taken together, these results elucidated a strong correlation between hyper-active glutamine metabolism and cisplatin resistance in colon cancer.
Figure 3.

Cisplatin-resistant CRC cells exhibit elevated glutamine metabolism
(A) Heatmap of differentially expressed genes from LoVo parental and CDDP R cells identified by RNA-seq. (B) MetaboAnalyst pathway enrichment analysis of metabolites in LoVo parental and CDDP R cells. (C) mRNA expression levels of glutamine metabolism enzymes and regulators were detected by qRT-PCR in LoVo parental and CDDP R cells. (D) Western blot results show protein expression of GLS1 and GLS2 in LoVo parental and CDDP R cells. (E and F) Glutamine uptake (E) and GLS activity (F) assays were performed in LoVo parental and CDDP R cells. (G) LoVo parental and CDDP R cells were cultured with regular medium or glutamine depletion medium, a clonogenic assay and (H) annexin V assay were performed. (I) LoVo CDDP R cells cultured with regular medium or glutamine depletion medium were treated with cisplatin at 10 and 20 μM for 48 h. (J) Cell viability and cell death were examined by a clonogenic assay and (H) annexin V assay. Columns include mean of three independent experiments; data are presented as mean ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
YTHDF1 promotes protein synthesis of GLS1 via direct targeting of the 3′ UTR of GLS1 mRNA
The above results describe YTHDF1-mediated cisplatin resistance and a positive association between glutamine metabolism and cisplatin resistance. It was known that YTHDF1 was a typical RNA-binding protein as an m6A reader, and that it post-transcriptionally regulated its target genes. To investigate whether YTHDF1 directly regulates glutamine metabolism through binding target mRNAs via the YTH domain, plasmid containing wild-type or YTH domain mutant YTHDF118 was transfected into colon cancer cells. As we expected, overexpression of YTHDF1 significantly stimulated glutamine uptake and GLS activity (Figures 4A and 4B). However, the glutamine uptake and GLS activity in cells with YTH domain mutant YTHDF1 overexpression were not affected. Moreover, the glutamine uptake and GLS activity were significantly attenuated in CRC cells with stable knockdown of YTHDF1 (Figures S3A and S3B). Since two GLS isoforms participated in glutamine metabolism, we identified GLS1 as the major molecule for glutamine uptake and GLS activity (Figures S4A and S4B). Results showed that GLS1 protein was upregulated but the transcription of GLS1 was not changed in cisplatin-resistant CRC cells (Figures 3C and 3D), indicating that YTHDF1 regulates translation rather than RNA abundance. We therefore asked whether GLS1 was a direct target of YTHDF1. Bioinformatics analysis illustrated a protein-RNA interaction between YTHDF1 and the 3′ UTR of GLS1 from starBase 2.0 online protein-RNA interaction analysis. Among the five predicted motifs that were potential targets of YTHDF1, one motif (Figure 4C) was validated by an electrophoretic mobility shift assay (EMSA) assay (Figures S5A and S5B) to have the strongest binding capacity with YTHDF1. We thus speculated that YTHDF1 upregulates GLS1 expression though enhancing either protein stability or translation of GLS1 by directly binding on the 3′ UTR of mRNA. To evaluate this, an RNA immunoprecipitation (RIP) assay was performed in LoVo and DLD-1 cells using an antibody specifically against YTHDF1. Consistent results from RT-PCR and qPCR showed that GLS1 mRNA was significantly enriched in YTHDF1 co-precipitated RNA fragments (Figures 4D and 4E) compared with the immunoglobulin G (IgG) control antibody. In addition, the YTHDF1-GLS1 mRNA interaction was further validated by an RNA pull-down assay. A biotin-labeled fragment of the GLS1 3′ UTR-containing binding motif was applied to immunoprecipitate putative binding proteins. Results demonstrated significant enrichment of YTHDF1 protein associating with the GLS1 3′ UTR compared with a scramble control (Figure 4F). To determine whether YTHDF1 upregulated GLS1 protein through its association, YTHDF1 was silenced in colon cancer cells that were subjected to a RIP assay. Consistently, cells with lower YTHDF1 immunoprecipitated a lower amount of GLS1 mRNA (Figures 4G and 4H). To further validate the binding of YTHDF1 on the predicted binding motif on GLS1, we constructed the predicted binding motif mutations on the GLS1 3′ UTR and performed a dual-luciferase reporter assay. Consistently, compared with control cells, the YTHDF1-silencing cells exhibited a significant increment of luciferase activity in the wild-type GLS1 3′ UTR reporter (Figures 4H and 4I). However, a less repressive effect on the binding motif mutant GLS1 3′ UTR was observed (Figures 4H and 4I), suggesting that YTHDF1 directly bonded on the predicted binding motif on the GLS1 3′ UTR. Furthermore, we evaluated the mechanisms for the YTHDF1-promoted GLS1 protein expression. Colon cancer cells were treated with protein translation inhibitor cycloheximide (CHX). Western blot results demonstrated that CHX treatments led to significant protein degradation of GLS1 (Figure 4K). However, the degradation of GLS1 protein turned out no difference in control or YTHDF1-silenced LoVo and DLD-1 cells (Figure 4K), indicating that YTHDF1 did not regulate protein stability of GLS1. Furthermore, cells were treated with MG132 to inhibit proteasome activity. Western blot results clearly showed that under protein degradation inhibition, colon cancer cells with YTHDF1 silencing displayed significantly decreased GLS1 levels compared with control cells (Figure 4L), suggesting that YTHDF1 promoted GLS1 protein synthesis. Taken together, the above results demonstrated that the YTHDF1-mediated GLS1 protein upregulation was through direct binding to the 3′ UTR of GLS1 mRNA, leading to translational promotion.
Figure 4.

YTHDF1 directly binds with GLS1 to promote its protein translation
(A and B) LoVo and DLD-1 cells were transfected with control, wild-type (WT) YTHDF1, or YTH domain mutant YTHDF1 for 48 h (A), and glutamine uptake and GLS activity (B) were detected. (C) Predicted YTHDF1 binding motif on the 3′ UTR of GLS1 mRNA. (D) RNA immunoprecipitation (RIP) was performed in LoVo and DLD-1 cells using anti-IgG control or anti-YTHDF1 antibody. (E and F) GLS1 abundance in the immunoprecipitated fraction was measured by agarose gel electrophoresis and (E) qRT-PCR. (F) An RNA pull-down assay was performed in LoVo and DLD-1 cells. The biotin-labeled 3′ UTR of WT or binding motif mutant GLS1 was incubated with proteins extracted from cells. The YTHDF1 protein, which was pulled down by the GLS1 binding motif, was detected by western blot. β-Actin was used as a negative control. (G) LoVo and DLD-1 cells were transfected with control shRNA or YTHDF1 shRNA for 48 h, and RIP experiments were performed using an anti-IgG control or anti-YTHDF1 antibody. GLS1 and β-actin mRNA abundance levels in the immunoprecipitated fraction were measured by agarose gel electrophoresis and (H) qRT-PCR. (I and J) Reporter constructs containing the WT or the binding motif mutant (Mut) GLS1 3′ UTR were co-transfected with control shRNA or GLS1 shRNA into LoVo and DLD-1 cells. Luciferase activities were measured using a Dual-Luciferase reporter assay kit. (K) LoVo and DLD-1 cells were treated with 10 μg/mL CHX for 0 and 6 h, and the relative GLS1 protein expression was measured by western blot. (L) LoVo and DLD-1 cells were treated with 50 nM MG-132 for 0 and 6 h, and the relative GLS1 protein expression was measured by western blot. β-Actin was used as an internal control. Data are presented as mean ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Inhibiting GLS effectively synergizes with cisplatin to induce colon cancer cell death
To investigate whether the above-described molecular pathway could benefit cisplatin-resistant colon cancer patients, GLS1 was blocked by either shRNA or a GLS specific inhibitor, BPTES.19 An MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay and clonogenic assay consistently demonstrated that GLS1-silenced DLD-1 and LoVo cells were more sensitive to cisplatin compared with control cells (Figures 5A–5D). From an MTT assay and annexin V assay, DLD-1 cells treated with low dosages (<IC50) of cisplatin plus GLS inhibitor significantly increased cytotoxicity to cisplatin (Figures 5E and 5F). Importantly, the combination index (CI) was analyzed to evaluate whether the combination of BPTES with cisplatin has additive or synergistic effects. As expected, the CI value of combined treatments with cisplatin (5, 10, or 20 μM) and BPTES (25 or 50 nM) showed a significantly synergistic effect (CI < 1) (Figure 5G). The similar synergistic effects by the combination of cisplatin (2, 4, or 8 μM) with BPTES (25 or 50 nM) treatments were also observed in LoVo cells (Figures 5H–5J). These consistent results demonstrated that blocking GLS1 effectively synergized with cisplatin to enhance the tumor-suppressive effects.
Figure 5.

Inhibiting GLS effectively synergizes with cisplatin to induce colon cancer cell death
(A) DLD-1 cells were transfected with control shRNA or sh-GLS1 for 48 h, followed by treatments of cisplatin at the indicated concentrations. Cell viability was determined by MTT and (B) clonogenic assays. (C) LoVo cells were transfected with control shRNA or sh-GLS1 for 48 h, followed by treatments of cisplatin at the indicated concentrations. Cell viability was determined by MTT and (D) clonogenic assays. (E) DLD-1 cells were treated with BPTES at 0, 25, or 50 nM plus cisplatin at the indicated concentrations. Cell viability and cell death were examined by MTT and (F) annexin V assays. (G) The combination index (CI) was calculated using CompuSyn software based on the results of (E) and (F). (H) LoVo cells were treated with BPTES at 0, 25, or 50 nM plus cisplatin at the indicated concentrations. Cell viability and cell death were examined by MTT and (I) annexin V assays. (J) The CI was calculated using CompuSyn software based on the results of (H) and (I). Data are presented as mean ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Blocking YTHDF1-mediated glutamine metabolism sensitizes colon cancer cells to cisplatin in vitro and in vivo
Given that YTHDF1 positively regulates GLS1 expression and contributes to cisplatin resistance of colon cancer cells, we then asked whether the YTHDF1-influenced cisplatin resistance was through the GLS1-glutamine metabolism pathway. DLD-1 cells were transfected with control or YTHDF1 overexpression plasmid alone or with BPTES treatment. Results from Figure 6A showed that YTHDF1 overexpression significantly promoted GLS activity, which could be effectively overcome by GLS inhibition. Moreover, MTT and annexin V assays consistently demonstrated that the YTHDF1-overexpressed DLD-1 cells displayed significantly increased resistance to cisplatin, which was further overridden by GLS inhibition (Figure 6B), suggesting that the YTHDF1-mediated GLS upregulation was required for cisplatin resistance. Similarly, overexpression of YTHDF1 activated the GLS, leading to acquired cisplatin resistance, and a phenotype was further reversed by GLS inhibition in LoVo cells. (Figures 6D–6F). In summary, these results strongly demonstrated that YTHDF1-promoted cisplatin resistance was through activating the GLS1-glutamine metabolism axis.
Figure 6.

YTHDF1-mediated cisplatin resistance is through promoting glutamine metabolism
(A) DLD-1 cells were transfected with control or YTHDF1 overexpression plasmid for 48 h. Cells were treated without or with BPTES for 24 h. The GLS activity was measured. (B) The above cells were treated with cisplatin at 0, 2.5, 5, 10, 20, or 40 μM for 48 h. Cell viability and death were assessed by an MTT assay and (C) annexin V assay, respectively. (D) LoVo cells were transfected with control or YTHDF1 overexpression plasmid for 48 h. Cells were treated without or with BPTES for 24 h. The GLS activity was measured. (E) The above cells were treated with cisplatin at 0, 2, 4, 8, 16, or 32 μM for 48 h. Cell viability and death were assessed by an MTT assay and (F) annexin V assay, respectively. Data are presented as mean ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Finally, we validated the above in vitro molecular pathway using a xenograft mouse model. To investigate whether the combination of YTHDF1 inhibition with cisplatin could significantly improve the survival rate, LoVo CDDP R cells with control or YTHDF1 silencing treatment were subcutaneously implanted into the mammary fat pads of nude mice. After 1 week, upon the establishment of xenograft tumors, mice were separated into four groups: control shRNA plus injection of control saline, YTHDF1 silencing plus control saline, control shRNA plus cisplatin, or YTHDF1 silencing plus cisplatin via intraperitoneal injection twice a week for 2 months. As expected, mice exhibited a high death rate with control or cisplatin alone treatments (Figure 7A). Mice that received YTHDF1 inhibition alone or cisplatin alone treatment displayed a slightly increased survival rate, but the combined treatments of YTHDF1 silencing plus cisplatin dramatically achieved a prolonged survival rate (Figure 7A). Accordingly, mice that underwent the combined treatments grew smaller tumors compared with control, YTHDF1 silencing alone, or cisplatin alone treatment (Figures 7B and 7C). Furthermore, died and survival mice were sacrificed, and tumor tissues were dissected for analysis of GLS1 protein expression. Consistently, tumor tissues from mice treated with YTHDF1 silencing alone showed significantly decreased GLS1 protein expression (Figure 7D). These xenograft mice results support the in vitro results that the YTHDF1-mediated cisplatin resistance of colon cancer was through upregulating GLS1.
Figure 7.

In vivo blocking YTHDF1-mediated glutamine metabolism sensitizes colon cancer cells to cisplatin
(A) LoVo CDDP R cells were transfected with control shRNA or YTHDF1 shRNA. Cells were subcutaneously injected into nude mice for developing xenograft tumors. Mice without or with YTHDF1 silencing xenograft tumors were grouped and treated with control saline or cisplatin via intraperitoneal injection twice a week. Mice survival rates and (B) tumor growth were examined. (C) A total of 60 xenograft tumors from the above-treated mice of each experiment group (15 for each group). (D) Xenograft tumors from mice were dissected, and the protein expression levels of GLS1 were examined by western blot and quantified. Data are presented as mean ± SD. ∗p < 0.05, ∗∗∗p < 0.001.
Discussion
Accumulating evidence has shown that m6A RNA methylation is a crucial biological process for tumor progressions.13, 14, 15 Previous reports have revealed that YTHDF1, the m6A RNA methylation reader, promotes diverse carcinogenesis.17 Recent studies have demonstrated that YTHDF1 positively regulates the tumorigenicity of colon cancer.20 Moreover, the cancer stem cell-like activity and cell cycle were promoted by YTHDF1 in human colorectal carcinoma.20 Another study reported that the oncogene c-Myc upregulates YTHDF1 expression in CRC, leading to activation of cancer cell proliferation and resistance to anticancer drugs such as fluorouracil and oxaliplatin.21 Importantly, an RNA-seq FPKM (fragments per kilobase of transcript per million mapped reads) analysis of 331 colorectal adenocarcinoma samples in The Cancer Genome Atlas database demonstrated that YTHDF1 was significantly upregulated in colon cancer, suggesting that YTHDF1 could be applied as a prognostic factor for colon cancer.22 Moreover, in non-small cell lung cancer, YTHDF1 was shown to be positively associated with cancer progress through regulating the translational efficiency of CDK2, CDK4, and cyclin D1.23 However, the precise roles of YTHDF1 in cisplatin resistance of colon cancer have not been elucidated. Consistent with previous reports, our present study reports that YTHDF1 was frequently amplified in CRC tissues and cells and contributed to cisplatin resistance, suggesting that YTHDF1 is a potential oncogene that might be a diagnostic marker for CRC patients.
Cisplatin acts through crosslinking with the purine bases on DNA to impair DNA repair, leading to inducement of cancer cell death. However, significant challenges such as drug resistance and considerable side effects limit its clinical applications.4,5 By establishing a cisplatin-resistant CRC cell line, we observed that YTHDF1 was positively associated with cisplatin resistance. Overexpression of YTHDF1 significantly de-sensitized colon cancer cells to cisplatin, prompting us to investigate a deeper understanding of the molecular mechanisms for the YTHDF1-promoted cisplatin resistance to develop alternative treatment strategies.
Cancer cells, in contrast to normal cells, exhibit aberrant metabolic characteristics, which were recognized as a new hallmark.6 Moreover, this unique feature of cancer cells emerged as an attractively therapeutic target for developing novel strategies against chemoresistant cancers.7 Studies described that CRC displayed deregulated glutamine metabolic profiling during tumorigenesis.8 However, the precise molecular mechanisms are still under investigation. Our results consistently demonstrated that cisplatin-resistant CRC cells were associated with a higher glutamine consumption rate as well as upregulated key regulators and enzymes of glutamine metabolism. Furthermore, cisplatin-resistant cells were more dependent on glutamine metabolism that under low glutamine supplement, the cell viability of cisplatin-resistant cells were significantly lower that parental cells. We observed depletion of glutamine-sensitized cisplatin-resistant cells, suggesting that blocking the glutamine metabolism pathway might effectively overcome cisplatin resistance.
Side effects of chemotherapeutic agents with high toxic dosages limited their clinical applications. Therefore, the combination of traditional chemotherapy with glutamine metabolism inhibitors might be an effective approach to improve chemotherapy outcomes. Based on the above-described YTHDF1-glutamine metabolism-cisplatin resistance axis, we illustrated a synergistically tumor-suppressive effect on cisplatin-resistant CRC cells by GLS inhibitor plus cisplatin treatments. Under BPTES treatments, relatively low toxic amounts of cisplatin significantly induced cell death, indicating that the combined strategy by glutamine metabolism inhibition and cisplatin could attenuate the high dosage-accompanied side effects of cisplatin in its clinical applications.
Emerging evidence demonstrated that metabolic key enzymes and regulators could be modulated by post-translational modifications, such as acetylation and methylation. To solve the YTHDF1-mediated cisplatin resistance in colon cancer, it is essential to uncover the molecular targets of YTHDF1 for developing specifically effective strategies. We thus applied screening strategies by comparison of the transcriptomes from CRC parental and cisplatin-resistant cells. Importantly, the predicted binding between YTHDF1 and its target, GLS1 mRNA, was validated by combining the RIP-PCR, RNA pull-down, and luciferase assays. Moreover, we demonstrated that YTHDF1 promoted GLS1 protein synthesis but did not prevent protein degradation of GLS1. From both in vitro and in vivo xenograft mice models, we illustrated that blocking YTHDF1 effectively overrode the cisplatin resistance of CRC cells, leading to opening new avenues that include targeting YTHDF1 as a promising strategy against chemoresistant cancers. However, our study still has limitations, including that the molecular mechanisms for the YTHDF1-regulated GLS1 protein translation through GLS1 m6A methylation require further investigation.
In summary, our study identifies that GLS1, a glutamine metabolism key enzyme, is the direct target of YTHDF1 in colon cancer cells. YTHDF1 promotes GLS1 translation and, consequently, contributes to cisplatin resistance of colon cancer cells. Thus, targeting YTHDF1 represents a potential therapeutic strategy for selectively inhibiting chemoresistant CRCs.
Materials and methods
Colon cancer patient sample collection
This study was approved by the Institutional Review Board of The First Affiliated Hospital of Zhejiang Chinese Medical University. Fresh human colon tumor specimens and their adjacent normal tissues were obtained from 50 patients during surgery in The First Affiliated Hospital of Zhejiang Chinese Medical University from June 2017 to August 2019. No patients received other cancer chemotherapies and/or radiotherapies prior to resection. All samples were examined by pathologists for the diagnosis of cancers. Written informed consent was provided from patients in this study. Tissues were frozen immediately in liquid nitrogen and stored at −80°C until use.
Cell culture and reagents
Human CRC cell lines LoVo, HT-29, DLD-1, HT-116, and SW480, as well as the normal colon epithelial cell line CRL-1790, were purchased from the American Type Culture Collection (ATCC). Cells were maintained in RPMI 1640 supplemented with 10% fetal bovine serum (FBS) (Sigma-Aldrich, Shanghai, China), 2 mM l-glutamine (Thermo Fisher Scientific), and 1% penicillin-streptomycin (Gibco-BRL, Grand Island, NY, USA) at 37°C with 5% CO2 in a humidified atmosphere. All cells are routinely confirmed as mycoplasma negative (MycoAlert). LoVo CDDP R cells were generated by stepwise treatment of cells with cisplatin for selecting resistant clones, which were further pooled for downstream experiments. Cisplatin resistance was examined every 3 months. Rabbit anti-GLS1 antibody (#56750), rabbit anti-YTHDF1 monoclonal antibody (#86463), and rabbit anti-β-actin (#4967) antibody were purchased from Cell Signaling Technology (Danvers, MA, USA). Rabbit anti-GLS2 polyclonal antibody (#PA5-78475) was purchased from Invitrogen (Carlsbad, CA, USA). Vector containing the wild-type open reading frame (ORF) clone of Homo sapiens protein YTHDF1 was purchased from Origene. sh-YTHDF1 and control shRNA were constructed by GenePharma (Shanghai, China); si-GLS1 and control small interfering RNA (siRNA) were constructed by Hanbio (Shanghai, China); and CHX, cisplatin, MG-132, and BPTES were purchased from Sigma-Aldrich (Shanghai, China).
Plasmid DNA, siRNA, and shRNA transfections
Transfections were performed in colon cancer cells using Lipofectamine 3000 reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. Cells (5 × 105 cells/well) were seeded in six-well plates for 24 h before transfection. When cells were achieved 70% confluence, overexpression plasmid, siRNA, or shRNA and their negative controls were transfected into cells for 48 h. Knockdown efficiency levels of siRNA and shRNA were validated by qRT-PCR. Plasmid DNA was transfected at 2 μg, and siRNAs were transfected at 50 nM. Transfections were performed in triplicate.
Bioinformatics analysis
The YTHDF1 binding motif on the 3′ UTR of GLS1 mRNA was predicted by starBase of ENCORI (http://starbase.sysu.edu.cn/).
RNA isolation and qRT-PCR
Total RNA extraction was performed using an RNeasy kit (#74134, QIAGEN) according to the manufacturer’s instructions. The quality and quantity of RNA samples were assessed by NanoDrop. 1 μg of total RNA was converted to complementary DNA (cDNA) using a high-capacity RNA-to-cDNA kit (#4387406, Thermo Fisher Scientific). qRT-PCR was performed with 1 μL of cDNA and TaqMan 2× universal PCR master mix (Thermo Fisher Scientific) by a real-time PCR system (Applied Biosystems). The thermal profile was set as follows: 95°C for 1 min and 40 cycles at 95°C for 15 s, 58°C for 20 s, and 72°C for 20 s. TaqMan primers were obtained from Life Technologies, that is, GLS1 (Hs00248163_m1) and GLS2 (Hs00998733_m1). Expressions were quantified to the housekeeping genes β-actin and calculated by the 2−ΔΔCt method. Experiments were performed in triplicate and repeated three times.
RNA-seq
Total RNA extraction was performed using an RNeasy kit (#74134, QIAGEN, Hilden, Germany) according to the manufacturer’s instructions. The quantity of RNA samples was assessed by NanoDrop. The integrity of RNA was verified by running on 1.5% agarose gel electrophoresis. 2 μg of total RNA of each sample was used for RNA-seq library preparation by a VAHTS stranded mRNA-seq library prep kit (Vazyme). Polyadenylated mRNAs were fragmented and then converted into double-strand cDNA followed by ligation into VAHTS RNA adapters (Vazyme). Purified ligation products were used for sequencing. The Illumina HiSeq 4000 system was used to collect sequencing data (Illumina, San Diego, CA, USA).
RIP
Total RNA extraction was performed using an RNeasy kit (#74134, QIAGEN, Hilden, Germany) according to the manufacturer’s instructions. The quantity of RNA samples was assessed by NanoDrop. RIP was conducted using a Magna RIP RNA-binding protein immunoprecipitation kit (Millipore, Bedford, MA, USA) according to the manufacturer’s instructions. Cells with wild-type YTHDF1 and mutant YTHDF1 were lysed by RIP lysis buffer. Anti-YTHDF1 or anti-IgG antibodies with A/G immunomagnetic beads were premixed in immunoprecipitation buffer and then added into cell lysates. After digestion with protease K, purified RNA samples were subjected to qPCR and RT-PCR to examine the enrichment of GLS1 mRNA fragments that bond with YTHDF1 protein. Experiments were repeated three times and performed in triplicate.
RNA pull-down assay
Total RNA extraction was performed using an RNeasy kit (#74134, QIAGEN, Hilden, Germany) according to the manufacturer’s instructions. Fragments of the GLS1 3′ UTR containing the predicted binding motif and the negative control (scramble control of binding motif) were in vitro transcribed using a biotin RNA labeling mix (Roche, Shanghai, China). Samples were treated with RNase-free DNase I and purified using an RNeasy mini kit (QIAGEN, Shanghai, China). The biotinylated RNA (50 pmol/mg) was incubated with total proteins extracted from colon cancer cells. The RNA-protein mixture was incubated with streptavidin agarose beads (Invitrogen, Carlsbad, CA, USA). After complete washing with NaCl/Pi, protein samples were eluted and detected by western blot. β-actin was used a negative control. Experiments were repeated three times.
Dual-luciferase reporter gene assay
The pGL3-reporter luciferase plasmid containing the wild-type 3′ UTR and the binding motif mutant 3′ UTR of GLS1 was constructed from GeneChem (Shanghai, China). Vectors were co-transfected into colon cancer cells using Lipofectamine 2000 (Thermo Fisher Scientific, Waltham, MA, USA). Forty-eight hours after transfection, luciferase activity was measured by a Dual-Luciferase reporter assay system (Promega, Fitchburg, WI, USA) according to the manufacturer’s instructions. Firefly luciferase activity was normalized to Renilla luciferase activity. Experiments were performed in triplicate and repeated three times.
IHC
The formalin-fixed, paraffin-embedded human colon normal tissues and tumor tissues were sectioned onto slides. After deparaffinization, rehydration, and quenching in 0.6% hydrogen peroxide/methanol for 15 min, slides were boiled for 20 min in 10 mM sodium citrate (pH 6.0) for antigen retrieval, followed by blocking with 5% serum and 1% BSA plus 0.5% Tween 20 for 1 h. Then, slides were incubated with primary antibodies in blocking buffer overnight at 4°C. After complete washing and incubation of biotinylated secondary antibodies, tissue sections were mounted for light microscopy analysis.
Glutamine uptake assay
Glutamine uptake was determined using a glutamine assay kit (colorimetric) (ab197011, Abcam) following the manufacturer’s instructions. According to the principle of glutamine conversion into glutamic acid and ammonia, the amount of glutamine was calculated by measuring the amount of ammonia. The relative glutamine uptake was calculated to the ratio of control cells and normalized by the protein amount of each group. Experiments were performed in triplicate and repeated three times.
GLS activity assay
GLS activity was determined using a GLS assay kit (#E-133, Biomedical Research Service Center, Buffalo, NY, USA) according to the manufacturer’s instructions and previous descriptions.24 The relative GLS activity was calculated to the ratio of control cells and normalized by the protein amount of each group. Experiments were performed in triplicate and repeated three times.
Clonogenic assay
Anchorage-dependent colon cancer cells were plated at a concentration of 1 × 105 cells/well in six-well plates for 24 h. After treatment, cells were cultured with fresh medium and incubated for an additional 6 days and then stained using a 5% crystal violet solution. The survival clones were measured under microscopy. Experiments were performed in triplicate.
Cell viability assays
Cell viability was determined by an MTT assay. Briefly, 5 × 103 cells were grown in 96-well plates and cultured for 24 h. After treatments, cells were incubated with MTT (5 mg/mL) for 4 h at 37°C. Then, medium was removed, and formazan crystals were solubilized with dimethyl sulfoxide (DMSO) (Thermo Fisher Scientific, Waltham, MA, USA). Optical density was determined by a Sunrise spectrophotometer at a wavelength of 570 nm. Relative viability of the experimental group was calculated to the ratio of control cells. Experiments were performed in triplicate and repeated three times.
Detection of cell death
Cells (1 × 105) were seeded in six-well plates for 24 h. Cell death was assessed by a fluorescein isothiocyanate (FITC)-annexin V/propidium iodide (PI) kit (#556547, BD Biosciences) according to the manufacturer’s instructions and was analyzed by a BD Accuri C6 flow cytometer (BD Biosciences, USA). Experiments were repeated three times.
Western blot
Cells or tissues were lysed with radioimmunoprecipitation assay (RIPA) lysis buffer (89900, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with protease inhibitor cocktail (Thermo Fisher Scientific, Waltham, MA, USA). Lysates were centrifuged at 14,000 rpm at 4°C for 10 min. Supernatants were collected. Protein concentrations were determined by a Bradford assay (Bio-Rad, Hercules, CA, USA). An equal amount protein of each experimental group was separated by 10% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a nitrocellulose membrane (Bio-Rad, Hercules, CA, USA). After blocking with 5% non-fat dry milk, the membranes were incubated with the primary antibodies in PBS with Tween 20 (PBST) with 5% non-fat dry milk overnight at 4°C. Membranes were completely washed using PBST and incubated with horseradish peroxidase-conjugated secondary antibody (1:3,000, Bio-Rad, Hercules, CA, USA). The immunoblots were visualized with western enhanced chemiluminescence (ECL) substrate (Thermo Scientific, Waltham, MA, USA). Three independent experiments were performed for each analysis.
Mice xenograft experiment
A total of 60 nude mice (BALB/c, 5 weeks old) were randomly allocated to experimental groups. Mice could access food and water freely and were exposed to a natural light-dark cycle. Mice were injected subcutaneously with LoVo (1 × 106) cells, which were stably transfected with the control shRNA or YTHDF1 shRNA. When the tumor size reached 100 mm3, mice were randomly separated into four groups (n = 15): control shRNA plus normal saline, YTHDF1 silencing with normal saline, control shRNA plus cisplatin (15 mg/kg), or YTHDF1 silencing with cisplatin (15 mg/kg). Survival rates of mice were counted each day. Tumor volumes were calculated using the following formula: (shortest diameter)2 × (longest diameter) × 0.5. After treatments, tumors from dead or survival mice were dissected for downstream analysis. Animal experiments were approved by the Animal Ethics Committee from the Institutional Review Board of The First Affiliated Hospital of Zhejiang Chinese Medical University and were in accordance with the European Communities Council Directive of November 24, 1986 (86/609/EEC).
Determination of the CI
The evaluation of synergistic effects by GLS inhibitor and cisplatin was conducted using the algorithm described by Fransson et al.25 Synergy is determined by calculating a CI. A CI of <1, 1, or >1 indicates synergistic, additive, or antagonistic effects, respectively.
Statistical analysis
Statistical analysis was performed using Prism 6.0 (GraphPad, La Jolla, CA, USA). Data are presented as means ± standard deviation (SD). Differences between two samples were analyzed by a Student’s t test or unpaired two-sided t test. Comparisons of more than two groups were performed using one-way ANOVA. Experiments were performed in triplicate and repeated three times. p <0.05 was considered to be statistically significant.
Acknowledgments
The authors express their gratitude to all of the medical doctors and to the research scientists from the Department of Gastroenterology, The First Affiliated Hospital of Zhejiang Chinese Medical University for their support during the conduct of this study. This study was supported by the Traditional Chinese Medicine Science and Technology Key Program of Zhejiang Province of China (no. 2021ZZ013).
Author contributions
P.C. and X.H. designed the present study; all authors performed the experiments; X.-q.L., L.-y.G., and X.L. analyzed the data; P.C. and X.-q.L. prepared the manuscript; and X.H. reviewed the manuscript. All authors read and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omto.2021.01.001.
Contributor Information
Shuo Zhang, Email: zhangshuotcm@163.com.
Xuan Huang, Email: huangxuan1976@163.com.
Supplemental information
References
- 1.Dekker E., Tanis P.J., Vleugels J.L.A., Kasi P.M., Wallace M.B. Colorectal cancer. Lancet. 2019;394:1467–1480. doi: 10.1016/S0140-6736(19)32319-0. [DOI] [PubMed] [Google Scholar]
- 2.Dienstmann R., Salazar R., Tabernero J. Personalizing colon cancer adjuvant therapy: selecting optimal treatments for individual patients. J. Clin. Oncol. 2015;33:1787–1796. doi: 10.1200/JCO.2014.60.0213. [DOI] [PubMed] [Google Scholar]
- 3.Meyers B.M., Cosby R., Quereshy F., Jonker D. Adjuvant chemotherapy for stage II and III colon cancer following complete resection: a Cancer Care Ontario systematic review. Clin. Oncol. (R. Coll. Radiol.) 2017;29:459–465. doi: 10.1016/j.clon.2017.03.001. [DOI] [PubMed] [Google Scholar]
- 4.Dasari S., Tchounwou P.B. Cisplatin in cancer therapy: molecular mechanisms of action. Eur. J. Pharmacol. 2014;740:364–378. doi: 10.1016/j.ejphar.2014.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Galluzzi L., Senovilla L., Vitale I., Michels J., Martins I., Kepp O., Castedo M., Kroemer G. Molecular mechanisms of cisplatin resistance. Oncogene. 2012;31:1869–1883. doi: 10.1038/onc.2011.384. [DOI] [PubMed] [Google Scholar]
- 6.Cluntun A.A., Lukey M.J., Cerione R.A., Locasale J.W. Glutamine metabolism in cancer: understanding the heterogeneity. Trends Cancer. 2017;3:169–180. doi: 10.1016/j.trecan.2017.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang L., Venneti S., Nagrath D. Glutaminolysis: a hallmark of cancer metabolism. Annu. Rev. Biomed. Eng. 2017;19:163–194. doi: 10.1146/annurev-bioeng-071516-044546. [DOI] [PubMed] [Google Scholar]
- 8.Jolfaie N.R., Mirzaie S., Ghiasvand R., Askari G., Miraghajani M. The effect of glutamine intake on complications of colorectal and colon cancer treatment: a systematic review. J. Res. Med. Sci. 2015;20:910–918. doi: 10.4103/1735-1995.170634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ling H.H., Pan Y.P., Fan C.W., Tseng W.K., Huang J.S., Wu T.H., Chou W.C., Wang C.H., Yeh K.Y., Chang P.H. Clinical significance of serum glutamine level in patients with colorectal cancer. Nutrients. 2019;11:898. doi: 10.3390/nu11040898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang T., Cai B., Ding M., Su Z., Liu Y., Shen L. c-Myc overexpression promotes oral cancer cell proliferation and migration by enhancing glutaminase and glutamine synthetase activity. Am. J. Med. Sci. 2019;358:235–242. doi: 10.1016/j.amjms.2019.05.014. [DOI] [PubMed] [Google Scholar]
- 11.Sarkadi B., Meszaros K., Krencz I., Canu L., Krokker L., Zakarias S., Barna G., Sebestyen A., Papay J., Hujber Z. Glutaminases as a novel target for SDHB-associated pheochromocytomas/paragangliomas. Cancers (Basel) 2020;12:599. doi: 10.3390/cancers12030599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Singleton D.C., Dechaume A.L., Murray P.M., Katt W.P., Baguley B.C., Leung E.Y. Pyruvate anaplerosis is a mechanism of resistance to pharmacological glutaminase inhibition in triple-receptor negative breast cancer. BMC Cancer. 2020;20:470. doi: 10.1186/s12885-020-06885-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dai D., Wang H., Zhu L., Jin H., Wang X. N6-methyladenosine links RNA metabolism to cancer progression. Cell Death Dis. 2018;9:124. doi: 10.1038/s41419-017-0129-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.He L., Li H., Wu A., Peng Y., Shu G., Yin G. Functions of N6-methyladenosine and its role in cancer. Mol. Cancer. 2019;18:176. doi: 10.1186/s12943-019-1109-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hong K. Emerging function of N6-methyladenosine in cancer. Oncol. Lett. 2018;16:5519–5524. doi: 10.3892/ol.2018.9395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao W., Cui Y., Liu L., Ma X., Qi X., Wang Y., Liu Z., Ma S., Liu J., Wu J. METTL3 facilitates oral squamous cell carcinoma tumorigenesis by enhancing c-Myc stability via YTHDF1-mediated m6A modification. Mol. Ther. Nucleic Acids. 2020;20:1–12. doi: 10.1016/j.omtn.2020.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu S., Li G., Li Q., Zhang Q., Zhuo L., Chen X., Zhai B., Sui X., Chen K., Xie T. The roles and mechanisms of YTH domain-containing proteins in cancer development and progression. Am. J. Cancer Res. 2020;10:1068–1084. [PMC free article] [PubMed] [Google Scholar]
- 18.Liu T., Wei Q., Jin J., Luo Q., Liu Y., Yang Y., Cheng C., Li L., Pi J., Si Y. The m6A reader YTHDF1 promotes ovarian cancer progression via augmenting EIF3C translation. Nucleic Acids Res. 2020;48:3816–3831. doi: 10.1093/nar/gkaa048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McDermott L., Koes D., Mohammed S., Iyer P., Boby M., Balasubramanian V., Geedy M., Katt W., Cerione R. GAC inhibitors with a 4-hydroxypiperidine spacer: requirements for potency. Bioorg. Med. Chem. Lett. 2019;29:126632. doi: 10.1016/j.bmcl.2019.126632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bai Y., Yang C., Wu R., Huang L., Song S., Li W., Yan P., Lin C., Li D., Zhang Y. YTHDF1 regulates tumorigenicity and cancer stem cell-like activity in human colorectal carcinoma. Front. Oncol. 2019;9:332. doi: 10.3389/fonc.2019.00332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nishizawa Y., Konno M., Asai A., Koseki J., Kawamoto K., Miyoshi N., Takahashi H., Nishida N., Haraguchi N., Sakai D. Oncogene c-Myc promotes epitranscriptome m6A reader YTHDF1 expression in colorectal cancer. Oncotarget. 2017;9:7476–7486. doi: 10.18632/oncotarget.23554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu T., Li C., Jin L., Li C., Wang L. The prognostic value of m6A RNA methylation regulators in colon adenocarcinoma. Med. Sci. Monit. 2019;25:9435–9445. doi: 10.12659/MSM.920381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shi Y., Fan S., Wu M., Zuo Z., Li X., Jiang L., Shen Q., Xu P., Zeng L., Zhou Y. YTHDF1 links hypoxia adaptation and non-small cell lung cancer progression. Nat. Commun. 2019;10:4892. doi: 10.1038/s41467-019-12801-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Y.Q., Wang H.L., Xu J., Tan J., Fu L.N., Wang J.L., Zou T.H., Sun D.F., Gao Q.Y., Chen Y.X., Fang J.Y. Sirtuin5 contributes to colorectal carcinogenesis by enhancing glutaminolysis in a deglutarylation-dependent manner. Nat. Commun. 2018;9:545. doi: 10.1038/s41467-018-02951-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fransson Å., Glaessgen D., Alfredsson J., Wiman K.G., Bajalica-Lagercrantz S., Mohell N. Strong synergy with APR-246 and DNA-damaging drugs in primary cancer cells from patients with TP53 mutant high-grade serous ovarian cancer. J. Ovarian Res. 2016;9:27. doi: 10.1186/s13048-016-0239-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.