

Abstract
Inhibitors of the proteasome have been extensively studied for their applications in the treatment of human diseases such as hematologic malignancies, autoimmune disorders, and viral infections. Many of the proteasome inhibitors reported in the literature target the non-primed site of proteasome’s substrate binding pocket. In this study, we designed, synthesized and characterized a series of novel α-keto phenylamide derivatives aimed at both the primed and non-primed sites of the proteasome. In these derivatives, different substituted phenyl groups at the head group targeting the primed site were incorporated in order to investigate their structure-activity relationship and optimize the potency of α-keto phenylamides. In addition, the biological effects of modifications at the cap moiety, P1, P2 and P3 side chain positions were explored. Many derivatives displayed highly potent biological activities in proteasome inhibition and anticancer activity against a panel of six cancer cell lines, which were further rationalized by molecular modeling analyses. Furthermore, a representative α-ketoamide derivative was tested and found to be active in inhibiting the cellular infection of SARS-CoV-2 which causes the COVID-19 pandemic. These results demonstrate that this new class of α-ketoamide derivatives are potent anticancer agents and provide experimental evidence of the anti-SARS-CoV-2 effect by one of them, thus suggesting a possible new lead to develop antiviral therapeutics for COVID-19.
Keywords: α-ketoamides, Cancer, SARS-CoV-2, COVID-19, Proteasome, Drug discovery
Graphical abstract

1. Introduction
The ubiquitin−proteasome system (UPS) plays essential roles in the turnover of numerous cellular proteins involved in cell cycle, signal transduction, antigen processing, protein quality control, etc [1]. The proteasome, an ATP dependent multi-subunit complex, is a central mediator of proteolysis in UPS. Diverse hybrid forms of proteasome have been identified. In general, the hybrid proteasome is composed of two parts: 20S core particle (CP) and regulatory particle (RP) including PA700 (or the 19S cap), PA28 (also known as 11S), and Blm10/PA200 protein. One 20S core particle binds with one or two regulatory particle to form singly (RP1–CP) or doubly (RP2–CP) hybrid proteasome, respectively [2,3].
26S proteasome is the most common form of proteasome, consisting of 20S core particle capped with two 19S regulatory particles [4]. The 19S regulatory particle is responsible for the ubiquitylated substrate recognition, deubiquitination, unfolding, and translocation of the unfolded chain into the core particle. The catalytic domain resides in the 20S core particle that is associated with the degradation of damaged proteins and cell signaling proteins. The 20S core particle consists of four stacked rings, with two outer rings composed of seven α subunits and two inner rings composed of seven β subunits [5,6]. The catalytic subunits β5, β2 and β1 reside in the inner β ring that exhibits chymotrypsin-like (CT-L), trypsin-like (T-L) and caspase-like (C-L) proteolytic activity, respectively [7]. In vertebrates, three isoforms of the 20S CP have been identified, including constitutive proteasome (cCP) which harbors β1c, β2c and β5c, immunoproteasome (iCP) which harbors β1i (LMP2), β2i (MECL-1) and β5i (LMP7), thymoproteasome (tCP) which harbors β1i, β2i and β5t [8,9]. The immunoproteasome is involved in antigen processing that influences cytokine production and T cell function [10]. Overexpression of immuno- and constitutive proteasome is observed in hematologic malignancies and autoimmune diseases. Different classes of proteasome inhibitors have been developed for clinical intervention of hematologic malignancies and autoimmune disorders [[11], [12], [13]].
Although many proteasome inhibitors including those clinically approved drugs such as bortezomib [14,15], carfilzomib [16], and ixazomib [17] have been developed that mostly target the non-primed substrate binding site of proteasome (Fig. 1 A), few proteasome inhibitors targeting both non-primed and primed sites or only the primed site of the substrate binding pocket have been reported. Among them, dipeptide α,β epoxyketone UK-101 (Fig. 1B) showing good selectivity for β1i subunit binds both non-primed and primed sites [18]. β-Lactone homobelactosin C primarily binds to the primed site exhibiting high potency to β5 subunit [19,20]. A recent study has shown that epoxyketone-based compounds targeting P1′ site have the efficacy against cell lines of de novo or acquired resistance to carfilzomib [21]. Thus, discovery of new compounds capable of binding the primed site remains an area to be explored further.
Fig. 1.

Schematic illustration of (A) Bortezomib recognizing only the non-primed site and ketoamide derivatives BSc2189 capable of binding to both primed and non-primed sites of the proteasome. (B) Chemical structures of ONX-0914, MG132 and UK101.
It has been reported that a ketoamide derivative BSc2189 (Fig. 1A) binds to the active site in the 20S proteasome in a reversible manner [[22], [23], [24]]. Its phenyl group at the head group occupies the primed substrate binding channel of 20S proteasome (Fig. 1A) [24]. Previous studies have utilized the primed site to improve the specificity and potency of protease inhibitors [[25], [26], [27]]. In this study, we incorporated different substitutions into α-keto phenylamide’s head group to examine their effects on the proteasome inhibitory potency. Additional modifications were also made on the cap moiety and the P1, P2 and P3 side chain positions of α-keto phenylamide to investigate the influence of these positions on bioactivity. These modification studies should yield insight into the structure-activity relationship (SAR) of α-ketoamide-based proteasome inhibitors which has not been reported before.
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a novel betacoronavirus which causes the ongoing Coronavirus Disease 2019 (COVID-19) pandemic [28]. Currently, antiviral treatment options for COVID-19 are limited. Thus, the development of effective drugs is an urgent challenge. Previous studies have shown that proteasome inhibitors can inhibit coronavirus replication by interfering with the early steps of the viral replication cycle [29,30]. For example, the proteasome inhibitor MG132 (Fig. 1B) and its analogs have been reported to have inhibitory activity against severe acute respiratory syndrome coronavirus (SARS-CoV) [31,32]. Mice infected with mouse hepatitis virus 1 and treated with MG132 had higher survival rate, less severe lung histopathological changes, and reduced lung virus replication, lung STAT phosphorylation, and expression of pneumoinflammatory cytokines. The anti-coronaviral effect of MG132 was suggested to be due to its inhibition of viral replication-dependent cysteine protease m-calpain [31]. Because of the high homology between SARS-CoV-2 and SARS-CoV [33,34], we hypothesized that proteasome inhibitors capable of blocking SARS-CoV may also be effective against SARS-CoV-2. To test this hypothesis, we examined the anti-SARS-CoV-2 activity of one of the α-ketoamide derivatives reported here, chosen based on its low toxicity compared with others in Vero E6 cells which was used for SARS-CoV-2 infection assay. This representative analog showed potent inhibition of SARS-CoV-2 infection, thus demonstrating experimentally that this and potentially some of the other α-ketoamide derivatives reported here may have the potential for the development of anti-SARS-CoV-2 agents.
2. Results
2.1. Synthesis of α-ketoamide derivatives
The general synthetic route is shown in Scheme 1, Scheme 2 . In general, amino acid methyl ester 1 was coupled with Boc protected amino acid to afford 2a-b or Cbz protected amino acid to afford 9a-c using HATU as the coupling reagent. The Boc group of 2a-b was removed using trifluoroacetic acid (TFA) and the resulting compound then coupled with commercially available carboxylic acid derivatives except for carboxylic acid 16 which was prepared in our lab (Scheme 2), affording compounds 4a-b. Compounds 4a-b or 9a-c were hydrolyzed to give 5a-b or 10a-c. Acid 5a-b with different cap moieties or acid 10a-c with Cbz cap moiety were coupled with L-leucinol in the presence of isobutyl chloroformate and N-methylmorpholine to yield intermediate 6a or 11a-c. 6a or 11a was oxidized into 7a or 12a using 2-iodoxybenzoic acid. Aldehyde 7a or 12a reacted with appropriate isonitrile by Passerini reaction method to generate 8a or 13a, which was subsequently oxidized to the desired target molecule with 2-iodoxybenzoic acid.
Scheme 1.

General synthetic procedure for α-ketoamide derivatives.
Reagents and conditions: (i) Boc-AA-OH or Cbz-AA-OH, HATU, HOBt, DIEA, THF, 0 °C to room temperature, 1 h; (ii) TFA, CH2Cl2, 0 °C, 3 h; (iii) carboxylic acid derivatives, HATU, HOBt, DIEA, THF, 0 °C to room temperature, 1 h; (iv) LiOH.H2O, THF, H2O, room temperature, 3 h; (v) L-Leucinol, isobutyl chloroformate, N-methylmorpholine, THF, −5 °C; (vi) 2-iodoxybenzoic acid, DMSO, room temperature, 4 h; (vii) pyridine, TFA, dry CH2Cl2, -15 °C–0 °C, 3 h; (viii) 2-iodoxybenzoic acid, DMSO, room temperature, 4–5 h; (ix) HCOOH, reflux, 3 h; (x) POCl3, DIEA, dry CH2Cl2, 0 °C to room temperature, 3 h.
Scheme 2.

General synthetic procedure for 5-(4-acetylphenyl)furan-2-carboxylic acid.
Reagents and conditions: (i) Pd(OAc)2, KOAc, DMAc, 130 °C, 4 h; (ii) LiOH.H2O, THF, H2O, room temperature, 3 h.
The isonitrile derivatives were prepared according to a slightly modified procedure reported in the literature [23]. Commercially available aniline derivatives reacted with HCOOH to yield N-formylated aniline derivatives which were converted into isonitrile derivatives using POCl3 and DIEA in dry dichloromethane, exemplified by synthesis of 14 (Scheme 1C).
The 5-aryl 2-furan carboxylic acid 16 was prepared according to the reported procedure [35], as illustrated in Scheme 2. Recently, an optimized reaction condition to prepare 2-arylated furan derivatives was reported [36].
2.2. Inhibition of β5 catalytic subunit in a cell-based chymotrypsin-like activity assay
All compounds were evaluated with a cell-based proteasome inhibition assay (Promega). Because the β5 catalytic subunit is the primary target of these proteasome inhibitors [4], all α-keto phenylamide derivatives were screened for their inhibition of chymotrypsin-like activity. The biological activity of these derivatives is presented and discussed below in different groups according to the structural modifications made at the N-cap position, P1, P2 and P3 positions, and the head group.
2.2.1. Modifications at the N-cap position
Initially, we designed 18 and a hybrid compound 17 which contains the peptide backbone of a β5i-selective inhibitor ONX-0914 [11] and the head group of BSc2189 [24]. 17 and 18 exhibited decreased chymotrypsin-like activity compared with ONX-0914 and BSc2189. The subsequent modifications of 18 focused on the N-cap position of the compound structure shown in Formula 1. A variety of cap groups were attempted (Table 1 ), including heterocycle (18), unbranched aliphatic alkyl groups (20), branched (21), cycloalkyl (22), and aromatic rings (compound 23, 24, 25, 26). The results indicated that compounds with substituted aromatic rings at the cap position exhibited more potent activity. For example, compounds 24, 25, 26 exhibited better potency against chymotrypsin-like activity than 21 and 22. 20 with a decanoyl group and 18 with a morpholinoacetyl group at the cap position had modest activity. Of note, 5-(4-acetylphenyl)-furan-2-carbonyl restored the activity of target compounds. Compound 26 retained similar bioactivity to ONX-0914 in the cell-based CT-L inhibition assay.
Table 1.
The cell-based CT-L inhibitory activity of cap moiety modified compounds.
| Compound | Cap moiety | CT-La, IC50 (nM) | Compound | Cap moiety | CT-La, IC50 (nM) |
|---|---|---|---|---|---|
| 18 (NI-04104) | 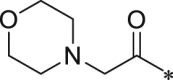 |
418.9 ± 82.0 | 24 (NI-04146) | 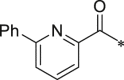 |
311.0 ± 19.7 |
| 19 (NI-01099) | 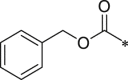 |
1026 ± 235.4 | 25 (NI-01114) | 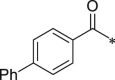 |
133.7 ± 26.9 |
| 20 (NI-04127) | 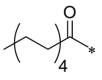 |
233.6 ± 17.4 | 26 (NI-02133) | 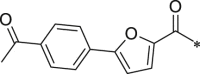 |
50.7 ± 4.5 |
| 21 (NI-04154) | 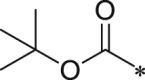 |
1753 ± 392.1 | ONX-0914 | 58.5 ± 4.3 | |
| 22 (NI-07014) | 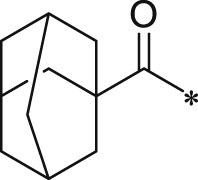 |
1356 ± 252.0 | BSc2189 | 34.5 ± 1.7 | |
| 23 (NI-02114) | 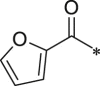 |
716.5 ± 139.2 |
The compounds were assayed in triplicate with the averaged IC50 ± SEM values shown.
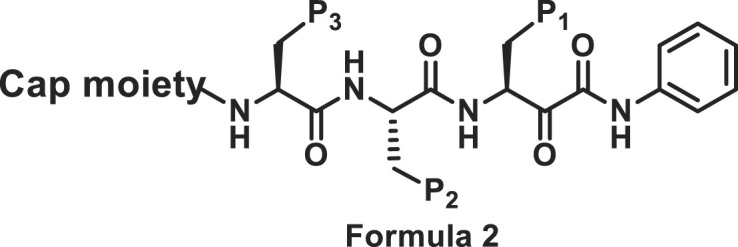
2.2.2. Modifications at P1, P2 and P3 positions
The sidechains on P1, P2 and P3 positions shown in Formula 2 were modified. Compound 17 with Phe at P1 position was slightly more potent towards chymotrypsin-like subunit than compound 18 with Leu at P1 position (Table 2 ).
Table 2.
The cell-based CT-L inhibitory activity of P1, P2 and P3 modified compounds.
| Compound | Cap moiety | P1 | P2 | P3 | CT-La IC50 (nM) |
|---|---|---|---|---|---|
| 17 (NI-01069) |  |
Ph | (4-CH3O)-Ph | H | 364.6 ± 42.2 |
| 18 (NI-04104) |  |
i-Pr | (4-CH3O)-Ph | H | 418.9 ± 82.0 |
| 19 (NI-01099) | 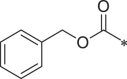 |
i-Pr | (4-CH3O)-Ph | H | 1026 ± 235.4 |
| 27 (NI-07028) | 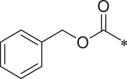 |
i-Pr | 1H-indol-3-yl | H | 155.7 ± 25.9 |
| 28 (NI-06032) | 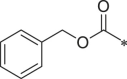 |
i-Pr | Ph | H | 2008 ± 219.7 |
| 29 (NI-07021) | 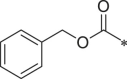 |
i-Pr | 1H-indol-3-yl | i-Pr | 66.5 ± 11.0 |
| 30 (NI-04162) | 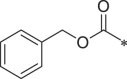 |
i-Pr | Ph | i-Pr | 230.8 ± 14.5 |
| 31 (NI-18088) | 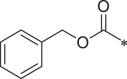 |
i-Pr | Ph | Cyclohexyl | 603.3 ± 26.7 |
| 32 (NI-18143) |  |
i-Pr | Ph | CH3O | 243.4 ± 32.8 |
| BSc2189 |  |
i-Pr | i-Pr | i-Pr | 34.5 ± 1.7 |
| ONX-0914 | 58.5 ± 4.3 |
The compounds were assayed in triplicate with the averaged IC50 ± SEM values shown.
Starting from a tripeptide scaffold with Cbz at the cap moiety, Ala at P3 and Leu at P1, different aromatic side chains were incorporated at P2 position. The results show that compound 27 with Trp at P2 is favorable to the biological activity. Compound 19 containing P2-Tyr(OMe) and compound 28 containing P2-Phe displayed decreased inhibitory potency. Starting from a tripeptide scaffold with Cbz in cap moiety, Leu at P3 and Leu at P1, different side chains were incorporated at P2 position. The order of the inhibitory potency of these compounds is: BSc2189 > 29 > 30. The results indicate that Leu at P2 position is preferred over Trp, Tyr(OMe) or Phe for β5 inhibitory potency.
In addition, the potency of proteasome inhibition of compounds bearing Ala or Leu at P3 position was determined. Compound with Leu at P3 (29, 30) showed increased activity compared to Ala at P3 (27, 28). These results indicated that P3 position may favor bulky side chains. Thus, a larger P3 side chain, cyclohexyl (31) was introduced at this site. Compound 31 showed 3-fold increase in potency relative to analog 28 with Ala at P3 site but ∼3-fold reduced activity compared with 30 with Leu at P3. Replacing Leu of 30 with Ser (OMe) (32) at P3 site retained the potency.
2.2.3. Modifications at the head group
The phenylamide group of BSc2189 could form π-alkyl interaction with Ser129 in β5’s primed site. In previous work by others, different electron donating groups such as methyl and electron withdrawing groups such as cyano were incorporated into the para position of phenylamide group [24], which found that compound BSc4999 with 2, 4-dimethyl phenylamide was about 2 fold more potent than BSc2189 with an unsubstituted phenylamide whereas most of other compounds showed decreased activity towards chymotrypsin-like subunit compared with BSc2189.
Here, we focused on using primed site to improve the potency. Phenyl group at the head position was deemed to be a suitable starting point to introduce various modifications. Various groups at the ortho and meta positions of phenylamide group were introduced based on different tripeptide scaffolds (Table 3 ).
Table 3.
The cell-based CT-L inhibitory activity of head group modified compounds.
| Compound | Cap moiety | P2 | P3 | Head group | CT-La IC50 (nM) |
|---|---|---|---|---|---|
| 33 (NI-01106) |  |
(4-CH3O)-Ph | H |  |
385.6 ± 36.9 |
| 34 (NI-04192) | 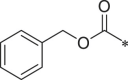 |
Ph | i-Pr | 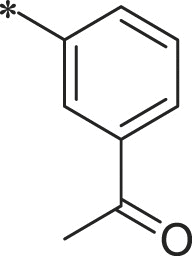 |
61.0 ± 3.1 |
| 35 (NI-09016) | 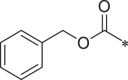 |
1H-indol-3-yl | i-Pr | 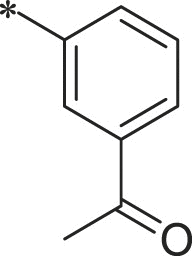 |
29.5 ± 3.1 |
| 36 (NI-18030) |  |
1H-indol-3-yl | i-Pr | 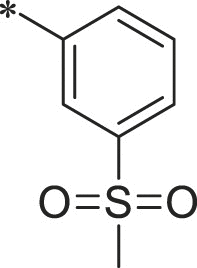 |
60.1 ± 10.0 |
| 37 (NI-09062) | 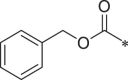 |
1H-indol-3-yl | i-Pr | 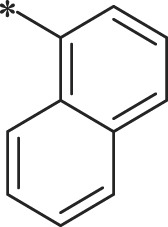 |
165.1 ± 7.6 |
| 38 (NI-23067) | 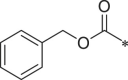 |
i-Pr | i-Pr | 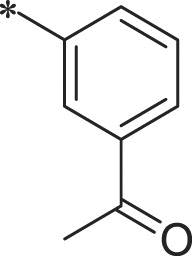 |
22.1 ± 1.2 |
| 39 (NI-18019) | 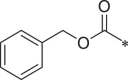 |
i-Pr | i-Pr | 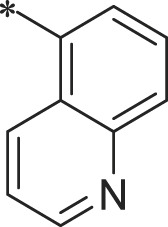 |
99.7 ± 24.7 |
| 40 (NI-18020) | 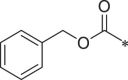 |
i-Pr | i-Pr | 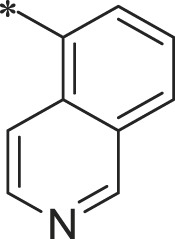 |
64.1 ± 16.6 |
| 41 (NI-09073) | 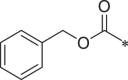 |
i-Pr | i-Pr | 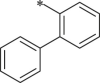 |
4701 ± 961 |
| 42 (NI-18073) | 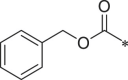 |
i-Pr | i-Pr | 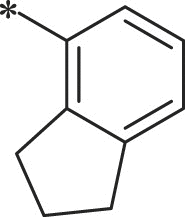 |
41.7 ± 2.5 |
| 43 (NI-18060) |  |
i-Pr | i-Pr | 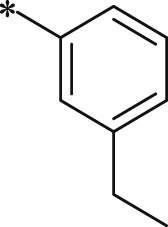 |
34.8 ± 2.6 |
| 44 (NI-09083) |  |
i-Pr | i-Pr | 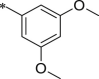 |
25.7 ± 3.1 |
| 45 (NI-18050) | 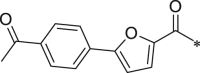 |
i-Pr | i-Pr | 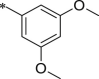 |
31.7 ± 4.9 |
| 46 (NI-15020) | 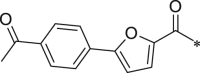 |
i-Pr | i-Pr | 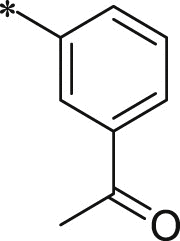 |
25.3 ± 3.8 |
| 47 (NI-18145) | 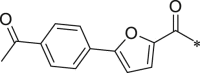 |
i-Pr | i-Pr | 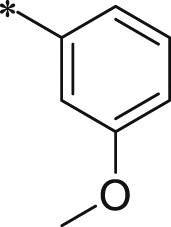 |
28.8 ± 3.4 |
| BSc2189 | 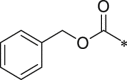 |
i-Pr | i-Pr | 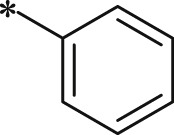 |
34.5 ± 1.7 |
| ONX-0914 | 58.5 ± 4.3 |

The compounds were assayed in triplicate with the averaged IC50 ± SEM values shown.
With 3-acetylphenyl substituent into the backbone of Cbz-Leu-Phe-Leu, Cbz-Leu-Trp-Leu and Cbz-Leu-Leu-Leu, compounds 34, 35 and 38 showed 1.5–4 fold increase in inhibitory activity than compounds 30, 29 and BSc2189 with unsubstituted phenyl. In addition, the data indicated that the effect of the head group modification on activity was also dependent of peptide backbone’s interaction with the non-primed site when the substituent group at the head position was the same. This was also demonstrated by 33 and 37. Compound 33 with 1-naphthyl at the head group showed ∼3 fold increase in inhibitory potency over compound 19 of unsubstituted phenyl whereas the same head group of 1-naphthyl in 37 resulted in 2 fold decrease in inhibitory potency over 29 of unsubstituted phenyl. Previous study showed that the compound with 1-naphthyl at the head group exhibited low nanomolar potency against the chymotryptic activity [37]. Incorporation of 3-methylsulfonylphenyl (36), 3,5-dimethoxylphenyl (44, 45) at the head group resulted in slightly improved activity compared with the corresponding unsubstituted phenyl compounds.
In order to enhance the interaction with hydrophobic residue Tyr168 of β5, a phenyl was introduced in the ortho position of the phenyl ring at the head group of the compound. However, this resulted in up to 150-fold reduced inhibitory activity in compound 41 possibly caused by the phenyl ring introduced in the ortho position being too bulky and giving rise to steric hindrance to the binding. The tripeptide with quinolinyl or isoquinolinyl at the head group showed less potent activity compared to BSc2189 which was synthesized as a control and tested together with other new compounds in the assays.
The results above seemed to suggest that the head group position is tolerant of introducing modification groups of suitable sizes and independent of the electron density on the phenyl ring. Thus, electron donating groups like 2, 3-dihydro-1H-indenyl (42) and 3-ethylphenyl (43) were introduced at the head position. The results showed that 42 and 43 had activity similar to BSc2189 with the IC50 values of 41.7 nM and 34.8 nM, respectively (Table 3).
The SAR results shown above suggested that a substituted aromatic ring at the cap moiety, Leu at the P1 position, Trp or Leu at the P2 position, Leu at the P3 position, and 3-acetylphenyl or 3,5-dimethoxyphenyl at the head group are favored for β5/CT-L inhibitory activity. Based on this pharmacophore, we further designed two hybrid molecules, 45 and 46 with all these features (Table 3). Both of them displayed potent inhibition of β5. In addition, the 3-methoxyphenyl at the head position (47) showed strong activity with an IC50 value of 28.8 nM against β5.
2.3. Inhibition of β1 and β2 in cell-based caspase-like and trypsin-like activity assays
In addition to β5, the activity of representative compounds, 35, 44 and 46 and control compound BSc2189 was determined against β1 and β2 in cell-based caspase-like and trypsin-like activity assay (Fig. 2 ). All of these compounds were more active against the β2 subunit than the β1 subunit. 44 and 46 had a low nanomolar IC50 for the trypsin-like subunit (β2). Recently others reported that dual inhibition of β2 and β5 activity could achieve maximum proteasome inhibition and was most effective in proteasome inhibitor-resistant multiple myeloma [38]. In light of this, 44 and 46 displaying activity in both β2 and β5 may merit further studies.
Fig. 2.

The cell-based caspase-like (β1, shown on the upper panel) and trypsin-like (β2, lower panel) inhibitory activity of compounds BSc2189, 35, 44 and 46.
2.4. Anticancer activity in a panel of six cancer cell lines
Based on the results of cell-based proteasome inhibition assays shown above in Table 1, Table 2, Table 3, a group of 7 compounds were selected for their more potent proteasome inhibitory activities than others and for representing modifications at different sites (the head group and cap moiety). These compounds were evaluated for anticancer activity in various cancer cell lines including RPMI8226, THP-1, Ramos (RA.1), SW480, HeLa, and MGC-803 using the MTS cytotoxicity assay (Table 4 ). We also determined the IC50 values of those compounds that exhibited ≥50% inhibition of RPMI8226 at 1 μM (Supplemental Table S1). The results indicated good correlation between proteasome inhibition potency and anticancer activity.
Table 4.
Cytotoxicity of compounds against tumor cell lines (IC50 ± SEM, nM)a.
| Compound | RPMI8226 | THP-1 | Ramos (RA 1) | SW480 | Hela | MGC-803 |
|---|---|---|---|---|---|---|
| 25 | 455.0 ± 42.1 | 567.9 ± 49.48 | 464.0 ± 9.53 | 2441 ± 123.9 | 8701 ± 1195 | 2017 ± 123.4 |
| 26 | 474.8 ± 53.3 | 475.4 ± 53.17 | 428.1 ± 7.28 | 2002 ± 213.2 | 3202 ± 47.62 | 1164 ± 4.74 |
| 35 | 90.99 ± 11.1 | 78.12 ± 1.09 | 38.35 ± 2.46 | 94.25 ± 4.77 | 448.9 ± 5.89 | 88.80 ± 5.01 |
| 43 | 40.41 ± 3.23 | 71.12 ± 2.02 | 45.73 ± 3.46 | 82.86 ± 4.67 | 211.8 ± 4.29 | 78.51 ± 7.46 |
| 44 | 37.57 ± 2.46 | 70.67 ± 6.11 | 26.92 ± 0.40 | 72.55 ± 7.54 | 120.0 ± 8.80 | 68.54 ± 7.04 |
| 46 | 38.11 ± 1.19 | 94.41 ± 1.45 | 26.13 ± 0.18 | 79.26 ± 1.87 | 260.4 ± 4.34 | 79.05 ± 12.59 |
| 47 | 43.4 ± 1.04 | 97.86 ± 2.79 | 38.33 ± 3.04 | 74.50 ± 3.13 | 238.93 ± 3.28 | 85.40 ± 6.73 |
| BSc2198 | 67.76 ± 5.79 | 88.07 ± 3.85 | 53.6 ± 2.21 | 106.3 ± 14.93 | 288.3 ± 25.22 | 118.8 ± 3.63 |
The IC50 was calculated with assay data measured at 7 concentrations while each value was determined from three independent experiments.
All tested compounds were highly effective on hematologic tumor cell lines RPMI8226, Ramos and THP-1. The IC50 of control compound BSc2189 was 288 nM in HeLa cells which was in consistent with its previously published value of 254 nM [23]. 46 was the most potent compound in both cell-based proteasome inhibition and cancer cell viability assays.
Compounds 25 and 26 were more effective on hematologic tumor cell lines RPMI8226, Ramos and THP-1 than solid tumor cell lines SW480, HeLa and MGC-803. On the other hand, 25 (THP-1 IC50: 567.9 nM, HeLa IC50: 8701 nM) and 26 (THP-1 IC50: 475.4 nM, HeLa IC50: 3202 nM) showed less cytotoxicity than ONX-0914 (THP-1 IC50: 202.4 nM, HeLa IC50: 590.5 nM) which may be related with their higher β5i selectivity [39,40].
2.5. Antiviral activity against SARS-CoV-2
After determining the potent anticancer activity of our new α-ketoamide derivatives, in response to the most recent COVID-19 global pandemic caused by SARS-CoV-2, we decided to test the hypothesis as described above that some of these compounds may possess anti-SARS-CoV-2 activity. We first tested the cytotoxicity of several representative derivatives in Vero E6 cells (Supplemental Table S2). Compound 17 was found to be the least toxic and subsequently tested in Caco-2 cells also. We selected compound 17 for antiviral study using a plaque reduction assay to determine the 50% antiviral effective dose (EC50) of the compound. In the plaque reduction assay, Vero E6 cells were seeded at 4 × 105 cells/well in 12-well tissue culture plates on the day before carrying out the assay. After 24 h of incubation, 50 plaque-forming units (PFU) of SARS-CoV-2 were added to the cell monolayer with or without the addition of the compound. The percentage of plaque inhibition relative to the control (i.e. without the addition of compound) wells were determined for each compound concentration. Remdesivir, a drug used clinically for treating COVID-19 was used as a positive control. Compound 17 displayed potent anti-SARS-CoV-2 activity in this plaque reduction assay with the EC50 of 1.28 μM (Fig. 3 ).
Fig. 3.

Anti-SARS-CoV-2 activity of compound 17in vitro. Upper panel: photos of plaque reduction assay using SARS-CoV-2-infected Vero E6 cells. Lower panel: compound 17 exhibits potent antiviral activity against SARS-CoV-2 with an EC50 of 1.28 μM as determined by plaque reduction assay, with Remdesivir as a positive control. The results are presented as means ± standard deviations.
3. Comparative molecular field analysis (CoMFA)
3.1. Data set for analysis
The SAR of the synthesized compounds was further studied by CoMFA using a selected set of 25 compounds with diverse chemical structures and distribution of bioactivities. The data set was divided into a training set (20 compounds) for model development and a test set (5 compounds) for model validation. Selection of the test set compounds was made based on structural and biological diversity. These compounds are shown in Supplemental Table S3. The calculation method is described in the method section.
3.2. Results of the CoMFA
The CoMFA model has good cross-validated correlation with a q2 value of 0.537 with 4 optimum components. The correlation coefficient r2 is 0.959, and the predicted correlation coefficients r2 pred is 0.814, which reveals that the CoMFA model has high predictive capability. The plots of predicted versus actual activity for all the data set are shown in Supplemental Figure S1. As to the steric and electrostatic field contribution, the former accounts for 0.714 while the latter 0.286, indicating that the steric factor contributes more to the binding affinities.
3.3. CoMFA contour maps analysis
The CoMFA contour map is shown in Fig. 4 . The cap position is highlighted in green color, which indicates that bulky substituents in this area may improve the activity. The bioactivity of 5-(4-acetylphenyl) furan containing 26 is much higher than furan containing 23. The yellow area at the P2 position indicates that the activity can be improved by decreasing the size of the methoxyl group of the phenyl core. The green area around the isopropyl group shows that it plays an important role in the contribution to bioactivities. The isopropyl group at P2 position has higher bioactivity than an aromatic group such as a phenyl or indole ring. The blue area at the warhead region suggests that a positive charge on the phenyl core may improve the activity.
Fig. 4.

The CoMFA contour map of the training set.
4. Molecular docking and dynamics simulations
4.1. Docking of compounds to 20S proteasome
Molecular docking simulation was performed to predict and visualize the possible binding modes of compound 46 to 20S proteasome. To evaluate the stability of receptor-ligand complexes, the docking pose closest to the experimentally determined binding mode of control BSc2189 (PDB: 4NO8) [23] were used to carry out molecular dynamics (MD) simulation to generate stable conformations. In the proteasome binding mode of compound 46 (Fig. 5 ), the amide group of the peptide backbone of the compound formed hydrogen bonds with proteasome′s residues Gly47, Ala49, and Thr21. Additional hydrogen bonds were observed between the acetyl group at the cap position of the compound and hydroxyl group of proteasome′s residue Tyr305. The furan group at the cap position and phenyl group at the head position of the compound formed H-π bond with proteasome’s residues Pro326 and Ser130. These results suggest that the interactions of the compound′s cap and head groups with proteasome may also contribute to high binding affinity of the compound towards its target.
Fig. 5.

The predicted complex structure of compound 46 (salmon stick) bound to human 20S proteasome (PDB ID: 5LEY) [41] generated from molecular dynamics simulation. Hydrogen bonds and H-π bonds are shown by red and green dashed lines.
4.2. Docking of compounds to m-calpain and SARS-CoV-2 main protease
To examine the possible mechanism for the observed anti-SARS-CoV-2 effect of compound 17, we performed molecular docking calculation of this compound with m-calpain because previous studies by others showed that a proteasome inhibitor MG132 blocked SARS-CoV virus replication by targeting m-calpain [31]. The binding modes of proteasome inhibitors MG132 and 17 as predicted by our docking calculations were shown in Fig. 6 A and B. Both the warhead α-ketoamide group of 17 and the warhead aldehyde group of MG132 formed a covalent bond with Cys105 of m-calpain while the backbone’s amide of both compounds formed hydrogen bond with Gly261 of m-calpain. The N-terminal morpholine cap moiety of 17 had weak van der Waals interaction with the protein while the N-terminal benzyl ring cap moiety of MG132 had stronger hydrophobic interaction with Trp214. The backbone’s carbonyl group of 17 formed hydrogen bond with Gly198 while the backbone’s carbonyl group of MG132 formed hydrogen bond with Trp106. Moreover, the methoxyl group at P2 position of 17 formed extra hydrogen bond with Arg337 of m-calpain. The carbonyl oxygen and amide at the warhead of 17 formed hydrogen bond with Gln99 and Gly261, respectively. The phenyl group at the warhead of 17 extended into the primed site of m-calpain. Although the reactive groups of BSc2189 and MG132 were not the same, the overall binding modes of these two compounds were basically consistent within the binding site of the receptor (Fig. 6C).
Fig. 6.

Predicted m-calpain (PDB ID: 3BOW) [44] bound structures of MG132 (A) and 17 (B); (C) Superposition of the structures of MG132 (green stick) and 17 (cyan stick) bound to m-calpain (wheat cartoon). (D) Predicted structure of 17 (cyan stick) bound to SARS-CoV-2 main protease (PDB ID: 6Y2F) [43].
Recent studies show that α-ketoamide derivatives such as boceprevir, narlaprevir [42], and 13b [43] act as potent SARS-CoV-2 main protease inhibitors. Therefore, we also examined the potential docking of compound 17 to the active site of SARS-CoV-2 main protease (Fig. 6D). Docking models indicated that α-keto oxygen and the amide oxygen of α-ketoamide head group formed two hydrogen bonds with His41 and Gly143 on SARS-CoV-2 main protease, which might suggest a possible advantage of α-ketoamide compared with other head group such as aldehyde and trans-α, β-unsaturated ethyl ester. The compound′s benzyl group at P1 position occupied S1 pocket formed by Glu166 and Phe140 on the main protease. The amide group of the compound formed a hydrogen bond with His41. The side chain of Tyr(OMe) at the P2 position of the compound didn′t properly insert into the hydrophobic S2 pocket but extended into another more flexible site, forming a hydrogen bond with Gln192. The N-terminal morpholine cap moiety of 17 did not occupy the small hydrophobic S4 pocket and showed a kinked conformation.
5. Discussion
Many previous studies have demonstrated that targeting the primed site can be an effective strategy for developing potent or selective inhibitors of proteases of viruses such as human immunodeficiency virus and hepatitis C virus [[25], [26], [27]]. Recently, several groups have reported α-keto phenylamide and epoxyketone based proteasome inhibitors targeting the primed site to increase potency or overcome resistance [21,45]. It was shown that the targeting of the primed site by 3-phenoxy phenyl could increase β5 inhibition of a compound. Here we studied the impact on bioactivity of different groups at the head position that interact with the primed site (Fig. 1A). Electron withdrawing groups, acetyl (34, 35, 38) and methylsulfonyl (36) incorporated into the meta-position of the phenyl ring at the head position led to 1.5–4 fold increase in inhibitory activity. Incorporation of an electron donating group methoxyl (44, 45, 47) also led to increase in inhibitory activity while doing so with an ethyl or cyclopentyl moiety resulted in similar inhibitory activity. These results indicate that the activity of ketoamide derivatives may not depend on the strength of the electron donating substituents at the head position. We reason that substituents targeting the primed site may affect the reaction rate between α-keto moiety and catalytic residue Thr1 or the lowest energy conformation of the inhibitory compound important for its potency. How the substituents targeting the primed site residues affect the potency and selectivity of the inhibitor needs to be further clarified.
Our data also show that compounds with the same moiety at the head position but attached to different peptide sequences display varying fold increase in inhibitory activity compared with compounds with an unsubstituted moiety at the head position. For example, 3-acetyl group at the head position gives rise to ∼4 fold increase in inhibitory activity of the compound with a peptide backbone Cbz-Leu-Phe-Leu, 2 fold increase for a peptide backbone Cbz-Leu-Trp-Leu, and 1.5 fold increase for a peptide backbone Cbz-Leu-Leu-Leu. Taken together, these results and those published previously by others demonstrate that the primed site, a region on the proteasome previously less explored compared to the non-primed site, modulates the activity of proteasome inhibitors and that incorporation of suitable substituent groups targeted at this site leads to better inhibitory activity. Besides the primed site, our studies also reveal new insights into the role of the non-primed site and effect on bioactivity of chemical modifications targeted at this site. The overall summary of SAR obtained from our studies is illustrated in Fig. 7 . In particular, biphenyl-4-carbonyl and 5-(4-acetylphenyl)furan-2-carbonyl at the cap position, and 3-methoxyphenyl, 3,5-dimethoxylphenyl, 3-acetylphenyl and 3-methylsulfonylphenyl at the head position are beneficial to compound’s inhibition of proteasome. Modifications at the side chains at P1, P2 and P3 positions also affect proteasome inhibitor’s activity.
Fig. 7.

Summary of the SAR of α-keto phenylamide derivatives.
It is worth noting that one of our α-ketoamide derivatives reported here, 17 was chosen as a representative compound to be tested in SARS-CoV-2 cellular infection assay and found to be active in inhibiting the viral infection. It has been previously reported that proteasome inhibitors such as MG132 can inhibit the replication of SARS-CoV likely via targeting and inhibiting m-calpain activity necessary for the viral replication cycle. Our molecular docking studies show that 17 binds m-calpain in a similar manner as MG132, which is consistent with the notion that proteasome inhibitors may intervene with the replication of SARS-CoV-2, which is highly homologous with SARS-CoV, by interfering with host cellular processes mediated by proteins like m-calpain that are essential for the virus life cycle. Recently, some α-ketoamide derivatives are reported to target SARS-CoV-2 main protease [42,43], which raises the possibility of an additional or alternative mechanism of 17 action on SARS-CoV-2. It awaits further studies to delineate the detailed mechanism of 17 and whether this compound or other α-ketoamide derivatives might exert anti-SARS-CoV-2 effect by targeting a host and/or a viral protein. Given the urgency and severity of the COVID-19 pandemic and need for effective prevention and treatment measures, the observed antiviral effect of our new α-ketoamide derivative against SARS-CoV-2 suggests that this may be a new lead for developing therapeutics against coronaviruses. If successfully developed, such therapeutics will add to the clinical arsenal for combinational therapy for the COVID-19 pandemic.
6. Conclusion
We have designed, synthesized, and characterized the biological activities and SAR of a new class of α-keto phenylamide derivatives. These novel compounds target both non-primed and primed sites of the proteasome and contain a variety of chemical modifications at the cap moiety, P1, P2 and P3 side chain positions, and the head group. Cell-based proteasome inhibition and anticancer activity assays have led to the identification of many compounds from this series that possess highly potent biological activity at the nanomolar concentrations. Among them, compounds 35, 44, 46 and 47 display most potent activities and may merit further studies of their potential as new anticancer agents. Another interesting and possibly important finding of our studies is that a representative compound 17 is found to be active in inhibiting the cellular infection of SARS-CoV-2 that causes the ongoing COVID-19 global pandemic, thus suggesting that the novel α-ketoamide derivatives reported here, besides their value for developing anticancer drugs, may offer a new lead in finding effective anti-SARS-CoV-2 therapeutics.
7. Experimental section
7.1. Chemistry
Commercially available reagents were used directly without further purification. The high resolution MS (HRMS) of target compounds were analyzed by using Waters Xevo G2 QTof. NMR spectra were obtained on the Bruker Ascend™ 400. The chemical shifts (δ) are reported in parts per million (ppm) using suitable deuterated NMR solvents in reference to tetramethyl silane (TMS) at 0 ppm. Multiplicities are defined as follows: s (singlet), brs (broad singlet), d (doublet), t (triplet), q (quartet), dd (doublet of doublets), and m (multiplets). The reaction was monitored by TLC (silica gel GF254). The compounds were purified by silica chromatography (silica gel 100–200 mesh or 200–300 mesh) or Prep-HPLC. HPLC Conditions, Column: XBridge Prep OBD™ C18 19 mm × 150 mm × 5 μm; detection: 220 nm; flow rate: 15 mL/min; run time: 30 min; mobile phase A: water (0.01% TFA); mobile phase B: acetonitrile (0.01% TFA).
The experimental details of compounds 35 and 46 are described below. The synthetic procedure of compounds 17–26, 45, 47 is similar to that of compound 46, as shown in Scheme 1A. The synthetic procedure of compounds 19, 27–34, 36–44 is similar to that of compound 35, as shown in Scheme 1B.
7.1.1. Procedure for the preparation of (S)-2-((S)-2-(5-(4-acetylphenyl)furan-2-carboxamido)-4-methylpentanamido)-4-methylpentanoic acid (5a)
To the mixture of H-Leu-OMe·HCl (12.7 g, 69.9 mmol, 1.0 equiv), Boc-Leu-OH (17.8 g, 76.9 mmol, 1.1 equiv) in THF (150 mL) was added HATU (29.22 g, 76.9 mmol, 1.1 equiv), HOBt (10.4 g, 76.9 mmol, 1.1 equiv). The reaction mixture was cooled to 0 °C, DIEA (3.5 equiv) was added. The temperature was increased to room temperature naturally and stirred for 1 h. After addition brine, the reaction mixture was extracted with ethyl acetate, the organic phase was washed with brine, dried over anhydrous Na2SO4, filtered, concentrated under vacuum. The crude product was purified by silica gel column chromatography (30% EtOAc/petroleum ether) to afford Boc-Leu-Leu-OMe 2a (20.8 g, 83% yield). 1H NMR (400 MHz, CDCl3) δ 6.50–6.41 (m, 1H), 4.97–4.82 (m, 1H), 4.67–4.58 (m, 1H), 4.19–4.03 (m, 1H), 3.74 (s, 3H), 1.73–1.64 (m, 4H), 1.63–1.48 (m, 2H), 1.46 (s, 9H), 0.99–0.90 (m, 12H).
Boc-Leu-Leu-OMe (20.8 g, 58.14 mmol, 1.0 equiv) was dissolved in CH2Cl2 (60 mL). The reaction solution was cooled to 0 °C and TFA (53 g, 465.1 mmol, 8.0 equiv) was added in portions. After the addition was completed, the reaction solution was stirred at 0 °C for 3 h. The reaction solution was evaporated in vacuum. The crude product was precipitated from ethyl ether and filtered to give 3a TFA salt (19.4 g, 94% yield) as off white solid. The crude product was used directly in the next step without further purification. 1H NMR (400 MHz, DMSO‑d 6) δ 8.84 (d, J = 7.5 Hz, 1H), 8.19 (s, 3H), 4.38–4.29 (m, 1H), 3.84–3.75 (m, 1H), 3.63 (s, 3H), 1.76–1.46 (m, 6H), 0.97–0.81 (m, 12H).
To a mixture of 3a TFA salt (8.6 g, 24.2 mmol, 1.0 equiv), compound 16 (6.2 g, 26.62 mmol, 1.1 equiv) in THF was added HATU (10.1 g, 26.62 mmol, 1.1 equiv), HOBt (3.6 g, 26.62 mmol, 1.1 equiv). The reaction solution was cooled to 0 °C and DIEA (12.5 g, 96.8 mmol, 4.0 equiv) was added. The temperature was increased to room temperature naturally and stirred for 1 h. The mixture reaction was partitioned between ethyl acetate and water. The aqueous layer was separated and extracted again with ethyl acetate. The combined organic layer was washed with brine, dried over anhydrous sodium sulfate, filtered, concentrated under vacuum. The crude product was purified by silica gel column chromatography to afford 4a (6.4 g, 56% yield) as light yellow powder.
Compound 4a (7.2 g, 15.3 mmol, 1.0 equiv) was dissolved in THF (80 mL), H2O (20 mL). The reaction solution was cooled to 0 °C. LiOH.H2O (1.9 g, 45.9 mmol, 3.0 equiv) was added. The reaction solution was stirred for 2 h at room temperature. TLC showed the reaction was completed. THF was removed under reduced pressure. The residue was diluted with water, then adjusted pH to 3 with 1 N HCl. The resulting mixture was extracted with ethyl acetate. The organic layers were combined, washed with brine, dried and concentrated under vacuum to give compound 5a (6.3 g, 90% yield) as light yellow powder. The product was used directly in the next step. 1H NMR (400 MHz, DMSO‑d 6) δ 12.48 (s, 1H), 8.57 (d, J = 8.5 Hz, 1H), 8.26 (d, J = 7.9 Hz, 1H), 8.12–8.01 (m, 4H), 7.33–7.29 (m, 2H), 4.64–4.53 (m, 1H), 4.29–4.13 (m, 1H), 2.62 (s, 3H), 1.78–1.44 (m, 6H), 0.98–0.77 (m, 12H).
7.1.2. Procedure for the preparation of (S)-2-((S)-3-(4-methoxyphenyl)-2-(2-morpholinoacetamido)propanamido)-4-methylpentanoic acid (5b)
To a solution of Boc-Tyr(OMe)–OH (3.6 g, 12.19 mmol, 1.0 equiv) in dry methanol was added SOCl2 (2.9 g, 24.38 mmol, 2.0 equiv) dropwise at 0 °C. The resulting solution was warmed to room temperature and stirred for 2 h. The reaction solution was concentrated under vacuum. The product was precipitated by the addition of ethyl ether and the solid was collected by filtration to give NH2-Tyr(OMe)–OH HCl salt 1b (2.72 g, 91% yield) as off white solid. 1H NMR (400 MHz, DMSO‑d 6) δ 8.81 (s, 3H), 7.17 (d, J = 8.5 Hz, 2H), 6.89 (d, J = 8.5 Hz, 2H), 4.20–4.13 (m, 1H), 3.74 (s, 3H), 3.66 (s, 3H), 3.23–3.14 (m, 1H), 3.12–3.03 (m, 1H).
To a solution of Boc-l-analine (2.1 g, 11.11 mmol, 1.0 equiv) in THF was added HATU (4.64 g, 12.22 mmol 1.1 equiv) and HOBt (1.65 g, 12.22 mmol, 1.1 equiv). The resulting solution was cooled to 0 °C. Compound 1b HCl salt (2.72 g, 11.11 mmol, 1.0 equiv) and DIEA (5.02 g, 38.9 mmol, 3.5 equiv) was added. The reaction solution was stirred at the same temperature for 1 h. Water was added and the mixture was extracted with ethyl acetate. The organic layer was separated and washed with brine, dried over anhydrous sodium sulfate, filtered, and concentrated under vacuum. The crude product was purified by silica gel column eluted with 1% CH3OH in CH2Cl2 to yield Boc-Ala-Tyr(OMe)–OMe 2b (3.7 g, 87% yield) as light yellow solid.
To a solution of 2b (3.7 g, 9.73 mmol, 1 equiv) in CH2Cl2 (10 mL) was added TFA (11.08 g, 97.3 mmol, 10 equiv) at 0 °C. The solution was stirred for 4 h at 0 °C. Then the solution was concentrated. The product was precipitated from ethyl ether. The precipitate was collected to give NH2-Ala-Tyr(OMe)–OMe TFA salt 3b as off white solid. 1H NMR (400 MHz, DMSO‑d 6) δ 9.09 (d, J = 7.4 Hz, 1H), 8.37 (s, 3H), 7.17 (d, J = 8.5 Hz, 2H), 6.89 (d, J = 8.5 Hz, 2H), 4.48–4.39 (m, 1H), 3.91–3.81 (m, 1H), 3.72 (s, 3H), 3.61 (s, 3H), 3.06–2.85 (m, 2H), 1.37 (t, J = 10.6 Hz, 3H).
To a solution of 2-morpholinoacetic acid (0.9 g, 6.21 mmol, 1.0 equiv) in THF was added HATU (2.6 g, 6.83 mmol, 1.1 equiv) and HOBt (0.9 g, 6.83 mmol, 1.1 equiv) subsequently. The reaction solution was cooled to 0 °C. Then 3b (2.6 g, 6.21 mmol, 1.0 equiv) was added. DIEA (3.52 g, 27.32 mmol, 4.0 equiv) was added dropwise. The reaction solution was stirred at the same temperature for 1 h. TLC showed the reaction was completed. The mixture reaction was partitioned between ethyl acetate and water. The aqueous layer was separated and extracted again with ethyl acetate. The combined organic layer was washed with brine, dried over anhydrous sodium sulfate, filtered, and concentrated under vacuum. The crude product was purified by silica gel column eluted with CH3OH: CH2Cl2 (30:1) to yield (S)-methyl 3-(4-methoxyphenyl)-2-((S)-2-(2-morpholinoacetamido)propanamido)propanoate 4b (1.9 g, 75% yield) as light yellow oil.
To a solution of 4b (1.9 g, 4.67 mmol, 1.0 equiv) in THF (15 mL) and H2O (5 mL), LiOH.H2O (0.2 g, 4.67 mmol, 1.0 equiv) was added at 0 °C. The reaction was stirred for 3 h at 0 °C. TLC showed the reaction was completed. Ethyl acetate was added, the organic layer was separated. The aqueous layer was adjusted pH to 5–6 with 1 N HCl. The crude product was purified by reverse phase silica gel eluted with 20% acetonitrile/H2O (1:4) and lyophilized to get compound 5b (1.5 g, 83% yield) as white solid. 1H NMR (400 MHz, DMSO‑d 6) δ 7.84 (d, J = 7.8 Hz, 1H), 7.44 (d, J = 7.0 Hz, 1H), 7.03 (d, J = 8.6 Hz, 2H), 6.72 (d, J = 8.6 Hz, 2H), 4.27–4.18 (m, 1H), 4.03–3.95 (m, 1H), 3.68 (s, 3H), 3.56–3.53 (m, 4H), 3.03–2.95 (m, 4H), 2.42–2.36 (m, 4H), 1.19 (d, J = 7.0 Hz, 3H).
7.1.3. Procedure for the preparation of Cbz-Leu-Trp-OH (10a), Cbz-Leu-Leu-OH (10b), Cbz-Leu-Phe-OH (10c)
To the solution of Cbz-Leu-OH (3.3 g, 12.4 mmol, 1.0 equiv) and H-Trp-OMe.HCl (3.16 g, 12.4 mmol, 1.0 equiv) in DMF (40 mL) was added HATU(5.2 g, 13.6 mmol, 1.1 equiv), HOBt (1.84 g, 13.6 mmol, 1.1 equiv). The reaction mixture was cooled to 0 °C, DIEA (5.6 g, 43.4 mmol, 3.5 equiv) was added. The temperature was increased to room temperature naturally and stirred for 1 h. After addition brine, the resulting mixture was extracted with ethyl acetate. The combined organic phase was washed with brine, dried over anhydrous Na2SO4, filtered, concentrated under vacuum. The crude product was purified by silica gel column chromatography (30% EtOAc/petroleum ether → 50% EtOAc/petroleum ether) to afford Cbz-Leu-Trp-OMe (5.3 g, yield 93%) as white solid. 1H NMR (400 MHz, DMSO‑d 6) δ 8.06 (s, 1H), 7.50 (d, J = 7.8 Hz, 1H), 7.37–7.26 (m, 6H), 7.19–7.12 (m, 1H), 7.11–7.04 (m, 1H), 6.94 (s, 1H), 6.59 (d, J = 7.3 Hz, 1H), 5.20–5.11 (m, 1H), 5.09–4.96 (m, 2H), 4.93–4.84 (m, 1H), 4.29–4.16 (m, 1H), 3.65 (s, 3H), 3.29 (d, J = 5.2 Hz, 2H), 1.68–1.52 (m, 2H), 1.50–1.38 (m, 1H), 0.92–076 (m, 6H).
Cbz-Leu-Trp-OMe (5.3 g, 11.4 mmol, 1.0 equiv) was dissolved in THF (60 mL), H2O (15 mL). The reaction solution was cooled to 0 °C. LiOH.H2O (1.4 g, 34.2 mmol, 3.0 equiv) was added. The reaction solution was stirred for 2 h at room temperature. TLC showed the reaction was completed. THF was removed under reduced pressure. The residue was diluted with water, then adjusted to pH 3 with 1 N HCl. The resulting mixture was extracted with ethyl acetate. The organic layers were combined, washed with brine, dried and concentrated under vacuum to give compound 10a (4.8 g, 94% yield) as light yellow powder. The product was used in the next step directly. 1H NMR (400 MHz, DMSO‑d 6) δ 10.86 (s, 1H), 8.05 (d, J = 7.6 Hz, 1H), 7.53 (d, J = 7.8 Hz, 1H), 7.44–7.22 (m, 7H), 7.17–7.12 (m, 1H), 7.10–7.02 (m, 1H), 7.01–6.93 (m, 1H), 5.15–4.86 (m, 2H), 4.55–4.43 (m, 1H), 4.14–3.99 (m, 1H), 3.23–2.99 (m, 2H), 1.67–1.33 (m, 3H), 0.92–0.74 (m, 6H).
Cbz-Leu-Leu-OH(10b) The synthetic procedure is similar to compound 10a using H-Leu-OMe·HCl as starting material. 1H NMR (400 MHz, CDCl3) δ 7.38–7.27 (m, 5H), 6.80–6.72 (m, 1H), 5.66–5.54 (m, 1H), 5.10 (s, 2H), 4.65–4.54 (m, 1H), 4.33–4.08 (m, 1H), 1.76–1.45 (m, 6H), 0.98–0.84 (m, 12H).
Cbz-Leu-Phe-OH(10c) The synthetic procedure is similar to compound 10a using H-Phe-OMe·HCl as starting material. 1H NMR (400 MHz, CDCl3) δ 8.66 (s, 1H), 7.45–7.29 (m, 5H), 7.25–7.19 (m, 3H), 7.16–7.11 (m, 2H), 7.07–6.96 (m, 1H), 5.73–5.58 (m, 1H), 5.22–5.03 (m, 2H), 4.92–4.77 (m, 1H), 4.39–4.08 (m, 1H), 3.26–3.13 (dd, J = 13.9, 5.4 Hz, 1H), 2.99 (dd, J = 13.9, 6.6 Hz, 1H), 1.73–1.35 (m, 3H), 0.95–0.80 (m, 6H).
7.1.4. General procedure for the preparation of 6a and 11a-c
To a solution of 5a or 10a-c (1.0 equiv) in THF (10 mL) was added NMM (3.0 equiv). The resulting solution was cooled to −5 °C. Isobutyl chloroformate (1.1 equiv) was added. The resulting solution was stirred at −5 °C for 30 min. Then L-leucinol (1.6 equiv) in THF (10 mL) was added dropwise. The reaction mixture was stirred at −5 to 0 °C for 2 h. Water was added and the mixture was extracted with ethyl acetate. The organic layer was separated and the aqueous layer was extracted with ethyl acetate again. The combined organic layer was washed with brine, dried over anhydrous sodium sulfate, filtered, concentrated under vacuum. The crude product was purified by silica gel column to yield 6a or 11a-c.
7.1.4.1. 5-(4-acetylphenyl)-N-((S)-1-(((S)-1-(((S)-1-hydroxy-4-methylpentan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)furan-2-carboxamide (6a)
Light yellow solid. Yield 41%. 1H NMR (400 MHz, CDCl3) δ 8.01 (d, J = 8.5 Hz, 2H), 7.81 (d, J = 8.4 Hz, 2H), 7.05–6.85 (m, 3H), 6.47 (d, J = 8.5 Hz, 1H), 4.64–4.52 (m, 1H), 4.43–4.31 (m, 1H), 4.09–3.99 (m, 1H), 3.70–3.62 (m, 1H), 3.54–3.44 (m, 1H), 2.62 (s, 3H), 1.86–1.51 (m, 9H), 1.03–0.85 (m, 18H).
7.1.4.2. ((S)-1-(((S)-1-(((S)-1-hydroxy-4-methylpentan-2-yl)amino)-3-(1H-indol-3-yl)-1-oxopropan-2-yl)amino)-4-methyl-1-oxopentan-2-yl) carbamic acid benzyl ester (11a)
White solid. Yield 34%. 1H NMR (400 MHz, DMSO‑d 6) δ 10.81 (s, 1H), 8.01 (d, J = 8.2 Hz, 1H), 7.61–7.48 (m, 2H), 7.43 (d, J = 8.2 Hz, 1H), 7.40–7.23 (m, 6H), 7.16–7.09 (m, 1H), 7.08–7.02 (m, 1H), 7.01–6.93 (m, 1H), 5.03 (q, J = 12.6 Hz, 2H), 4.62–4.46 (m, 2H), 4.08–3.98 (m, 1H), 3.84–3.71 (m, 1H), 3.28–3.18 (m, 1H), 3.17–3.03 (m, 2H), 2.98–2.84 (m, 1H), 1.63–1.47 (m, 2H), 1.44–1.16 (m, 4H), 0.88–0.77 (m, 12H).
7.1.4.3. ((S)-1-(((S)-1-(((S)-1-hydroxy-4-methylpentan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)amino)-4-methyl-1-oxopentan-2-yl) carbamic acid benzyl ester (11b)
White solid. Yield 52%. 1H NMR (400 MHz, CDCl3) δ 7.39–7.29 (m, 5H), 6.85–6.75 (m, 1H), 6.66 (d, J = 8.3 Hz, 1H), 5.57–5.46 (m, 1H), 5.16–5.04 (m, 2H), 4.45–4.36 (m, 1H), 4.25–4.12 (m, 1H), 4.07–3.95 (m, 1H), 3.64 (dd, J = 11.2, 3.5 Hz, 1H), 3.52 (dd, J = 11.2, 5.6 Hz, 1H), 2.97 (s, 1H), 1.78–1.46 (m, 7H), 1.45–1.27 (m, 2H), 0.94–0.82 (m, 18H).
7.1.4.4. ((S)-1-(((S)-1-(((S)-1-hydroxy-4-methylpentan-2-yl)amino)-1-oxo-3-phenylpropan-2-yl)amino)-4-methyl-1-oxopentan-2-yl) carbamic acid benzyl ester (11c)
White solid. Yield 43%. 1H NMR (400 MHz, CDCl3) δ 7.39–7.30 (m, 5H), 7.29–7.26 (m, 1H), 7.25–7.24 (m, 1H), 7.23–7.15 (m, 3H), 6.94–6.85 (m, 1H), 6.36–6.19 (m, 1H), 5.40–5.30 (m, 1H), 5.15–4.92 (m, 2H), 4.62 (q, J = 7.4 Hz, 1H), 4.20–4.09 (m, 1H), 4.02–3.90 (m, 1H), 3.52–3.32 (m, 2H), 3.18–2.99 (m, 2H), 2.41 (s, 2H), 1.65–1.39 (m, 4H), 1.38–1.17 (m, 2H), 0.91–0.83 (m, 12H).
7.1.5. General procedure for the preparation of 7a and 12a
To a solution of 6a or 11a (1 equiv) in DMSO, IBX (1.5 equiv) was added. The resulting solution was stirred at room temperature for 4 h. The reaction mixture was partitioned between ethyl acetate (30 mL) and saturated NaHCO3 (30 mL). The organic layer was separated and the aqueous layer was extracted with ethyl acetate (2 × 30 mL). The combined organic layer was washed with saturated NaHCO3 and brine, dried over anhydrous sodium sulfate, filtered, concentrated under vacuum. The crude product was purified by silica gel column to yield 7a or 12a.
7.1.5.1. (S)-2-((5-(4-acetylphenyl)furan-2-yl)amino)-4-methyl-N-((S)-4-methyl-1-(((S)-4-methyl-1-oxopentan-2-yl)amino)-1-oxopentan-2-yl)pentanamide (7a)
White solid. Yield 72%.1H NMR (400 MHz, DMSO‑d 6) δ 9.38 (s, 1H), 8.60 (d, J = 8.2 Hz, 1H), 8.27 (d, J = 7.3 Hz, 1H), 8.21–8.13 (m, 1H), 8.12–7.97 (m, 4H), 7.36–7.24 (m, 2H), 4.68–4.46 (m, 1H), 4.43–4.27 (m, 1H), 4.18–4.07 (m, 1H), 2.62 (s, 3H), 1.75–1.35 (m, 9H), 0.97–0.72 (m, 18H).
7.1.5.2. ((S)-1-(((S)-3-(1H-indol-2-yl)-1-(((S)-4-methyl-1-oxopentan-2-yl)amino)-1-oxopropan-2-yl)amino)-4-methyl-1-oxopentan-2-yl) carbamic acid benzyl ester (12a)
Off white solid. Yield 68%. 1H NMR (400 MHz, DMSO‑d 6) δ 10.86 (s, 1H), 9.14 (s, 1H), 8.34 (d, J = 7.3 Hz, 1H), 8.10 (d, J = 8.0 Hz, 1H), 7.58 (d, J = 7.8 Hz, 1H), 7.44 (d, J = 8.2 Hz, 1H), 7.40–7.25 (m, 6H), 7.16 (d, J = 2.0 Hz, 1H), 7.09–6.93 (m, 2H), 5.08–4.95 (m, 2H), 4.68–4.54 (m, 1H), 4.11–3.99 (m, 2H), 3.22–3.07 (m, 1H), 3.06–2.86 (m, 1H), 1.65–1.25 (m, 6H), 0.92–0.70 (m, 12H).
7.1.6. Procedure for the preparation of 1-(3-isocyanophenyl)ethanone (14)
The mixture of 3-aminoacetophenone (6.0 g, 44.4 mmol, 1.0 equiv) and HCOOH (6.1 g, 133.2 mmol, 2.5 equiv) was stirred for 3 h at 60 °C. After completion of the reaction, CH2Cl2 was added. The organic layer was washed with cold water, saturated aqueous NaHCO3 and brine, then dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The crude N-(3-acetylphenyl)formamide was used in the next step without further purification.
N-(3-acetylphenyl)formamide (44.4 mmol, 1.0 equiv) and DIEA (17.2 g, 133.2 mmol, 3.0 equiv) are dissolved in 100 mL dry CH2Cl2. The reaction solution was cooled to 0 °C. POCl3 (8.2 g, 53.3 mmol, 1.1 equiv) was added dropwise at 0 °C. The temperature of reaction was increased to room temperature naturally and stirred for 3 h CH2Cl2 was added. The organic layer was washed with cold water, saturated aqueous NaHCO3 and brine. The organic layer was separated, dried over Na2SO4, filtered and concentrated under reduced pressure at room temperature. The crude product was purified by silica gel column chromatography to afford 1-(3-isocyanophenyl)ethanone 14 as light yellow solid (5.9 g, 92% yield).1H NMR (400 MHz, CDCl3) δ 8.01–7.93 (m, 2H), 7.61–7.51 (m, 2H), 2.63 (s, 3H). Due to the strong smell of isonitrile, all operations were conducted in a well-ventilated hood.
7.1.7. Procedure for the preparation of 5-(4-acetylphenyl)furan-2-carboxylic acid (16)
To a solution of 4-bromoacetophenone (15.0 g, 75.3 mmol, 1.0 equiv), methyl 2-furoate (126.1 g, 301.2 mmol, 4.0 equiv), KOAc (22.2 g, 225.9 mmol, 3.0 equiv) in dimethylacetamide (150 mL) was added Pd(OAc)2 (0.253 g, 1.13 mmol, 0.015 equiv) under nitrogen atmosphere. The resulting mixture was stirred at 130 °C for 4 h under nitrogen atmosphere. Then saturated NH4Cl was added and extracted with CH2Cl2. The organic layer was washed with 0.1 N HCl (2 × ), brine, and concentrated under reduced pressure. The crude product was applied onto a silica gel column with ethyl acetate/petroleum ether (1:10) to give methyl 5-(4-acetylphenyl)-furan-2-carboxylate 15 (10.1 g, 55% yield) as light yellow solid. 1H NMR (400 MHz, CDCl3) δ 8.02 (d, J = 8.3 Hz, 2H), 7.87 (d, J = 8.3 Hz, 2H), 7.29–7.27 (m, 1H), 6.92–6.85 (m, 1H), 3.94 (s, 3H), 2.63 (s, 3H).
Methyl 5-(4-acetylphenyl)-furan-2-carboxylate 15 (10.1 g, 41.4 mmol, 1.0 equiv) was dissolved in THF (100 mL), H2O (25 mL). LiOH.H2O (5.1 g, 124.2 mmol, 3.0 equiv) was added. The reaction solution was stirred for 2 h at room temperature. CH2Cl2 and water was added. The water layer was separated, then adjusted pH to 3 with 2 N HCl. The resulting mixture was extracted with ethyl acetate. The organic layers were combined, washed with brine, dried and concentrated under vacuum to give compound 16 (8.5 g, 89% yield) as off white solid.
7.1.8. General procedure for the preparation of 35 and 46
To a solution of 7a or 12a (1.0 equiv) and 14 (1.5 equiv) in dry CH2Cl2 (2 mL), trifluoroacetic acid (2.0 equiv) was added dropwise at −5 °C. The reaction mixture was allowed to stir for 2 h at 0 °C. Then pyridine (4.0 equiv) was added at 0 °C. The resulting solution was stirred at 0 °C for 1 h CH2Cl2 was added, the pH value of the reaction was adjusted to 5–6 with 1 N HCl in ice bath. The aqueous layer was separated, the organic layer was washed with water (30 mL) and brine (30 mL). The combined organic layer was dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The crude product was applied onto a silica gel column with ethyl acetate/petroleum ether (1 : 5) to get product as white solid.
To a solution of compound 8a or 13a (1.0 equiv) in DMSO was added IBX (2.0 equiv). The resulting solution was stirred at room temperature for 5 h. Saturated NaHCO3 solution was added and extracted with ethyl acetate (3 × 30 mL). The combined organic layer was washed with saturated NaHCO3 solution, water, brine, dried over anhydrous sodium sulfate, filtered, concentrated under vacuum. The crude product was precipitated from ethyl acetate to give final product 35 or 46.
7.1.8.1. 5-(4-acetylphenyl)-N-((S)-1-(((S)-1-(((S)-1-((3-acetylphenyl)amino)-5-methyl-1,2-dioxohexan-3-yl)amino)-4-methyl-1-oxopentan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)furan-2-carboxamide (46)
White solid. Yield 35%. 1H NMR (400 MHz, CDCl3) δ 8.93 (s, 1H), 8.19 (s, 1H), 8.08–7.93 (m, 3H), 7.86–7.72 (m, 3H), 7.48 (t, J = 7.9 Hz, 1H), 6.94 (d, J = 7.9 Hz, 1H), 6.89 (d, J = 3.5 Hz, 1H), 6.85 (d, J = 6.9 Hz, 1H), 6.70 (d, J = 7.1 Hz, 1H), 5.49–5.38 (m, 1H), 4.73–4.62 (m, 1H), 4.55–4.46 (m, 1H), 2.71–2.52 (m, 6H), 1.86–1.67 (m, 6H), 1.64–1.47 (m, 6H), 1.10–0.72 (m, 18H). 13C NMR (101 MHz, CDCl3) δ 197.78, 197.30, 196.95, 172.20, 172.10, 158.28, 157.45, 154.73, 147.4, 137.99, 137.19, 136.80, 133.54, 129.62, 129.08, 125.30, 124.59, 119.75, 117.34, 109.54, 53.05, 51.73, 41.30, 40.76, 40.24, 26.85, 26.73, 25.44, 25.01, 24.81, 23.30, 22.99, 22.78, 22.40, 22.34, 21.59. HRMS (ESI): m/z (M + Na)+ calcd for C40H50N4NaO8, 737.8368; found,737.3535.
7.1.8.2. ((S)-1-(((S)-1-(((S)-1-((3-acetylphenyl)amino)-5-methyl-1,2-dioxohexan-3-yl)amino)-3-(1H-indol-3-yl)-1-oxopropan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)carbamic acid benzyl ester (35)
White solid. Yield 21%. 1H NMR (400 MHz, CDCl3) δ 8.84 (s, 1H), 8.34 (s, 1H), 8.16 (s, 1H), 7.96–7.84 (m, 1H), 7.81–7.69 (m, 1H), 7.64–7.43 (m, 2H), 7.42–7.27 (m, 4H), 7.15–6.94 (m, 3H), 7.84–6.61 (m, 1H), 5.35–5.21 (m, 1H), 5.11–4.93 (m, 2H), 4.83–4.72 (m, 1H), 4.22–4.04 (m, 1H), 3.33–3.05 (m, 1H), 2.64 (s, 3H), 1.60–1.51 (m, 2H), 1.50–1.16 (m, 4H), 1.06–0.65 (m, 12H). 13C NMR (101 MHz, DMSO‑d 6) δ 197.52, 196.78, 172.13, 171.91, 159.70, 155.89, 137.99, 137.33, 137.02, 135.99, 135.86, 129.25, 128.36, 127.78, 127.68, 127.31, 124.90, 123.37, 120.84, 119.82, 118.32, 118.16, 111.23, 109.93, 65.40, 53.16, 52.56, 52.13, 40.67, 38.32, 27.69, 26.79, 24.54, 24.12, 23.13, 23.03, 21.42, 21.18. HRMS (ESI): m/z (M + H)+ calcd for C40H48N5O7,710.8384; found, 710.3558.
7.1.8.3. (S)-3-((S)-3-(4-methoxyphenyl)-2-((S)-2-(2-morpholinoacetamido)propanamido) propanamido)-2-oxo-N,4-diphenylbutanamide (17)
White solid. Yield 10%. 1H NMR (400 MHz, CDCl3) δ 8.67 (s, 1H), 7.72–7.60 (m, 2H), 7.45–7.34 (m, 2H), 7.25–7.16 (m, 4H), 7.11–6.99 (m, 3H), 6.84–6.72 (m, 2H), 6.69–6.50 (m, 1H), 5.67–5.48 (m, 1H), 4.65–4.47 (m, 1H), 4.40–4.27 (m, 1H), 3.92–3.58 (m, 7H), 3.38–3.22 (m, 1H), 3.19–2.83 (m, 5H), 2.80–2.33 (m, 4H), 1.35–1.24 (m, 3H). HRMS (ESI): m/z (M + H)+ calcd for C35H42N5O7, 644.7373; found, 644.3080.
7.1.8.4. (S)-3-((S)-3-(4-methoxyphenyl)-2-((S)-2-(2-morpholinoacetamido)propanamido) propanamido)-5-methyl-2-oxo-N-phenylhexanamide (18)
White solid. Yield 60%. 1H NMR (400 MHz, CDCl3) δ 8.67 (s, 1H), 7.71–7.47 (m, 3H), 7.41–7.32 (m, 2H), 7.24–7.06 (m, 3H), 6.88–6.67 (m, 3H), 6.66–6.49 (m, 1H), 5.41–5.28 (m, 1H), 4.71–4.56 (m, 1H), 4.48–4.24 (m, 1H), 3.82–3.69 (m, 7H), 3.19–2.92 (m, 4H), 2.72–2.41 (s, 4H), 1.75–1.67 (m, 1H), 1.63–1.54 (m, 1H), 1.50–1.42 (m, 1H), 1.39–1.32 (m, 3H), 1.01–0.95 (m, 3H), 0.94–0.89 (m, 3H). HRMS (ESI): m/z (M + H)+ calcd for C32H44 N5O7, 610.7211; found, 610.3252.
7.1.8.5. ((S)-1-(((S)-3-(4-methoxyphenyl)-1-(((S)-5-methyl-1,2-dioxo-1-(phenylamino)hexan-3-yl)amino)-1-oxopropan-2-yl)amino)-1-oxopropan-2-yl)carbamic acid benzyl ester (19)
White solid. Yield 54%. 1H NMR (400 MHz, CDCl3) δ 8.62 (s, 1H), 7.68–7.58 (m, 2H), 7.41–7.32 (m, 5H), 7.23–7.15 (m, 1H), 7.11 (d, J = 8.5 Hz, 2H), 6.86–6.74 (m, 2H), 6.63–6.41 (m, 2H), 5.39–5.28 (m, 1H), 5.18–4.94 (m, 3H), 4.70–4.55 (m, 1H), 4.24–4.10 (m, 1H), 3.77–3.65 (m, 3H), 3.15–2.89 (m, 2H), 1.77–1.66 (m, 1H), 1.61–1.52 (m, 1H), 1.49–1.43 (s, 1H), 1.38–1.30 (m, 3H), 1.04–0.86 (m, 6H). HRMS (ESI): m/z (M + Na)+ calcd for C34H40N4NaO7, 639.6938; found, 639.2794.
7.1.8.6. N-((S)-1-(((S)-3-(4-methoxyphenyl)-1-(((S)-5-methyl-1,2-dioxo-1-(phenylamino)hexan-3-yl)amino)-1-oxopropan-2-yl)amino)-1-oxopropan-2-yl)decanamide (20)
White solid. Yield 56%. 1H NMR (400 MHz, CDCl3) δ 8.63 (s, 1H), 7.63 (d, J = 7.8 Hz, 2H), 7.37 (t, J = 7.9 Hz, 2H), 7.21–7.15 (m, 1H), 7.14–7.08 (m, 2H), 6.79 (d, J = 8.6 Hz, 2H), 6.58 (dd, J = 21.0, 7.7 Hz, 2H), 5.96–5.79 (m, 1H), 5.39–5.28 (m, 1H), 4.70–4.53 (m, 1H), 4.52–4.35 (m, 1H), 3.72 (s, 3H), 3.15–2.90 (m, 2H), 2.13 (t, J = 7.4 Hz, 2H), 1.77–1.65 (m, 2H), 1.59–1.52 (m, 2H), 1.49–1.41 (m, 1H), 1.37–1.32 (m, 3H), 1.31–1.22 (m, 12H), 1.01–0.96 (m, 3H), 0.93–0.84 (m, 6H). HRMS (ESI): m/z (M + H)+ calcd for C36H53N4O6, 637.8292; found, 637.3986.
7.1.8.7. ((S)-1-(((S)-3-(4-methoxyphenyl)-1-(((S)-5-methyl-1,2-dioxo-1-(phenylamino)hexan-3-yl)amino)-1-oxopropan-2-yl)amino)-1-oxopropan-2-yl)carbamic acid tert-butyl ester (21)
White solid. Yield 60%. 1H NMR (400 MHz, CDCl3) δ 8.69–8.62 (m, 1H), 7.66–7.61 (m, 1H), 7.40 (t, J = 7.9 Hz, 2H), 7.21 (t, J = 7.4 Hz, 1H), 7.14 (d, J = 8.6 Hz, 2H), 6.87–6.77 (m, 2H), 6.61–6.54 (m, 1H), 5.40–5.32 (m, 1H), 4.87–4.74 (m, 1H), 4.69–4.58 (m, 1H), 4.15–3.99 (m, 1H), 3.83–3.71 (m, 3H), 3.20–3.12 (m, 1H), 3.00–2.87 (m, 1H), 1.75–1.52 (m, 3H), 1.51–1.29 (m, 12H), 1.02–0.96 (m, 3H), 0.91–0.88 (m, 3H). 13C NMR (101 MHz, CDCl3) δ 196.16, 172.47, 170.77, 158.81, 156.80, 155.76, 136.38, 130.48, 129.33, 128.26, 125.51, 119.99, 114.24, 80.66, 55.31, 54.30, 52.87, 50.97, 40.33, 36.89, 28.36, 25.16, 23.27, 21.54, 18.20. HRMS (ESI): m/z (M + Na)+ calcd for C31H42N4NaO7, 605.6775; found, 605.2941.
7.1.8.8. N-((S)-1-(((S)-3-(4-methoxyphenyl)-1-(((S)-5-methyl-1,2-dioxo-1-(phenylamino)hexan-3-yl)amino)-1-oxopropan-2-yl)amino)-1-oxopropan-2-yl)adamantane-1-carboxamide (22)
White solid. Yield 25%. 1H NMR (400 MHz, CDCl3) δ 8.66 (s, 1H), 7.62 (d, J = 8.1 Hz, 2H), 7.37 (t, J = 7.8 Hz, 2H), 7.18 (t, J = 7.4 Hz, 1H), 7.10 (d, J = 8.3 Hz, 2H), 6.84–6.71 (m, 3H), 6.47–6.37 (m, 1H), 5.99–5.85 (m, 1H), 5.49–5.35 (m, 1H), 4.71–4.61 (m, 1H), 4.39–4.26 (m, 1H), 3.72 (s, 3H), 3.33–3.19 (m, 1H), 2.93–2.78 (m, 1H), 2.23–2.06 (m, 2H), 2.05–1.96 (m, 3H), 1.78–1.55 (m, 12H), 1.54–1.46 (m, 1H), 1.45–1.34 (m, 3H), 1.08–0.97 (m, 3H), 0.94–0.82 (m, 3H). HRMS (ESI): m/z (M + H)+ calcd for C37H49N4O6, 645.8082; found, 645.3650.
7.1.8.9. N-((S)-1-(((S)-3-(4-methoxyphenyl)-1-(((S)-5-methyl-1,2-dioxo-1-(phenylamino)hexan-3-yl)amino)-1-oxopropan-2-yl)amino)-1-oxopropan-2-yl)furan-2-carboxamide (23)
White solid. Yield 43%.1H NMR (400 MHz, CDCl3) δ 8.66 (s, 1H), 7.63 (d, J = 7.9 Hz, 2H), 7.53–7.46 (m, 1H), 7.37 (t, J = 7.8 Hz, 2H), 7.25–7.03 (m, 4H), 6.84–6.59 (m, 5H), 6.58–6.47 (m, 1H), 5.40–5.32 (m, 1H), 4.68–4.52 (m, 2H), 3.74–3.61 (m, 3H), 3.18–2.89 (m, 2H), 1.81–1.38 (m, 6H), 1.05–0.78 (m, 6H). HRMS (ESI): m/z (M + H)+ calcd for C31H37N4O7, 577.6481; found, 577.2662.
7.1.8.10. N-((S)-1-(((S)-3-(4-methoxyphenyl)-1-(((S)-5-methyl-1,2-dioxo-1-(phenylamino)hexan-3-yl)amino)-1-oxopropan-2-yl)amino)-1-oxopropan-2-yl)-6-phenylpicolinamide (24)
White solid. Yield 15%. 1H NMR (400 MHz, CDCl3) δ 8.68 (s, 1H), 8.42 (d, J = 6.6 Hz, 1H), 8.13–7.84 (m, 5H), 7.72–7.46 (m, 5H), 7.42–7.31 (m, 2H), 7.22–7.10 (m, 1H), 7.08–6.98 (m, 1H), 6.89–6.72 (m, 2H), 6.63–6.45 (m, 2H), 5.45–5.26 (m, 1H), 4.85–4.46 (m, 2H), 3.81–3.42 (m, 3H), 3.19–2.91 (m, 2H), 1.80–1.43 (m, 6H), 1.07–0.78 (d, J = 96.3 Hz, 6H). HRMS (ESI): m/z (M + H)+ calcd for C38H42N5O6, 664.7700; found, 664.3145.
7.1.8.11. N-((S)-1-(((S)-3-(4-methoxyphenyl)-1-(((S)-5-methyl-1,2-dioxo-1-(phenylamino)hexan-3-yl)amino)-1-oxopropan-2-yl)amino)-1-oxopropan-2-yl)-[1,1′-biphenyl]-4-carboxamide (25)
White solid. Yield 32%.1H NMR (400 MHz, CDCl3) δ 8.84 (s, 1H), 7.89–7.75 (m, 2H), 7.74–7.30 (m, 7H), 7.26–7.20 (m, 2H), 7.19–7.01 (m, 3H), 7.01–6.87 (m, 1H), 5.42–5.22 (m, 1H), 4.75–4.51 (m, 2H), 3.73 (s, 3H), 3.19–2.93 (m, 2H), 1.41–1.75 (m, 6H), 0.75–1.07 (m, 6H). HRMS (ESI): m/z (M + H)+ calcd for C39H43N4O6,663.7819; found, 663.3195.
7.1.8.12. N-((S)-1-(((S)-3-(4-methoxyphenyl)-1-(((S)-5-methyl-1,2-dioxo-1-(phenylamino)hexan-3-yl)amino)-1-oxopropan-2-yl)amino)-1-oxopropan-2-yl)5-(4-acetylphenyl)furan-2-carboxamide (26)
White solid. Yield 61%. 1H NMR (400 MHz, CDCl3) δ 8.63 (s, 1H), 8.03 (d, J = 8.5 Hz, 2H), 7.81 (d, J = 8.5 Hz, 2H), 7.63 (d, J = 7.7 Hz, 2H), 7.44–7.33 (m, 2H), 7.25–7.16 (m, 2H), 7.14–7.06 (m, 2H), 6.90 (d, J = 3.6 Hz, 1H), 6.83–6.77 (m, 1H), 6.75–6.65 (m, 3H), 6.56–6.43 (m, 1H), 5.41–5.31 (m, 1H), 4.72–4.57 (m, 2H), 3.67–3.53 (m, 3H), 3.14–2.94 (m, 2H), 2.64 (s, 3H), 1.78–1.69 (m, 1H), 1.66–1.55 (m, 2H), 1.53–1.46 (m, 3H), 1.03–0.97 (m, 3H), 0.96–0.89 (m, 3H). 13C NMR (101 MHz, DMSO) δ 197.22, 196.95, 171.78, 171.27, 159.38, 157.72, 157.28, 153.50, 147.64, 137.49, 136.15, 133.24, 130.18, 129.42, 128.86, 128.73, 124.57, 124.34, 120.41, 116.26, 113.35, 109.97, 54.75, 53.75, 52.02, 48.22, 38.32, 36.48, 26.74, 24.51, 23.05, 21.14, 17.48. HRMS (ESI): m/z (M + H)+ calcd for C39H43N4O8, 695.7807; found, 695.3109.
7.1.8.13. ((S)-1-(((S)-3-(1H-indol-3-yl)-1-(((S)-5-methyl-1,2-dioxo-1-(phenylamino)hexan-3-yl)amino)-1-oxopropan-2-yl)amino)-1-oxopropan-2-yl)carbamic acid benzyl ester (27)
White solid. Yield 20%.1H NMR (400 MHz, CDCl3) δ 8.63–8.44 (m, 1H), 8.00–7.83 (m, 1H), 7.71–7.56 (m, 2H), 7.50–7.30 (m, 6H), 7.29–7.26 (m, 1H), 7.25–7.24 (m, 1H), 7.23–7.01 (m, 3H), 6.76–6.56 (m, 1H), 6.49–6.35 (m, 1H), 5.36–5.23 (m, 1H), 5.11–4.92 (m, 2H), 4.84–4.66 (m, 1H), 4.26–4.07 (m, 1H), 3.43–3.26 (m, 1H), 3.23–3.06 (m, 1H), 1.73–1.56 (m, 2H), 1.54–1.42 (m, 1H), 1.39–1.27 (m, 3H), 0.93 (d, J = 6.5 Hz, 3H), 0.89–0.80 (m, 3H). HRMS (ESI): m/z (M + H)+ calcd for C35H40N5O6, 626.7220; found, 626.2984.
7.1.8.14. ((S)-1-(((S)-1-(((S)-5-methyl-1,2-dioxo-1-(phenylamino)hexan-3-yl)amino)-1-oxo-3-phenylpropan-2-yl)amino)-1-oxopropan-2-yl)carbamic acid benzyl ester (28)
White solid. Yield 3%. 1H NMR (400 MHz, CDCl3) δ 8.62 (s, 1H), 7.67–7.56 (m, 1H), 7.44–7.30 (m, 5H), 7.30–7.26 (m, 4H), 7.22–7.14 (m, 3H), 6.62–6.39 (m, 2H), 5.41–5.29 (m, 1H), 5.12–4.96 (m, 2H), 4.77–4.60 (m, 1H), 4.22–4.07 (m, 1H), 3.22–2.97 (m, 2H), 1.76–1.52 (m, 3H), 1.37–1.27 (m, 3H), 1.06–0.84 (m, 6H). HRMS (ESI): m/z (M + H)+ calcd for C33H39N4O6, 587.6860; found, 587.2872.
7.1.8.15. ((S)-1-(((S)-3-(1H-indol-3-yl)-1-(((S)-5-methyl-1,2-dioxo-1-(phenylamino)hexan-3-yl)amino)-1-oxopropan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)carbamic acid benzyl ester (29)
White solid. Yield 48%.1H NMR (400 MHz, DMSO‑d 6) δ 10.89–10.77 (m, 1H), 10.67–10.58 (m, 1H), 8.50–8.31 (m, 1H), 8.28–7.95 (m, 1H), 7.85–7.76 (m, 2H), 7.62–7.49 (m, 1H), 7.48–7.21 (m, 8H), 7.19–7.08 (m, 2H), 7.07–7.00 (m, 1H), 6.99–6.90 (m, 1H), 5.22–5.12 (m, 1H), 5.10–4.92 (m, 2H), 4.75–4.53 (m, 1H), 4.09–3.92 (m, 1H), 3.18–3.03 (m, 1H), 2.96–2.84 (m, 1H), 1.79–1.16 (m, 6H), 0.98–0.60 (m, 12H). HRMS (ESI): m/z (M + H)+ calcd for C38H46N5O6, 668.8017; found, 668.3454.
7.1.8.16. ((S)-4-methyl-1-(((S)-1-(((S)-5-methyl-1,2-dioxo-1-(phenylamino)hexan-3-yl)amino)-1-oxo-3-phenylpropan-2-yl)amino)-1-oxopentan-2-yl)carbamic acid benzyl ester (30)
White solid. Yield 48%. 1H NMR (400 MHz, CDCl3) δ 8.64 (s, 1H), 7.67–7.53 (m, 2H), 7.43–7.28 (m, 7H), 7.26–7.23 (m, 1H), 7.22–7.11 (m, 4H), 6.80–6.26 (m, 2H), 5.44–5.29 (m, 1H), 5.11–4.97 (m, 2H), 4.82–4.61 (m, 1H), 4.19–3.94 (m, 1H), 3.25–2.92 (m, 2H), 1.80–1.49 (m, 4H), 1.48–1.30 (m, 2H), 1.02–0.75 (m, 12H). HRMS (ESI): m/z (M + Na)+ calcd for C36H44N4NaO6, 651.7475; found, 651.3157.
7.1.8.17. ((2S)-3-cyclohexyl-1-(((2S)-1-((5-methyl-1,2-dioxo-1-(phenylamino)hexan-3-yl)amino)-1-oxo-3-phenylpropan-2-yl)amino)-1-oxopropan-2-yl)carbamic acid benzyl ester (31)
White solid. Yield 39%. 1H NMR (400 MHz, CDCl3) δ 8.61 (s, 1H), 7.62 (d, J = 7.8 Hz, 2H), 7.43–7.29 (m, 6H), 7.25–7.22 (m, 2H), 7.21–7.13 (m, 3H), 6.59–6.43 (m, 2H), 5.40–5.29 (m, 1H), 5.12–4.94 (m, 2H), 4.72–4.60 (m, 1H), 4.21–4.07 (m, 1H), 3.21–2.95 (m, 2H), 1.77–1.51 (m, 8H), 1.49–1.35 (m, 2H), 1.34–1.08 (m, 4H), 1.03–0.80 (m, 7H). 13C NMR (101 MHz, CDCl3) δ 195.95, 172.05, 170.47, 156.51, 156.28, 136.31, 136.17, 135.91, 129.27, 129.23, 128.68, 128.62, 128.37, 128.09, 127.04, 125.44, 119.86, 67.29, 54.13, 53.24, 52.87, 40.11, 39.58, 37.71, 33.98, 33.64, 32.39, 26.30, 26.12, 25.94, 25.15, 23.17, 21.43. HRMS (ESI): m/z (M + H)+ calcd for C39H49N4O6, 669.8296; found, 669.3641.
7.1.8.18. ((S)-3-methoxy-1-(((S)-1-(((S)-5-methyl-1,2-dioxo-1-(phenylamino)hexan-3-yl)amino)-1-oxo-3-phenylpropan-2-yl)amino)-1-oxopropan-2-yl) carbamic acid benzyl ester (32)
White solid. Yield 5%. 1H NMR (400 MHz, CDCl3) δ 8.68–8.57 (m, 1H), 7.62 (d, J = 8.3 Hz, 2H), 7.48–7.30 (m, 6H), 7.25–7.09 (m, 4H), 6.97–6.61 (m, 2H), 6.57–6.37 (m, 1H), 5.66–5.48 (m, 1H), 5.45–5.29 (m, 1H), 5.17–4.95 (m, 2H), 4.83–4.61 (m, 1H), 4.34–4.18 (m, 1H), 3.84–3.62 (m, 1H), 3.56–3.35 (m, 1H), 3.33–3.24 (m, 2H), 3.23–2.94 (m, 2H), 1.77–1.64 (m, 1H), 1.61–1.48 (m, 1H), 1.47–1.36 (s, 1H), 1.08–0.69 (m, 6H). HRMS (ESI): m/z (M + Na)+ calcd for C34H40N4NaO7, 639.6938; found, 639.2814.
7.1.8.19. (S)-3-((S)-3-(4-methoxyphenyl)-2-((S)-2-(2-phenylacetamido)propanamido)propanamido)-5-methyl-N-(naphthalen-1-yl)-2-oxohexanamide (33)
White solid. Yield 35%. 1H NMR (400 MHz, CDCl3) δ 9.24 (d, J = 6.2 Hz, 1H), 8.19 (t, J = 8.2 Hz, 1H), 7.95–7.81 (m, 2H), 7.77–7.70 (m, 1H), 7.63–7.45 (m, 3H), 7.40–7.28 (m, 5H), 7.17–7.08 (m, 2H), 6.81 (t, J = 8.8 Hz, 2H), 6.70–6.46 (m, 2H), 5.45–5.35 (m, 1H), 5.16–4.95 (m, 3H), 4.74–4.60 (m, 1H), 4.23–4.07 (m, 1H), 3.70 (d, J = 25.8 Hz, 3H), 3.16–2.92 (m, 2H), 1.82–1.69 (m, 1H), 1.66–1.44 (m, 2H), 1.33 (t, J = 7.8 Hz, 3H), 1.05–0.88 (m, 6H). HRMS (ESI): m/z (M + Na)+ calcd for C38H42N4NaO7, 689.7524; found, 689.2951.
7.1.8.20. ((S)-1-(((S)-1-(((S)-1-((3-acetylphenyl)amino)-5-methyl-1,2-dioxohexan-3-yl) amino)-1-oxo-3-phenylpropan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)carbamic acid benzyl ester (34)
White solid. Yield 65%. 1H NMR (400 MHz, CDCl3) δ 8.77 (s, 1H), 8.18 (s, 1H), 7.96–7.87 (m, 1H), 7.82–7.73 (m, 1H), 7.53–7.44 (m, 1H), 7.41–7.30 (m, 4H), 7.28–7.26 (m, 1H), 7.25–7.22 (m, 2H), 7.21–7.14 (m, 2H), 6.62–6.51 (m, 1H), 5.39–5.28 (m, 1H), 5.12–4.96 (m, 2H), 4.73–4.62 (m, 1H), 4.16–4.06 (m, 1H), 3.17–2.99 (m, 2H), 2.63 (s, 3H), 1.74–1.53 (m, 4H), 1.50–1.36 (m, 2H), 1.05–0.78 (m, 12H). HRMS (ESI): m/z (M + H)+ calcd for C38H47N4O7, 671.8024; found, 671.3445.
7.1.8.21. ((S)-1-(((S)-3-(1H-indol-3-yl)-1-(((S)-5-methyl-1-((3-(methylsulfonyl)phenyl)amino)-1,2-dioxohexan-3-yl)amino)-1-oxopropan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)carbamic acid benzyl ester (36)
White solid. Yield 11%. 1H NMR (400 MHz, DMSO‑d 6) δ 11.02 (s, 1H), 10.80 (s, 1H), 8.51–8.44 (m, 2H), 8.07 (d, J = 8.1 Hz, 1H), 7.99 (d, J = 8.1 Hz, 1H), 7.74–7.60 (m, 2H), 7.50 (d, J = 7.8 Hz, 1H), 7.40–7.27 (m, 6H), 7.17–7.10 (m, 1H), 7.08–7.00 (m, 1H), 6.99–6.89 (m, 1H), 5.17–5.08 (m, 1H), 5.06–4.94 (m, 2H), 4.69–4.58 (m, 1H), 4.06–3.96 (m, 1H), 3.18 (s, 3H), 3.14–3.05 (m, 1H), 2.95–2.86 (m, 1H), 1.76–1.45 (m, 4H), 1.36–1.18 (m, 2H), 0.96–0.68 (m, 12H). HRMS (ESI): m/z (M + H)+ calcd for C39H48N5O8S, 746.8921; found, 746.3225.
7.1.8.22. ((S)-1-(((S)-3-(1H-indol-3-yl)-1-(((S)-5-methyl-1-(naphthalen-1-ylamino)-1,2-dioxohexan-3-yl)amino)-1-oxopropan-2-yl)amino)-4-methyl-1-oxopentan-2-yl) carbamic acid benzyl ester (37)
White solid. Yield 32%. 1H NMR (400 MHz, CDCl3) δ 9.17 (d, J = 14.8 Hz, 1H), 8.26–8.15 (m, 1H), 7.97–7.82 (m, 3H), 7.78–7.72 (m, 1H), 7.68–7.46 (m, 5H), 7.44–7.29 (m, 5H), 7.18–7.03 (m, 3H), 6.78–6.43 (m, 1H), 5.44–5.31 (m, 1H), 5.09–4.94 (m, 2H), 4.92–4.69 (m, 1H), 4.21–4.04 (m, 1H), 3.45–3.28 (m, 1H), 3.24–3.09 (m, 1H), 1.76–1.49 (m, 9H), 0.99–0.94 (m, 3H), 0.93–0.84 (m, 9H). HRMS (ESI): m/z (M + H)+ calcd for C42H48N5O6, 718.8604; found, 718.3611.
7.1.8.23. ((S)-1-(((S)-1-(((S)-1-((3-acetylphenyl)amino)-5-methyl-1,2-dioxohexan-3-yl)amino)-4-methyl-1-oxopentan-2-yl)amino)-4-methyl-1-oxopentan-2-yl) carbamic acid benzyl ester (38)
White solid. Yield 21%. 1H NMR (400 MHz, DMSO‑d 6) δ 10.81 (s, 1H), 8.50–8.38 (m, 2H), 8.06–7.95 (m, 1H), 7.91 (d, J = 8.3 Hz, 1H), 7.75 (d, J = 7.8 Hz, 1H), 7.51 (t, J = 7.9 Hz, 1H), 7.44–7.24 (m, 6H), 5.09–4.93 (m, 3H), 4.43–4.33 (m, 1H), 4.09–3.98 (m, 1H), 2.57 (s, 3H), 1.78–1.66 (m, 1H), 1.64–1.47 (m, 4H), 1.46–1.29 (m, 4H), 1.01–0.61 (m, 18H). HRMS (ESI): m/z (M + H)+ calcd for C35H49N4O7, 637.7862; found, 637.3611.
7.1.8.24. ((S)-4-methyl-1-(((S)-4-methyl-1-(((S)-5-methyl-1,2-dioxo-1-(quinolin-5-ylamino)hexan-3-yl)amino)-1-oxopentan-2-yl)amino)-1-oxopentan-2-yl)carbamic acid benzyl ester (39)
White solid. Yield 27%. 1H NMR (400 MHz, CDCl3) δ 9.18 (s, 1H), 9.04–8.91 (m, 1H), 8.47–8.29 (m, 1H), 8.25–8.08 (m, 2H), 7.87–7.72 (m, 1H), 7.65–7.50 (m, 1H), 7.39–7.31 (m, 3H), 7.30–7.27 (m, 1H), 7.20–6.88 (m, 1H), 6.48–6.29 (m, 1H), 5.39–5.28 (m, 1H), 5.17–5.06 (m, 2H), 4.57–4.42 (m, 1H), 4.22–4.06 (m, 1H), 1.83–1.47 (m, 9H), 1.13–0.65 (m, 18H). HRMS (ESI): m/z (M + H)+ calcd for C36H48N5O6, 646.7962; found, 646.3607.
7.1.8.25. ((S)-1-(((S)-1-(((S)-1-(isoquinolin-5-ylamino)-5-methyl-1,2-dioxohexan-3-yl)amino)-4-methyl-1-oxopentan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)carbamic acid benzyl ester (40)
White solid. Yield 16%. 1H NMR (400 MHz, CDCl3) δ 9.33 (s, 1H), 9.23 (s, 1H), 8.67–8.55 (m, 1H), 8.52–8.41 (m, 1H), 7.96–7.87 (m, 1H), 7.80–7.63 (m, 2H), 7.40–7.27 (m, 4H), 7.20–7.02 (m, 1H), 6.97–6.84 (m, 1H), 6.44–6.30 (m, 1H), 5.43–5.32 (m, 1H), 5.15–5.04 (m, 2H), 4.55–4.43 (m, 1H), 4.20–4.07 (m, 1H), 1.88–1.58 (m, 9H), 1.13–0.58 (m, 18H). HRMS (ESI): m/z (M + H)+ calcd for C36H48N5O6, 646.7962; found, 646.3597.
7.1.8.26. ((S)-1-(((S)-1-(((S)-1-([1,1′-biphenyl]-2-ylamino)-5-methyl-1,2-dioxohexan-3-yl)amino)-4-methyl-1-oxopentan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)carbamic acid benzyl ester (41)
White solid. Yield 32%. 1H NMR (400 MHz, CDCl3) δ 8.89 (s, 1H), 8.52–8.43 (m, 1H), 7.55–7.27 (m, 11H), 7.26–7.18 (m, 1H), 6.96–6.60 (m, 1H), 6.41–6.21 (m, 1H), 5.46–5.35 (m, 1H), 5.14–5.05 (m, 2H), 4.54–4.36 (m, 1H), 4.21–4.05 (m, 1H), 1.77–1.63 (m, 6H), 1.52–1.41 (m, 3H), 1.06–0.76 (m, 18H). HRMS (ESI): m/z (M + Na)+ calcd for C39H50N4NaO6, 693.8273; found, 693.3616.
7.1.8.27. ((S)-1-(((S)-1-(((S)-1-((2,3-dihydro-1H-inden-4-yl)amino)-5-methyl-1,2-dioxohexan-3-yl)amino)-4-methyl-1-oxopentan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)carbamic acid benzyl ester (42)
White solid. Yield 6%. 1H NMR (400 MHz, CDCl3) δ 8.52 (d, J = 6.1 Hz, 1H), 7.93 (d, J = 7.9 Hz, 1H), 7.40–7.28 (m, 4H), 7.22–7.13 (m, 1H), 7.09–7.03 (m, 1H), 6.95–6.70 (m, 1H), 6.38–6.27 (m, 1H), 5.45–5.33 (m, 1H), 5.15–5.06 (m, 2H), 4.53–4.40 (m, 1H), 4.22–4.09 (m, 1H), 3.01–2.77 (m, 4H), 2.19–2.06 (m, 2H), 1.87–1.48 (m, 9H), 1.11–0.68 (m, 18H). HRMS (ESI): m/z (M + Na)+ calcd for C36H50N4NaO6, 657.7952; found, 657.3691.
7.1.8.28. ((S)-1-(((S)-1-(((S)-1-((3-ethylphenyl)amino)-5-methyl-1,2-dioxohexan-3-yl)amino)-4-methyl-1-oxopentan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)carbamic acid benzyl ester (43)
White solid. Yield 12%. 1H NMR (400 MHz, CDCl3) δ 8.68 (s, 1H), 7.55–7.41 (m, 2H), 7.40–7.36 (m, 1H), 7.35–7.31 (m, 2H), 7.31–7.24 (m, 1H), 7.04 (d, J = 7.5 Hz, 1H), 6.94–6.85 (m, 1H), 6.66–6.51 (m, 1H), 5.46–5.28 (m, 2H), 5.13 (s, 2H), 4.58–4.46 (m, 1H), 4.30–4.16 (m, 1H), 2.67 (q, J = 7.5 Hz, 2H), 1.85–1.47 (m, 9H), 1.25 (t, J = 7.6 Hz, 3H), 1.11–0.85 (m, 18H). 13C NMR (101 MHz, CDCl3) δ 196.79, 172.49, 171.76, 156.86, 156.46, 145.73, 136.29, 136.16, 129.23, 128.71, 128.42, 128.19, 125.19, 119.51, 117.40, 67.36, 53.79, 53.02, 51.70, 41.34, 40.63, 40.21, 28.98, 25.40, 24.85, 23.36, 23.04, 22.88, 22.25, 22.08, 21.51, 15.62. HRMS (ESI): m/z (M + H)+ calcd for C35H51N4O6, 623.8026; found, 623.3800.
7.1.8.29. ((S)-1-(((S)-1-(((S)-1-((3,5-dimethoxyphenyl)amino)-5-methyl-1,2-dioxohexan-3-yl)amino)-4-methyl-1-oxopentan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)carbamic acid benzyl ester (44)
White solid. Yield 65%. 1H NMR (400 MHz, CDCl3) δ 8.59 (s, 1H), 7.41–7.29 (m, 5H), 6.85 (d, J = 2.2 Hz, 2H), 6.77–6.70 (m, 1H), 6.45–6.33 (m, 1H), 6.31–6.27 (m, 1H), 5.46–5.32 (m, 1H), 5.16–5.07 (m, 2H), 4.51–4.39 (m, 1H), 4.23–4.09 (m, 1H), 3.80 (s, 6H), 1.81–1.62 (m, 2H), 1.55–1.44 (m, 2H), 1.04–0.79 (m, 18H). HRMS (ESI): m/z (M + Na)+ calcd for C35H50N4NaO8, 677.78327; found,677.35193.
7.1.8.30. N-((S)-1-(((S)-1-(((S)-1-((3,5-dimethoxyphenyl)amino)-5-methyl-1,2-dioxohexan-3-yl)amino)-4-methyl-1-oxopentan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)5-(4-acetylphenyl)-furan-2-carboxamide (45)
White solid. Yield 6%. 1H NMR (400 MHz, CDCl3) δ 8.59 (s, 1H), 8.02 (d, J = 8.4 Hz, 2H), 7.80 (d, J = 8.4 Hz, 2H), 7.25–7.23 (m, 1H), 6.89 (d, J = 3.6 Hz, 1H), 6.86–6.83 (m, 2H), 6.82–6.77 (m, 1H), 6.68 (d, J = 7.3 Hz, 1H), 6.57–6.51 (m, 1H), 6.31–6.27 (m, 1H), 5.46–5.38 (m, 1H), 4.70–4.62 (m, 1H), 4.52–4.43 (m, 1H), 3.79 (s, 6H), 2.64 (s, 3H), 1.82–1.64 (m, 7H), 1.58–1.48 (m, 2H), 1.10–0.81 (m, 18H). HRMS (ESI): m/z (M + H)+ calcd for C40H53N4O9, 733.8702; found, 733.3821.
7.1.8.31. N-((S)-1-(((S)-1-(((S)-1-((3-methoxyphenyl)amino)-5-methyl-1,2-dioxohexan-3-yl)amino)-4-methyl-1-oxopentan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)5-(4-acetylphenyl)-furan-2-carboxamide (47)
White solid. Yield 11%. 1H NMR (400 MHz, CDCl3) δ 8.62 (s, 1H), 8.02 (d, J = 8.4 Hz, 2H), 7.81 (d, J = 8.5 Hz, 2H), 7.37–7.32 (m, 1H), 7.29–7.26 (m, 1H), 7.25–7.23 (m, 1H), 7.15–7.07 (m, 1H), 6.94–6.85 (m, 2H), 6.76–6.71 (m, 1H), 6.70–6.65 (m, 1H), 6.64–6.55 (m, 1H), 5.49–5.38 (m, 1H), 4.71–4.62 (m, 1H), 4.52–4.42 (m, 1H), 3.82 (s, 3H), 2.64 (s, 3H), 1.86–1.67 (m, 6H), 1.65–1.47 (m, 3H), 1.10–0.73 (m, 18H). HRMS (ESI): m/z (M + Na)+ calcd for C39H50N4NaO8, 725.8261; found, 725.3524.
7.2. Biological assays
7.2.1. Proteasome-Glo™ chymotrypsin-like, trypsin-like and caspase-like cell-based assays
The cell based proteasome inhibition were assayed following the promega protocol. 1250H226 cells were plated in 45 μL/well in a 96-well plate. Cells were incubated at 37 °C, 5% CO2 overnight. And then incubated with 5 μL/well various inhibitor concentrations for 105 min with two repetitions of each. 50 μL/well of Proteasome-Glo™ cell-based reagent containing luminogenic substrate Suc-LLVY-aminoluciferin for chymotrypsin-like, Z-LRR-aminoluciferin for trypsin-like, and Z-nLPnLD-aminoluciferin for caspase-like activity was added. Incubate at room temperature for 10 min. The luminescence was measured with a plate-reader. The IC50 values were calculated by GraphPad Prism software.
7.2.2. Anticancer cell assay
RPMI8226, Ramos, THP-1 were grown in RPMI 1640 supplemented with 10% fetal bovine serum at 37 °C in 5% CO2. MGC-803, HeLa, SW480 were grown in DMEM supplemented with 10% fetal bovine serum at 37 °C in 5% CO2.
A standard MTS assay was used to measure cancer cell growth. A 100 μL RPMI8226 (1∗104/well), THP-1(5∗104/well), Ramos (5∗104/well), MGC-803 (5∗103/well), HeLa (5∗103/well), SW480 cells (5∗103/well) were seeded into 96-well plates. After treated with tested compounds for 72 h, an amount of 20 μL of a solution of MTS was added to each well and incubated for another 3 h at 37 °C. Optical density was determined at 490 nm by microplate reader. The assays were done in triplicate with triplicate wells per experiment. The IC50 values were calculated by GraphPad Prism software.
7.2.3. Plaque reduction assay
Plaque reduction assay was performed to plot the 50% antiviral effective dose (EC50) as we described previously with slight modifications [[46], [47], [48]]. Briefly, Vero E6 cells were seeded at 4 × 105 cells/well in 12-well tissue culture plates on the day before carrying out the assay. After 24 h of incubation, 50 plaque-forming units (PFU) of SARS-CoV-2 were added to the cell monolayer with or without the addition of drug compounds and the plates were further incubated for 1 h at 37 °C in 5% CO2 before removal of unbound viral particles by aspiration of the media and washing once with DMEM. Monolayers were then overlaid with media containing 1% low melting agarose (Cambrex Corporation, New Jersey, USA) in DMEM and appropriate concentrations of individual compound, inverted and incubated as above for another 72 h. The wells were then fixed with 10% formaldehyde (BDH, Merck, Darmstadt, Germany) overnight. After removal of the agarose plugs, the monolayers were stained with 0.7% crystal violet (BDH, Merck) and the plaques counted. The percentage of plaque inhibition relative to the control (i.e. without the addition of compound) wells were determined for each drug compound concentration. The EC50 was calculated using Sigma plot (SPSS) in an Excel add-in ED50V10. The plaque reduction assay experiments were performed in triplicate and repeated twice for confirmation.
7.2.4. Cell viability assay
Before performing the above described plaque reduction assay, the CellTiterGlo luminescent assay (Promega Corporation, Madison, WI, USA) was conducted to detect the cytotoxicity of the selected compound as we described previously [49]. Briefly, Vero E6 cells and/or Caco-2 cells (2 × 104 cells/well) were incubated with the serially diluted compound for 48 h, followed by addition of substrate and measurement of luminance 10 min according to manufacturer’s protocol. The 50% cytotoxic concentrations (CC50) of the drug were calculated by GraphPad Prism software.
7.3. Molecular modeling
7.3.1. CoMFA model
The CoMFA model is built by SYBYL_X 2.0 suite software (Tripos Inc., USA). The set of compounds (25 in total) are minimized with the Tripos force field and Gasteiger-Hückel charge. The conformation of compound BSc2198 is used as the template, the other 24 compounds are aligned by fitting the three chiral carbon atoms. CoMFA descriptors calculation and partial least-squares (PLS) model fitting are conducted in the QSAR module of SYBYL suite. SAMPLS with leave-one-out (LOO) are carried out to calculate the cross-validation value q2. The conventional correlation coefficient r2 is then calculated with no validation using the optimum number of component. In order to improve reduce ‘noise’, a minimum filter value of 2.00 kcal/mol is used.
7.3.2. Molecular docking
The covalent docking studies are performed with GOLD version 2020.1. The crystal of human 20S proteasome complex with oprozomib (PDB: 5LEY) [41] is used for the docking studies. The initial structure of protein and ligands for docking are prepared using the software SYBYL_X 2.0. A radius of 10 Å from the centroid of the co-crystallized ligand is regarded as the docking site. The GA runs are 50, the scoring function is ChemScore, and the other parameters are keep the default. The carbonyl group of the α-Ketoamide is the link atom that binds to the hydroxyl oxygen of Thr1.
The crystal structure of m-calpain (PDB: 3BOW) [44] was used for 17 and MG132 docking studies. The initial structure of protein and ligands for docking were prepared using the software SYBYL_X 2.0. The residue Ser105 of m-calpain was mutated to Cys105 before docking. The hydrogen atom attached to the sulfur atom of Cys105 was removed. The carbonyl group (C O) in the aldehyde group of MG132 or in the α-Ketoamide of our proteasome inhibitors was replaced by carbon-sulfur (C–S) and carbon-oxygen (C–O). The sulfur atoms of Cys105 and MG132 or our proteasome inhibitors were set as the protein link atom and ligand link atom, respectively. The binding site was defined as 12 Å around the sulfur atom of Cys105. GoldScore scoring function was used and the number of GA runs was 50. The side chains of Cys105 was set to be flexible with ‘crystal mode’, and the other parameters were set as defaults.
The crystal structure of SARS-CoV-2 main protease (PDB: 6Y2F) [43]was used for compound 17 docking studies. The binding site was defined as 10 Å around the sulfur atom of Cys145. ChemScore scoring function was used and the number of GA runs was 50 and the other parameters were set as defaults.
7.3.3. Molecular dynamics simulation
The covalent docking complexes of human 20S proteasome with 46 are used as the initial structures in the MD simulation. The calculations are carried out with MOLARIS package version 9.15. The simulation systems are solvated by a standard MOLARIS surface constrained all-atom solvent (SCAAS) model centered around the ligand. This involved a simulation sphere of 18 Å with a 2 Å surface constraint region surrounded by a 2 Å Langevin dipole surface. The MD simulations are carried out with the ENZYMIX force field. All complexes are relaxed for 0.5 ns, with 1 fs time step. The simulations are conducted at 30 K.
It should be noted that much more quantitative docking calculations can be performed in the future, combining the empirical valence bond (EVB) method and binding calculations. This is in particular feasible now following a very recent EVB study of the mechanism of the proteasome [50].
Author contributions
J.W. performed chemical syntheses and wrote the manuscript; B.L., L.N. participated in chemical syntheses; Y.C., H.Y. performed the proteasome inhibition and cancer cell toxicity experiments; J.F.W.C., S.Y. performed anti-SARS-CoV-2 assays; J.Z., Y.W., M.W., L.S.H., J.A. and A.W. performed the molecular modeling studies and data analysis; K.Y.Y., A.C., Z.H, and Y. X. oversaw the project and revised the manuscript.
Funding sources
This work was supported by the grant from Guangdong province (grant no. 00201503).
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ejmech.2021.113267.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Hershko A., Ciechanover A. The ubiquitin system. Annu. Rev. Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 2.Livneh I., Cohen-Kaplan V., Cohen-Rosenzweig C., Avni N., Ciechanover A. The life cycle of the 26S proteasome: from birth, through regulation and function, and onto its death. Cell Res. 2016;26:869–885. doi: 10.1038/cr.2016.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rousseau A., Bertolotti A. Regulation of proteasome assembly and activity in health and disease. Nat. Rev. Mol. Cell Biol. 2018;19:697–712. doi: 10.1038/s41580-018-0040-z. [DOI] [PubMed] [Google Scholar]
- 4.Cromm P.M., Crews C.M. The proteasome in modern drug discovery: second life of a highly valuable drug target. ACS Cent. Sci. 2017;3:830–838. doi: 10.1021/acscentsci.7b00252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Groll M., Ditzel L., Löwe J., Stock D., Bochtler M., Bartunik H.D., Huber R. Structure of 20S proteasome from yeast at 2.4Å resolution. Nature. 1997;386:463–471. doi: 10.1038/386463a0. [DOI] [PubMed] [Google Scholar]
- 6.Löwe J., Stock D Fau - Jap B., Jap B Fau - Zwickl P., Zwickl P Fau - Baumeister W., Baumeister W Fau - Huber R., Huber R. Crystal structure of the 20S proteasome from the archaeon T. acidophilum at 3.4 A resolution. Science. 1995;268:533–539. doi: 10.1126/science.7725097. [DOI] [PubMed] [Google Scholar]
- 7.Orlowski M. The multicatalytic proteinase complex, a major extralysosomal proteolytic system. Biochemistry. 1990;29:10289–10297. doi: 10.1021/bi00497a001. [DOI] [PubMed] [Google Scholar]
- 8.Isabel S., David V. The mechanistic links between proteasome activity, aging and agerelated diseases. Curr. Genom. 2014;15:38–51. doi: 10.2174/138920291501140306113344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Murata S., Sasaki K., Kishimoto T., Niwa S.-i., Hayashi H., Takahama Y., Tanaka K. Regulation of CD8+ T cell development by thymus-specific proteasomes. Science. 2007;316:1349–1353. doi: 10.1126/science.1141915. [DOI] [PubMed] [Google Scholar]
- 10.Groettrup M., Kirk C.J., Basler M. Proteasomes in immune cells: more than peptide producers? Nat. Rev. Immunol. 2010;10:73–78. doi: 10.1038/nri2687. [DOI] [PubMed] [Google Scholar]
- 11.Muchamuel T., Basler M., Aujay M.A., Suzuki E., Kalim K.W., Lauer C., Sylvain C., Ring E.R., Shields J., Jiang J., Shwonek P., Parlati F., Demo S.D., Bennett M.K., Kirk C.J., Groettrup M. A selective inhibitor of the immunoproteasome subunit LMP7 blocks cytokine production and attenuates progression of experimental arthritis. Nat. Med. 2009;15:781–787. doi: 10.1038/nm.1978. [DOI] [PubMed] [Google Scholar]
- 12.Huber E.M., Groll M. Inhibitors for the immuno- and constitutive proteasome: current and future trends in drug development. Angew. Chem. Int. Ed. 2012;51:8708–8720. doi: 10.1002/anie.201201616. [DOI] [PubMed] [Google Scholar]
- 13.Ettari R., Zappalà M., Grasso S., Musolino C., Innao V., Allegra A. Immunoproteasome-selective and non-selective inhibitors: a promising approach for the treatment of multiple myeloma. Pharmacol. Ther. 2018;182:176–192. doi: 10.1016/j.pharmthera.2017.09.001. [DOI] [PubMed] [Google Scholar]
- 14.LeBlanc R., Catley L.P., Hideshima T., Lentzsch S., Mitsiades C.S., Mitsiades N., Neuberg D., Goloubeva O., Pien C.S., Adams J., Gupta D., Richardson P.G., Munshi N.C., Anderson K.C. Proteasome inhibitor PS-341 inhibits human myeloma cell growth in vivo and prolongs survival in a murine model. Canc. Res. 2002;62:4996–5000. [PubMed] [Google Scholar]
- 15.Adams J., Behnke M., Chen S., Cruickshank A.A., Dick L.R., Grenier L., Klunder J.M., Ma Y.-T., Plamondon L., Stein R.L. Potent and selective inhibitors of the proteasome: dipeptidyl boronic acids. Bioorg. Med. Chem. Lett. 1998;8:333–338. doi: 10.1016/s0960-894x(98)00029-8. [DOI] [PubMed] [Google Scholar]
- 16.Kuhn D.J., Chen Q., Voorhees P.M., Strader J.S., Shenk K.D., Sun C.M., Demo S.D., Bennett M.K., van Leeuwen F.W.B., Chanan-Khan A.A., Orlowski R.Z. Potent activity of carfilzomib, a novel, irreversible inhibitor of the ubiquitin-proteasome pathway, against preclinical models of multiple myeloma. Blood. 2007;110:3281–3290. doi: 10.1182/blood-2007-01-065888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chauhan D., Tian Z., Zhou B., Kuhn D., Orlowski R., Raje N., Richardson P., Anderson K.C. In vitro and in vivo selective antitumor activity of a novel orally bioavailable proteasome inhibitor MLN9708 against multiple myeloma cells. Clin. Canc. Res. 2011;17:5311–5321. doi: 10.1158/1078-0432.CCR-11-0476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ho Y.K., Bargagna-Mohan P., Wehenkel M., Mohan R., Kim K.-B. LMP2-Specific inhibitors: chemical genetic tools for proteasome biology. Chem. Biol. 2007;14:419–430. doi: 10.1016/j.chembiol.2007.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Groll M., Larionov O.V., Huber R., de Meijere A. Inhibitor-binding mode of homobelactosin C to proteasomes: new insights into class I MHC ligand generation. Proc. Natl. Acad. Sci. U.S.A. 2006;103:4576–4579. doi: 10.1073/pnas.0600647103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gräwert M.A., Groll M. Exploiting nature’s rich source of proteasome inhibitors as starting points in drug development. Chem. Commun. 2012;48:1364–1378. doi: 10.1039/c1cc15273d. [DOI] [PubMed] [Google Scholar]
- 21.Lee M.J., Bhattarai D., Yoo J., Miller Z., Park J.E., Lee S., Lee W., Driscoll J.J., Kim K.B. Development of novel epoxyketone-based proteasome inhibitors as a strategy to overcome cancer resistance to carfilzomib and bortezomib. J. Med. Chem. 2019;62:4444–4455. doi: 10.1021/acs.jmedchem.8b01943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Braun H.A., Umbreen S., Groll M., Kuckelkorn U., Mlynarczuk I., Wigand M.E., Drung I., Kloetzel P.M., Schmidt B. Tripeptide mimetics inhibit the 20 S proteasome by covalent bonding to the active threonines. J. Biol. Chem. 2005;280:28394–28401. doi: 10.1074/jbc.M502453200. [DOI] [PubMed] [Google Scholar]
- 23.Stein M.L., Cui H., Beck P., Dubiella C., Voss C., Krüger A., Schmidt B., Groll M. Systematic comparison of peptidic proteasome inhibitors highlights the α-ketoamide electrophile as an auspicious reversible lead motif. Angew. Chem. Int. Ed. 2014;53:1679–1683. doi: 10.1002/anie.201308984. [DOI] [PubMed] [Google Scholar]
- 24.Voss C., Scholz C., Knorr S., Beck P., Stein M.L., Zall A., Kuckelkorn U., Kloetzel P.M., Groll M., Hamacher K., Schmidt B. alpha-Keto phenylamides as P1’-extended proteasome inhibitors. ChemMedChem. 2014;9:2557–2564. doi: 10.1002/cmdc.201402244. [DOI] [PubMed] [Google Scholar]
- 25.Ingallinella P., Fattori D., Altamura S., Steinkühler C., Koch U., Cicero D., Bazzo R., Cortese R., Bianchi E., Pessi A. Prime site binding inhibitors of a serine Protease: NS3/4A of hepatitis C virus. Biochemistry. 2002;41:5483–5492. doi: 10.1021/bi025603x. [DOI] [PubMed] [Google Scholar]
- 26.Ganesan R., Jelakovic S., Campbell A.J., Li Z.Z., Asgian J.L., Powers J.C., Grütter M.G. Exploring the S4 and S1 prime subsite specificities in caspase-3 with aza-peptide epoxide inhibitors, Biochemistry. 2006;45:9059–9067. doi: 10.1021/bi060364p. [DOI] [PubMed] [Google Scholar]
- 27.Rusere L.N., Lockbaum G.J., Lee S.-K., Henes M., Kosovrasti K., Spielvogel E., Nalivaika E.A., Swanstrom R., Yilmaz N.K., Schiffer C.A., Ali A. HIV-1 protease inhibitors incorporating stereochemically defined P2′ ligands to optimize hydrogen bonding in the substrate envelope. J. Med. Chem. 2019;62:8062–8079. doi: 10.1021/acs.jmedchem.9b00838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chan J.F.-W., Yuan S., Kok K.-H., To K.K.-W., Chu H., Yang J., Xing F., Liu J., Yip C.C.-Y., Poon R.W.-S., Tsoi H.-W., Lo S.K.-F., Chan K.-H., Poon V.K.-M., Chan W.-M., Ip J.D., Cai J.-P., Cheng V.C.-C., Chen H., Hui C.K.-M., Yuen K.-Y. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster. Lancet. 2020;395:514–523. doi: 10.1016/S0140-6736(20)30154-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ma X.Z., Bartczak A., Zhang J., Khattar R., Chen L., Liu M.F., Edwards A., Levy G., McGilvray I.D. Proteasome inhibition in vivo promotes survival in a lethal murine model of severe acute respiratory syndrome. J. Virol. 2010;84:12419–12428. doi: 10.1128/JVI.01219-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Raaben M., Posthuma C.C., Verheije M.H., te Lintelo E.G., Kikkert M., Drijfhout J.W., Snijder E.J., Rottier P.J., de Haan C.A. The ubiquitin-proteasome system plays an important role during various stages of the coronavirus infection cycle. J. Virol. 2010;84:7869–7879. doi: 10.1128/JVI.00485-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schneider M., Ackermann K., Stuart M., Wex C., Protzer U., Schätzl H.M., Gilch S. Severe acute respiratory syndrome coronavirus replication is severely impaired by MG132 due to proteasome-independent inhibition of M-calpain. J. Virol. 2012;86:10112–10122. doi: 10.1128/JVI.01001-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barnard D.L., Hubbard J., Fau - Burton Vd, Burton J Fau - Smee D.F., Smee Df Fau - Morrey J.D., Morrey Jd Fau - Otto M.J., Otto Mj Fau - Sidwell R.W., Sidwell R.W. Inhibition of severe acute respiratory syndrome-associated coronavirus (SARSCoV) by calpain inhibitors and beta-D-N4-hydroxycytidine. Antivir. Chem. Chemother. 2004;15:15–22. doi: 10.1177/095632020401500102. [DOI] [PubMed] [Google Scholar]
- 33.Zhou P., Yang X.-L., Wang X.-G., Hu B., Zhang L., Zhang W., Si H.-R., Zhu Y., Li B., Huang C.-L., Chen H.-D., Chen J., Luo Y., Guo H., Jiang R.-D., Liu M.-Q., Chen Y., Shen X.-R., Wang X., Zheng X.-S., Zhao K., Chen Q.-J., Deng F., Liu L.-L., Yan B., Zhan F.-X., Wang Y.-Y., Xiao G.-F., Shi Z.-L. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chan J.F.-W., Kok K.-H., Zhu Z., Chu H., To K.K.-W., Yuan S., Yuen K.-Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microb. Infect. 2020;9:221–236. doi: 10.1080/22221751.2020.1719902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fu H.Y., Doucet H. Methyl 2-furoate: an alternative reagent to furan for palladium-catalysed direct arylation. Eur. J. Org Chem. 2011:7163–7173. 2011. [Google Scholar]
- 36.Yilmaz V.T., Icsel C., Turgut O.R., Aygun M., Evren E., Ozdemir I. Synthesis, structures and catalytic activity of Pd(II) saccharinate complexes with monophosphines in direct arylation of five-membered heteroarenes with aryl bromides. Inorg. Chim. Acta. 2020;500:11920. [Google Scholar]
- 37.Pacifico S., Ferretti V., Albanese V., Fantinati A., Gallerani E., Nicoli F., Gavioli R., Zamberlan F., Preti D., Marastoni M. Synthesis and biological activity of peptide α-ketoamide derivatives as proteasome inhibitors. ACS Med. Chem. Lett. 2019;10:1086–1092. doi: 10.1021/acsmedchemlett.9b00233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Besse A., Besse L., Kraus M., Mendez-Lopez M., Bader J., Xin B.-T., de Bruin G., Maurits E., Overkleeft H.S., Driessen C. Proteasome inhibition in multiple myeloma: head-to-head comparison of currently available proteasome inhibitors. Cell Chem. Biol. 2019;26:340–351. doi: 10.1016/j.chembiol.2018.11.007. [DOI] [PubMed] [Google Scholar]
- 39.Dubiella C., Cui H., Gersch M., Brouwer A.J., Sieber S.A., Kruger A., Liskamp R.M., Groll M. Selective inhibition of the immunoproteasome by ligand-induced crosslinking of the active site. Angew. Chem. Int. Ed. 2014;53:11969–11973. doi: 10.1002/anie.201406964. [DOI] [PubMed] [Google Scholar]
- 40.Ladi E., Everett C., Stivala C.E., Daniels B.E., Durk M.R., Harris S.F., Huestis M.P., Purkey H.E., Staben S.T., Augustin M., Blaesse M., Steinbacher S., Eidenschenk C., Pappu R., Siu M. Design and evaluation of highly selective human immunoproteasome inhibitors reveal a compensatory process that preserves immune cell viability. J. Med. Chem. 2019;62:7032–7041. doi: 10.1021/acs.jmedchem.9b00509. [DOI] [PubMed] [Google Scholar]
- 41.Schrader J., Henneberg F., Mata R.A., Tittmann K., Schneider T.R., Stark H., Bourenkov G., Chari A. The inhibition mechanism of human 20S proteasomes enables next-generation inhibitor design. Science. 2016;353:594–598. doi: 10.1126/science.aaf8993. [DOI] [PubMed] [Google Scholar]
- 42.Ma C., Sacco M.D., Hurst B., Townsend J.A., Hu Y., Szeto T., Zhang X., Tarbet B., Marty M.T., Chen Y., Wang J. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020;30:678–692. doi: 10.1038/s41422-020-0356-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., Becker S., Rox K., Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020;368:409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hanna R.A., Campbell R.L., Davies P.L. Calcium-bound structure of calpain and its mechanism of inhibition by calpastatin. Nature. 2008;456:409–412. doi: 10.1038/nature07451. [DOI] [PubMed] [Google Scholar]
- 45.Stubba D., Bensinger D., Steinbacher J., Proskurjakov L., Salcedo Gomez A., Schmidt U., Roth S., Schmitz K., Schmidt B. Cell-based optimization of covalent reversible ketoamide inhibitors bridging the unprimed to the primed site of the proteasome beta5 subunit. ChemMedChem. 2019;14:2005–2022. doi: 10.1002/cmdc.201900472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yuan S., Chu H., Chan J.F., Ye Z.W., Wen L., Yan B., Lai P.M., Tee K.M., Huang J., Chen D., Li C., Zhao X., Yang D., Chiu M.C., Yip C., Poon V.K., Chan C.C., Sze K.H., Zhou J., Chan I.H., Kok K.H., To K.K., Kao R.Y., Lau J.Y., Jin D.Y., Perlman S., Yuen K.Y. SREBP-dependent lipidomic reprogramming as a broad-spectrum antiviral target. Nat. Commun. 2019;10:120. doi: 10.1038/s41467-018-08015-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yuan S., Chan C.C.-Y., Chik K.K.-H., Tsang J.O.-L., Liang R., Cao J., Tang K., Cai J.-P., Ye Z.-W., Yin F., To K.K.-W., Chu H., Jin D.-Y., Hung I.F.-N., Yuen K.-Y., Chan J.F.-W. Broad-Spectrum host-based antivirals targeting the interferon and lipogenesis pathways as potential treatment options for the pandemic coronavirus disease 2019 (COVID-19) Viruses. 2020;12:628. doi: 10.3390/v12060628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yuan S., Chan J.F.W., Chik K.K.H., Chan C.C.Y., Tsang J.O.L., Liang R., Cao J., Tang K., Chen L.-L., Wen K., Cai J.-P., Ye Z.-W., Lu G., Chu H., Jin D.-Y., Yuen K.-Y. Discovery of the FDA-approved drugs bexarotene, cetilistat, diiodohydroxyquinoline, and abiraterone as potential COVID-19 treatments with a robust two-tier screening system. Pharmacol. Res. 2020;159:104960. doi: 10.1016/j.phrs.2020.104960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chan J.F.W., Chan K.-H., Kao R.Y.T., To K.K.W., Zheng B.-J., Li C.P.Y., Li P.T.W., Dai J., Mok F.K.Y., Chen H., Hayden F.G., Yuen K.-Y. Broad-spectrum antivirals for the emerging Middle East respiratory syndrome coronavirus. J. Infect. 2013;67:606–616. doi: 10.1016/j.jinf.2013.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Saha A., Oanca G., Mondal D., Warshel A. Exploring the proteolysis mechanism of the proteasomes. J. Phys. Chem. B. 2020;124:5626–5635. doi: 10.1021/acs.jpcb.0c04435. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.