

Abstract
UV-damaged DNA binding protein (UV-DDB) is a heterodimeric complex, composed of DDB1 and DDB2, and is involved in global genome nucleotide excision repair. Mutations in DDB2 are associated with xeroderma pigmentosum complementation group E. UV-DDB forms a ubiquitin E3 ligase complex with cullin-4A and RBX that helps to relax chromatin around UV-induced photoproducts through the ubiquitination of histone H2A. After providing a brief historical perspective on UV-DDB, we review our current knowledge of the structure and function of this intriguing repair protein. Finally, this article discusses emerging data suggesting that UV-DDB may have other non-canonical roles in base excision repair and the etiology of cancer.
1. Introduction.
Life has evolved a series of pathways to remove specific types of DNA damage. Nucleotide excision repair (NER) is dedicated to the removal of a wide variety of helix distorting lesions, including: ultraviolet (UV) photolesions, cyclobutane pyrimidine dimers (CPD) and 6–4 photoproducts (6–4PP); bulky adducts formed by chemical carcinogens, such as polycyclic aromatic hydrocarbons; and certain DNA lesions formed from chemotherapeutic agents, like cisplatin. NER consists of two sub-pathways: global genome NER (GG-NER) and transcription-coupled NER (TC-NER). GG-NER detects and repairs DNA damage occurring in the entire genome, while TC-NER recognizes and repairs lesions in the transcribed DNA strand of active genes, reviewed in [1] and [2], respectively.
GG-NER of UV-induced photoproducts is initiated by the UV-damaged DNA binding protein (UV-DDB), which is a heterodimeric protein consisting of DDB1 (127kDa) and DDB2 (48kDa). UV-DDB is part of a larger complex containing cullin-4A/4B and RBX1 that possess E3 ligase activity, and associates with chromatin in response to UV radiation [3, 4], see Fig. 1. UV-DDB ubiquitinates histones to destabilize the nucleosome, thereby allowing downstream repair proteins, such as XPC, to access the lesion [5, 6] (Fig. 1). Defects in NER proteins are associated with a rare autosomal recessive disorder called xeroderma pigmentosum (XP), characterized by extreme sensitivity to sunlight-induced skin pigmentation changes and increased risk of skin cancer [7]. In particular, mutations in DDB2 are associated with xeroderma pigmentosum complementation group E (XP-E) [7].
Fig. 1.

Canonical role of UV-DDB in global genome nucleotide excision repair of UV-induced photoproducts. Two major UV-induced photoproducts, (A) cyclobutane pyrimidine dimer (CPD), PDB code: 1N4E; and (B) 6–4 pyrimidine-pyrimidone product, (6–4PP), PDB code: 3EI1. (C) UV-DDB (DDB1, green; DDB2, red) forms a complex with CUL4A (tan) and RBX (pink) to form an E3 ubiquitin ligase. Step 1, the UV-DDB-CUL4A-RBX complex- E3 ligase (PDB code: 4A0K, [54]) recognizes UV-induced photoproducts (orange) in the context of chromatin. Step 2, the E3 ligase ubiquitinates (black star) histone H2A. Step 3, the ubiquitinated nucleosome is evicted, and DDB2 is auto-poly-ubiquitinated decreasing its binding affinity for the UV-photoproduct, [30], allowing the recruitment of XPC (blue), which is mono-ubiquitinated stabilizing its interaction with the UV-photoproduct.
After reviewing early experiments characterizing UV-DDB, this article explores structure and function studies that have given molecular insights into how UV-DDB participates in NER. We then describe a new non-canonical role of UV-DDB in base excision repair [8]. Finally, we describe provocative studies suggesting how loss of DDB2 may function in cancer development.
2. Historical perspective of UV-DDB: evidence for its canonical role in NER.
2.1. Early characterization of UV-DDB.
Chu and Chang, in 1988, used electrophoretic mobility shift assays (EMSA) to analyze proteins in HeLa cell extracts that bind avidly to UV-irradiated DNA [9]. They reported two slowly migrating protein-DNA complexes with high affinity to damaged DNA as compared to undamaged DNA. These bands were observed in extracts from all XP complementation groups (A-G), except XP-E. The absence of these bands in XP-E was corrected by the addition of extracts from other XP groups, indicating that this factor was specifically involved in recognition of UV damage. Similar studies were performed by Protic and Levine at about the same time [10]. Interestingly, a comparable DNA binding factor was purified from human placenta in the mid-1970s by Feldberg and Grossman [11]. In the early 1990s, the UV-damaged DNA binding factor, which was later shown to consist of DDB1 and DDB2, was purified from primate cell extracts [12–15]. However, there was considerable heterogeneity in the activity of these preparations.
A defect in UV-DDB binding activity in XP-E provided correlational, but not causal evidence, therefore the Linn and Holland laboratories investigated the ability of purified UV-DDB to correct the DNA repair defect in XP-E cells by microinjection [16]. Using an unscheduled DNA synthesis (UDS) assay, labeled thymidine incorporation was quantified after exposing XP-E cells (with and without injected UV-DDB) to UVC. Injection of UV-DDB protein corrected the repair deficiency in two XP-E patient cells (XP2RO and XP82TO) by ~2-fold. This stimulation of UDS was not observed in cells from other XP groups, providing direct evidence that loss of UV-DDB causes repair defects in XP-E cells [17, 18]. Shortly after, Linn’s laboratory isolated the human genes encoding DDB1 and DDB2 subunits and revealed that DDB2 is mutated in XP-E cells Chu and colleagues further studied UV-DDB in hamster cells [19], in which, similarly to XP-E cells [7, 20], the UV-DDB binding activity is deficient and inactive DDB1 is expressed. Consequently, transfection of hamster cell lines with the human DDB2 cDNA led to activation of DDB1, resulting in DNA binding activity of UV-DDB.
2.2. DDB2 mRNA is regulated by p53.
The loss of the human tumor suppressor gene p53 is one of the most common genetic alterations (41%) in human cancers [21]. In human cells, the transcription factor, p53, is stabilized in response to DNA damage, which in turn induces transcription of downstream genes, leading to cell cycle arrest and apoptosis [22]. Interestingly, like XP-E cells, p53−/− human cells are deficient in GG-NER of UV photoproducts [23, 24]. Hwang and Chu compared p53+/+ and p53−/− human fibroblast lines to demonstrate that p53 is required to induce DDB2 mRNA levels after UV and ionizing radiation (IR) [25]. The increase in DDB2 protein levels after UV damage was observed between 16–24 hours, consistent with a gradual accumulation of stable p53 protein. In contrast, DDB1 levels were not dependent on p53. The human DDB2 gene was found to have a p53 binding site on the 5’-UTR sequence [26]. Surprisingly, this regulatory region is not conserved in mice, suggesting that the mouse Ddb2 gene is not p53-dependent. Consistent with this notion, when treated with UV light, neither WT nor p53−/− mouse fibroblast lines displayed an increase in DDB2 mRNA levels. Furthermore, most mouse cells express very low levels of DDB2 and consequently have much reduced GG-NER of CPD compared to human cells. As Hanawalt and colleagues have pointed out, since the regulation of GG-NER in mice and humans is fundamentally different, it is important to be cautious when using mouse models to recapitulate certain human cancer conditions [27].
2.3. Contrasting studies suggest differential recognition of CPD and 6–4PP by UV-DDB.
Several in vitro and in vivo studies show differential stimulation of CPD and 6–4PP repair by UV-DDB [25]. While XP-E patients are deficient in GG-NER of CPDs, they also display delayed repair of 6–4PP [28]. Hence, UV-DDB might partially be required for 6–4PP repair in certain parts of the genome, thereby rendering studies in live cells or in reconstituted nucleosomes an essential step in delineating the action mechanism of UV-DDB.
To better understand the role of UV-DDB in the different repair rates for CPD and 6–4PP, the Ford and Yasui laboratories established CPD or 6–4PP specific photolyase-expressing cell lines in XP-A cells, allowing them to determine the relative affinity of UV-DDB for either of these two photoproducts in cells [29]. By exposing cells to UVC through a 3-micron filter (local UV damage) and reactivating a photoproduct specific photolyase, each type of lesion could be observed in isolation. Using this technique, it was observed that while DDB2 recognized both CPD and 6–4PP, XPC, a damage verification protein in GG-NER, only appeared to recognize 6–4PP. Overexpression of DDB2 enabled CPD recognition by XPC, suggesting that for XPC to recognize CPDs embedded in chromatin, UV-DDB must process the nucleosome, as shown by Lan et. al. [30]. Another in vivo study compared normal and XP-E fibroblasts after local UV damage induction to show a delay in the removal of 6–4PP by ~50% in XP-E cells and a delay in the recruitment of other NER factors to the lesion, indicating that UV-DDB partially stimulates 6–4PP repair in cells [31]. However, no difference in 6–4PP repair between normal and XP-E cells was observed when UVC was exposed globally to the entire plate of cells. This suggests that UV-DDB can enhance 6–4PP repair when the number of lesions is sufficiently low. It was calculated that a 30J/m2 local UV exposure induces roughly the same number of lesions as UV-DDB molecules (~105 [13]), while a global UV exposure of the same dose to the entire plate of cells induces 7-fold more 6–4PPs and therefore exceeds the number of UV-DDB molecules.
2.4. UV-DDB is an E3 ligase and ubiquitinates histone H2A.
Cellular experiments demonstrated a puzzling finding of UV-DDB levels after UV-induced DNA damage. Rapic-Otrin et. al. demonstrated that UV-DDB associates tightly with chromatin, and only DDB2 is concomitantly degraded within 3 hours of UV exposure in a dose-dependent manner [32]. They also showed that in XP-E cells with mutated DDB2, which binds poorly to UV-irradiated DNA, degradation of DDB2 does not occur. Further studies with a proteasome inhibitor, NIP-L3-VS blocked degradation of poly-ubiquitinated DDB2. This treatment also helped stabilize p53 by inhibiting MDM2-mediated degradation of p53. Other DNA damaging agents, such as IR did not show depletion of DDB2, but due to IR-induced stabilization of p53, DDB2 expression was induced as early as 12 hours after damage and was stably and significantly increased at 24 and 48 hours after IR. Finally, this study showed that UV-DDB interacts with the histone acetyl transferase, p300, via DDB1. Sugasawa and co-workers, in a study published in 2005, confirmed that UV-DDB is a ubiquitin E3 ligase, consisting of cullin4A (CUL4A) and Roc1 (RBX) that auto-ubiquitinates itself and also ubiquitinates XPC [33]. They went on to show that while mono-ubiquitinated XPC is stabilized at sites of UV-induced DNA damage, the binding of UV-DDB is destabilized, due to poly-ubiquitination of DDB2. This work led to the concept that UV-DDB needs to poly-ubiquitinate itself to allow proper hand off of UV-induced photoproducts to XPC during GG-NER, Fig. 1C [4, 34, 35]. Both CUL4A and CUL4B have been shown to interact with UV-DDB and support ubiquitination of H2A [36].
In a series of experiments, Lan et. al. showed that UV-DDB-CUL4B E3 ligase can specifically bind to mononucleosomes containing UV damage and mono-ubiquitinate histone H2A and H3 [30]. Mono-ubiquitinated H2A at Lys119 and Lys120 helps facilitate the destabilization of nucleosome containing UV-induced photoproducts, as mutating these residues to Arg prevented the dissociation of poly-ubiquitinated DDB2 from the UV damage containing nucleosome. Finally, they showed that while UV-DDB can mono-ubiquitinate H3, H3 ubiquitination is not necessary for UV-DDB-mediated destabilization of the nucleosome.
2.5. UV-DDB-dependent regulation of NER.
During the purification and characterization of proteins necessary and sufficient to reconstitute NER on naked DNA, Wood and coworkers found that UV-DDB stimulated the incorporation of radiolabeled nucleotides into UV damaged plasmids by around 2-fold, suggesting that UV-DDB might play “an accessory, but not a core role in NER” [37]. Using a fully reconstituted NER system consisting of seven purified proteins and 136 base pair (bp) DNA substrates containing either a CPD or 6–4PP lesion, Matsunaga and coworkers demonstrated that UV-DDB could stimulate excision of CPD by 5–17-fold in a reaction containing 571 fmol of UV-DDB, but only displayed a ~2 fold stimulation for 6–4PP even when used at a significantly reduced amount (0.57 – 5.7 fmol) [38]. Interestingly, higher concentrations of UV-DDB were inhibitory for 6–4PP excision, probably due to UV-DDB’s higher affinity for 6–4PP as compared to CPD, which could form abnormally tight complexes and potentially block subsequent steps in the repair process. Furthermore, using the locally induced UV damage method (described above), the authors showed that FLAG-tagged p48 (DDB2) translocated to the UV-irradiated regions immediately after irradiation, suggesting that DDB2 participates in the early steps of repair. This recruitment was observed even in cells lacking other NER components (XP-A, XP-C and XP-F cells), strongly supporting the idea that UV-DDB is upstream of other DNA repair enzymes and that its binding acts as a “first responder” at UV photoproducts. Seidman and colleagues showed that UV-DDB was also an early responder at sites of monoadducts caused by angelicin plus 365 nm light, but not at psoralen crosslinks [39]. This result is also quite interesting, indicating that UV-DDB might have a broader substrate repertoire than previously identified, which may include some forms of chemical base damage.
Hoeijmakers and Vermeulen’s group have pioneered the use of fluorescence recovery after photobleaching (FRAP) to study UV-DDB in response to UV damage in cells [40]. This was done by photobleaching a strip spanning the nucleus and monitoring the fluorescence recovery in a region of untreated and UVC-treated cells. Using this method, they determined that the binding kinetics of mCherry-tagged DDB1 is similar to DDB2 or CUL4A, demonstrating that these proteins are recruited to the UV damage sites as one large complex. Moreover, when either DDB2 or CUL4A was depleted by siRNA, UV-induced immobilization of DDB1 at the damaged region was dependent on DDB2, but not on CUL4A. Finally, they showed that the UV-induced immobilization of DDB1 is observed for up to two hours in XP-A and XP-C cells, and by four hours is reduced to that of non-irradiated cells, suggesting that this dissociation of DDB1 is not due to repair progression, but is due to the degradation of DDB2 by the 26S proteasome.
To further characterize the regulation of NER by UV-DDB, Sugasawa and colleagues used FRAP to monitor XPC-GFP in the presence or absence of DDB2 [41]. Presence of UVC abrogated the recovery of fluorescence, signifying that XPC-GFP is trapped on UV lesions. Furthermore, overexpression of DDB2 reduced the fluorescence recovery of XPC, suggesting that DDB2 either facilitates recruitment of XPC to lesions or allows it to remain longer at the lesions. To examine this, they used a 266 nm laser to locally induce UVC damage and showed that recruitment of XPC-GFP was delayed in the absence of DDB2. It is important to note that this study was performed using low doses of UVC (5–10J/m2), therefore the number of lesions generated was probably lower than the number of UV-DDB molecules. At higher doses of UVC in which the initial photoproduct density exceeds the number of UV-DDB molecules, the repair process might occur in two phases: a fast phase, stimulated by UV-DDB and a slower phase, corresponding to direct binding of the lesion by XPC. However, XPC has been suggested to be at lower concentrations in the cell than UV-DDB [42], and repair kinetics could be altered by DNA condensation into heterochromatin.
2.6. DDB2 can directly change the core histone density at UV-induced DNA lesions.
A number of chromatin remodelers have been shown to play an important role in the regulation of nucleotide excision repair (reviewed in [43]). Dantuma and colleagues identified a novel role for DDB2 in unfolding of higher order chromatin structures at the sites of UVC damage [44]. Using a LacR-tagged DDB2 construct in various cell lines consisting of integrated LacO arrays, it was shown that tethering of LacR-DDB2 can significantly reduce density of GFP-tagged H1, H2A and H4. Furthermore, the decondensation of chromatin by DDB2 was independent of DDB1-CUL4A E3 ligase activity and dependent on ATP hydrolysis, indicating that ATP-dependent chromatin remodeling factors might be involved in the process. Also, this process required poly(ADP-ribose) polymerase (PARP1) activity, which has been linked to chromatin remodeling in the context of double-strand breaks [45, 46].
Using FRAP in cells stably expressing SNAP-tagged histone 3.3, Polo and Almouzni demonstrated that DDB2 is necessary and sufficient for changing the histone density at locally induced UV damage sites, causing a local redistribution of SNAP-H3.3 [47]. Knockdown of chromatin remodeling factors, ALC1 and INO80, did not affect the histone dynamics, suggesting that DDB2 binding is upstream of any chromatin remodeling activity. Furthermore, in contrast to the previous study discussed above [44], redistribution of histone H3.3 at sites of UV damage was PARP1-independent. How PARP is mechanistically linked to chromatin remodeling during NER is still unclear and requires further investigation, see also section 4.1 below.
3. Molecular Architecture of UV-DDB: clues to damage recognition.
3.1. Structure of UV-DDB.
Human DDB1 and zebrafish DDB2 lacking the N-terminus were co-crystalized with either a 14 bp DNA containing a 6–4 photoproduct or a 16 bp DNA containing an abasic site analog, tetrahydrofuran (THF), and their structures were resolved to 2.8 and 2.3 Å, respectively, by Thoma and coworkers [48], Fig. 2A & B. Later, Yeh and coworkers solved a co-crystal structure of full-length human UV-DDB bound to a THF in a 25 bp DNA that was resolved to 2.85 Å [49]. This new structure revealed two interesting features. First, the N-terminal 66 residues of DDB2, folded into three helical segments as a triangular paddle. Second, the crystal revealed a dimer of UV-DDB (DDB1-DDB2)2 bound to two separate DNA helices. This study also used electron microscopy (EM) and atomic force microscopy (AFM) to show that UV-DDB binds DNA both in a monomeric (DDB1-DDB2) and a dimeric (DDB1-DDB2)2 form, the latter containing two DNA molecules in each complex. Together these UV-DDB structures from both laboratories revealed extensive contacts between DDB2 and the DNA strands around the damaged site and provided important clues on how UV-DDB can recognize different types of damage.
Fig. 2.

Structure of UV-DDB. (A) Molecular model of UV-DDB-CUL4A-RBX complex bound to a nucleosome. DDB1 (green); DDB2 (red); CUL4A (tan); RBX (pink); histones (blue); nucleosome DNA (gray); 6–4 photoproduct (orange). Built from PDB codes: 4A0K and 6R8Y, [54] & [61]. (B) Damage recognition interface of DDB2 (red ribbon) with tetrahydrofuran, THF (yellow)-containing DNA (gray) with adjacent 3’ base flipped out (orange). This flipped out base is stabilized by W203 (green) and an FQH knuckle (dark blue) fills the void made by the two flipped out bases. Important salt contacts with positively charged amino acids (cyan) with phosphate backbone are shown. K244 when mutated to E causes XP-E. PDB codes: 4E54 & 4E5Z, see [49].
DDB2 is organized as a seven-bladed WD40 β-propeller in which the hairpins of repeats 4–7 make extensive contacts around the damaged site. Both structures showed that the 6–4 photoproduct and the THF with the 3’ adjacent base were flipped into a hydrophobic pocket in DDB2 and the 3’ base was stabilized by a stacking interaction with Trp203, Fig. 2B. These flipped out bases leave a two-base gap in the DNA duplex, which is filled by Phe334 (F), Gln335 (Q), and His336 (H) that form a beta-hairpin knuckle-like structure that is inserted through the minor groove. The structures also revealed an extensive set of salt-bridges between the Arg112, Lys132, and Lys244 on the damaged strand, and the Arg332 and Arg370 contacting the non-damaged strand, Fig. 2B, also see Movie 1. While the Thoma structure revealed a 40° bend at the damaged site, the Yeh structures and AFM analysis did not reveal such a sharp bend for UV-DDB complexes [49, 50].
3.2. XP-E mutations give insights into DDB2 function.
Of the 15 confirmed patients with XP-E, eight amino acid changes, including several frameshift/truncation mutants, altered splice mutants and deletions affecting both alleles, have been discovered, Table 1, Fig. 3, and Movie 2. Strikingly, an XP-E patient (XP82TO) was shown to have a Lys244 to Glu mutation and this single variant was shown to be sufficient to cause loss in photolesion recognition and subsequent repair [51]. As discussed below (section 3.5), this mutant causes increased sliding on the DNA [50]. Several other DDB2 variants from XP-E patients (Arg239Ile, Asp307Tyr, Thr305Asn, Pro357Leu) map to the DDB2-DNA interface and would be expected to decrease or eliminate UV-DDB DNA binding activity. One compound mutation (a heterozygous mutation with different mutations on each allele), Asn349Δ/Leu350Pro, as well as the Arg273His variant are localized within the DWD box motif, mutations of which impair the interaction with DDB1, thus rendering the DDB2 directed ubiquitination inactive [52, 53].
Table 1.
Characterization of XP-E causing mutations
| Patient / Cell line | Allele 1 Amino Acid | Allele 2 Amino Acid♦ | Cancer | UDS | HCR | Biochemistry | Cellular Imaging |
|---|---|---|---|---|---|---|---|
| XP1GO | Thr305Asn | BCC, SCC, M [20] | NR | Reduced [20] | NR | NR | |
| XP37BE& | Arg273His | BCC, SCC [20] |
NR | Reduced [20] |
*DDB1 and Cul4A not detected by co-IP [91, 92] |
No binding to local UV damage; *Part of the DDB-CUL4A-ROC complex [42] |
|
| XP66BE& | Arg273His | M [20] | NR | Reduced [20] |
*DDB1 and Cul4A not detected by co-IP [91, 92] |
No binding to local UV damage; *Part of the DDB-CUL4A-ROC complex [42] |
|
| XP408BE/ GM01389/ GM01646 | Leu350Pro | Asn349del | BCC, SCC, M [20] | 50% [51, 93] |
Reduced [20] | EMSA, no binding activity (E) [51, 93]; DDB1and CUL4A not detected by co-IP [51, 94];no mono-Ub-H2A on chromatin after UVC [94] |
No binding to local UV damage; Fails to recruit DDB1 and Cul4A [44] |
| XP2RO/ GM02415$ | Arg273His | BCC [20, 95] | 40–60% [51, 93, 96] |
NR | EMSA, no binding activity (E) [97] *DDB1 and Cul4A not detected by co-IP [91, 92] |
No binding to local UV damage; *Part of the DDB-CUL4A-ROC complex [42] |
|
| XP3RO/GM02450$ | Arg273His | BCC [20, 95] |
40–60% [51, 96] |
NR |
*DDB1 and Cul4A not detected by co-IP [91, 92] |
No binding to local UV damage; *Part of the DDB-CUL4A-ROC complex [42]; |
|
| XP82TO | Lys244Glu | None (as of 2011) [20, 95] | 44% [51, 96] |
NR | EMSA, no binding activity (E) [28, 93, 97]; Partial binding activity detectable (P) [91]; no histone ubiquitination in nucleosome [91] |
No binding to local UV damage [44, 91]; Slides on DNA in the absence of Mg2+ [50] | |
| XP23PV | Leu235_Lys3 41del | BCC [20, 51] | 65% [51, 93] |
NR | EMSA, no binding activity (E) [51, 93] | No UV-induced chromatin decondensation [44] | |
| XP25PV | Asp307Tyr No change |
BCC, SCC [20, 51] |
50% [51, 93] |
NR | EMSA, no binding activity (E) [51, 93]; DDB1 not detected by co-IP [51] |
No binding to local UV damage; No recruitment of DDB1 and Cul4A [44] |
|
| XP27PV | Lys244X Trp236Valfs10 Leu235_ Lys341del |
BCC, SCC, M [20, 51] | 48% [51] |
NR | EMSA, no binding activity (E) [51, 93]; | NR | |
| Ops1 | Arg313X | BCC, SCC, M [20, 28, 95] |
99–138% [28] |
NR | EMSA, no binding activity (E) [28] DDB1 and Cul4A not detected by co-IP [91] |
NR | |
| XP115BR | Met383fs | None (as of 2016) [7] |
~50% [7] |
NR | NR | NR | |
| XP105BR | Pro357Leu | Arg239Ile | BCC, SCC, M [7] |
~50% [7] |
NR | NR | NR |
| XP98BR | Trp54X | BCC, SCC, M [7] |
~50% [7] |
NR | NR | NR | |
| XP100BR | Splice | BCC [7] |
~50% [7] |
NR | NR | NR |
Abbreviations: BCC, Basal cell carcinoma; SCC, Squamous cell carcinoma; M, Melanoma, UDS, Unscheduled DNA synthesis assay; HCR, Host cell reactivation; Biochemistry includes electrophoretic mobility shift assays (EMSA), western blotting and Co-IP; E: Cell extracts; P: Purified protein; Cellular Imaging examines both recruitment to local damage and FRAP experiments. NR: not reported
Siblings;
Second cousins;
empty boxes for no allele 2 amino acid indicate homozygous mutation;
contrasting results from biochemistry and cellular studies
Fig. 3.
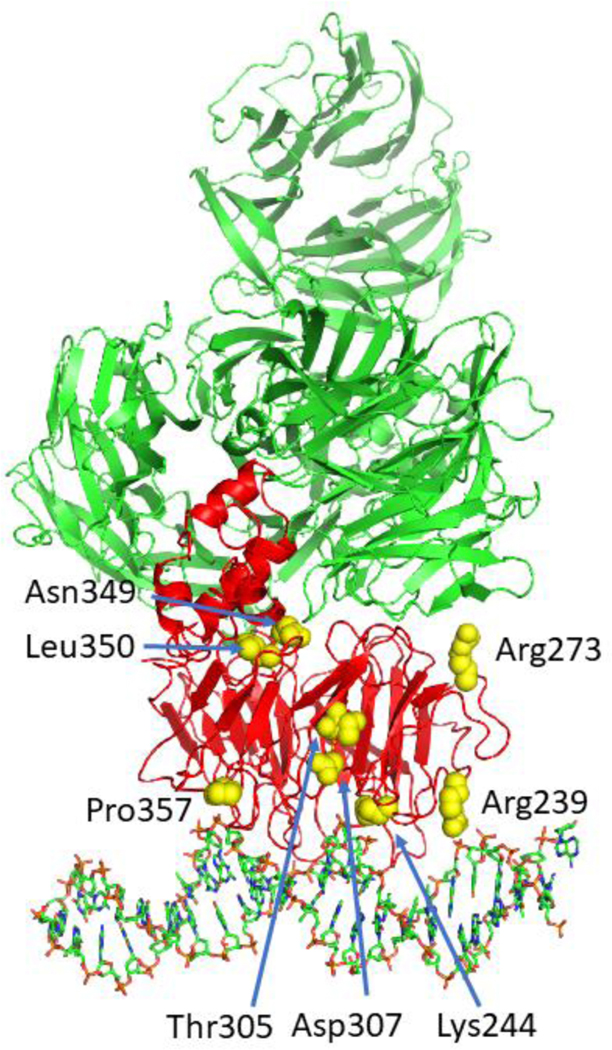
Positions of mutated amino acids in DDB2 that cause XP-E. Shown on the structure of DDB2 (red) are amino acids (yellow) that are altered from point mutations shown in Table 1. All other mutations resulting in frameshifts, stop codons and large deletions are not shown on this structure. Note that patient GM01389 apparently showed a compound mutation in one allele involving a Leu350Pro substitution and deletion of Asn349 in the other allele. Thus, both amino acids are shown in yellow in the DDB2 structure.
3.3. Structural aspects of UV-DDB-CUL4-RBX E3 ligase.
Thoma and coworkers have solved the molecular architecture of UV-DDB bound to CUL4-RBX (CRL4) which forms a CRL4DDB2 E3 ligase [54]. There are over 200 different CRLs in human cells that fall into five distinct families (CRL1,2,3,4A/B, and 5). CUL4A/B containing CRLs belong to a family of E3 ubiquitin ligases which regulate a wide range of cellular processes through dedicated substrate receptors (DCAFs). CRL4CSA and CRL4DDB2 help target this ubiquitin E3 ligase to DNA during TC-NER and GG-NER, respectively.
3.4. Cop9 signalosome as a chaperone for UV-DDB.
In the absence of DNA damage, cellular CRL4DDB2 is bound to the COP9 signalosome complex (CSN) [55], see a UV-DDB interactome in Fig. 4. The CSN is an eight subunit isopeptidase complex that through its CSN5 subunit, proteolytically removes a ubiquitin-like moiety, NEDD8 from the cullins. NEDD8 modification serves as a regulator of cullin activity. When NEDD8 is attached to CUL4, its E3 ligase is in an active mode. However, when bound by CSN complex, UV-DDB-CUL4 is rendered inactive through deneddylation. Structural studies by the Thoma laboratory revealed that DNA binding by DDB2 antagonizes its interaction with CSN3, helping to displace the CSN complex and allowing continued neddylation and activation of the E3 ligase [56]. These structural studies confirmed the earlier work by Nakatani and coworkers who showed that CSN differentially regulates CRL4CSA and CRL4DDB2 during nucleotide excision repair [57]. Knockdown of CSN5 with siRNA in BJ1 fibroblasts decreased GG-NER by 55% and TC-NER by 43%. Despite this pioneering work on these complexes in 2003, it is still not clear why removal of a negative regulator of these two E3 ligases paradoxically slows both forms of repair, rather than enhances repair. It is possible that the COP9 signalosome helps chaperone spurious DNA binding by DDB2, which has high affinity for non-damaged DNA. In this regard, it is interesting to note that van Driel and coworkers used FRAP analysis to show that the 3D diffusion of EGFP-labeled CUL4A and EYFP-labeled DDB2 were consistent with a complex of ~ 500 kDa, strongly supporting that this complex is being bound by the COP9 signalosome [42].
Fig. 4.
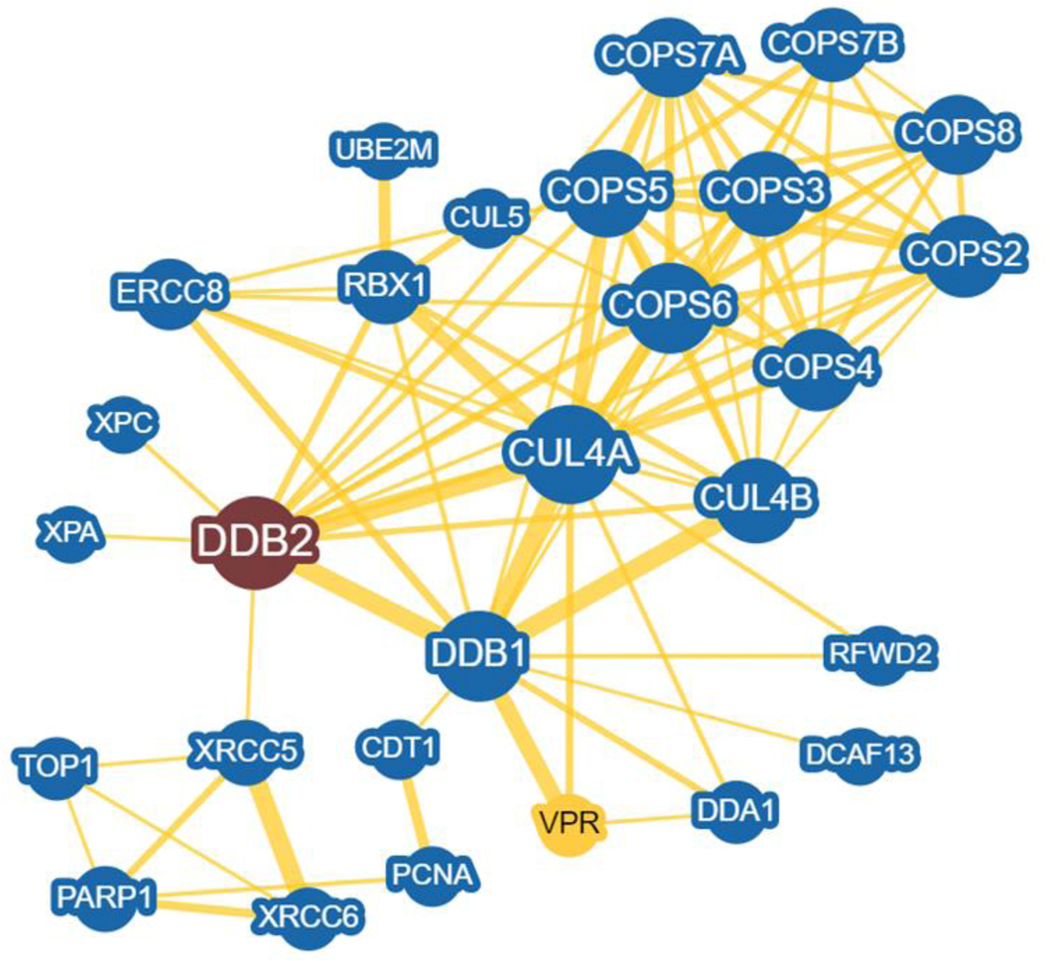
DDB2 interactome prepared in BioGRID. Human DDB2 was selected and the minimum evidence was set to 5. The interactome network centered on DDB2 (red) was created using an arbor layout. Nodes in blue = 28 are DDB1 and/or DDB2 interacting proteins in human cells. Some low interacting nodes were trimmed out of the network. These interactors are shown in yellow for direct physical evidence. Greater node size represents increased connectivity and thicker edge sizes represent increased evidence supporting the association. VPR node is in yellow and is a major HIV protein that interacts with DDB1 to help direct the ubiquitination and degradation of key cellular proteins to aid in HIV infection.
3.5. Single-molecule analysis of UV-DDB reveals 3D searching and dimerization on DNA.
As described above, early work on UV-DDB damage recognition using EMSAs indicated a major band consistent with a heterodimeric complex of DDB1 and DDB2 bound to DNA, but a more slowly migrating band is often observed at higher molecular weights consistent with either multiple DNA binding events or the formation of two UV-DDB complexes binding together. In order to investigate this phenomena in greater detail, Ghodke in our group examined UV-DDB interaction on defined substrates by two single-molecule approaches, AFM and fluorescence microscopy of quantum-dot labeled proteins on a DNA tightrope optical platform [50, 58]. AFM provides volume data on protein-DNA complexes in such a way that the volume is directly proportional to the molecular weight of the complex and therefore the oligomeric state [58]. Using AFM, Ghodke was able to show that both WT-UV-DDB and a DDB2-Lys244Glu variant were able to dimerize forming (DDB1-DDB2)2 complexes on DNA in which they bound to two DNA molecules. This Lys244Glu variant of DDB2 found in patient XP82TO was previously shown to bind weakly to DNA [51], see also Table 1 and Fig. 3. Using a fluorescence microscopy imaging platform consisting of UV-irradiated lambda DNA suspended on 5μm poly-L-lysine coated beads and Quantum-dot labeled UV-DDB, Ghodke followed association and dissociation events of both WT and DDB2 Lys244Glu variant UV-DDB in real-time. He found that while WT UV-DDB performed a 3D search, rapidly binding and releasing from DNA without sliding (see Movie 3), the XP-E mutant, variant Lys244Glu slid on DNA and was able to form dimers (see Movie 4). The dissociation rates of WT UV-DDB consisted of time intervals of ~ 1, 10, and 100 seconds. This led to the concept that UV-DDB uses “conformational proofreading” in which binding to a damaged site helps to alter the conformation of both the DNA and the protein to increase stability and therefore, the length of the interaction of UV-DDB on DNA [59]. The Lys244Glu variant was not able to bind specifically to damage sites and showed little or no long-lived pausing on DNA. Thus, it would appear that the loss of a single positively charged amino acid residue interacting with DNA is sufficient to cause UV-DDB to slide on DNA. As described below in section 5, we have found that WT UV-DDB has limited linear diffusion on DNA in the presence of Mg2+.
3.6. Interaction of UV-DDB with nucleosomes containing site-specific lesions.
Based on Lan et. al. [30], and later work by Kurumizaka, Sugasawa and Thoma [60], UV-DDB was found to bind specifically to nucleosomes containing DNA photoproducts and was proposed to be the first responder to UV-induced DNA damage in chromatin. In a major tour-de-force study, Thoma and coworkers used cryo-EM to examine UV-DDB’s interactions with a nucleosome containing defined damage sites at various positions along the nucleosome [61]. Due to the inherent wrapping of the DNA around the histone octamer, it might be expected that lesions pointing more inward would not be accessible to UV-DDB. However, UV-DDB was shown to shift the DNA register, altering the nucleosome architecture as much as by three base pairs to allow access to occluded sites.
4. Roles of post-translational modifications (PTM) in regulating UV-DDB-mediated repair.
4.1. PARylation.
UV-DDB is known to interact with a number of proteins during GG-NER, including the histone acetyl transferase p300 and the Cul4A-RBX1 complex that ubiquitinates histones (H2A, H3, H4), DDB2 and XPC in response to UV radiation [34]. Additionally, Mullenders and colleagues identified a novel role for PARP1 in PolyADP-ribosylation (PARylation) of DDB2 and subsequent recruitment of SWI/SNF chromatin remodeler, ALC1 to UV damaged DNA [62]. DDB2-associated proteins were analyzed by chromatin immunoprecipitation and mass spectrometry, revealing that PARP1 bound both DDB1 and DDB2. Consistent with the PARylation of DDB2, rapid recruitment of GFP-ALC1 to sites of UV lesions was observed and found to be DDB2-dependent. Furthermore, PAR was detected by immunofluorescence at sites of UV damage and colocalized with TFIIH. Although DDB1 was also shown to bind to PARP1, whether it is PARylated or acts in association with PARP1 was not pursued and requires further investigation.
Using FRAP, it was observed that DDB2 had a prolonged retention time on the DNA in the absence of Poly(ADP-ribose) glycohydrolase (PARG), suggesting that PARylation might stabilize the protein at the lesion site. Moreover, inhibition of PARP activity resulted in suppressed PARylation but increased ubiquitination of DDB2, indicating that PARylation may prevent DDB2 auto-ubiquitination and subsequent degradation. In contrast to the previous study, PARP1 inhibition led to extended retention of DDB2 on the damaged DNA as measured by immunoblotting [63]. It would be interesting to determine whether PARylation of UV-DDB helps prevent auto-ubiquitination and whether PARP inhibition causes more rapid degradation of UV-DDB through the 26S proteasome. This study also verified that DDB2 is PARylated after UV damage using dot blots and further showed that XPC recruitment is delayed in the absence of PARP1-mediated PARylation of DDB2 [63].
4.2. SUMOylation.
Small ubiquitin-related modifiers (SUMOs) can regulate a variety of cellular processes including transcriptional regulation, signal transduction and maintenance of genome integrity by causing rapid changes in protein-protein interactions. SUMO is covalently attached to proteins through a cascade similar to that of ubiquitination. Using co-immunoprecipitation, Iijima and colleagues showed that DDB2 is SUMOylated post-UV damage and PIASy (protein inhibitor of activated STST proteins) was the major SUMO E3 ligase involved [64]. Furthermore, knockdown of PIASy impaired the repair of CPDs, but not 6–4PP, as measured by ELISA. Future cellular studies showing a DDB2-dependent recruitment of SUMO to sites of UV damage will help clarify this PTM of UV-DDB in NER.
To this end, another study using immunoprecipitation showed DDB2 SUMOylation after UV damage and further screened the DDB2 sequence to identify the sites of SUMOylation [65]. Three lysine residues (Lys5, Lys77 and Lys309) were identified and mutated to arginine. Lys309Arg completely abolished the DDB2 modification upon UV damage. Moreover, when expressed in cells, this mutant was deficient in the removal of CPDs as well as in the recruitment of XPC as compared to the WT, indicating that the SUMOylation at Lys309 is functionally significant.
5. Novel role of UV-DDB in the repair of oxidative DNA damage.
Our genomic DNA is constantly exposed to oxidative stress in the form of reactive oxygen species (ROS) from mitochondria and pro-inflammatory processes, or from exogenous sources like UVA/B and IR. This can lead to direct oxidation of the DNA bases such as 8-oxoguanine (8-oxoG). Repair of this lesion is initiated by 8-oxoG glycosylase (OGG1), which removes the oxidized base through hydrolysis of the glycosidic bond, creating an apurinic/apyrimidinic (AP) site. This is recognized by an AP endonuclease (APE1), which incises the damaged strand, leaving a single nucleotide gap with a 3’-OH primer. DNA polymerase β (pol β) synthesizes DNA from the 3’-OH end. DNA ligase III and x-ray cross-complementing factor 1 (XRCC1) help ligate and seal the nick [66].
The earliest evidence of recognition of AP sites by UV-DDB was shown by Chu and colleagues using an electrophoresis DNA binding assay [67]. A 148 bp DNA probe was end-labeled with 32P and damaged by UV to induce CPD and 6–4PP. Unlabeled DNA containing AP sites was used as a competitor. Partially purified UV-DDB from HeLa cell extracts was run on a gel along with the DNA. While the DNA containing AP sites was able to inhibit UV-DDB binding to UV damaged DNA, the affinity was 17-fold lower for the AP sites.
More direct evidence of recognition of AP sites by UV-DDB was established separately by Fujiwara [68], as well as Wood and colleagues by using recombinant purified UV-DDB and DNA substrates with a site-specific CPD, 6–4PP and AP site [69]. The latter group observed a 6-fold higher affinity for CPD, 83-fold higher affinity for 6–4PP and a 46-fold higher affinity for an AP site as compared to undamaged DNA. A mismatch substrate was also tested with a 50-fold higher affinity as compared to undamaged DNA. This broad substrate specificity is probably because UV-DDB does not detect the damage site directly but recognizes helix-distorting lesions that can be flipped into the binding site of DDB2 (Fig. 2B).
Recently, our lab has established a novel role for UV-DDB in the repair of oxidative DNA damage [8]. Using a combination of biochemical, single-molecule and cellular studies, we demonstrated that UV-DDB can recognize and aid in the repair of 8-oxoG lesions. We further showed that when the EMSA are done in the presence of Mg2+ in the binding buffer, gel, and running buffers, UV-DDB has enhanced specificity for both abasic sites and 8-oxoG. As shown in Table 2, while Mg2+ greatly diminishes binding to a non-damaged 37 bp duplex by 26-fold from 42 nM to 1100 nM, Mg2+ only decreased binding to abasic site and 8-oxoG:C base pairs by 4–5-fold, greatly enhancing the specificity window of UV-DDB. We next showed that UV-DDB stimulated OGG1 and APE1 activity on 8-oxoG:C and abasic site containing substrates by ~3-fold and ~9-fold, respectively. Using a single-molecule DNA tightrope assay where damaged DNA is suspended between 5μm poly-L-lysine coated beads and quantum-dot labeled purified proteins are observed in real time [58], UV-DDB was found to undergo limited linear diffusion in the presence of Mg2+ in the flow cell as compared to strict 3D searching on DNA with little or no linear diffusion in the absence of Mg2+, Movie 5. We also showed that UV-DDB could form transient complexes with OGG1 and APE1 on the damaged site [8]. Moreover, UV-DDB facilitated the dissociation of these proteins from the damaged site, suggesting that UV-DDB can help turnover OGG1 and APE1 from the abasic site. Also, hTERT-immortalized fibroblasts that had greatly reduced DDB2 expression were more sensitive to potassium bromate, an oxidizing agent that forms predominantly 8-oxoG lesions in the DNA.
Table 2.
Magnesium increases the binding specificity of UV-DDB.
| Substrate | No Mg2+ | 5 mM Mg2+ | Fold Difference |
|---|---|---|---|
| 37 bp duplex | Kd1 (nM) | Kd1 (nM) | |
| THF | 0.9 ± 0.2 | 3.9 ± 0.5 | 4.3 |
| CPD | 4.5 ± 0.3 | 30.4 ± 2.4 | 6.8 |
| 8-oxoG:C | 35.3 ± 2.1 | 159.6 ± 12.4 | 4.5 |
| undamaged | 42.3 ± 2.6 | 1108 ± 95.5 | 26.2 |
CPD, cyclobutane pyrimidine dimer (thymine-thymine); THF, tetrahydrofuran.
1, Kd = equilibrium dissociation constant determined by EMSA from three separate experiments.
Finally, a novel chemoptogenetic approach was used to target 8-oxoG lesions specifically to the telomeres [70, 71]. This system uses a telomeric repeat-binding factor 1 (TRF1) fused to a fluorogen activating protein (FAP) that binds avidly to malachite green (MG-2I) dye. This TRF1-FAP-dye combination, when excited by far red wavelength (660nm) generates singlet oxygen, forming 8-oxoG lesions in telomeric DNA. Using this technique, we demonstrated that UV-DDB is recruited to 8-oxoG lesions immediately after damage. Moreover, OGG1 co-localizes with UV-DDB, but is recruited more slowly, suggesting that UV-DDB may be the first responder to base damage within chromatin. Based on these data and structural studies by Thoma’s group, we propose that UV-DDB plays two roles in the repair of 8-oxoG, Fig. 5: 1) UV-DDB recognizes the lesion directly in the nucleosome and aids in the recruitment of downstream proteins; 2) UV-DDB helps turnover OGG1 and APE1 from the abasic site to allow for coordinated and unperturbed repair. Future studies will characterize the downstream repair pathway that is being regulated by UV-DDB in 8-oxoG repair. Furthermore, it will be interesting to adapt this approach to follow the formation and repair of 8-oxoG lesions in different regions of the genome and analyze the potential differences in repair kinetics depending on the chromatin structure. Finally, since this study also demonstrated that UV-DDB stimulated MUTY removal of A across from 8-oxoG, it will be interesting to study whether UV-DDB can stimulate the nine other known mammalian glycosylases during BER [72].
Fig. 5.

New non-canonical role of UV-DDB in base excision repair (BER) of oxidative lesions. (A) 8-oxoG, a major form of base damage induced by oxidative stress. (B) Tetrahydrofuran (THF), a stable abasic site analog. (C) A working model of how UV-DDB stimulates BER removal of 8-oxoG. For details see section 5 and ref [8]. UV-DDB is the first responder at 8-oxoG sites in nuclear DNA. Biochemical and single-molecule evidence showed that UV-DDB stimulates OGG1 removal of 8-oxoG by 3-fold and APE1 by 8-fold through UV-DDB-mediated dissociation. UV-DDB stimulated the DNA polβ gap filling step of BER in vitro by 30-fold. Figure adapted from [8].
6. Provocative role of DDB2 in cancer.
6.1. Internal cancers in XP patients and potential role of DDB2 in human cancers.
While XP patients show extremely high levels of skin cancer, 14 of the 726 XP patients had brain, breast or respiratory track neoplasms [73]. More recent analysis of 31 patients indicated that four of them also displayed elevated levels of internal cancers, including T-lymphoblastic leukemia, myelodysplasia, kidney adenocarcinoma, cervical cancer and thyroid adenocarcinoma [74]. Furthermore, several XP patients were reported to die from their internal malignancies [75]. Certain complementation groups also show neurological disorders, suggesting a potential role of NER proteins in the removal of oxidative DNA lesions [7]. The number of patients reported with XP-E is extremely rare. While internal cancers have been found in XP patients, only one XP-E patient in the NIH series [described in reference 20] has shown an internal cancer – papillary thyroid carcinoma (Kenneth Kraemer, personal communication). Since thyroid cancers are not infrequent in the general population, it is not clear whether thyroid cancer in this XP-E patient is due to the loss of DDB2 activity. Additionally, XP-E patients develop a larger number of skin lesions, including melanoma, despite having later onset of skin cancers compared to XP patients in other complementation groups [7, 76]. The Cancer Genome Atlas (TCGA) data indicates that endometrial, cervical and breast cancer patients with higher levels of DDB2 mRNA expression show significantly better long-term survival (Fig. 6A).
Fig. 6.

Association of decreased DDB2 mRNA expression with increased cancer risk. (A) The Human Protein Atlas was queried for DDB2 under the Pathology tab. The prognostic summary reached high significance for patient survival in three cancer types: endometrial, cervical, and breast. Low mRNA expression (blue) indicated a poor prognosis as compared to high (pink) mRNA expression levels of DDB2. Other cancers sites showing a similar trend using Kplot.com [98], included, kidney renal clear and papillary cell, liver, lung, sarcoma, stomach, and thyroid. Linn and colleagues showed that DDB2 heterozygous and knockout mice have decreased overall survival (B) and tumor-free survival (C). From [77] with permission.
6.2. DDB2-deficient mice are more prone to UV-induced skin cancer and die prematurely due to spontaneous internal cancers.
Four independent studies have reported increased cancer risk and tumor formation in DDB2-deficient mice [77–80] exposed to UV light. A protective role for DDB2 against tumor formation was first shown in 2005 by Raychaudhuri and coworkers in which all sixteen DDB2 knockout mice developed skin tumors after 38 weeks of UVB irradiation compared to only two of fifteen wildtype and two of sixteen heterozygotes [80]. This was confirmed by Linn and coworkers using sample sizes of 40 – 77 for wildtype, heterozygote and DDB2 knockout mice treated with a dose of 2,500J/m2 of UV-B five times a week for up to 35 weeks [78]. These DDB2 knockout mice not only developed skin tumors within 13 weeks of treatment compared to 19 weeks in heterozygotes and wildtype mice, but also developed a significantly larger number of skin tumors with an average of 13 in the knockouts as compared to 0.3 in the heterozygotes and 0.08 in the wildtype mice [78]. In a similar study, the Mullender laboratory demonstrated that mice with increased DDB2 expression in skin keratinocytes (Lys14-DDB2) had increased tumor-free survival and smaller tumor sizes compared to wildtype and heterozygote mice when exposed to 500J/m2 of UVB [79]. To provide more impactful results, it would have been interesting to show that knocking out XPA in these genetic backgrounds completely abrogated the effect of DDB2 overexpression in the avoidance of UV-induced skin cancer, which would demonstrate a direct role of DDB2 in enhanced GG-NER.
In addition to DDB2’s role in cancer protection when exposed to UV irradiation, DDB2 also plays a remarkable role in protecting against spontaneous internal tumors in mice, Fig. 6B & C [77]. DDB2 knockout (KO) mice were shown to have lower overall survival and developed spontaneous tumors with a tumor-free survival of 25.3 months compared to 28.5 or 33.4 months for their heterozygote or wildtype counterparts. These mice also had a higher tumor incidence of 46% compared to 37% for heterozygotes and 25.6% for wildtype. The spontaneous tumors included adenocarcinoma of the lung and mammary gland as well as several forms of sarcoma [77]. Together these studies suggest a direct role of DDB2 in delaying internal cancers that arise spontaneously in mice. These mice data combined with our recent observation for a role of UV-DDB in stimulating base excision repair of oxidative lesions suggest that perhaps ROS arising from pro-inflammatory conditions may be sufficient to drive tumors in DDB2-deficient mice. In this regard, it would be of interest to look at the mutation signatures in the spontaneous tumors of DDB2 KO mice.
6.3. The effect of Cul4A knockout on DDB2 in cancer prevention.
As described earlier, DDB2 is regulated by Cul4A/B ubiquitin E3 ligase. Auto-ubiquitination of DDB2 with multiple ubiquitin chains causes degradation by the 26S proteasome, potentially constraining UV-DDB’s ability to promote GG-NER. Hence, contrary to the established role for the CRL4DDB2 in GG-NER (Figure 1C), it might be speculated that abrogating Cul4A ubiquitin ligase may increase DDB2 stability and enhance DNA repair, leading to cancer protection. This hypothesis was directly tested in Cul4A knockout mice, which exhibited elevated CPD repair and delayed proliferation [81]. Additionally, 13 out of 14 Cul4A knockout mice when exposed to UVB irradiation remained cancer-free compared to their 19 wildtype counterparts that all had squamous cell carcinoma within 48 weeks [81]. This study indicates that DDB2’s tumor suppression is restricted by Cul4A ubiquitin ligase and that targeting Cul4A ubiquitin ligase may serve as a novel therapy to increase DNA repair capacity.
6.4. Potential tumor suppressor: cell survival and cancer protection.
It has been indicated that DDB2 may have a wider role beyond GG-NER and act as a tumor suppressor, such that its deficiency results in increased cancer risk and tumors as described above, also recently reviewed [82]. One way DDB2 may act as a tumor suppressor is through promotion of apoptosis in damaged cells. To this end, Linn and coworkers reported a higher viable cell count from mouse embryonic fibroblasts (MEFs) isolated from DDB2 knockout mice compared to their heterozygote and wildtype mice counterparts, up to 120 hours post-UVC irradiation [78]. They also observed a significant decrease in caspase-3 activity in these knockout MEFs post-UVB irradiation. However, there are opposing results suggesting that DDB2 does not play a direct role in apoptosis. In 2005, Mullenders and coworkers showed that enhanced DDB2 expression in mouse dermal fibroblasts resulted in no significant change in apoptotic activity compared to knockout and wildtype mice after exposure to UVC irradiation [79]. The lack of apoptosis in this latter study could be due to a number of factors including: the small dose of UV irradiation, 8J/m2 of UVC, that may not have yielded sufficient lesions to see distinct differences between the knockout and wildtype cells; the differences in cells tested; and the different techniques employed for caspase-3 [78, 79].
6.5. Potential oncogene in breast cancer.
In contrast to TCGA data, which indicates that increased DDB2 mRNA expression is correlated with higher survival for breast cancer patients, Becuwe and coworkers describe DDB2 as a possible oncogene in breast cancer [83]. They found that knocking down DDB2 in MCF7 cells resulted in reduced colony formation by ~2-fold and overexpressing DDB2 in MDA-MB231 cells resulted in increased colony formation by ~3.7-fold. Additionally, DDB2 knockdown MCF7 cells had delayed cell cycle progression in flow cytometry experiments. Although these data may suggest DDB2 acts as a potential oncogene in breast cancer, future work should seek to confirm these findings in other breast cancer cell lines.
6.6. Tumor formation and metastasis.
Additional experiments in mice have suggested that loss of DDB2 facilitates metastatic colon cancer [84, 85]. Specifically, mice injected with HCT116 colon cancer cells deficient in DDB2 had larger tumor masses than those injected with HCT116 cells with wildtype DDB2 [84]. Similarly, it was demonstrated that DDB2 knockout mice treated with azoxymethane/dextran sulfate, a colon cancer carcinogen, had a higher abundance of tumors and a larger tumor mass compared to wildtype animals [85]. Staining of tumors in DDB2 KO mice indicated reduced expression of RNF43, a suppressor of Wnt signaling and displayed increased Cdx1 mRNA expression, a Wnt target gene. Furthermore, CHIP-seq data suggested that DDB2, perhaps with DDB1, binds to the upstream regulatory region of the RNF43 promoter and its knockdown in HT-29 colorectal cancer resulted in decreased ubiquitination of the Wnt receptor, thereby increasing Wnt signaling [85]. More experiments are necessary to confirm DDB2’s role in Wnt signal regulation, such as UV-DDB’s binding affinity to the promoter sequence of the Rnf43 gene.
In addition to DDB2’s role in tumor formation, independent of its E3 ligase function, DDB2 has been suggested to inhibit epithelial to mesenchymal transition (EMT) in colon cancer [84]. Interestingly, there is a negative correlation between the grade of colorectal cancer and expression of DDB2 and E-cadherin, an epithelial cell marker. This was further investigated by examining the morphology and mesenchymal/epithelial markers in HCT116 cells expressing normal or low levels of DDB2: DDB2-deficient cells had mesenchymal-like features such as elongated morphology, increased vimentin, a mesenchymal marker, and decreased E-cadherin expression. Similar findings were demonstrated in a metastatic colorectal cancer cell line, SW620 compared to an early stage colorectal cancer cell line from the same patient, SW480 [84]. Furthermore, when colorectal cancer tumors expressing normal or low levels of DDB2 were transplanted or injected into mice and examined after 4 weeks, there was a higher incidence of lung and liver metastasis in mice that received DDB2-deficient cells [84]. These findings possibly indicate a role for DDB2 in suppressing metastasis via inhibition of EMT. Future studies should examine mice for metastatic sites in other organs and over longer lengths of time. Since cancer cells often use EMT to transition to metastasis, DDB2’s role in EMT may suggest a function of DDB2 in preventing metastasis.
7. Outlook.
UV-DDB, as part of CUL4-RBX E3 ligase, plays an essential role in the removal of UV-induced photoproducts in the context of chromatin during global genome NER. Recent work from our lab suggests that UV-DDB plays a non-canonical role in BER for the removal of 8-oxoG by the stimulation of OGG1 and APE1 [8]. Mammalian cells contain 11 glycosylases and it will be of great interest to determine whether UV-DDB can stimulate these enzymes during the removal of other forms of base damage [72]. More studies are needed to understand the complete damage repertoire of UV-DDB both in naked DNA and in the context of nucleosomes. Since UV-DDB has high affinity for undamaged DNA, it is possible that its binding to non-damaged sites in the genome affects chromatin structure and influences gene expression of critical cellular genes that are altered during cellular transformation. The increase in spontaneous tumors in DDB2 knockout mice [77, 80], and their high sensitivity to UV-induced skin tumors [78], as well as the more surprising sensitivity to the azoxymethane/DSS model of colon cancer [85], needs to be studied in greater detail. If UV-DDB aids in the removal of alkylation damage in chromatin, then it is possible this latter result is due to a decrease in the removal of potentially mutagenic methylated bases. Many DNA glycosylases appear to be inhibited in their ability to remove damaged bases in the context of nucleosomes [86–90]. Since UV-DDB has been found to bind to abasic sites embedded in nucleosomes in any orientation [60] and also alter the register of DNA lesions on nucleosomes [61], it is possible that UV-DDB binding directly to damage sites or through its associated E3 ligase activity may work to facilitate the removal of damaged bases during BER. The future holds great promise for investigation of this remarkable protein.
Note Added in Proof:
While this manuscript was under consideration, three new cases of XP-E were reported. Two Chinese patients with the same homozygous deletion in DDB2 resulting in a complete loss of the protein (Yang R, Kong Q, Duan Y, Li W, Sang H. BMC Med Genet. 2020 Mar 30;21(1):67.) The second case resulted from a mutation found in a youth in Brazil resulting in a terminal truncation of the protein at residue 335 (Santiago KM, Castro LP, Neto JPD, de Nóbrega AF, Pinto CAL, Ashton-Prolla P, Pinto E Vairo F, de Medeiros PFV, Ribeiro EM, Ribeiro BFR, do Valle FF, Doriqui MJR, Leite CHB, Rocha RM, Moura LMS, Munford V, Galante PAF, Menck CFM, Rogatto SR, Achatz MI. J Eur Acad Dermatol Venereol. 2020 Apr 1.)
Supplementary Material
Acknowledgements:
We thank Dr. Emily Beckwitt for creating movie 1. We apologize in advance for any work which, due to page limitations, was not directly cited here. This work was supported by NIH grants R01ES019566 (BVH), and R01ES028686 (BVH), and 2P30CA047904 to UPMC Hillman Cancer Center.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- [1].Scharer OD, Nucleotide excision repair in eukaryotes, Cold Spring Harbor perspectives in biology, 5 (2013) a012609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Hanawalt PC, Spivak G, Transcription-coupled DNA repair: two decades of progress and surprises, Nat Rev Mol Cell Biol, 9 (2008) 958–970. [DOI] [PubMed] [Google Scholar]
- [3].Kusakabe M, Onishi Y, Tada H, Kurihara F, Kusao K, Furukawa M, Iwai S, Yokoi M, Sakai W, Sugasawa K, Mechanism and regulation of DNA damage recognition in nucleotide excision repair, Genes and environment : the official journal of the Japanese Environmental Mutagen Society, 41 (2019) 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Sugasawa K, Regulation of damage recognition in mammalian global genomic nucleotide excision repair, Mutation research, 685 (2010) 29–37. [DOI] [PubMed] [Google Scholar]
- [5].Wakasugi M, Sancar A, Assembly, subunit composition, and footprint of human DNA repair excision nuclease, Proc Natl Acad Sci U S A, 95 (1998) 6669–6674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Riedl T, Hanaoka F, Egly JM, The comings and goings of nucleotide excision repair factors on damaged DNA, The EMBO journal, 22 (2003) 5293–5303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Fassihi H, Sethi M, Fawcett H, Wing J, Chandler N, Mohammed S, Craythorne E, Morley AM, Lim R, Turner S, Henshaw T, Garrood I, Giunti P, Hedderly T, Abiona A, Naik H, Harrop G, McGibbon D, Jaspers NG, Botta E, Nardo T, Stefanini M, Young AR, Sarkany RP, Lehmann AR, Deep phenotyping of 89 xeroderma pigmentosum patients reveals unexpected heterogeneity dependent on the precise molecular defect, Proc Natl Acad Sci U S A, 113 (2016) E1236–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Jang S, Kumar N, Beckwitt EC, Kong M, Fouquerel E, Rapic-Otrin V, Prasad R, Watkins SC, Khuu C, Majumdar C, David SS, Wilson SH, Bruchez MP, Opresko PL, Van Houten B, Damage sensor role of UV-DDB during base excision repair, Nat Struct Mol Biol, 26 (2019) 695–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Chu G, Chang E, Xeroderma pigmentosum group E cells lack a nuclear factor that binds to damaged DNA, Science, 242 (1988) 564–567. [DOI] [PubMed] [Google Scholar]
- [10].Protic M, Hirschfeld S, Tsang AP, Wagner M, Dixon K, Levine AS, Induction of a novel damage-specific DNA binding protein correlates with enhanced DNA repair in primate cells, Molecular toxicology, 2 (1989) 255–270. [PubMed] [Google Scholar]
- [11].Feldberg RS, Grossman L, A DNA binding protein from human placenta specific for ultraviolet damaged DNA, Biochemistry, 15 (1976) 2402–2408. [DOI] [PubMed] [Google Scholar]
- [12].Hwang BJ, Chu G, Purification and characterization of a human protein that binds to damaged DNA, Biochemistry, 32 (1993) 1657–1666. [DOI] [PubMed] [Google Scholar]
- [13].Keeney S, Chang GJ, Linn S, Characterization of a human DNA damage binding protein implicated in xeroderma pigmentosum E, J Biol Chem, 268 (1993) 21293–21300. [PubMed] [Google Scholar]
- [14].van Assendelft GB, Rigney EM, Hickson ID, Purification of a HeLa cell nuclear protein that binds selectively to DNA irradiated with ultra-violet light, Nucleic Acids Res, 21 (1993) 3399–3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Abramic M, Levine AS, Protic M, Purification of an ultraviolet-inducible, damage-specific DNA-binding protein from primate cells, J Biol Chem, 266 (1991) 22493–22500. [PubMed] [Google Scholar]
- [16].Keeney S, Eker AP, Brody T, Vermeulen W, Bootsma D, Hoeijmakers JH, Linn S, Correction of the DNA repair defect in xeroderma pigmentosum group E by injection of a DNA damage-binding protein, Proc Natl Acad Sci U S A, 91 (1994) 4053–4056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Dualan R, Brody T, Keeney S, Nichols AF, Admon A, Linn S, Chromosomal localization and cDNA cloning of the genes (DDB1 and DDB2) for the p127 and p48 subunits of a human damage-specific DNA binding protein, Genomics, 29 (1995) 62–69. [DOI] [PubMed] [Google Scholar]
- [18].Nichols AF, Ong P, Linn S, Mutations specific to the xeroderma pigmentosum group E Ddb- phenotype, J Biol Chem, 271 (1996) 24317–24320. [DOI] [PubMed] [Google Scholar]
- [19].Hwang BJ, Toering S, Francke U, Chu G, p48 Activates a UV-damaged-DNA binding factor and is defective in xeroderma pigmentosum group E cells that lack binding activity, Mol Cell Biol, 18 (1998) 4391–4399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Oh KS, Emmert S, Tamura D, DiGiovanna JJ, Kraemer KH, Multiple skin cancers in adults with mutations in the XP-E (DDB2) DNA repair gene, The Journal of investigative dermatology, 131 (2011) 785–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, Srinivasan P, Gao J, Chakravarty D, Devlin SM, Hellmann MD, Barron DA, Schram AM, Hameed M, Dogan S, Ross DS, Hechtman JF, DeLair DF, Yao J, Mandelker DL, Cheng DT, Chandramohan R, Mohanty AS, Ptashkin RN, Jayakumaran G, Prasad M, Syed MH, Rema AB, Liu ZY, Nafa K, Borsu L, Sadowska J, Casanova J, Bacares R, Kiecka IJ, Razumova A, Son JB, Stewart L, Baldi T, Mullaney KA, Al-Ahmadie H, Vakiani E, Abeshouse AA, Penson AV, Jonsson P, Camacho N, Chang MT, Won HH, Gross BE, Kundra R, Heins ZJ, Chen HW, Phillips S, Zhang H, Wang J, Ochoa A, Wills J, Eubank M, Thomas SB, Gardos SM, Reales DN, Galle J, Durany R, Cambria R, Abida W, Cercek A, Feldman DR, Gounder MM, Hakimi AA, Harding JJ, Iyer G, Janjigian YY, Jordan EJ, Kelly CM, Lowery MA, Morris LGT, Omuro AM, Raj N, Razavi P, Shoushtari AN, Shukla N, Soumerai TE, Varghese AM, Yaeger R, Coleman J, Bochner B, Riely GJ, Saltz LB, Scher HI, Sabbatini PJ, Robson ME, Klimstra DS, Taylor BS, Baselga J, Schultz N, Hyman DM, Arcila ME, Solit DB, Ladanyi M, Berger MF, Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients, Nat Med, 23 (2017) 703–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zilfou JT, Lowe SW, Tumor suppressive functions of p53, Cold Spring Harbor perspectives in biology, 1 (2009) a001883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Ford JM, Hanawalt PC, Li-Fraumeni syndrome fibroblasts homozygous for p53 mutations are deficient in global DNA repair but exhibit normal transcription-coupled repair and enhanced UV resistance, Proc Natl Acad Sci U S A, 92 (1995) 8876–8880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ford JM, Hanawalt PC, Expression of wild-type p53 is required for efficient global genomic nucleotide excision repair in UV-irradiated human fibroblasts, J Biol Chem, 272 (1997) 28073–28080. [DOI] [PubMed] [Google Scholar]
- [25].Hwang BJ, Ford JM, Hanawalt PC, Chu G, Expression of the p48 xeroderma pigmentosum gene is p53-dependent and is involved in global genomic repair, Proc Natl Acad Sci U S A, 96 (1999) 424–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Tan T, Chu G, p53 Binds and activates the xeroderma pigmentosum DDB2 gene in humans but not mice, Mol Cell Biol, 22 (2002) 3247–3254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hanawalt PC, Ford JM, Lloyd DR, Functional characterization of global genomic DNA repair and its implications for cancer, Mutation research, 544 (2003) 107–114. [DOI] [PubMed] [Google Scholar]
- [28].Itoh T, Mori T, Ohkubo H, Yamaizumi M, A newly identified patient with clinical xeroderma pigmentosum phenotype has a non-sense mutation in the DDB2 gene and incomplete repair in (6–4) photoproducts, The Journal of investigative dermatology, 113 (1999) 251–257. [DOI] [PubMed] [Google Scholar]
- [29].Fitch ME, Nakajima S, Yasui A, Ford JM, In vivo recruitment of XPC to UV-induced cyclobutane pyrimidine dimers by the DDB2 gene product, J Biol Chem, 278 (2003) 46906–46910. [DOI] [PubMed] [Google Scholar]
- [30].Lan L, Nakajima S, Kapetanaki MG, Hsieh CL, Fagerburg M, Thickman K, Rodriguez-Collazo P, Leuba SH, Levine AS, Rapic-Otrin V, Monoubiquitinated histone H2A destabilizes photolesion-containing nucleosomes with concomitant release of UV-damaged DNA-binding protein E3 ligase, J Biol Chem, 287 (2012) 12036–12049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Moser J, Volker M, Kool H, Alekseev S, Vrieling H, Yasui A, van Zeeland AA, Mullenders LH, The UV-damaged DNA binding protein mediates efficient targeting of the nucleotide excision repair complex to UV-induced photo lesions, DNA repair, 4 (2005) 571–582. [DOI] [PubMed] [Google Scholar]
- [32].Rapic-Otrin V, McLenigan MP, Bisi DC, Gonzalez M, Levine AS, Sequential binding of UV DNA damage binding factor and degradation of the p48 subunit as early events after UV irradiation, Nucleic Acids Res, 30 (2002) 2588–2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Sugasawa K, Okuda Y, Saijo M, Nishi R, Matsuda N, Chu G, Mori T, Iwai S, Tanaka K, Tanaka K, Hanaoka F, UV-induced ubiquitylation of XPC protein mediated by UV-DDB-ubiquitin ligase complex, Cell, 121 (2005) 387–400. [DOI] [PubMed] [Google Scholar]
- [34].Sugasawa K, UV-DDB: a molecular machine linking DNA repair with ubiquitination, DNA repair, 8 (2009) 969–972. [DOI] [PubMed] [Google Scholar]
- [35].Sugasawa K, The CUL4 enigma: culling DNA repair factors, Mol Cell, 34 (2009) 403–404. [DOI] [PubMed] [Google Scholar]
- [36].Guerrero-Santoro J, Kapetanaki MG, Hsieh CL, Gorbachinsky I, Levine AS, Rapic-Otrin V, The cullin 4B-based UV-damaged DNA-binding protein ligase binds to UV-damaged chromatin and ubiquitinates histone H2A, Cancer research, 68 (2008) 5014–5022. [DOI] [PubMed] [Google Scholar]
- [37].Aboussekhra A, Biggerstaff M, Shivji MK, Vilpo JA, Moncollin V, Podust VN, Protic M, Hubscher U, Egly JM, Wood RD, Mammalian DNA nucleotide excision repair reconstituted with purified protein components, Cell, 80 (1995) 859–868. [DOI] [PubMed] [Google Scholar]
- [38].Wakasugi M, Kawashima A, Morioka H, Linn S, Sancar A, Mori T, Nikaido O, Matsunaga T, DDB accumulates at DNA damage sites immediately after UV irradiation and directly stimulates nucleotide excision repair, J Biol Chem, 277 (2002) 1637–1640. [DOI] [PubMed] [Google Scholar]
- [39].Muniandy PA, Thapa D, Thazhathveetil AK, Liu ST, Seidman MM, Repair of laser-localized DNA interstrand cross-links in G1 phase mammalian cells, J Biol Chem, 284 (2009) 27908–27917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Alekseev S, Luijsterburg MS, Pines A, Geverts B, Mari PO, Giglia-Mari G, Lans H, Houtsmuller AB, Mullenders LH, Hoeijmakers JH, Vermeulen W, Cellular concentrations of DDB2 regulate dynamic binding of DDB1 at UV-induced DNA damage, Mol Cell Biol, 28 (2008) 7402–7413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Nishi R, Alekseev S, Dinant C, Hoogstraten D, Houtsmuller AB, Hoeijmakers JH, Vermeulen W, Hanaoka F, Sugasawa K, UV-DDB-dependent regulation of nucleotide excision repair kinetics in living cells, DNA repair, 8 (2009) 767–776. [DOI] [PubMed] [Google Scholar]
- [42].Luijsterburg MS, Goedhart J, Moser J, Kool H, Geverts B, Houtsmuller AB, Mullenders LH, Vermeulen W, van Driel R, Dynamic in vivo interaction of DDB2 E3 ubiquitin ligase with UV-damaged DNA is independent of damage-recognition protein XPC, Journal of cell science, 120 (2007) 2706–2716. [DOI] [PubMed] [Google Scholar]
- [43].Czaja W, Mao P, Smerdon MJ, The emerging roles of ATP-dependent chromatin remodeling enzymes in nucleotide excision repair, Int J Mol Sci, 13 (2012) 11954–11973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Luijsterburg MS, Lindh M, Acs K, Vrouwe MG, Pines A, van Attikum H, Mullenders LH, Dantuma NP, DDB2 promotes chromatin decondensation at UV-induced DNA damage, The Journal of cell biology, 197 (2012) 267–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Chou DM, Adamson B, Dephoure NE, Tan X, Nottke AC, Hurov KE, Gygi SP, Colaiacovo MP, Elledge SJ, A chromatin localization screen reveals poly (ADP ribose)-regulated recruitment of the repressive polycomb and NuRD complexes to sites of DNA damage, Proc Natl Acad Sci U S A, 107 (2010) 18475–18480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Ahel D, Horejsi Z, Wiechens N, Polo SE, Garcia-Wilson E, Ahel I, Flynn H, Skehel M, West SC, Jackson SP, Owen-Hughes T, Boulton SJ, Poly(ADP-ribose)-dependent regulation of DNA repair by the chromatin remodeling enzyme ALC1, Science, 325 (2009) 1240–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Adam S, Dabin J, Chevallier O, Leroy O, Baldeyron C, Corpet A, Lomonte P, Renaud O, Almouzni G, Polo SE, Real-Time Tracking of Parental Histones Reveals Their Contribution to Chromatin Integrity Following DNA Damage, Mol Cell, 64 (2016) 65–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Scrima A, Konickova R, Czyzewski BK, Kawasaki Y, Jeffrey PD, Groisman R, Nakatani Y, Iwai S, Pavletich NP, Thoma NH, Structural basis of UV DNA-damage recognition by the DDB1-DDB2 complex, Cell, 135 (2008) 1213–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Yeh JI, Levine AS, Du S, Chinte U, Ghodke H, Wang H, Shi H, Hsieh CL, Conway JF, Van Houten B, Rapic-Otrin V, Damaged DNA induced UV-damaged DNA-binding protein (UV-DDB) dimerization and its roles in chromatinized DNA repair, Proc Natl Acad Sci U S A, 109 (2012) E2737–2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Ghodke H, Wang H, Hsieh CL, Woldemeskel S, Watkins SC, Rapic-Otrin V, Van Houten B, Single-molecule analysis reveals human UV-damaged DNA-binding protein (UV-DDB) dimerizes on DNA via multiple kinetic intermediates, Proc Natl Acad Sci U S A, 111 (2014) E1862–1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Rapic-Otrin V, Navazza V, Nardo T, Botta E, McLenigan M, Bisi DC, Levine AS, Stefanini M, True XP group E patients have a defective UV-damaged DNA binding protein complex and mutations in DDB2 which reveal the functional domains of its p48 product, Human molecular genetics, 12 (2003) 1507–1522. [DOI] [PubMed] [Google Scholar]
- [52].He YJ, McCall CM, Hu J, Zeng Y, Xiong Y, DDB1 functions as a linker to recruit receptor WD40 proteins to CUL4-ROC1 ubiquitin ligases, Genes Dev, 20 (2006) 2949–2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Jin J, Arias EE, Chen J, Harper JW, Walter JC, A family of diverse Cul4-Ddb1-interacting proteins includes Cdt2, which is required for S phase destruction of the replication factor Cdt1, Mol Cell, 23 (2006) 709–721. [DOI] [PubMed] [Google Scholar]
- [54].Fischer ES, Scrima A, Bohm K, Matsumoto S, Lingaraju GM, Faty M, Yasuda T, Cavadini S, Wakasugi M, Hanaoka F, Iwai S, Gut H, Sugasawa K, Thoma NH, The molecular basis of CRL4DDB2/CSA ubiquitin ligase architecture, targeting, and activation, Cell, 147 (2011) 1024–1039. [DOI] [PubMed] [Google Scholar]
- [55].Hannss R, Dubiel W, COP9 signalosome function in the DDR, FEBS letters, 585 (2011) 2845–2852. [DOI] [PubMed] [Google Scholar]
- [56].Cavadini S, Fischer ES, Bunker RD, Potenza A, Lingaraju GM, Goldie KN, Mohamed WI, Faty M, Petzold G, Beckwith RE, Tichkule RB, Hassiepen U, Abdulrahman W, Pantelic RS, Matsumoto S, Sugasawa K, Stahlberg H, Thoma NH, Cullin-RING ubiquitin E3 ligase regulation by the COP9 signalosome, Nature, 531 (2016) 598–603. [DOI] [PubMed] [Google Scholar]
- [57].Groisman R, Polanowska J, Kuraoka I, Sawada J, Saijo M, Drapkin R, Kisselev AF, Tanaka K, Nakatani Y, The ubiquitin ligase activity in the DDB2 and CSA complexes is differentially regulated by the COP9 signalosome in response to DNA damage, Cell, 113 (2003) 357–367. [DOI] [PubMed] [Google Scholar]
- [58].Kong M, Beckwitt EC, Springall L, Kad NM, Van Houten B, Single-Molecule Methods for Nucleotide Excision Repair: Building a System to Watch Repair in Real Time, Methods in enzymology, 592 (2017) 213–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Savir Y, Tlusty T, Conformational proofreading: the impact of conformational changes on the specificity of molecular recognition, PloS one, 2 (2007) e468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Osakabe A, Tachiwana H, Kagawa W, Horikoshi N, Matsumoto S, Hasegawa M, Matsumoto N, Toga T, Yamamoto J, Hanaoka F, Thoma NH, Sugasawa K, Iwai S, Kurumizaka H, Structural basis of pyrimidine-pyrimidone (6–4) photoproduct recognition by UV-DDB in the nucleosome, Sci Rep, 5 (2015) 16330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Matsumoto S, Cavadini S, Bunker RD, Grand RS, Potenza A, Rabl J, Yamamoto J, Schenk AD, Schubeler D, Iwai S, Sugasawa K, Kurumizaka H, Thoma NH, DNA damage detection in nucleosomes involves DNA register shifting, Nature, 571 (2019) 79–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Pines A, Vrouwe MG, Marteijn JA, Typas D, Luijsterburg MS, Cansoy M, Hensbergen P, Deelder A, de Groot A, Matsumoto S, Sugasawa K, Thoma N, Vermeulen W, Vrieling H, Mullenders L, PARP1 promotes nucleotide excision repair through DDB2 stabilization and recruitment of ALC1, The Journal of cell biology, 199 (2012) 235–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Robu M, Shah RG, Petitclerc N, Brind’Amour J, Kandan-Kulangara F, Shah GM, Role of poly(ADP-ribose) polymerase-1 in the removal of UV-induced DNA lesions by nucleotide excision repair, Proc Natl Acad Sci U S A, 110 (2013) 1658–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Tsuge M, Masuda Y, Kaneoka H, Kidani S, Miyake K, Iijima S, SUMOylation of damaged DNA-binding protein DDB2, Biochemical and biophysical research communications, 438 (2013) 26–31. [DOI] [PubMed] [Google Scholar]
- [65].Han C, Zhao R, Kroger J, He J, Wani G, Wang QE, Wani AA, UV radiation-induced SUMOylation of DDB2 regulates nucleotide excision repair, Carcinogenesis, 38 (2017) 976–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Beard WA, Horton JK, Prasad R, Wilson SH, Eukaryotic Base Excision Repair: New Approaches Shine Light on Mechanism, Annual review of biochemistry, 88 (2019) 137–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Payne A, Chu G, Xeroderma pigmentosum group E binding factor recognizes a broad spectrum of DNA damage, Mutation research, 310 (1994) 89–102. [DOI] [PubMed] [Google Scholar]
- [68].Fujiwara Y, Masutani C, Mizukoshi T, Kondo J, Hanaoka F, Iwai S, Characterization of DNA recognition by the human UV-damaged DNA-binding protein, J Biol Chem, 274 (1999) 20027–20033. [DOI] [PubMed] [Google Scholar]
- [69].Wittschieben BO, Iwai S, Wood RD, DDB1-DDB2 (xeroderma pigmentosum group E) protein complex recognizes a cyclobutane pyrimidine dimer, mismatches, apurinic/apyrimidinic sites, and compound lesions in DNA, J Biol Chem, 280 (2005) 39982–39989. [DOI] [PubMed] [Google Scholar]
- [70].He J, Wang Y, Missinato MA, Onuoha E, Perkins LA, Watkins SC, St Croix CM, Tsang M, Bruchez MP, A genetically targetable near-infrared photosensitizer, Nat Methods, 13 (2016) 263–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Fouquerel E, Barnes RP, Uttam S, Watkins SC, Bruchez MP, Opresko PL, Targeted and Persistent 8-Oxoguanine Base Damage at Telomeres Promotes Telomere Loss and Crisis, Mol Cell, 75 (2019) 117–130 e116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Jacobs AL, Schar P, DNA glycosylases: in DNA repair and beyond, Chromosoma, 121 (2012) 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Kraemer KH, Lee MM, Scotto J, DNA repair protects against cutaneous and internal neoplasia: evidence from xeroderma pigmentosum, Carcinogenesis, 5 (1984) 511–514. [DOI] [PubMed] [Google Scholar]
- [74].Hadj-Rabia S, Oriot D, Soufir N, Dufresne H, Bourrat E, Mallet S, Poulhalon N, Ezzedine K, Grandchamp B, Taieb A, Catteau B, Sarasin A, Bodemer C, Unexpected extradermatological findings in 31 patients with xeroderma pigmentosum type C, The British journal of dermatology, 168 (2013) 1109–1113. [DOI] [PubMed] [Google Scholar]
- [75].Lai JP, Liu YC, Alimchandani M, Liu Q, Aung PP, Matsuda K, Lee CC, Tsokos M, Hewitt S, Rushing EJ, Tamura D, Levens DL, Digiovanna JJ, Fine HA, Patronas N, Khan SG, Kleiner DE, Oberholtzer JC, Quezado MM, Kraemer KH, The influence of DNA repair on neurological degeneration, cachexia, skin cancer and internal neoplasms: autopsy report of four xeroderma pigmentosum patients (XP-A, XP-C and XP-D), Acta neuropathologica communications, 1 (2013) 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Oh KS, Imoto K, Emmert S, Tamura D, DiGiovanna JJ, Kraemer KH, Nucleotide excision repair proteins rapidly accumulate but fail to persist in human XP-E (DDB2 mutant) cells, Photochem Photobiol, 87 (2011) 729–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Itoh T, Iwashita S, Cohen MB, Meyerholz DK, Linn S, Ddb2 is a haploinsufficient tumor suppressor and controls spontaneous germ cell apoptosis, Human molecular genetics, 16 (2007) 1578–1586. [DOI] [PubMed] [Google Scholar]
- [78].Itoh T, Cado D, Kamide R, Linn S, DDB2 gene disruption leads to skin tumors and resistance to apoptosis after exposure to ultraviolet light but not a chemical carcinogen, Proc Natl Acad Sci U S A, 101 (2004) 2052–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Alekseev S, Kool H, Rebel H, Fousteri M, Moser J, Backendorf C, de Gruijl FR, Vrieling H, Mullenders LH, Enhanced DDB2 expression protects mice from carcinogenic effects of chronic UV-B irradiation, Cancer research, 65 (2005) 10298–10306. [DOI] [PubMed] [Google Scholar]
- [80].Yoon T, Chakrabortty A, Franks R, Valli T, Kiyokawa H, Raychaudhuri P, Tumor-prone phenotype of the DDB2-deficient mice, Oncogene, 24 (2005) 469–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Liu L, Lee S, Zhang J, Peters SB, Hannah J, Zhang Y, Yin Y, Koff A, Ma L, Zhou P, CUL4A abrogation augments DNA damage response and protection against skin carcinogenesis, Mol Cell, 34 (2009) 451–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Gilson P, Drouot G, Witz A, Merlin JL, Becuwe P, Harle A, Emerging Roles of DDB2 in Cancer, Int J Mol Sci, 20 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Kattan Z, Marchal S, Brunner E, Ramacci C, Leroux A, Merlin JL, Domenjoud L, Dauca M, Becuwe P, Damaged DNA binding protein 2 plays a role in breast cancer cell growth, PloS one, 3 (2008) e2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Roy N, Bommi PV, Bhat UG, Bhattacharjee S, Elangovan I, Li J, Patra KC, Kopanja D, Blunier A, Benya R, Bagchi S, Raychaudhuri P, DDB2 suppresses epithelial-to-mesenchymal transition in colon cancer, Cancer research, 73 (2013) 3771–3782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Huang S, Fantini D, Merrill BJ, Bagchi S, Guzman G, Raychaudhuri P, DDB2 Is a Novel Regulator of Wnt Signaling in Colon Cancer, Cancer research, 77 (2017) 6562–6575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Bilotti K, Tarantino ME, Delaney S, Human Oxoguanine Glycosylase 1 Removes Solution Accessible 8-Oxo-7,8-dihydroguanine Lesions from Globally Substituted Nucleosomes Except in the Dyad Region, Biochemistry, 57 (2018) 1436–1439. [DOI] [PubMed] [Google Scholar]
- [87].Kennedy EE, Caffrey PJ, Delaney S, Initiating base excision repair in chromatin, DNA repair, 71 (2018) 87–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Kennedy EE, Li C, Delaney S, Global Repair Profile of Human Alkyladenine DNA Glycosylase on Nucleosomes Reveals DNA Packaging Effects, ACS chemical biology, 14 (2019) 1687–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Li C, Delaney S, Challenges for base excision repair enzymes: Acquiring access to damaged DNA in chromatin, The Enzymes, 45 (2019) 27–57. [DOI] [PubMed] [Google Scholar]
- [90].Tarantino ME, Dow BJ, Drohat AC, Delaney S, Nucleosomes and the three glycosylases: High, medium, and low levels of excision by the uracil DNA glycosylase superfamily, DNA repair, 72 (2018) 56–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Takedachi A, Saijo M, Tanaka K, DDB2 complex-mediated ubiquitylation around DNA damage is oppositely regulated by XPC and Ku and contributes to the recruitment of XPA, Mol Cell Biol, 30 (2010) 2708–2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Matsuda N, Azuma K, Saijo M, Iemura S, Hioki Y, Natsume T, Chiba T, Tanaka K, Tanaka K, DDB2, the xeroderma pigmentosum group E gene product, is directly ubiquitylated by Cullin 4A-based ubiquitin ligase complex, DNA repair, 4 (2005) 537–545. [DOI] [PubMed] [Google Scholar]
- [93].Rapic Otrin V, Kuraoka I, Nardo T, McLenigan M, Eker AP, Stefanini M, Levine AS, Wood RD, Relationship of the xeroderma pigmentosum group E DNA repair defect to the chromatin and DNA binding proteins UV-DDB and replication protein A, Mol Cell Biol, 18 (1998) 3182–3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Kapetanaki MG, Guerrero-Santoro J, Bisi DC, Hsieh CL, Rapic-Otrin V, Levine AS, The DDB1-CUL4ADDB2 ubiquitin ligase is deficient in xeroderma pigmentosum group E and targets histone H2A at UV-damaged DNA sites, Proc Natl Acad Sci U S A, 103 (2006) 2588–2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Tang J, Chu G, Xeroderma pigmentosum complementation group E and UV-damaged DNA-binding protein, DNA repair, 1 (2002) 601–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Kondo S, Fukuro S, Mamada A, Kawada A, Satoh Y, Fujiwara Y, Assignment of three patients with xeroderma pigmentosum to complementation group E and their characteristics, The Journal of investigative dermatology, 90 (1988) 152–157. [DOI] [PubMed] [Google Scholar]
- [97].Nichols AF, Itoh T, Graham JA, Liu W, Yamaizumi M, Linn S, Human damage-specific DNA-binding protein p48. Characterization of XPE mutations and regulation following UV irradiation, J Biol Chem, 275 (2000) 21422–21428. [DOI] [PubMed] [Google Scholar]
- [98].Nagy A, Lanczky A, Menyhart O, Gyorffy B, Validation of miRNA prognostic power in hepatocellular carcinoma using expression data of independent datasets, Sci Rep, 8 (2018) 9227. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.