

Abstract
Hepatotoxicity is a key concern in the clinical translation of nanotherapeutics because preclinical studies have consistently shown that nanotherapeutics accumulate extensively in the liver. However, clinical stage nanotherapeutics have not shown increased hepatotoxicity. Factors that can contribute to the hepatotoxicity of nanotherapeutics beyond the intrinsic hepatotoxicity of nanoparticles (NPs) are poorly understood. Because of this knowledge gap, clinical translation efforts have avoided hepatotoxic molecules. Herein, by examining the hepatotoxicity of nanoformulations of known hepatotoxic compounds, we demonstrated that nanotherapeutics are associated with lower hepatotoxicity than their small molecule counterparts. We then found that the reduced hepatotoxicity is related to the uptake of nanotherapeutics by macrophages in the liver. These findings can facilitate further development and clinical translation of nanotherapeutics.
Keywords: nanotoxicity, hepatotoxicity, clinical translation, nanomedicine, macrophage uptake
Nanoparticles (NPs) as drug delivery vehicles hold great promise in improving the treatment of human diseases, such as cancers and infections.[1–5] A key concern in clinical translation of nanomedicine has been potential toxicities, especially hepatotoxicity.[6–8] This is because NPs have high biodistribution to the liver with parenteral delivery.[9,10] Due to the extensive liver distribution of nanotherapeutics,[11,12] there has been considerable concern regarding the hepatotoxicity of NP drugs.[10,13] However, many nanotherapeutics have been approved for clinical use and none of these have shown increased hepatotoxicity thus far.[14–16] Moreover, factors that contribute to the hepatotoxicity of nanotherapeutics beyond the intrinsic hepatotoxicity of NPs have not been studied.[17] Because of this knowledge gap, clinical translation efforts have avoided hepatotoxic molecules. Recently, we observed that NP delivery of hepatotoxic wortmannin actually reduced wortmannin hepatotoxicity in preclinical studies.[3] Based on this observation, we aimed to examine NP’s hepatotoxicity in more detail.
To examine hepatotoxicity associated with NP delivery, we engineered NP formulations of several hepatotoxic compounds including chlorpromazine (CPZ),[18] 7-ethyl-10-hydroxycamptothecin (SN-38)[19] and wortmannin (Wtmn).[20] Polymeric micelles were employed because polymeric NPs are generally utilized for clinical formulations of hydrophobic/amphiphilic compounds such as CPZ, SN-38 and Wtmn. In this study, the NPs were formulated using poly (lactic-co-glycolic acid) as core material for drug encapsulation and the lipid mixture of DSPE-PEG (1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[carboxy(polyethylene glycol) and lecithin as shell material for the control of drug release (Figure 1).[21,22] Characterization of the NPs demonstrated spherical morphology with an average hydrodynamic diameter of 120.8±1.9 nm for CPZ-loaded NP, 107.5±1.1 nm for Wtmn-loaded NP and 43.4±2.6 nm for SN-38-loaded NP (Figure S1 and Table S1, Supporting Information). NPs exhibited a similar negative charged surface of −37.0±0.3 mV for CPZ-loaded NP, −39.7±1.8 mV for Wtmn-loaded NP and −32.5±8.8 mV for SN-38-loaded NP; and a comparable drug loading efficiency of 5.5±0.3% for CPZ-loaded NP, 2.4±0.3% for Wtmn-loaded NP and 3.3±0.3% for SN-38-loaded NP (Table S1, Supporting Information). We characterized Wtmn NP stored at 4 °C for 24 h and 48 h. We found that they had no significant change in size, PDI and Zeta potential, indicating that the NP is quite stable (Table S2. Supporting Information). The release percentage of NPs was 37.5% for CPZ-loaded NP, 66.8% for Wtinn-loaded NP and 67.5% for SN-38-loaded NP within 24 h (Figure S2, Supporting Information).
Figure 1.
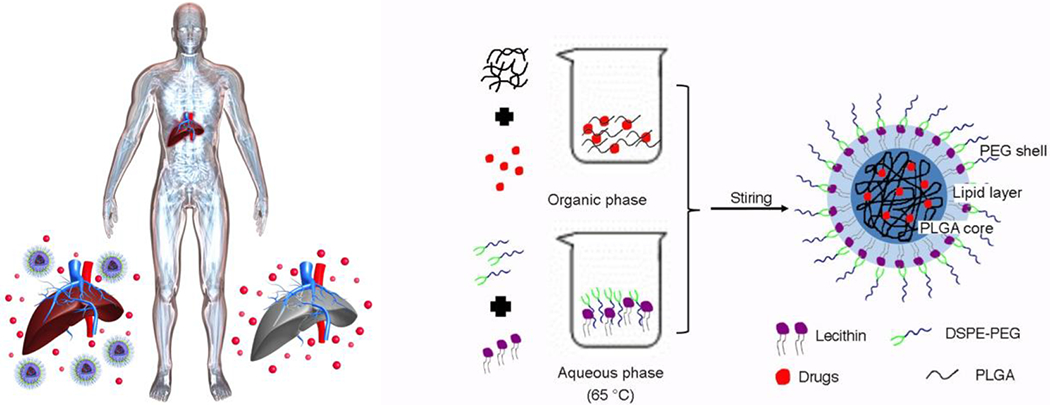
Schematic showing that nanotherapeutics are associated with lower hepatotoxicity than their small molecule counterparts, and the description of the components and formulation of core-shell nanoparticles (NPs).
We first compared the hepatotoxicity between nanotherapeutics and their small molecule counterparts. CD-1 mice were treated with nanotherapeutics and their small molecule counterparts including CPZ (Figure 2a,b), Wtmn (Figure 2c,d) and SN-38 (Figure 2e,f) at one-half the maximum tolerated dose (½ MTD) of small molecules (Table S3, Supporting Information). The dose was chosen because higher dose caused death to partial mice in our pilot study. Hepatotoxicity was assessed by liver enzymes alanine transaminase (ALT) and aspartate transaminase (AST) 6 h, 12 h and 24 h post treatment.[23] We found that the liver enzymes from nanotherapeutic-treated mice were significantly lower than that of small molecules for most of the time points, e.g. at 24 h post treatment, the ALT of NP vs. Free was 25.2±1.5 vs. 50.4±7.6 U/L for CPZ, 22.0±3.2 vs. 33.3±4.9 U/L for Wtmn and 15.7±2.4 vs. 20.3±2.2 U/L for SN-38; the AST of NP vs. Free was 82.6±3.2 vs. 122.6±8.7 U/L for CPZ, 69.5±7.4 vs. 57.8±6.5 U/L for Wtmn and 44.4±8.7 vs. 70.4±6.1 U/L for SN-38 (Data represent mean±SEM, n=8-10) (Table S4, Supporting Information). The results were further confirmed on C57BL/6 mice (Figure S3, Supporting Information) and CB6F1 mice (Figure S4, Supporting Information) with free CPZ and CPZ-loaded NP, e.g. at 24 h post treatment, the ALT of NP vs. Free was 28.8±4.8 vs. 29.9±15.3 U/L for C57BL/6 mice, 17.6±4.3 vs. 23.1±5.8 U/L for CB6F1 mice; the AST of NP vs. Free was 100.2±17.1 vs. 121.3±34.1 U/L for C57BL/6 mice, 60.9±1.2 vs. 90.0±19.5 U/L for CB6F1 mice (Data represent mean±SEM, n=8-10) (Table S5, Supporting Information). We also tested the ALT and AST of mice treated by blank carrier with the same dose as the highest one used above and found no significant difference from the untreated mice (Figure S5, Supporting Information). The ALT and AST of mice treated by CPZ and Wtmn for longer time points were also analyzed, where most of them decreased to normal level and showed no significant difference from the untreated mice as well as from each other (Figure S6 and S7, Supporting information). These data reveal that NP delivery actually reduced or at least not increased the hepatotoxicity of CPZ, SN-38 and Wtmn regardless of mouse species.
Figure 2. Nanotherapeutics have lower hepatotoxicity than their small molecule counterparts in vivo.
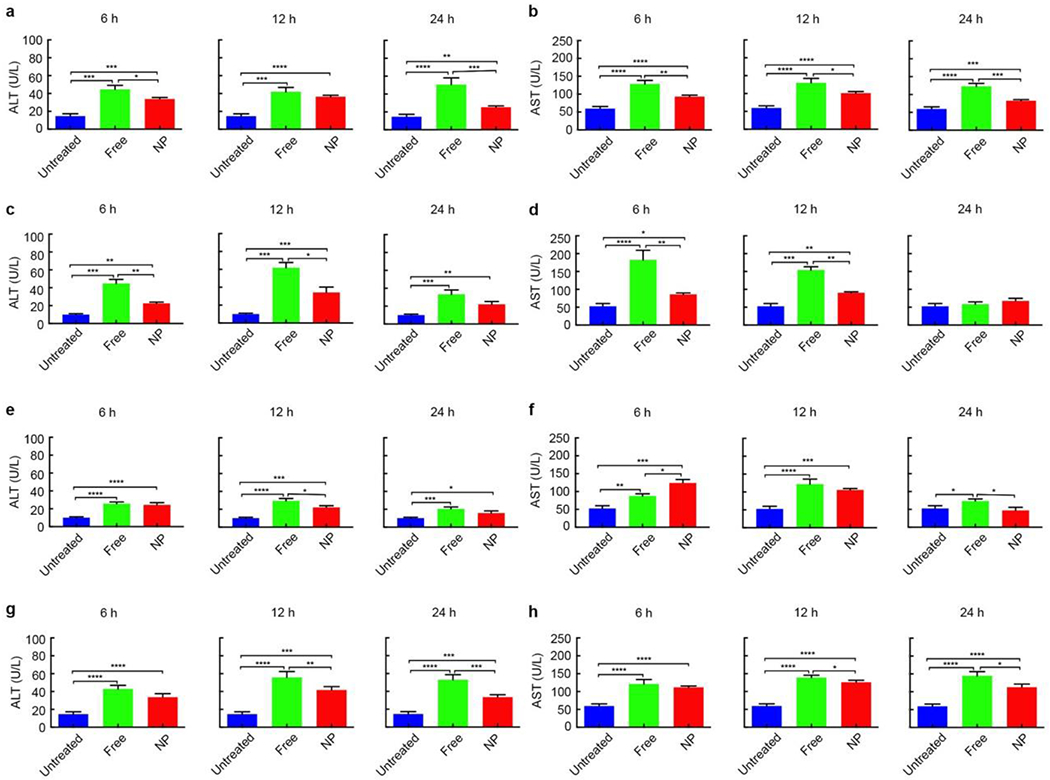
The hepatotoxicity of single-dose CPZ-loaded NPs and its small molecule counterparts (a, b), single-dose Wtmn-loaded NPs and its small molecule counterparts (c, d), single-dose SN-38-loaded NPs and its small molecule counterparts (e, f), and multi-dose (mice were injected twice (each at ½ MTD) intravenously at an interval of 24 h) CPZ-loaded NPs and its small molecule counterparts (g, h) on CD-1 mice 6 h, 12 h and 24 h post treatments. Hepatotoxicity was assessed by liver enzymes alanine transaminase (ALT) (a, c, e, g) and aspartate transaminase (AST) (b, d, f, h). Data represent mean ± SEM. (n = 8-10). Statistical significance was assessed using the Mann Whitney test. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
To further assess liver damage, we examined the expression of heme oxygenase-1 (HO-1) and manganese superoxide dismutase (Mn-SOD) by immunohistochemistry (IHC) staining in the liver samples from mice that had 24 h and 72 h treatment of single dose (½ MTD) free CPZ and CPZ-loaded NPs or free Wtmn and Wtmn-loaded NPs.[24] Both of these markers will increase in response to oxidative stress.[25,26] The results showed that NPs-treated groups had much less positive staining for both HO-1 (Figure 3a,e) and Mn-SOD (Figure 3b,f) than non-formulated free drugs-treated groups. The positive staining ratio in the livers among untreated, free drugs and NPs arms was quantified as 1:22.9:12.7 at 24 h and 1:25.2:18.1 at 72 h for HO-1 treated by free CPZ and CPZ-loaded NP (Figure 3c and Table S6, Supporting Information); 1:32.7:5.8 at 24 h and 1:16.6:1.8 at 72 h for Mn-SOD treated by free CPZ and CPZ-loaded NP (Figure 3d and Table S6, Supporting Information); 1:14.7:5.8 at 24 h and 1:17.1:15.4 at 72 h for HO-1 treated by free Wtmn and Wtmn-loaded NP (Figure 3g and Table S6, Supporting Information); and 1:57.8:7.7 at 24 h and 1:32.9:34.2 at 72 h for Mn-SOD treated by free Wtmn and Wtmn-loaded NP (Figure 3h and Table S6, Supporting Information). The less expression of these oxidative stress markers in most NP-treated groups indicated that nanotherapeutics led to less liver damage than their small molecule counterparts.
Figure 3. Immunohistochemistry (IHC) staining of the livers from CD-1 mice 24 h and 72 h after treatment by NPs and their small molecule counterparts.
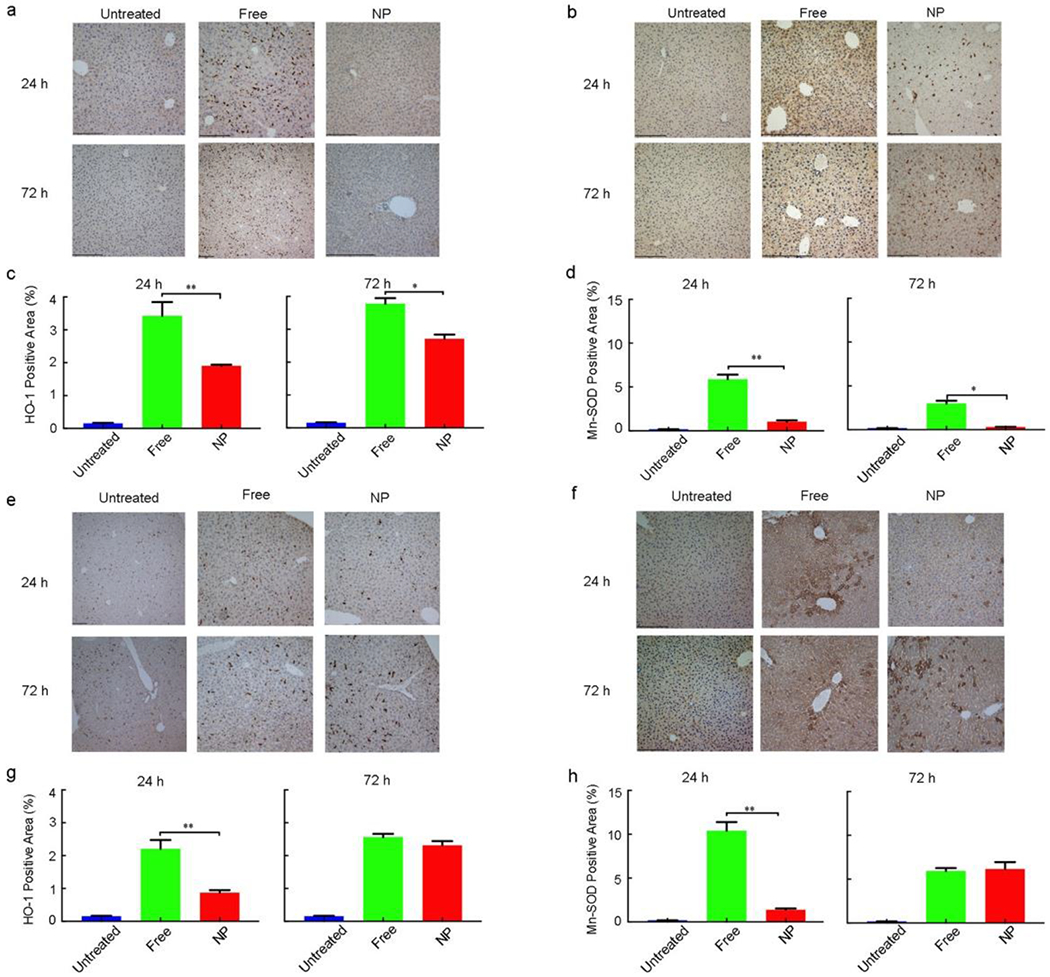
a, HO-1 staining of the livers from mice treated with free CPZ and CPZ-loaded NPs. b, Mn-SOD staining of the livers treated with free CPZ and CPZ-loaded NPs. c, d, Quantitative analysis of positive staining percentage in (a) and (b), respectively. Scale bar = 480 μm. Data represent mean ± SEM (n = 5). Statistical significance was assessed using the Mann Whitney test. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. e, HO-1 staining of the livers from mice treated with free Wtmn and Wtmn-loaded NPs. f, Mn-SOD staining of the livers treated with free Wtmn and Wtmn-loaded NPs. g, h, Quantitative analysis of positive staining percentage in (e) and (f), respectively. Scale bar = 480 μm. Data represent mean ± SEM (n = 5). Statistical significance was assessed using the Mann Whitney test. *p < 0.05, **p < 0.01, ***p < 0.001.
To determine whether nanotherapeutics decrease the hepatotoxicity of their small molecule counterparts after multiple dose treatment, free CPZ and CPZ-loaded NPs were injected twice (each at ½ MTD) intravenously at an interval of 24 h into CD-1 mice (Figure 2g,h) , the ALT of NP vs. Free was 33.6±3.9 vs. 42.7±4.2 U/L at 6 h post treatment, 41.6±3.9 vs. 55.7±6.7 U/L at 12 h post treatment, and 33.5±2.7 vs. 52.9±5.7 U/L at 24 h post treatment; the AST of NP vs. Free was 111.5±3.2 vs. 120.9±12.7 U/L at 6 h post treatment, 125.7±5.8 vs. 138.9±6.5 U/L at 12 h post treatment, and 112.5±9.1 vs. 144.5±11.8 U/L at 24 h post treatment (Data represent mean±SEM, n=8-10) (Table S4, Supporting Information). The lower ALT and AST in NP-treated mice demonstrated that NP drug delivery reduced the hepatotoxicity of their small molecule counterparts after multiple dose treatment (Table S4, Supporting Information).
Taken together, these data suggest that nanotherapeutics led to decreased hepatotoxicity for their non-formulated small molecule drugs. Considering the increased liver accumulation of nanoformulations, this phenomenon needs further investigation to understand the key factor that determines nanotherapeutics’ hepatotoxicity.
Factor that can affect the hepatotoxicity of nanotherapeutics’ is associated with uptake by macrophages in the liver. This is because NPs show higher uptake efficiency through their size-dependent endocytotic uptake mechanism, which is a key difference to small molecules. To study how macrophage uptake can affect hepatotoxicity, we first examined it in vitro. We isolated primary hepatocytes from CD-1 mice and cultured in liver biomatrix-coated plates using the method reported in our previous paper.[27] Non-formulated free CPZ and CPZ-loaded NP or non-formulated free Wtmn and Wtmn-loaded NP were placed in a Slide-ALyzer MINI dialysis microtube with or without incubation with murine macrophage cells (RAW 264.7) (Figure S8, Supporting Information). We found that the cell viability of primary hepatocytes treated by both CPZ-loaded NP and Wtmn-loaded NP significantly increased after incubation with macrophage cells (Figure 4a,b and Table S7, Supporting Information). The increase of cell viability was from 58.6±1.9% to 89.0±2.6% at 6 h and from 51.9±1.7% to 69.1±1.8% at 24 h for CPZ-loaded NP-treated arm, i.e. an increase of 52% and 33% respectively; from 46.5±0.7% to 69.9±3.2% at 6 h and from 38.9±1.7% to 50.2±1.7% at 24 h for Wtmn-loaded NP-treated arm, i.e. an increase of 50% and 29% respectively. Meanwhile the cell viability of primary hepatocytes treated by free CPZ and free Wtmn only increased slightly after incubation with macrophage cells, i.e. an increase of 5% at 6 h and 19% at 24 h for free CPZ; and 8% at 6 h and 3% at 24 h for free Wtmn. These data indicate that macrophage uptake could be a key factor in determining the reduced hepatotoxicity of nanotherapeutics.
Figure 4. Macrophage uptake reduces the hepatotoxicity of nanotherapeutics.
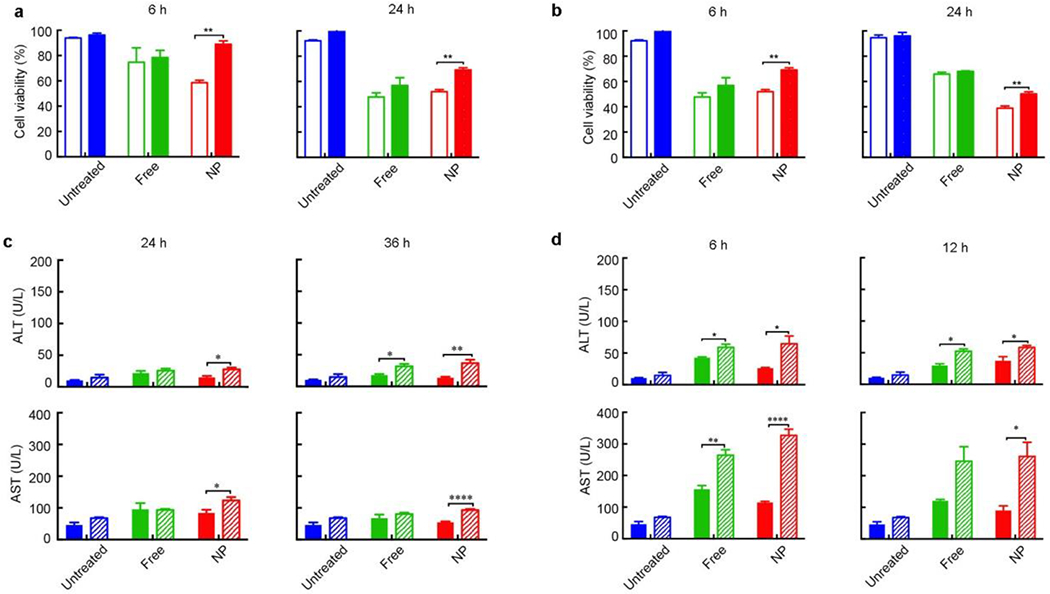
a, b, In vitro cytotoxicity of primary hepatocytes cultured in liver matrix coated plates. The hepatocytes were treated with free CPZ and CPZ-loaded NPs (NP) (a) or free Wtmn and Wtmn-loaded NPs (b) without (hollow columns) or with (solid columns) macrophage incubation. The cytotoxicity was analyzed by MTS assay 6 h and 24 h after treatments. Data represent mean ± SEM. (n = 6). Statistical significance was assessed using the Mann Whitney test. *p < 0.05, **p < 0.01. c, d, In vivo hepatotoxicity in CD-1 mice treated with free CPZ and CPZ-loaded NPs (c) or free Wtmn and Wtmn-loaded NPs (d). The mice were either without (solid columns) or with (stripe columns) macrophage depletion. The hepatotoxicity was analyzed by ALT and AST 24 h and 36 h after treatment with free CPZ and CPZ-loaded NPs, or 6 h and 12 h after treatment with free Wtmn and Wtmn-loaded NPs. Data represent mean ± SEM. (n = 8-10). Statistical significance was assessed using the Mann Whitney test. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
To validate our in vitro results, we evaluated the hepatotoxicity of free drugs and NPs in vivo with or without depletion of macrophages.[28] Macrophages were depleted by Clodronate liposomes and the depletion lasted for 144 h (Figure S9a,b, Supporting Information). ALT and AST values increased due to injection of the Clodronate liposomes and returned to normal levels 96 h after injection (Figure S9c,d, Supporting Information). Therefore, NPs were administrated 96 h after Clodronate liposomes injection to ensure that ALT and AST were not significantly affected by macrophage depletion. Generally, we found that AST and ALT values increased in all NP-treated groups after the depletion of macrophages (Figure 4c,d). We calculated the increase of ALT and AST after the depletion of macrophages (Tables S8 and S9, Supporting Information). The ALT significantly increased from 15.0±2.5 to 27.8±2.7 U/L at 24 h and 13.6±1.6 to 36.8±5.4 U/L at 36 h for CPZ-loaded NP-treated arm, i.e. an increase of 85% and 170% respectively; and the AST significantly increased from 84.2±10.0 to 123.6±10.6 U/L at 24 h and 54.6±2.4 to 93.0±3.6 U/L at 36 h for CPZ-loaded NP-treated arm, i.e. an increase of 47% and 70% respectively. As a control, the increase of ALT was only 18% at 24 h and 78% at 36 h for free CPZ-treated arm. The increase of AST was only −2% at 24 h and 19% at 36 h for free CPZ-treated arm (Table S8, Supporting Information). Similarly, the ALT significantly increased from 26.0±1.2 to 64.6±12.4 U/L at 6 h and 37.2±6.6 to 58.4±3.0 U/L at 12 h for Wtmn-loaded NP-treated arm, i.e. an increase of 148% and 57% respectively; and the AST significantly increased from 114.6±3.9 to 327.0±19.4 U/L at 6 h and 90.0±14.8 to 261.2±44.8 U/L at 12 h for Wtmn-loaded NP-treated arm, i.e. an increase of 185% and 190% respectively. As a control, the increase of ALT was 38% at 6 h and 76% at 12 h for free Wtmn-treated arm. The increase of AST was 69% at 6 h and 104% at 12 h for free Wtmn-treated arm (Table S9, Supporting Information). Noticeably, Wtmn-loaded NP showed a faster drug release rate than CPZ-loaded NP. Therefore, Wtmn-loaded NPs exhibited significantly different hepatotoxicity before and after the depletion of macrophages at earlier time point than CPZ-loaded NPs. It is also worth pointing out that before the depletion of macrophages, most mice treated by NPs showed lower ALT and AST than those treated by non-formulated free. On the contrary, after the depletion of macrophages, they showed higher ALT and AST than those treated by non-formulated free drugs.
The liver damage before and after the depletion of macrophages was further validated by IHC staining, where HO-1 and Mn-SOD were stained and quantified in the livers of CD-1 mice with or without macrophage depletion 24 h and 72 h post treatment by non-formulated free drugs and NPs (Figure 5 and Table S10, Supporting Information). The positive staining of HO-1 after the depletion of macrophages showed a significant increase of 292% at 24 h and 27% at 72 h for CPZ-loaded NP-treated arm. As a control, the increase was 46% at 24 h and 9% at 72 h for free CPZ-treated arm. The positive staining of Mn-SOD after the depletion of macrophages showed a significant increase of 893% at 24 h and 403% at 72 h for CPZ-loaded NP-treated arm. As a control, the increase was 54% at 24 h and 48% at 72 h for free CPZ-treated arm. Similarly, the increase of HO-1 was 114% at 24 h and 1.4% at 72 h for Wtmn-loaded NP-treated arm; 15% at 24 h and 10% at 72 h for free Wtmn-treated arm. The increase of Mn-SOD was 447% at 24 h and 56% at 72 h for Wtmn-loaded NP-treated arm; −3% at 24 h and 15% at 72 h for free Wtmn-treated arm (Table S10, Supporting Information). The increased expression of HO-1 and Mn-SOD after the depletion of macrophages indicated elevated liver damage. Our data showed that such increase of these oxidative stress markers was more significant in most NP-treated groups than that in non-formulated free drug-treated groups, indicating that macrophage uptake had more impact on the hepatotoxicity of nanotherapeutics.
Figure 5. Immunohistochemistry (IHC) staining of the livers from CD-1 mice without (solid columns) or with (stripe columns) macrophage depletion 24 h and 72 h after treatment with NPs and their small molecule counterparts.
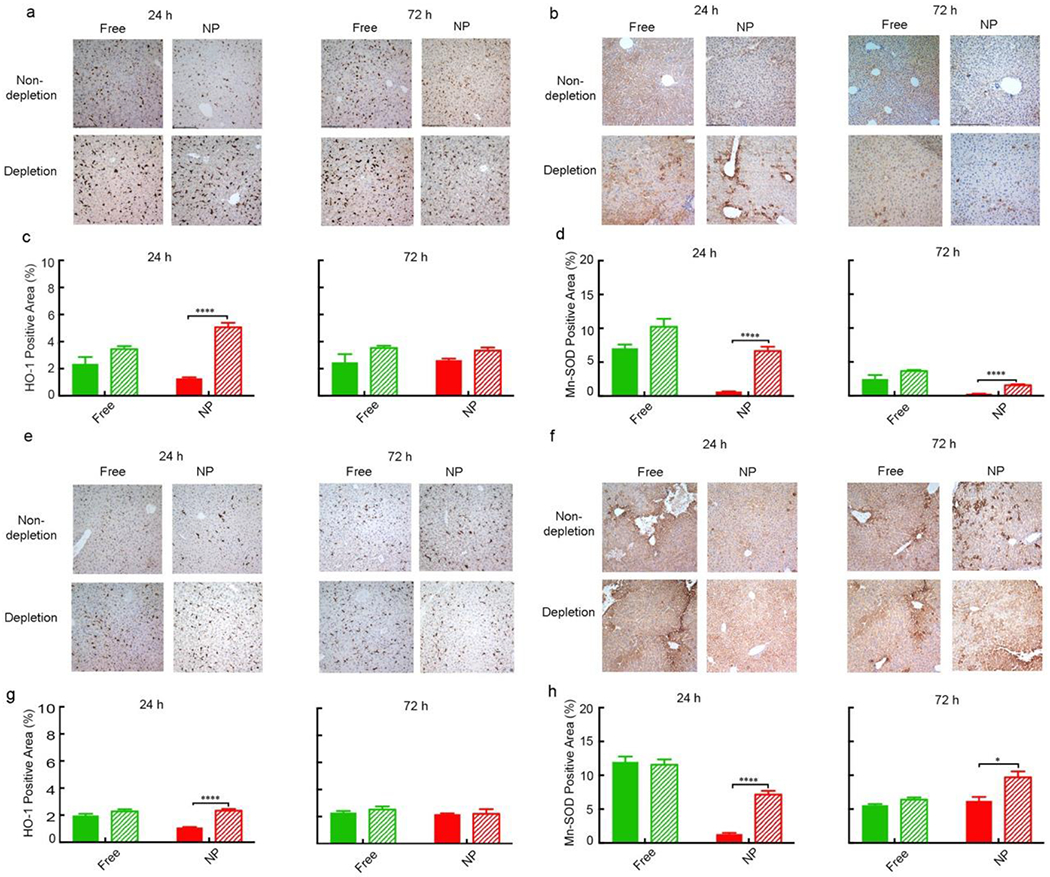
a, HO-1 staining of the livers treated with free CPZ and CPZ-loaded NP. b, Mn-SOD staining of the livers treated with free CPZ and CPZ-loaded NP. c, d, Quantitative analysis of positive staining percentage in (a) and (b), respectively. Scale bar= 480 μm. Data represent mean ± SEM (n = 5). Statistical significance was assessed using the Mann Whitney test. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. e, HO-1 staining of the livers treated with free Wtmn and Wtmn-loaded NPs. f, Mn-SOD staining of the livers treated with free Wtmn and Wtmn-loaded NPs. g, h, Quantitative analysis of positive staining percentage in (e) and (f), respectively. Scale bar= 480 μm. Data represent mean ± SEM (n = 5). Statistical significance was assessed using the Mann Whitney test. *p < 0.05, **p < 0.01, ***p < 0.001.
Taken together, nanotherapeutics showed lower hepatotoxicity than their small molecule counterparts. However, the hepatotoxicity of nanotherapeutics significantly increased after the depletion of macrophages. The increase of hepatotoxicity in non-formulated free drug-treated groups was not as significant as that in NP-treated groups, which led to the hepatotoxicity of nanotherapeutics became comparable or even higher than their small molecule counterparts after the depletion of macrophages. Therefore, we concluded that macrophage uptake is one of the key factors in the reduction of hepatotoxicity of nanotherapeutics.
Recently, some papers reported that using Intralipid 20% to temporarily and partially blunt the reticuloendothelial system (RES) could reduce the toxicity of platinum-containing nanodrugs in liver, spleen and kidney. The reason of the reduced toxicity was proposed as Intralipid competes with the nanodrugs to be taken up by RES, thus reduces the amount of drugs delivered to RES and their toxicity in the RES organs. They found that the liver accumulation of nanodrugs showed a decrease of 24.9% at 5 h, a decrease of 20.4% at 24 h, and an increase of 8.7% at 72 h.[29–31] However, in our study, complete depletion of macrophage by clodronate liposomes increases the hepatotoxicity of nanotherapeutics with increased ALT and AST levels. The reason might be that the existence of macrophages allows the uptake of nanotherapeutics by macrophages in the liver, which competes with the uptake by hepatocytes and leads to a decreased hepatotoxicity of known hepatotoxicity compounds. Therefore, one should be careful when blunting RES system during administration of nanotherapeutics. The optimal level and time point for RES blunting should be determined to combine nanotherapeutics and RES reduced agents.
In summary, we showed that NP delivery of hepatotoxic compounds can reduce hepatotoxicity, despite the extensive liver distribution. Importantly, we demonstrated that macrophage uptake is the key factor in determining the hepatotoxicity of nanotherapeutics. Our findings bridge an important knowledge gap in clinical nanomedicine and drug delivery. More importantly, our findings suggest an important new clinical application for NP drug delivery: to enable clinical translation of hepatotoxic compounds. Since many compounds are abandoned during development or withdrawn from the market after approval due to hepatotoxicity, our finding may provide a novel and much-needed strategy to overcome hepatotoxicity of therapeutics.
Experimental Section
Materials
Poly (D, L-lactide-coglycolide) (PLGA, Mw: ~60 K, 75:25) was purchased from Polyscitech®. Soybean lecithin was obtained from Alfa Aesar. DSPE-PEG2000-COOH[1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-carbox(polyethy-lene glycol) 2000] was obtained from Avanti Polar Lipids, Inc. Wortmannin (Wtmn) and 7-Ethyl-10-hydroxy-camptothecin (SN-38) were purchased from SelleckChem. Chlorpromazine (CPZ) was obtained from Sigma-Aldrich. Clodrosome® (5mg mL−1) was purchased from Encapsula NanoSciences (Brentwood, TN). Williams’ Medium E and cell maintenance supplements (Combo kit CM4000) were obtained from Gibco (Frederick, MD). MTS [(3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxy phenyl)-2-(4-sulfophenyl)-2H-tetrazo lium)] was obtained from Promega. HO-1 and Mn-SOD, F4/80 antibodies were purchased from Cell Signaling.
NP preparation
The PLGA-core, PEG-DSPE-lecithin shell NPs were synthesized using a nanoprecipitation method.[21] Drugs (Wtmn, CPZ or SN38) and PLGA were dissolved in acetonitrile with a final concentration of 1 mg mL−1 for drugs and 10 mg mL−1 for PLGA. The aqueous phase was prepared by dissolving lecithin and DSPE-PEG-COOH (7:3, molar ratio) in 4% ethanol with a final concentration of 1 mg mL−1 and preheated for 15 min at 65 °C. The ratio of lipids (lecithin and DSPE-PEG-COOH) to polymer was 100%. For SN-38 encapsulated NPs, the organic phase was sonicated and heated at 37 °C to facilitate dissolution. The organic phase was added dropwise into the aqueous phase. The volume ratio of organic phase and aqueous phase was 1:2.5, and the solution was stirred under vacuum for 3 h to completely evaporate the acetonitrile. The obtained NPs were washed twice with deionized water using Amicon™ Ultra (100 KDa) centrifuge tubes.
Characterization of NPs
Hydrodynamic diameters, polydispersity (PDI) and zeta potentials (ζ) of NPs were determined by Zetasizer Nano ZS system (Malvern Instruments Ltd., Malvern, UK). Transmission electron microscopy (TEM) images were recorded using Zeiss EM 910 for morphology analysis.
Drug loading efficiency was determined using HPLC. Briefly, 50 μL of NPs were dissolved in 200 μL acetonitrile, vortexed for 1 min, and sonicated for 10 min. The obtained solution (10 μL) was analyzed by HPLC and drug load was calculated using a standard concentration curve. All measurements were determined in triplicate.
In vitro drug release
In vitro release profiles were assayed using Slide-Alyzer mini dialysis microtubes (molecular weight cutoff 10 kDa, Pierce). Briefly, 3 mL of NP solutions (1 mg mL−1) were equally split into 30 dialysis microtubes and dialyzed against purified H2O at 37 °C under mild stirring (100 rpm min−1). An aliquot (80 μL) of solution was taken from the microtube at predetermined time points and mixed with 240 μL of acetonitrile. 10 μL of the mixture was analyzed by HPLC to quantify drug concentrations. The amount of released drug was calculated from the weight difference between total loaded drug and remaining drug in the microtubes. Data at each time point are obtained from three independent experiments.
Cell lines and culture
Murine macrophage cell lines Raw 264.7 were obtained from ATCC. Raw 264.7 cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) (Gibco) supplemented with 10% fetal bovine serum (Mediatech) and penicillin (100 U mL−1)/streptomycin (100 μg mL−1, Mediatech).
Primary hepatocytes were isolated from CD-1 mice using a perfusion-based isolation method.[32] Isolation started by cannulating the portal vein. The vasculature was perfused with 70 mL of preheated (37 °C) HBSS supplemented with 0.5 mM EDTAto remove blood, followed by perfusion with 90 mL of preheated low-glucose DMEM (1 g L−1) supplemented with type I collagenase (100 CDU mL−1), HEPES (15 mM), penicillin (100 U mL−1) and streptomycin (100 μg mL−1) (Mediatech). Finally, hepatocytes were harvested by centrifugation at 50 g for 2 min and washed twice with 25 mL of Williams’ medium E. Hepatocytes were counted with a hemocytometer and seeded into liver matrix (300 μg protein per well) coated 24-well plates at a density of 2 × 104well−1 on Day 0. Liver matrix was prepared from mice using a perfusion process and sterilized as previously reported.27 Hepatocytes were cultured in Williams’ medium E and the medium was replaced every three days.
In vitro toxicity
Primary hepatocytes were isolated and seeded into liver-matrix-coated 24-well plates (300 μg protein per well) at a density of 2 × 104 well−1 in Williams’ medium E on Day 0, and were treated with NPs on Day 5. Working concentrations of Wtmn and CPZ were 0.5 μM and 25 μM, respectively. In one experimental group, NPs (100 μL) were added to cell culture medium and placed in a Slide-ALyzer MINI dialysis microtube with a molecular weight cut-off of 2 kDa. The dialysis microtubes were placed above the hepatocyte culture plates and were gently stirred on a shaker inside the cell culture incubator. In another group, NPs (100 μL) were incubated with RAW 264.7 cells for 3 hours. RAW 264.7 cells were harvested, washed and placed in a Slide-ALyzer MINI dialysis microtube with a molecular weight cut-off of 2 kDa. The dialysis microtubes were placed above the hepatocyte culture plates and were gently stirred on a shaker inside the cell culture incubator. Cell viability of hepatocytes was determined using MTS [(3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium)] after 6 h and 24 h of treatments. The experiments were repeated three times.
Animal study
For animal studies, CD-1 (Charles River Labs), CB6F1 (Charles River Labs) and C57/BL6 (Jackson Labs) female mice with 20 g body weight were used. All animal work was approved and monitored by the University of North Carolina Animal Care and Use Committee.
Sample size is calculated based on our preliminary data with Wtmn and CPZ NPs. We calculated an effect size of 1.821. The nonparametric analog of this effect size can be stated in terms of p1=Pr (X<Y), or an observation in Group X is less than an observation in Group Y when H1 is true. The null hypothesis being tested is p1=0.5. For effect size 1.821, p1=0.099. A sample size of 8 in each group will have 80% power to detect a probability of 0.099 that an observation in Group X is less than an observation in Group Y, using a Wilcoxon (Mann-Whitney) rank-sum test, with a 0.05 two-sided significance level.
Macrophage depletion
CD-1 mice were given 200 μL of either clodronate or control liposome (5 μg mL−1) through tail vein injection.[28] Blood samples (100 μL) were collected at indicated time points (6, 12 and 24 h). Serum was separated for aspartate transaminase (AST) and alanine transaminase (ALT) assays by centrifuging at 3000 rpm for 10 min using serum blood collection tubes (BD).[33] Livers were collected and fixed with 10% neutral buffered formalin (Thermo Scientific) for 72 h and underwent F4/80 staining by Animal Clinical Laboratory Core Facility at UNC School of Medicine to assess the macrophage depletion.
In vivo hepatotoxicity
CD-1 mice were injected intravenously with free (non-NP formulation) CPZ, Wtmn and SN-38 at different doses. The amount of injected drugs was normalized by body weight. For in vivo hepatotoxicity assay, 1/2 MTD of free drugs or NP-formulated drugs were administrated through tail vein injection (n=8-10; 15 mg kg−1 for CPZ, 5 mg kg−1 for SN38 and 0.35 mg kg−1 for Wtmn). Serum samples were obtained at designated time points for AST and ALT test. In multiple-dose treatment, CPZ-loaded NPs were injected twice (each at ½ MTD of small molecules) intravenously at an interval of 24 h into CD-1 mice. Serum samples were collected at 6 h, 12 h and 24 h after the second injection. Mice were sacrificed after blood collection, and livers were collected and fixed with 10% neutral buffered formalin for 72 h prior to immunohistochemistry.
Immunohistochemistry study
Livers were fixed in 10% neutral buffered formalin for 72 h at room temperature, paraffin embedded and cut into 4 μm sections. Immunohistochemistry staining were performed on paraffin-embedded sections. Prior to staining, Paraffin specimens were dewaxed (67 °C, 2 h), rehydrated, heated in a microwave oven, and rinsed with Tris buffer solution (pH = 6.0) for antigen retrieval. Endogenous peroxidase was blocked by 1% hydrogen peroxide, epitope unmasking was performed by immersing sections in antigen retrieval solution (0.01 mol L−1) and heating for 10 min in a microwave oven. Normal goat serum was utilized for nonspecific binding. Primary antibodies with appropriate dilutions using mouse anti-F4/80 (1:25), anti-HO-1(1:300) and anti-Mn-SOD (1:250) were incubated overnight at 4 °C. Slides were washed with Tris buffer, followed by incubation with secondary antibodies for 30 min at room temperature. The densitometry of the staining signal was measured and quantified using ImageJ analysis software, version 2.0.0.
Statistical analysis
Statistical analyses were performed using Prism GraphPad software, version 7.0a. Multiplicity adjusted P values are reported. One-way ANOVA or Student’s t-test was employed to compare the data between different groups; P < 0.05 was considered to be significant.
Supplementary Material
Acknowledgements
The authors would like to thank Stephanie Montgomery, Dawud Hilliard and Ling Wang from Animal Histopathology & Lab Medicine Core for their assistance in hepatotoxicity analysis and histology studies. The authors would like to thank Victoria J. Madden and Kristen K. White from Microscopy Services Laboratory (MSL) for their assistance in TEM and IHC photographing. The authors would like to thank Alain Valdivia, Mark Ross and Charlene Santos for their assistance in mice tail vein injection. This work was supported by funding from the National Institutes of Health/National Cancer Institute R01GM130590, U54CA198999, R01 CA178748 and Department of Defense Congressionally Directed Medical Research Programs-Peer Reviewed Cancer Research Program Idea Award CA150391 (A.Z.W.). DF and KLRB are supported by NIH R35 GM122576 (KLRB). Feifei Yang is supported by funding from CAMS Initiative for Innovative Medicine (CAMS-I2M 2017-I2M-1-013) and CSC scholarship for studying in the University of North Carolina at Chapel Hill.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Competing interests: The authors declare no competing financial interests.
Data availability statement
All relevant data within the text and Supporting Information are available from the corresponding authors on request.
Animal study ethics statement
All the animal work was approved and monitored by the University of North Carolina Animal Care and Use Committee.
Contributor Information
Feifei Yang, Institute of Medicinal Plant Development (IMPLAD), Chinese Academy of Medical Sciences & Peking Union Medical College, Haidian District, Beijing, 100193, P.R. China; Laboratory of Nano- and Translational Medicine, Lineberger Comprehensive Cancer Center, Carolina Center for Cancer Nanotechnology Excellence, Carolina Institute of Nanomedicine, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, USA; Department of Radiation Oncology, University of North Carolina at Chapel Hill, Chapel Hill, NC, 27599, USA.
Yusra Medik, Laboratory of Nano- and Translational Medicine, Lineberger Comprehensive Cancer Center, Carolina Center for Cancer Nanotechnology Excellence, Carolina Institute of Nanomedicine, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, USA; Department of Radiation Oncology, University of North Carolina at Chapel Hill, Chapel Hill, NC, 27599, USA.
Liantao Li, Laboratory of Nano- and Translational Medicine, Lineberger Comprehensive Cancer Center, Carolina Center for Cancer Nanotechnology Excellence, Carolina Institute of Nanomedicine, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, USA; Department of Radiation Oncology, University of North Carolina at Chapel Hill, Chapel Hill, NC, 27599, USA; Jiangsu Center for the Collaboration and Innovation of Cancer Biotherapy, Cancer Institute, Xuzhou Medical University, Xuzhou, Jiangsu 221004, China.
Xi Tian, Laboratory of Nano- and Translational Medicine, Lineberger Comprehensive Cancer Center, Carolina Center for Cancer Nanotechnology Excellence, Carolina Institute of Nanomedicine, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, USA; Department of Radiation Oncology, University of North Carolina at Chapel Hill, Chapel Hill, NC, 27599, USA.
Dong Fu, UNC Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC, 27599, USA.
Kim L.R. Brouwer, UNC Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC, 27599, USA
Kyle Wagner, Laboratory of Nano- and Translational Medicine, Lineberger Comprehensive Cancer Center, Carolina Center for Cancer Nanotechnology Excellence, Carolina Institute of Nanomedicine, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, USA; Department of Radiation Oncology, University of North Carolina at Chapel Hill, Chapel Hill, NC, 27599, USA.
Bo Sun, Laboratory of Nano- and Translational Medicine, Lineberger Comprehensive Cancer Center, Carolina Center for Cancer Nanotechnology Excellence, Carolina Institute of Nanomedicine, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, USA; Department of Radiation Oncology, University of North Carolina at Chapel Hill, Chapel Hill, NC, 27599, USA.
Hossein Sendi, Laboratory of Nano- and Translational Medicine, Lineberger Comprehensive Cancer Center, Carolina Center for Cancer Nanotechnology Excellence, Carolina Institute of Nanomedicine, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, USA; Department of Radiation Oncology, University of North Carolina at Chapel Hill, Chapel Hill, NC, 27599, USA.
Yu Mi, Laboratory of Nano- and Translational Medicine, Lineberger Comprehensive Cancer Center, Carolina Center for Cancer Nanotechnology Excellence, Carolina Institute of Nanomedicine, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, USA; Department of Radiation Oncology, University of North Carolina at Chapel Hill, Chapel Hill, NC, 27599, USA.
Andrew Z. Wang, Laboratory of Nano- and Translational Medicine, Lineberger Comprehensive Cancer Center, Carolina Center for Cancer Nanotechnology Excellence, Carolina Institute of Nanomedicine, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, USA; Department of Radiation Oncology, University of North Carolina at Chapel Hill, Chapel Hill, NC, 27599, USA.
References
- [1].Gaitanis A, Staal S, Methods. Mol. Biol 2010, 624, 385. [DOI] [PubMed] [Google Scholar]
- [2].Brannon-Peppas L, Blanchette JO, Adv. Drug. Deliv. Rev 2004, 56, 1649. [DOI] [PubMed] [Google Scholar]
- [3].Karve S, Werner ME, Sukumar R, Cummings ND, Copp JA, Wang EC, Li C, Sethi M, Chen RC, Pacold ME, Wang AZ, Proc. Natl. Acad. Sci. U S A. 2012, 109, 8230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Zhu Q, Talton J, Zhang G, Cunningham T, Wang Z, Waters RC, Kirk J, Eppler B, Klinman DM, Sui Y, Gagnon S, Belyakov IM, Mumper RJ, Berzofsky JA, Nat. Med 2012, 18, 1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Pornpattananangkul D, Zhang L, Olson S, Aryal S, Obonyo M, Vecchio K, Huang CM, Zhang L, J. Am. Chem. Soc 2011, 133, 4132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Nel A, Xia T, Mädler L, Li N, Science. 2006, 311, 622. [DOI] [PubMed] [Google Scholar]
- [7].Zhang YN, Poon W, Tavares AJ, McGilvray ID, Chan WCW, J. Control. Release. 2016, 240, 332. [DOI] [PubMed] [Google Scholar]
- [8].Love SA, Maurer-Jones MA, Thompson JW, Lin YS, Haynes CL, Annu. Rev. Anal. Chem 2012, 5, 181. [DOI] [PubMed] [Google Scholar]
- [9].Tsoi KM, MacParland SA, Ma XZ, Spetzler VN, Echeverri J, Ouyang B, Fadel SM, Sykes EA, Goldaracena N, Kaths JM, Conneely JB, Alman BA, Selzner M, Ostrowski MA, Adeyi OA, Zilman A, McGilvray ID, Chan WC, Nat. Mater 2016, 15, 1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Soepenberg O, de Jonge MJ, Sparreboom A, de Bruin P, Eskens FA, de Heus G, Wanders J, Cheverton P, Ducharme MP, Verweij J, Clin. Cancer. Res 2005, 11, 703. [PubMed] [Google Scholar]
- [11].Hume DA, Curr. Opin. Immunol 2006, 18, 49. [DOI] [PubMed] [Google Scholar]
- [12].Peracchia MT, Vauthier C, Desmaële D, Gulik A, Dedieu JC, Demoy M, d’Angelo J, Couvreur P, Pharm. Res 1998, 15, 550. [DOI] [PubMed] [Google Scholar]
- [13].Matsumura Y, Hamaguchi T, Ura T, Muro K, Yamada Y, Shimada Y, Shirao K, Okusaka T, Ueno H, Ikeda M, Watanabe N, Br. J. Cancer. 2004, 91, 1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wang AZ, Langer R, Farokhzad OC, Annu. Rev. Med 2012, 63, 185. [DOI] [PubMed] [Google Scholar]
- [15].Meerum Terwogt JM, ten Bokkel Huinink WW, Schellens JH, Schot M, Mandjes IA, Zurlo MG, Rocchetti M, Rosing H, Koopman FJ, Beijnen JH, Anticancer. Drugs. 2001, 12, 315. [DOI] [PubMed] [Google Scholar]
- [16].Boddy AV, Plummer ER, Todd R, Sludden J, Griffin M, Robson L, Cassidy J, Bissett D, Bernareggi A, Verrill MW, Calvert AH, Clin. Cancer. Res 2005, 11, 7834. [DOI] [PubMed] [Google Scholar]
- [17].Li X, Kondoh M, Watari A, Hasezaki T, Isoda K, Tsutsumi Y, Yagi K, Pharmazie 2011, 66, 282. [PubMed] [Google Scholar]
- [18].Gandhi A, Guo T, Ghose R, J. Toxicol. Sci 2010, 35, 163. [DOI] [PubMed] [Google Scholar]
- [19].Cai Z, Yang J, Shu X, Xiong X, J. BUON 2014, 19, 350. [PubMed] [Google Scholar]
- [20].Workman P, Clarke PA, Raynaud FI, van Montfort RL, Cancer. Res 2010, 70, 2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Caster JM, Yu SK, Patel AN, Newman NJ, Lee ZJ, Warner SB, Wagner KT, Roche KC, Tian X, Min Y, Wang AZ, Nanomedicine 2017, 13, 1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Chan JM, Zhang L, Yuet KP, Liao G, Rhee JW, Langer R, Farokhzad OC, Biomaterials 2009, 30, 1627. [DOI] [PubMed] [Google Scholar]
- [23].Hyder MA, Hasan M, Mohieldein A, J. Clin. Diagn. Res 2016, 10, BC01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Alfaifi M, Eom YW, Newsome PN, Baik SK, J. Hepatol 2018, 68, 1272. [DOI] [PubMed] [Google Scholar]
- [25].Finkel T, Holbrook NJ, Nature 2000, 408, 239. [DOI] [PubMed] [Google Scholar]
- [26].Yachie A, Niida Y, Wada T, Igarashi N, Kaneda H, Toma T, Ohta K, Kasahara Y, Koizumi S, J. Clin. Invest 1999, 103, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Tian X, Werner ME, Roche KC, Hanson AD, Foote HP, Yu SK, Warner SB, Copp JA, Lara H, Wauthier EL, Caster JM, Herring LE, Zhang L, Tepper JE, Hsu DS, Zhang T, Reid LM, Wang AZ, Nat. Biomed. Eng 2018, 2, 443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Sturm E, Havinga R, Baller JF, Wolters H, van Rooijen N, Kamps JA, Verkade HJ, Karpen SJ, Kuipers F, J. Hepatol 2005, 42, 102. [DOI] [PubMed] [Google Scholar]
- [29].Liu L, Ye Q, Lu M, Lo YC, Hsu YH, Wei MC, Chen YH, Lo SC, Wang SJ, Bain DJ, Ho C. Sci. Rep 2015, 5, 10881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Liu L, Ye Q, Lu M, Chen ST, Tseng HW, Lo YC, Ho C. Sci. Rep 2017, 7, 16106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Liu L, Hitchens TK, Ye Q, Wu Y, Barbe B, Prior DE, Li WF, Yeh FC, Foley LM, Bain DJ, Ho C. Biochim. Biophys. Acta 2013, 1830, 3447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Shulman M, Nahmias Y, Methods. Mol. Biol 2013, 945, 287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Sethi M, Sukumar R, Karve S, Werner ME, Wang EC, Moore DT, Kowalczyk SR, Zhang L, Wang AZ, Nanoscale 2014, 6, 2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.