

Abstract
Targets in the protein docking experiment CAPRI generally present new challenges and contribute to new developments in methodology. In rounds 38-45 of CAPRI (Critical Assessment of PRedicted Interactions) template-based methods (TBMs) have been used with success for modeling complexes for which good templates were available. For weak or ambiguous templates however, integrating free docking and template-based methods becomes necessary. We demonstrate that free docking using ClusPro can reproduce some interfaces suggested by weak or ambiguous templates while not reproducing others, and thereby can help to choose the template resulting in the best model. In other cases free docking may reveal if none of the available templates is likely to provide a near-native interface. We discuss the potential advantages of combining template-based modeling with traditional free docking. Results are also presented to demonstrate that purely template-based modeling by the ClusPro TBM server can yield fairly accurate models for targets with at least one productive template. The new server is freely available for non-commercial use at https://tbm.cluspro.org.
Keywords: Protein-protein complexes, protein-peptide complexes, homology modeling, template selection, ambiguous templates, docking server
Introduction
Protein-protein interactions are important for understanding cellular function and organization. Mechanistic interpretation of these interactions frequently requires atom-level details, ideally obtained by X-ray crystallography. However, since experimental structure determination for complexes can be very difficult, a number of computational docking methods have been developed for generating complex structures from the structures of the component proteins.1,2 Our protein-protein docking server ClusPro, first introduced in 2004,3–5 was substantially improved by adding the docking program PIPER in 2006.6 The server is heavily used today; it has over 13,000 registered users (registration is not required), and has performed over 300,000 docking calculations so far, on the average of over 5,000 per month. Docked structures generated by ClusPro have been reported in around 600 publications.
Between 2015 and 2018 we added four new options to ClusPro.7 These options were considering pairwise interaction data as restraints;8 accounting for Small Angle X-ray Scattering (SAXS) data in docking;9 performing discrimination between biological and crystallographic dimers;10 and adding PeptiDock, a method of docking flexible peptides to proteins.11 As will be described in this report, the most important addition in 2018/2019 has been the option of using template-based methods (TBMs) in ClusPro, thus starting the calculations from sequences rather than structures of the component proteins.5 Adding this last option became particularly important after merging CAPRI with CASP (Critical Assessment of Structure Prediction). Although this option of ClusPro is very new and has not yet been extensively tested, it was already used in the CAPRI/CASP13 protein structure prediction experiment.12
While several groups have a long record of developing template-based methods (TBMs) for docking,13–16 ClusPro used only free docking but still performed well in earlier CAPRI rounds. However, in the latest rounds it became very clear that an increasing fraction of targets could be modeled substantially better by TBMs than by classical docking,17–19 and hence adding TBMs to ClusPro has become a priority. In particular, rounds 38-45 of CAPRI included five targets that were easily solved by many predictor groups using TBMs. Adding TBMs to ClusPro and its applications to some easy targets have been described previously,12 and hence in this paper we focus on the more challenging targets of rounds 38-45 of CAPRI. These included two targets that had no templates available for the complexes and hence required homology modeling of individual subunits, and then docking of the homology models. As we will argue, this problem is still largely unsolved, and no predictor group submitted even acceptable predictions. Between the easy targets amenable to TBMs and the very difficult ones that require docking of homology models laid a number of targets for which homologous templates were present, but homology modeling was neither straightforward, or outright inapplicable. In another target the problem was to dock a short flexible peptide to a protein. This problem has been recently addressed by several groups.11,20–22 Here we describe our attempts to deal with all these targets using a combination of template-based modeling and free docking. We hope that this discussion will be useful for drawing the appropriate line between the domains of applicability for template-based and free docking methods. While the focus on this paper is on the integration of the two approaches, we also report on the recent development of the ClusPro TBM server and its performance on the CAPRI 38-45 targets.
Methods
Free docking
The basic free docking capabilities of the ClusPro server are based on a two-stage approach which hasn’t changed much since its original conception.3,4 In the first stage, all possible rigid body orientations of two rigid proteins are sampled exhaustively to produce 1000 low-energy poses. The sampling is performed using the Fast Fourier Transform based docking program PIPER using a translational step of 1.0 Å and a pre-generated set of 70000 rotations, approximately corresponding to 5 degree rotational steps.6,23 To score the sampled conformations, ClusPro uses a physics-inspired scoring function which includes Van der Waals- and electrostatics-based terms as well as the knowledge-based DARS potential.24 In the second stage, the top-scoring poses are clustered together with a 10 Å RMSD clustering radius, clusters are ranked by population and cluster centers are selected as final models. These final models are subjected to local energy minimization to resolve any clashes and are then reported to the user.23 During the latest CAPRI rounds, we relied on the HHpred server25,26 to generate the homology models for the subunit structures whenever such structures were not available.
Information-driven docking
Over the years, the basic ClusPro docking protocol was enhanced with various additions, including the ability to handle arbitrary geometric7,8 and certain symmetry-induced restraints, to perform filtering based on small angle X-ray scattering (SAXS) data,27 and to use specific scoring that substantially improved the prediction of antibody-antigen complexes.28 In rounds 38-45 of the CAPRI experiment, we repeatedly took advantage of these new options and leveraged the various available information sources to guide server predictions. In most cases, we relied on the available structural templates to derive geometric restraints for the expected interfacial residues. The procedure was largely manual and involved selecting 1-3 residue pairs that are in contact in the template interface, and then converting them into the restraints for the corresponding residues in the target complex. The restraining distances corresponded to the minimal distance between any pair of atoms in the two residues and were set to values in the range of 4-8 Å. The resulting restraints were submitted to ClusPro together with subunit structures to generate the models.
Peptide-protein docking
As will be described, round 38 featured one target, T121, representing a protein-peptide complex determined by NMR. Participants were provided with sequences for both parts of the complex. The availability of known motifs within the target peptide sequence made T121 an ideal candidate for our peptide-docking protocol implemented in the ClusPro PeptiDock server.11 The protocol is based on the observation that a known motif can be used to adequately sample peptide conformational space by extracting representative fragments from larger structures in the Protein Data Bank (PDB).29 The most common conformations are selected by population after an initial clustering step of the extracted fragments that can number in the thousands. Each of these selected conformers is then globally docked to the protein structure using PIPER,6 with top-scoring results pooled together, using a final clustering radius of 3.5 Å. The protocol was developed using a subset of the PeptiDB v2 peptide-protein docking benchmark set30 and validated on a set of additional complexes. Evaluation of these predicted complexes demonstrated consistent placement of the peptide within 4.0 Å backbone RMSD of the native peptide structure.11
Template-based modeling in ClusPro
While the approach based on restraint-guided free docking proved to be an efficient way of handling template information, it became increasingly obvious that, at least in its current shape, it has the downside of being very labor-intensive. This was due to the fact that all the essential steps, including the identification of templates, the building of docking-suitable homology models of the monomers, and the derivation of restraints, were largely manual procedures. Additionally, the latest CAPRI rounds have demonstrated that the number of targets that can be directly modeled based on homology makes up a significant fraction of the total. Indeed, at least four of the eight targets representing protein-protein complexes could be modeled by a TBM using homologous complexes as templates. The share of such “TBM-amenable” cases was even higher in the recent joint CAPRI-CASP experiment.5 In addition, we recognized that template-based docking generally yields higher accuracy models if good template structures are available. Motivated by these observations, we recently enhanced the ClusPro server with an automated template-based modeling functionality, and reported the resulting protocol.12 The method is fully automated and bypasses the docking step entirely, instead fully relying on the template structure. Here we briefly describe the basic features of this protocol, as well as some very recent development motivated by our CAPRI participation.
Our template-based modeling protocol ClusPro TBM is fairly straightforward. It takes the sequences of the subunits and the expected stoichiometry of the target complex, and identifies potential templates for the assembly in the PDB using HHsearch.31 Once templates containing homologs for all subunits have been identified, the method performs a “smart” stoichiometry check, testing whether the template can accommodate the required number of copies of each subunit type. Some non-trivial cases, including the use of homomeric templates for heteromeric targets or the use of single chain templates for multimers, can be handled within this framework. Finally, models of the complex subunits are built with MODELLER32 and aligned to the template structure to build models of the whole complex.
PDB100 database and clustering
The above protocol had some obvious weak points. The most important limitation was its reliance on the pdb70 database of protein sequences, which only contains the cluster centers of sequences present in the PDB database, clustered at 70% sequence identity.33 While the pdb70 database is sufficient for homology modeling of individual proteins, protein assemblies are often much more sensitive to sequence changes than protein folds, and thus a wealth of potential templates can get lost during clustering, especially in the case of heteromeric complexes. To remediate these deficiencies, a new HHsearch database, dubbed pdb100, was generated. Since we wanted to perform a comprehensive search over all the protein structures available in the PDB database, the sequence clustering step was skipped, and the Hidden Markov Models and multiple sequence alignments (MSAs) were generated for all the entries available in the PDB at the time. Aside from skipping the clustering with MMSeqs2,34 the database preparation protocol was the same as the one used for pdb70. First, a local mirror of the PDB database was created, and, for each entry, the sequence in FASTA format was extracted. Then, for each FASTA, an MSA was built using HHblits33 with the UniProt 20 database (dated February 2016). The secondary structure analysis step was skipped due to time constraints. The MSAs were used to build hidden Markov models using hhmake. The resulting database could then be used with HHsearch to extract all homologous templates present in the PDB.
The goal of creating the comprehensive pdb100 database was to allow HHsearch to extract all possible templates for the target complex, without losses caused by the clustering of individual proteins. However, the search in the extended database often produced many structurally redundant hits. Therefore, when models with correct sequence and stoichiometry were produced from the template proteins found by HHsearch, the final set contained many identical assemblies. We performed a simple clustering step to remove this redundancy. Clustering was done by computing Cα RMSD values between all pairs of models and finding those with the largest number of neighbors within 10.0 Å RMSD. This procedure takes into account the complex stoichiometry. For example, if the complex stoichiometry is A2B2, there exist several ways to align the chains of one model onto the other model. Clustering takes care of this ambiguity by attempting all possible alignments and choosing the one with minimal RMSD.
Results
Rounds 38-45 of the CAPRI competition included 16 targets, representing eight protein-protein, three protein-peptide, and five protein-sugar complexes. Two of the three protein-peptide targets were based on the same complex, so there were a total of eight distinct protein-protein and two protein-peptide complexes involved. The protein-sugar targets represented interactions of the same protein with a series of sugars of different length. We participated in the predictions of all targets, both as the team running the automated server ClusPro and as a human predictor group, and were among the top performers in both categories. Here, we provide a short analysis of the recent rounds and of our performance.
Of the 11 protein-protein and protein-peptide targets, five (T125, T133, T134, T135 and T136) represented relatively straightforward exercises in template-based modeling for which the majority of participants, including us, were able to produce acceptable or medium quality models. Another two (T123 and T124) were free docking targets that required modeling of the component proteins. The modeling turned out to be very difficult, and no predictor group submitted any acceptable or better prediction. The remaining three protein-protein targets (T122, T131, T132) and one protein-peptide target (T121) represented different, but closely related aspects of the template selection problem. With the increasing role of template-based methods in modeling of protein-protein interactions, these targets, in our opinion, provide examples that help to better understand the limits of applying purely template-based methods and to explore how such methods can be combined with free docking to get improved results.
Target T122: Cytokine receptor complex
Target T122 represented the assembly of the cytokine receptor complex, comprised by receptor monomer IL23R and a cytokine heterodimer IL23, formed by the IL23A and IL12B subunits. Since the organizers provided the participants with the structure of the IL23A-IL12B complex, the problem was essentially reduced to finding the correct orientation of the IL23R subunit relative to the IL23A-IL12B subcomplex. For this target, a number of template assemblies were available (e.g., 1I1R, 1P9M, 2D9Q, and 2Q7N). In the majority of these template structures, multiple copies of IL23R and IL23A homologs were present in the biological assemblies, with multiple interfaces formed between IL23R- and IL23A-like molecules. We found that two distinct interaction interfaces could be inferred from these templates (see Figure 1), and it is worth noting that across the majority of templates one of these interfaces was consistently easy to miss if only a single asymmetric unit was considered. We believe this simple fact explains why this target received such a small number of acceptable quality predictions from participants.
Figure 1:
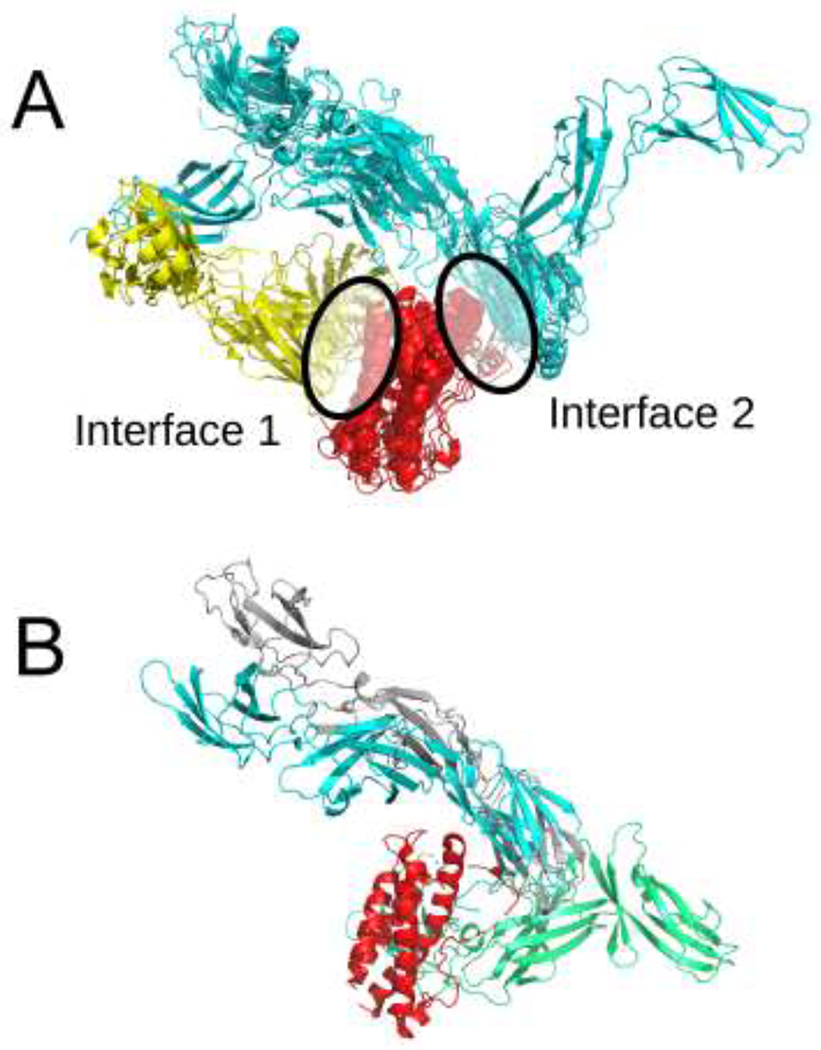
A) Two classes of interaction between IL23R and IL23A homologs observed among the structures in the PDB. The IL23A homologs are shown in red, IL23R homologs corresponding to two different interaction types are shown in yellow and cyan. B) The best model among the top 10 predictions in our human submission. The model is in gray, the structure of the native complex (PDB ID 5MZV) is in red (IL23A), green (IL12B) and cyan (IL23R).
As a part of our server submission, we performed the docking of the IL23A-IL12B subcomplex with a homology model of IL23R using two sets of restraints corresponding to the two potential interfaces, denoted as type 1 and type 2, derived from homologous complexes, and executed an additional free docking run for validation. We found that the type 1 interface is not reproduced by free docking, while a model with the type 2 interface was ranked 3rd. Additionally, we found that even with restraints applied, models with interfaces similar to type 1 tended to deviate from the template structures unless the restraints were specified in a way that completely precluded any movement of IL23R. Together, these observations motivated us to submit the models restrained to the type 2 interface. The top-ranked model in our submission was scored as having acceptable quality.
As a human group, we used MODELLER to prepare the homology models of the complex based on the four templates we identified, and combined those with manually selected models generated by free docking. The manual selection was used to ensure that the models occupy the type 2 interface. Our best model, which also happened to be the best submitted model in terms of L-RMSD and the fraction of native contacts among all participants, came from the set of models obtained by free docking.
Targets T131 and T132: Host-pathogen protein complexes
The two related targets T131 and T132 represented interactions between the human cell adhesion protein CEACAM1 with the Helicobacter pylori cell adhesion proteins HopQ type I and HopQ type II, respectively. Multiple structures of HopQ proteins and close homologs were available in the PDB (5F7K, 5F7Y, 5F93, and 5LP2). These structures caught our attention because many of them were crystallized with an Ig-like domain, a class of proteins to which CEACAM1 also belongs. During the server prediction stage, we thus assumed that the CEACAM1-HopQ interaction should follow the same pattern, and performed restrained docking of CEACAM1 homology models to HopQ type I/II homology models (or structures, whenever available). Similarly to the case of T122, we also performed free docking of the subunits, with a notable result being that none of the models recapitulated the interaction seen in the template. In this case, however, we discarded the free docking results and submitted the structures obtained by restrained docking as our server predictions for the two targets. None of the submitted models turned out to be correct.
For our human submission, we prepared homology models of the assemblies based on the same templates using MODELLER. However, for target T131 we also added models obtained by free docking to our submission package. It turned out to be the only submission by all predictor groups that contained a near-native model in the top 10 predictions, and it was actually a medium quality model according to the CAPRI assessors. No model from free docking was added to our submission for target T132, and it included no acceptable prediction.
Target T121: Protein-peptide complex
Capri target T121 represented an interaction between the P. aeruginosa TolA periplasmic domain and a 13-residue peptide from the C-terminal region of the TolB protein. For this target, While no clear-cut templates involving protein-peptide interactions were available for this complex, we found that many protein-protein and domain-domain interactions in which TolA homologs participated involved an antiparallel beta sheet stacking interaction (e.g., PDB IDs 1TOL, 2X9A, and 1U07), suggesting it to be the most likely binding mode for this target.For our server submission, we performed global protein-peptide docking using the recently developed ClusPro-PeptiDock automated protocol.11 The protocol accepts a structure of the protein, as well as the sequence and a sequence motif of the peptide, and produces structural models of the complex. Our manual involvement was limited to the selection of these inputs. For the protein structure we constructed a homology model of the TolA domain based on PDB ID 1TOL. As the ClusPro-PeptiDock server puts a limit on the peptide length, we had to restrict our modeling attempts to the seven-residue fragment of the original peptide.
Since we only modeled 7 of the 13 residues of the peptide, our submissions did not meet the CAPRI sequence identity criterion, and were not included in the final ranking. However, the models were still evaluated, and seven of the ten, including the one ranked first, had a backbone RMSD under 4 Å, corresponding to an acceptable quality model. The best of these models, ranked number 10 in our submission package, had a backbone RMSD of 2.8 Å, which brings it to the level of medium quality models. To our surprise, those models featured a binding mode that was in stark contrast to our original assumptions based on available templates. As a human team, we experimented with different methods for refinement of the complex, using the full length peptide, and our submission included a medium quality model. It should be noted that only two medium quality models were submitted by predictors for this target, both using the Cluspro protocol for the global docking step. The details describing these two approaches to refinement of the protein-peptide complexes are discussed in a separate paper focused on peptide docking, also published in this issue.
Further development and validation of the ClusPro TBM server
At the time of CAPRI rounds 38-45, some aspects of our ClusPro TBM server were still under development. Due to a number of more recent changes, including the switch to pdb100 from pdb70, we decided to test the ability of the server to produce near-native models of the protein-protein targets T122, T125, T133 and T136. These targets were selected because each of them had at least one productive template available. During the testing procedure the native PDBs where excluded from the template library using the exclusion list implemented in the server. Here we briefly describe the results of this analysis.
For target T122, which involved the construction of a 3-component complex of IL23R, IL12B and IL23A, we focused on modeling the interaction between IL23R and IL23A, as the structure for the IL23A-IL12B complex was available during the competition. ClusPro TBM was able to locate the templates corresponding to both types of interfaces described in the section dedicated to our approach to modeling this target during CAPRI. A near-native model based on PDB ID 2Q7N was ranked third.
Target T125 featured an interaction between a single dimer of the extracellular domain of LLT1 with two dimers of the extracellular domain of its inhibitor NKR-P1. The currently available native structure of this complex (PDB ID 5MGT) has only automatic biological assembly assignment generated by PISA.35 This assignment only includes the homodimeric interfaces between copies of LLT1 and NKR-P1 (heterodimeric interfaces are not assigned as biological). Therefore we attempted to reproduce the two types of PISA-assigned homodimeric assemblies, as well as the heterodimeric interface present in the asymmetric unit, and performed several ClusPro TBM runs using different values of the stoichiometry requirement parameter. In particular we attempted to model the A2, B2, A1B1, A2B2, and A2B4 stoichiometries, where the AnBm motif specifies the number of subunits of LLT1 (A) and NKR-P1 (B) in the generated models. The top models returned by the A2 and B2 submissions corresponded to the homodimeric assemblies of LLT1 and NKR-P1 found in the native structure. The A1B1 submission produced a model ranked 12 recapitulating the heterodimeric interactions between chains B and D in PDB ID 5MGT. Our submissions featuring higher-order stoichiometry did not produce any near-native models. This result suggests that partial templates could be combined to produce the structure of the whole assembly, and efficient identification and utilization of such partial templates should become one of the avenues for the future development of the ClusPro TBM server.
Target T133 represented the modeling of redesigned version of the Colicin E2 DNase – IM2 complex. A number of structures for the original complex were available (one was even pointed out by the organizers at the time of the competition), and ClusPro TBM produced a near-native model ranked 1 as expected. We note, however, that the current version of the server is not capable of capturing the moderate conformational changes, relative to the structure of the original complex, arising from a few interface residue mutations introduced in the design process. Finally, target T136 represented a homodecamer assembly of the Pseudomonas aeruginosa decarboxylase LdcA. Although the native structure is not available at the moment, and thus the quantitative analysis cannot be performed, all the models produced by ClusPro TBM feature the characteristic D5 symmetry, and overall arrangement found in related proteins (e.g., in E. Coli AdiA and Ldcl).
Discussion
Starting in 2014, CAPRI has teamed up with CASP to run joint prediction experiments, evaluated at CASP11 (2014), CASP12 (2016), and CASP13 (2018) meetings. Accordingly, a substantial fraction of the CAPRI/CASP and CAPRI targets involve modeling of structures of protein complexes from sequences rather than structures of the component proteins. In the most general case this is a difficult problem that requires the integration of template-based modeling of individual subunits, with docking calculations, or relying entirely on template-based modeling of the whole complex. The merging of CASP and CAPRI was useful for both experiments. For CAPRI the gain has been the increased number of targets, although a majority of new targets were homo-oligomers rather than complexes formed by two different proteins. In fact, it is clear that determining the structure of protein-protein complexes by X-ray crystallography is still difficult, resulting in a limited number of available targets. There is hope that this will change with the increased usage of cryogenic electron microscopy (cryo-EM) methods that are suitable for determining the structures of large protein assemblies without need for crystallization. For CASP the gain of adding the prediction of protein assemblies has been a new motivation for the use of protein modeling tools, since protein-protein and protein-peptide interactions generally provide functional information. However, this gain was limited by the already mentioned high fraction of homo-oligomeric targets.
The need for predicting protein complex structures from sequences meant new challenges and required changes in docking methodology. On the positive side, it demonstrated the advantages of homology modeling, since traditional free docking generally failed to compete with template-based modeling in terms of accuracy for targets with high quality templates available. Indeed, homology modeling tools have been perfected over many years, and can yield highly accurate predictions, particularly for homodimers. We have recently compared the prediction of 15 homodimeric targets from CASP11 and CASP12 using the two approaches.5 Although the numbers of acceptable predictions were almost the same, template-based modeling resulted in three times more medium quality or better predictions than did direct free docking.5 Similar trends also apply to CASP13 targets.12 Accordingly, CASP predictor groups experienced in homology modeling were among the top performers in the assembly prediction category of CASP as well as in CAPRI.17–19,36 On the negative side, the high percentage of easy targets, involving mostly homodimers, created the impression that template-based methods can be used for the majority of docking problems and hence there is no need for focusing on the further development of free docking methods. As we already mentioned, in rounds 38-45 of CAPRI, of the 11 protein-protein and protein-peptide targets, five (T125, T133, T134, T135, and T136) were easy targets with good templates available, and participants were able to produce acceptable or medium quality models using homology modeling tools. Our ClusPro TBM server also performed reasonably well as shown in the results.
The other seven targets, however, emphasize the remaining significant challenges. First, templates were available only for component proteins in targets T123 and T124, but not for the complexes. Thus, solving these problems required docking of homology models. We previously argued that this is a difficult and largely unsolved problem, since methods developed for docking X-ray structures may not be optimal for docking homology models.7 We even put together a benchmark set of homology models for the development of specific docking methods.37 However, the success of template-based modeling in CASP/CAPRI seemed to remove the urgency of such development. The results of CAPRI rounds 38-45 again emphasize that docking homology models is still a problem to be dealt with. In fact, targets T123 and T124 turned out to be very difficult, and no predictor group submitted any acceptable or better prediction.
We focused on the protein-protein targets T122, T131, and T132 that have shown different but closely related aspects of the template selection problem, and emphasized the need for combining template-based methods with direct docking. As described, templates for target T122 left us with ambiguity in the form of two very different putative interfaces. We solved this problem by trying to reproduce these interfaces by direct docking, both with and without specified restraints. These re-docking experiments have shown that only one of the interfaces could be reproduced, and this insight lead to an acceptable quality model. In contrast, templates for targets T131 and T132 suggested a well-defined arrangement which, however, was not reproduced by free docking, and this questioned the quality of the templates. We did take advantage of this finding and added models from direct docking to our manual submission for target T131, resulting in a medium quality model. Similarly, in target T121 free docking of the peptide component significantly preferred an arrangement not seen in any of the templates, and the docking results turned out to be correct, despite the fact that input preparation was based on templates.
Conclusion
In this paper we describe how template based modeling can be combined with free docking for target complexes that either have multiple putative templates with different interfaces, or all available templates are uncertain for some reason, usually due to limited sequence similarity. The strategy we suggest in such cases is building models based on all templates, separating the component proteins, and re-docking them as they would be X-ray structures. Generating a large cluster of docked structures close to one of the original models increases the probability that the particular model is near-native, and that the template leading to it is the best choice. Conversely, it is likely that none of the templates is acceptable if the free docking does not reproduce any of the models. In such cases direct docking is the method of choice, but it is clear that the predictions are less reliable and most likely less accurate than the ones that would be based on verified templates. Our results suggest that the re-docking strategy can be very useful if there is any doubt concerning the suitability of the templates, which is frequently the case. It is also possible to use restraints with different strengths in order to understand the biasing forces that are needed to stabilize specific interfaces.
While we emphasized the advantages of combining template-based modeling with traditional free docking, we also presented validation results to demonstrate that direct application of ClusPro TBM server yields accurate models for targets that have at least one productive template. These results were obtained by the current version of the server freely available for non-commercial use at https://tbm.cluspro.org. We note that the server and the methods described here do not include a refinement stage. Although current papers demonstrate that the quality of homology models can be improved by refinement tools, primarily based on repeated molecular dynamics simulations,38,39 at this time the approach is computationally too expensive to be implemented as a public server.
Figure 2:
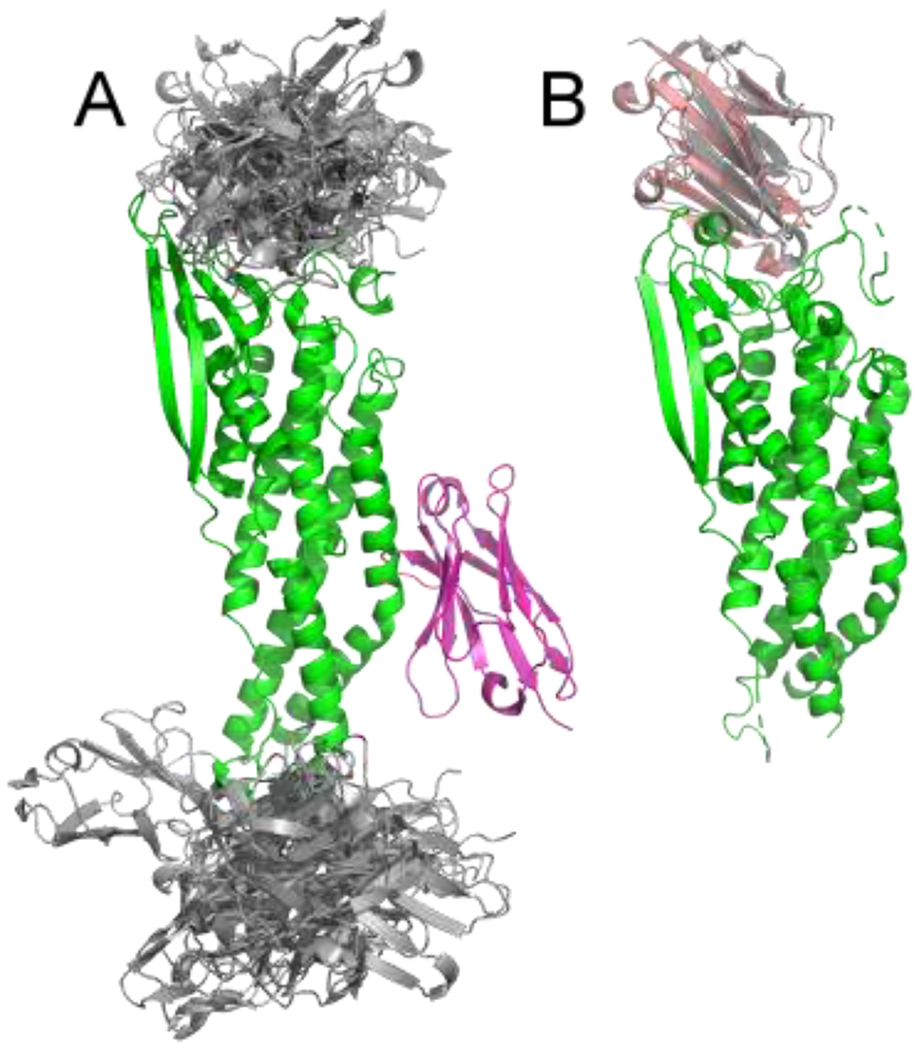
Free docking and template-based modeling for T131. A) None of the top 20 free docking models (shown in gray) validated the template-based orientation of the CEACAM1 protein (magenta) relative to HopQ (green). B) Free docking model ranked 3 (shown in gray) closely matches the orientation of CEACAM1 (wheat) relative to HopQ (green) in the native structure of the complex (PDB ID 6GBG).
Acknowledgements
This investigation was supported by grants DBI1759277 and AF1645512 from the National Science Foundation, and R35GM118078 and R21GM127952 from the National Institute of General Medical Sciences.
References
- 1.Smith GR, Sternberg MJ. Prediction of protein-protein interactions by docking methods. Curr Opin Struct Biol. 2002;12(1):28–35. [DOI] [PubMed] [Google Scholar]
- 2.Vajda S, Vakser IA, Sternberg MJ, Janin J. Modeling of protein interactions in genomes. Proteins. 2002;47(4):444–446. [DOI] [PubMed] [Google Scholar]
- 3.Comeau SR, Gatchell DW, Vajda S, Camacho CJ. ClusPro: a fully automated algorithm for protein-protein docking. Nucleic Acids Res. 2004;32(Web Server issue):W96–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Comeau SR, Gatchell DW, Vajda S, Camacho CJ. ClusPro: an automated docking and discrimination method for the prediction of protein complexes. Bioinformatics. 2004;20(1):45–50. [DOI] [PubMed] [Google Scholar]
- 5.Porter KA, Desta I, Kozakov D, Vajda S. What method to use for protein-protein docking? Curr Opin Struct Biol. 2019;55:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kozakov D, Brenke R, Comeau SR, Vajda S. PIPER: an FFT-based protein docking program with pairwise potentials. Proteins. 2006;65(2):392–406. [DOI] [PubMed] [Google Scholar]
- 7.Vajda S, Yueh C, Beglov D, et al. New additions to the ClusPro server motivated by CAPRI. Proteins. 2017;85(3):435–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xia B, Vajda S, Kozakov D. Accounting for pairwise distance restraints in FFT-based protein-protein docking. Bioinformatics. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang S Methods for SAXS-Based Structure Determination of Biomolecular Complexes. Adv Mater. 2014;26(46):7902–7910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yueh C, Hall DR, Xia B, Padhorny D, Kozakov D, Vajda S. ClusPro-DC: Dimer Classification by the Cluspro Server for Protein-Protein Docking. J Mol Biol. 2017;429(3):372–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Porter KA, Xia B, Beglov D, et al. ClusPro PeptiDock: Efficient global docking of peptide recognition motifs using FFT. Bioinformatics. 2017; 33(20), 3299–3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Porter KA, Padhorny D, Desta I, et al. Template‐Based Modeling by ClusPro in CASP13 and the Potential for Using Co-evolutionary Information in Docking. Proteins. 2019; DOI: 10.1002/prot.25808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sinha R, Kundrotas PJ, Vakser IA. Docking by structural similarity at protein-protein interfaces. Proteins. 2010;78(15):3235–3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kundrotas PJ, Zhu ZW, Janin J, Vakser IA. Templates are available to model nearly all complexes of structurally characterized proteins. Proc Nat Acad Sci USA. 2012;109(24):9438–9441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sinha R, Kundrotas PJ, Vakser IA. Protein docking by the interface structure similarity: how much structure is needed? PloS One. 2012;7(2):e31349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Keskin O, Nussinov R, Gursoy A. Prism: Protein-Protein Interaction Prediction by Structural Matching. Func Proteomics: Meth Protoc. 2008;484:505–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lensink MF, Velankar S, Kryshtafovych A, et al. Prediction of homo- and hetero-protein complexes by protein docking and template-based modeling: a CASP-CAPRI experiment. Proteins. 2016;84:323–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lensink MF, Velankar S, Wodak SJ. Modeling protein-protein and protein-peptide complexes: CAPRI 6th edition. Proteins. 2017;85(3):359–377. [DOI] [PubMed] [Google Scholar]
- 19.Lensink MF, Velankar S, Baek M, Heo L, Seok C, Wodak SJ. The challenge of modeling protein assemblies: the CASP12-CAPRI experiment. Proteins. 2018;86 Suppl 1:257–273. [DOI] [PubMed] [Google Scholar]
- 20.Yan C, Xu X, Zou X. Fully blind docking at the atomic level for protein-peptide complex structure prediction. Structure. 2016;24(10):1842–1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schindler CEM, de Vries SJ, Zacharias M. Fully blind peptide-protein docking with pepATTRACT. Structure. 2015;23(8):1507–1515. [DOI] [PubMed] [Google Scholar]
- 22.Alam N, Goldstein O, Xia B, Porter KA, Kozakov D, Schueler-Furman O. High-resolution global peptide-protein docking using fragments-based PIPER-FlexPepDock. PLoS Comput Biol. 2017;13(12):e1005905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kozakov D, Hall DR, Xia B, et al. The ClusPro web server for protein-protein docking. Nat Protoc. 2017;12(2):255–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chuang GY, Kozakov D, Brenke R, Comeau SR, Vajda S. DARS (Decoys As the Reference State) potentials for protein-protein docking. Biophys J. 2008;95(9):4217–4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hildebrand A, Remmert M, Biegert A, Soding J. Fast and accurate automatic structure prediction with HHpred. Proteins. 2009;77 Suppl 9:128–132. [DOI] [PubMed] [Google Scholar]
- 26.Soding J, Biegert A, Lupas AN. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005;33(Web Server issue):W244–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xia B, Mamonov A, Leysen S, et al. Accounting for observed small angle X-ray scattering profile in the protein-protein docking server cluspro. J Comp Chem. 2015;36(20):1568–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kozakov D, Schueler-Furman O, Vajda S. Discrimination of near-native structures in protein-protein docking by testing the stability of local minima. Proteins. 2008;72(3):993–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Berman HM, Westbrook J, Feng Z, et al. The Protein Data Bank. Nucleic Acids Res. 2000;28(1):235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lavi A, Ngan CH, Movshovitz-Attias D, et al. Detection of peptide-binding sites on protein surfaces: the first step toward the modeling and targeting of peptide-mediated interactions. Proteins. 2013;81(12):2096–2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nordstrom KJ, Sallman Almen M, Edstam MM, Fredriksson R, Schioth HB. Independent HHsearch, Needleman--Wunsch-based, and motif analyses reveal the overall hierarchy for most of the G protein-coupled receptor families. Mol Biol Evol. 2011;28(9):2471–2480. [DOI] [PubMed] [Google Scholar]
- 32.Fiser A, Sali A. Modeller: generation and refinement of homology-based protein structure models. Meth Enzymol. 2003;374:461–491. [DOI] [PubMed] [Google Scholar]
- 33.Remmert M, Biegert A, Hauser A, Soding J. HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat Methods. 2011;9(2):173–175. [DOI] [PubMed] [Google Scholar]
- 34.Steinegger M, Soding J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat Biotechnol. 2017;35(11):1026–1028. [DOI] [PubMed] [Google Scholar]
- 35.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372(3):774–797. [DOI] [PubMed] [Google Scholar]
- 36.Wodak SJ, Janin J. Modeling protein assemblies: Critical Assessment of Predicted Interactions (CAPRI) 15 years hence.: 6TH CAPRI evaluation meeting April 17-19 Tel-Aviv, Israel., Proteins. 2017;85(3):357–358. [DOI] [PubMed] [Google Scholar]
- 37.Bohnuud T, Luo L, Wodak SJ, et al. A benchmark testing ground for integrating homology modeling and protein docking. Proteins. 2017;85(1):10–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Terashi G, Kihara D. Protein structure model refinement in CASP12 using short and long molecular dynamics simulations in implicit solvent. Proteins. 2018;86 Suppl 1:189–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heo L, Arbour CF, Feig M. Driven to near-experimental accuracy by refinement via molecular dynamics simulations. Proteins. 2019. Jun 14. [DOI] [PMC free article] [PubMed] [Google Scholar]