

To the Editor:
Respiratory virus infections, including influenza, are a top 10 cause of human mortality, and new therapeutics are needed for limiting infectious pulmonary injury. Existing antivirals have a limited spectrum of activities and can have limited therapeutic windows for efficacy in reducing tissue damage. Thus, new broad-acting therapeutics are needed to combat existing and emergent respiratory viruses. A host-directed strategy that limits tissue damage from virus-induced inflammation, while also promoting lung regeneration and repair capacity, would hold promise for preserving airway integrity until a respiratory virus is cleared by the immune system. Herein, we demonstrate this precise therapeutic profile for an antiinflammatory tissue repair protein, MG53 (mitsugumin 53), in lethal influenza virus infections of mice.
MG53 is a member of the TRIM (tripartite motif) family of proteins and is also known as TRIM72. It plays an essential role in cell membrane repair (1), and MG53 knockout (mg53−/−) mice develop pulmonary and cardiac pathology due to cumulative cell membrane damage (2–4). We recently reported that mg53−/− mice experience worsened morbidity and delayed recovery compared with wild-type mice in a nonlethal infection with influenza virus, correlating with increased inflammatory pathology in the lungs of the mutant animals (4). Consistent with these findings, MG53 possesses an antiinflammatory role in dampening NF-κB responses (4). Mice engineered to express elevated levels of circulating MG53 have an increased lifespan and preserved function of multiple organs as they age (5). Additionally, exogenous delivery of recombinant human (rh) MG53 protein protected against heart and lung injuries in rodents and larger animal models and did not induce adverse effects during long-term systemic administration (6, 7).
Given the detrimental effects that we observed upon influenza virus infection of mg53−/− mice (4), we conversely explored the potential benefits of rhMG53 in lethal influenza virus infection. According to protocols approved by the Ohio State University Institutional Animal Care and Use Committee, C57BL/6J wild-type mice (female, 8–10 wk of age) were intranasally infected with 100 TCID50 of influenza virus strain A/PR/8/34 (H1N1). Female mice were chosen for our studies because they experience more severe influenza virus infections than male mice, thus providing a stringent model for examining rhMG53 effects. Infected saline-treated controls displayed aggressive loss of body weight and died within 9 days after infection (n = 12, Figures 1A and 1B). In sharp contrast, 92% of mice (11/12 mice from two independent experiments) survived when they received tail vein injection of rhMG53 (2 mg/kg, daily for 7 d) beginning 24 hours after viral infection (Figure 1B). The rhMG53-treated mice also lost significantly less weight than control animals (Figure 1A) and recovered to normal body weight by day 14 after infection (Figure 1B). Furthermore, rhMG53-treated mice remained ambulant and active throughout the course of infection.
Figure 1.
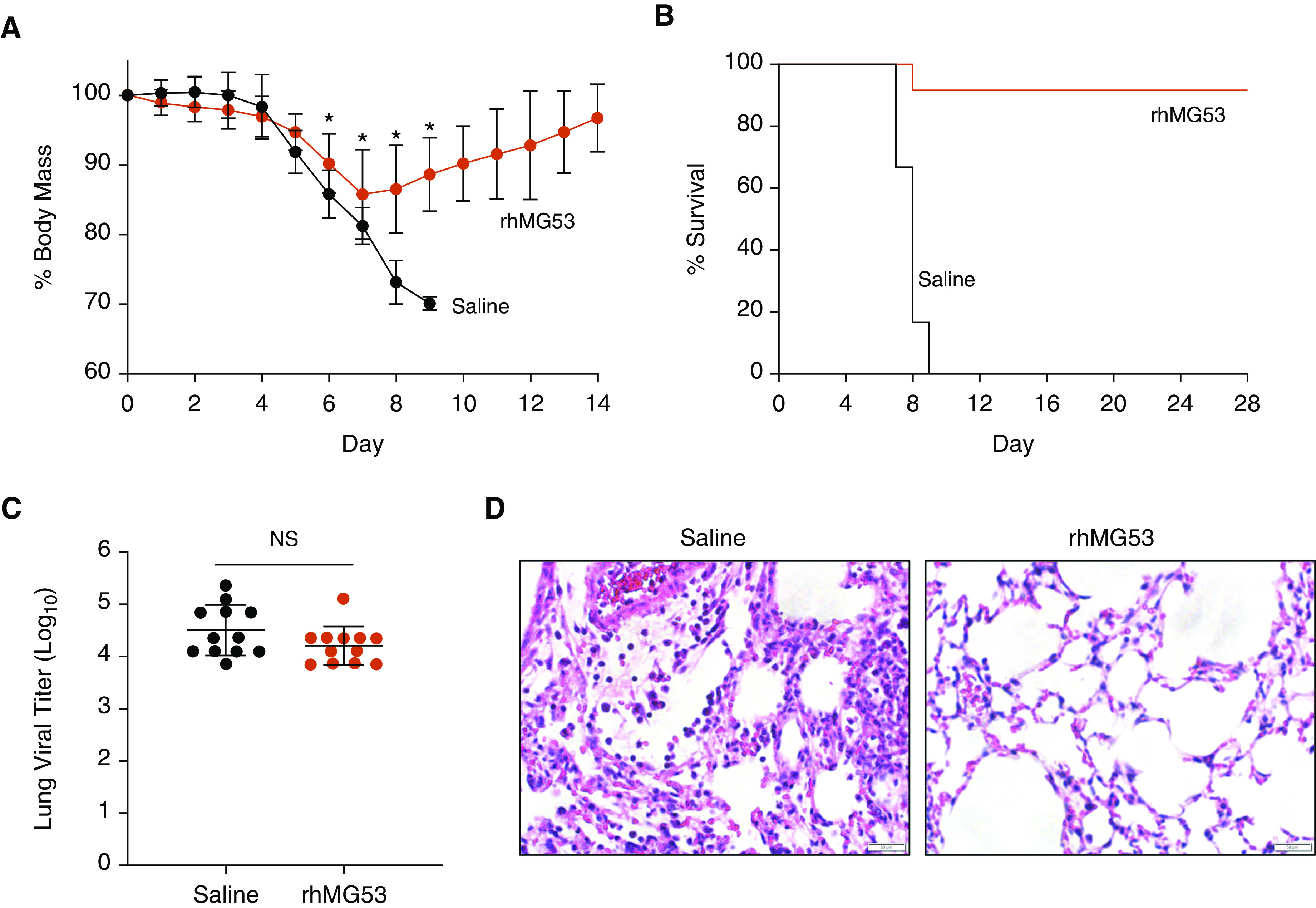
rhMG53 prevents death of mice infected with a lethal dose of influenza virus. Mice were intranasally infected with influenza A virus H1N1 strain PR8 (100 TCID50). Twenty-four hours after infection, mice were injected with rhMG53 (2 mg/kg) or saline daily. Mice were examined daily for (A) weight loss and (B) survival (n = 12 per group, two independent experiments). Points in A depict mean values and error bars represent SD. *P < 0.001 by unpaired t test. (C) In parallel, cohorts of infected mice were killed at 7 days after infection for measurement of virus titers in the lung via TCID50 assay using MDCK cells (n = 12, two independent experiments). Each point represents an individual mouse and bars represent mean values. Error bars represent SD. (D) Representative hematoxylin and eosin staining of lung sections derived from saline- or rhMG53-treated mice at 7 days after infection. Scale bars, 50 μm. NS = not significant by unpaired t test; rhMG53 = recombinant human mitsugumin 53.
To gain mechanistic insights into the protective effects of rhMG53 during viral infection, we examined cohorts of saline control and rhMG53-treated mice at 7 days after infection to measure viral titers as well as parameters of lung inflammation and histopathology. Despite their striking differences in mortality, viral titers were similar between saline control and rhMG53-treated mice, indicating that rhMG53 is not directly antiviral (Figure 1C). However, histological evaluation revealed remarkable preservation of lung alveolar structure in the rhMG53 treatment group (Figure 1D), quantified by significant decreases in the average lung injury score (saline 0.75 ± 0.07 vs. rhMG53 0.56 ± 0.1, P = 0.0009, unpaired t test, n = 12 per group) (8).
Because inflammatory cytokine storm is associated with tissue damage during viral infections, we examined levels of three critical inflammatory cytokines, IFN-β, IL-6, and IL-1β, by ELISA (R&D Systems). Each of these inflammatory cytokines was significantly lower on average in lung tissue from rhMG53-treated mice compared with saline controls (Figures 2A–2C), suggesting reduced inflammation may underlie the survival of the rhMG53 treatment. Interestingly, reduced levels of IL-1β in rhMG53-treated lung tissue suggests decreased inflammasome activation. NLRP3 inflammasome signaling and resulting pyroptotic cell death has been shown to mediate lung injury following influenza virus infection (9). As such, NLRP3 levels were elevated during influenza virus infection in saline control mice but were significantly reduced by rhMG53 treatment, comparable to levels observed in uninfected mice (Figure 2D). On average, NLRP3/GAPDH levels were significantly reduced from 2.2 ± 0.5 (virus/saline) to 1.13 ± 0.4 (virus/rhMG53) (P < 0.0001, ANOVA with Tukey’s multiple comparisons test, n = 12 per virus group).
Figure 2.
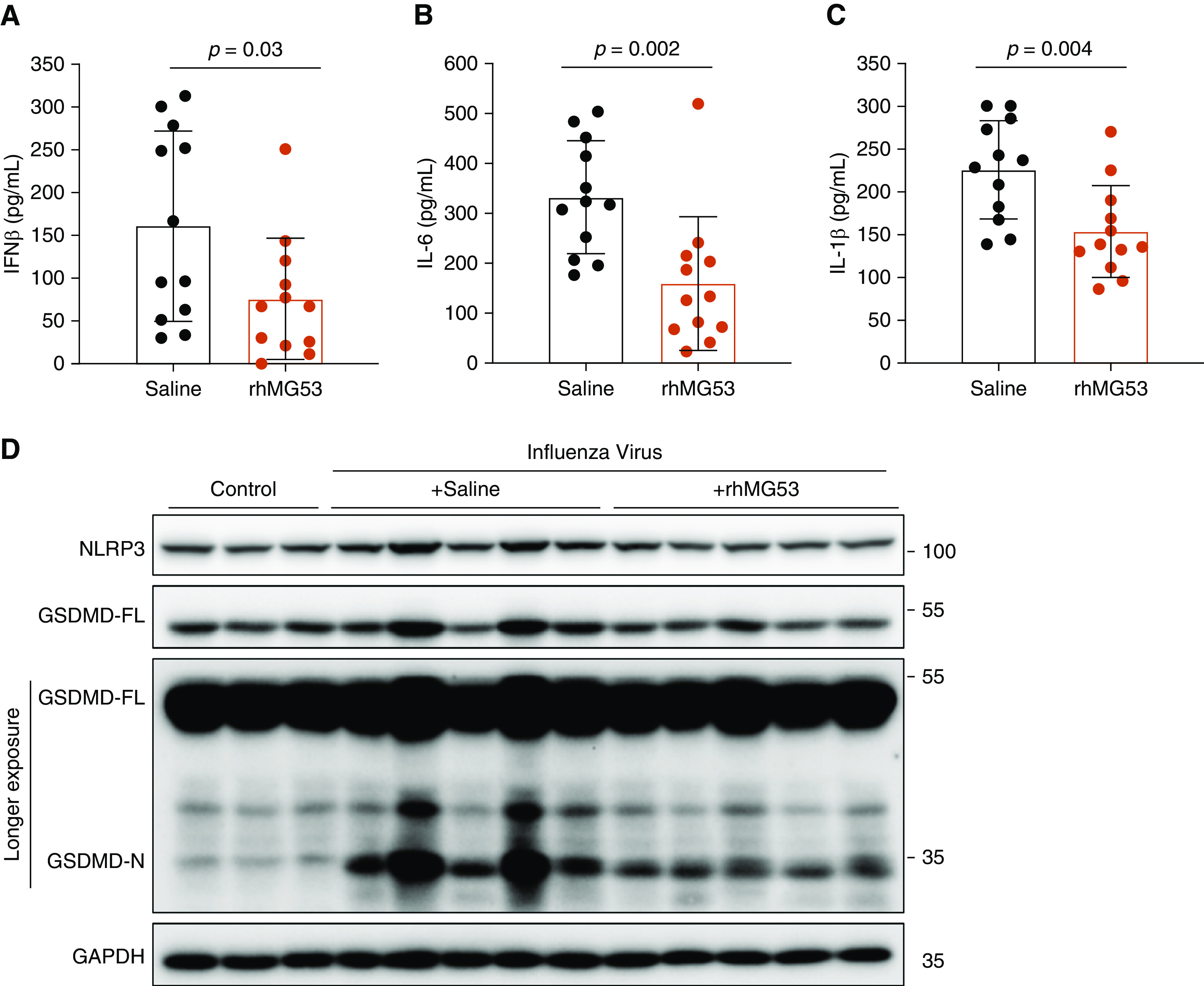
rhMG53 suppresses cytokine storm and mitigates pyroptosis in influenza virus–infected lung tissue. (A–C) ELISA quantification of IFN-β (A), IL-6 (B), and IL-1β (C) from lungs of influenza virus–infected mice treated with saline or rhMG53 and killed 7 days after infection. Each point represents an individual mouse and bars represent mean values. Error bars represent SD. P values determined by unpaired t test. (D) Representative Western blots of NLRP3 and GSDMD in lung tissues derived from uninfected mice (control), mice infected with influenza virus (Day 7) receiving saline, and mice infected with influenza virus (Day 7) receiving rhMG53 treatment. GAPDH blotting served as a loading control. Each lane is a sample from an individual mouse. GSDMD = gasdermin-D; GSDMD-FL = full length GSDMD; GSDMD-N = N-terminal fragment of GSDMD; rhMG53 = recombinant human mitsugumin 53.
To determine if pyroptotic cell death was affected by rhMG53, we examined GSDMD (gasdermin-D) levels. GSDMD is a cleavage target of caspases downstream of inflammasome activation, and the amino-terminal portion of GSDMD, GSDMD-N, forms pores at the cell membrane, causing cell lysis and the release of inflammatory cytoplasmic contents (10). Strikingly, rhMG53 treatment significantly reduced GSDMD-N levels in lungs when compared with saline control mice (Figure 2D). The normalized GSDMD-N/GAPDH levels were reduced from 31 ± 18 (virus/saline) to 10 ± 5 (virus/rhMG53) (P < 0.005, ANOVA with Tukey’s multiple comparisons test, n = 12 per virus group). We conclude that rhMG53 dampens inflammasome activation during influenza virus infection, revealing a new mechanism by which MG53 preserves lung integrity during inflammatory insults.
Collectively, our studies demonstrate a near-absolute survival benefit for administration of rhMG53 following lethal respiratory infection by influenza virus. The therapeutic efficacy of rhMG53 is linked to protection from virus-induced lung injury via mitigation of cytokine storm and inhibition of pyroptosis, major contributors to airway damage. The beneficial effect of rhMG53 is independent of an impact on viral titers, representing a major conceptual departure from current influenza therapeutics that directly target the virus. The rhMG53 biologic may therefore provide a frontline therapy for organ preservation in influenza virus infection and for other pathogen-associated infections for which therapeutics or vaccines are unavailable. This host-directed therapy offers the potential to dramatically reduce or prevent mortality during seasonal and pandemic influenza outbreaks. In addition, when confronted with other novel pathogenic viruses, such as severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), rhMG53 may offer a treatment option where one does not currently exist. Thus, our findings provide the first proof of concept in support of further clinical development of rhMG53 as a biologic to treat inflammation-driven infectious diseases.
Supplementary Material
Acknowledgments
Acknowledgment
The authors thank Dr. Eugene Oltz and Dr. Sandy Snyder for critical reading of the manuscript.
Footnotes
Supported by NIH grants AI130110, AI151230, and AI142256 to J.S.Y., HL143000 to B.A.W., and AR061385, AR070752, DK106394, and HL138570 to J.M., as well as by U.S. Department of Defense grant W81XWH-18-1-0787 to J.M. A.D.K. was supported by an NIH T32 fellowship funded by NIH grant GM068412 and the Ohio State University Presidential Fellowship. Z.L. is a visiting scholar at Ohio State University and received partial support from the Chinese Scholarship Council.
Author Contributions: J.M. and J.S.Y. conceived the study. A.D.K., Z.L., Z.B., X.Z., H.L., and C.C. performed experiments and analyzed data. B.A.W., T.T., C.C., J.M., and J.S.Y. provided supervisory project leadership and oversight. T.T., J.M., and J.S.Y. drafted the manuscript with substantive editorial input from all authors. All authors approved the final submission.
Originally Published in Press as DOI: 10.1164/rccm.202007-2908LE on October 8, 2020
Author disclosures are available with the text of this letter at www.atsjournals.org.
References
- 1.Cai C, Masumiya H, Weisleder N, Matsuda N, Nishi M, Hwang M, et al. MG53 nucleates assembly of cell membrane repair machinery. Nat Cell Biol. 2009;11:56–64. doi: 10.1038/ncb1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cao CM, Zhang Y, Weisleder N, Ferrante C, Wang X, Lv F, et al. MG53 constitutes a primary determinant of cardiac ischemic preconditioning. Circulation. 2010;121:2565–2574. doi: 10.1161/CIRCULATIONAHA.110.954628. [DOI] [PubMed] [Google Scholar]
- 3.Wang X, Xie W, Zhang Y, Lin P, Han L, Han P, et al. Cardioprotection of ischemia/reperfusion injury by cholesterol-dependent MG53-mediated membrane repair. Circ Res. 2010;107:76–83. doi: 10.1161/CIRCRESAHA.109.215822. [DOI] [PubMed] [Google Scholar]
- 4.Sermersheim M, Kenney AD, Lin PH, McMichael TM, Cai C, Gumpper K, et al. MG53 suppresses interferon-β and inflammation via regulation of ryanodine receptor-mediated intracellular calcium signaling. Nat Commun. 2020;11:3624. doi: 10.1038/s41467-020-17177-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bian Z, Wang Q, Zhou X, Tan T, Park KH, Kramer HF, et al. Sustained elevation of MG53 in the bloodstream increases tissue regenerative capacity without compromising metabolic function. Nat Commun. 2019;10:4659. doi: 10.1038/s41467-019-12483-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jia Y, Chen K, Lin P, Lieber G, Nishi M, Yan R, et al. Treatment of acute lung injury by targeting MG53-mediated cell membrane repair. Nat Commun. 2014;5:4387. doi: 10.1038/ncomms5387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weisleder N, Takizawa N, Lin P, Wang X, Cao C, Zhang Y, et al. Recombinant MG53 protein modulates therapeutic cell membrane repair in treatment of muscular dystrophy. Sci Transl Med. 2012;4:139ra85. doi: 10.1126/scitranslmed.3003921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matute-Bello G, Downey G, Moore BB, Groshong SD, Matthay MA, Slutsky AS, et al. Acute Lung Injury in Animals Study Group. An official American Thoracic Society workshop report: features and measurements of experimental acute lung injury in animals. Am J Respir Cell Mol Biol. 2011;44:725–738. doi: 10.1165/rcmb.2009-0210ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuriakose T, Man SM, Malireddi RK, Karki R, Kesavardhana S, Place DE, et al. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci Immunol. 2016;1:aag2045. doi: 10.1126/sciimmunol.aag2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535:111–116. doi: 10.1038/nature18590. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.