

Abstract
Rationale:
Cerebrovascular function is critical for brain health, and endogenous vascular-protective pathways may provide therapeutic targets for neurological disorders. Sphingosine 1-phosphate (S1P) signaling coordinates vascular functions in other organs, and S1P receptor-1 (S1P1) modulators including fingolimod show promise for the treatment of ischemic and hemorrhagic stroke. However, S1P1 also coordinates lymphocyte trafficking, and lymphocytes are currently viewed as the principal therapeutic target for S1P1 modulation in stroke.
Objective:
To address roles and mechanisms of engagement of endothelial cell (EC) S1P1 in the naïve and ischemic brain and its potential as a target for cerebrovascular therapy.
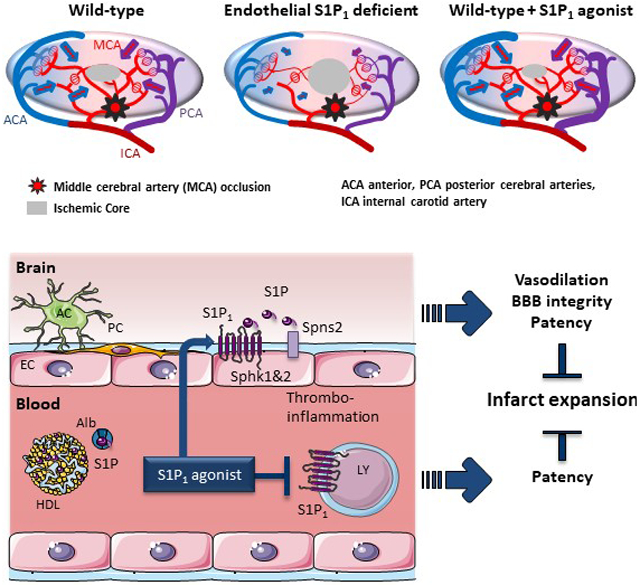
Methods and Results:
Using spatial modulation of S1P provision and signaling, we demonstrate a critical vascular protective role for endothelial S1P1 in the mouse brain. With an S1P1 signaling reporter, we reveal that abluminal polarization shields S1P1 from circulating endogenous and synthetic ligands after maturation of the blood-neural barrier, restricting homeostatic signaling to a subset of arteriolar ECs. S1P1 signaling sustains hallmark endothelial functions in the naïve brain, and expands during ischemia by engagement of cell-autonomous S1P provision. Disrupting this pathway by EC-selective deficiency in S1P production, export, or the S1P1 receptor substantially exacerbates brain injury in permanent and transient models of ischemic stroke. By contrast, profound lymphopenia induced by loss of lymphocyte S1P1 provides modest protection only in the context of reperfusion. In the ischemic brain, EC S1P1 supports blood-brain barrier (BBB) function, microvascular patency, and the rerouting of blood to hypo-perfused brain tissue through collateral anastomoses. Selective S1P1 agonism counteracts cortical infarct expansion after middle cerebral artery occlusion by engaging the endothelial receptor pool after BBB penetration.
Conclusions:
This study provides genetic evidence to support a pivotal role for the endothelium in maintaining perfusion and microvascular patency in the ischemic penumbra that is coordinated by S1P signaling and can be harnessed for neuroprotection with BBB-penetrating S1P1 agonists.
Keywords: Endothelial function, stroke, sphingosine-1-phosphate, Fingolimod, lipid metabolites, collateral circulation, neuroprotective agent
Subject Terms: Cerebrovascular Disease/Stroke, Hemodynamics, Ischemic Stroke, Pharmacology, Vascular Biology
Graphical Abstract
INTRODUCTION
Ischemic stroke is one of the most prevalent causes of death and morbidity worldwide and represents a major societal and economic burden (https://www.who.int).1 Despite a number of clinical trials with neuroprotective therapeutics, treatment options remain limited to thrombolysis and mechanical recanalization.2 Novel safe and affordable adjunct treatment strategies are therefore needed.
Ischemic stroke is caused by thrombotic or embolic occlusion of a large cerebral artery. This produces an core of necrotic tissue immediately downstream of the occlusion site surrounded by an ischemic penumbra that can be rescued if adequate perfusion is sustained.1, 3 The speed by which the core encroaches upon the penumbra depends on the efficiency of retrograde blood supply through cortical collateral anastomoses extended across the borders with neighboring cerebral arterial territories. Although governed primarily by the number and size of existing anastomoses, blood rerouting also depends on the dilatory capacity, integrity, and patency of the regional vasculature.4 Improving microvascular function to counteract the expansion of the infarct core and improve the efficacy and safety of anterograde reperfusion may therefore provide a therapeutic opportunity.5 Strategies explored to this end include the inhibition of lymphocyte-driven inflammatory thrombosis and the stimulation of endothelial cell (EC) function to promote local redistribution of blood flow, reinforce the blood-brain barrier (BBB), and suppress inflammation and coagulation in affected territories.5, 6
S1P is a signaling lipid with critical roles in both immune and vascular function exerted by five cognate G protein-coupled receptors (GPCRs), S1P1-5.7 Lymphocytes depend on S1P1-mediated S1P sensing to egress from lymphoid organs, and both inhibition and activation of lymphocyte S1P1 induces profound lymphopenia.8 S1P1 is also amongst the most abundant EC GPCRs, and selective constitutive or temporal deletion of S1pr1 (encoding S1P1) in mouse ECs impairs embryonic and postnatal angiogenesis, vascular integrity, and flow-mediated vasodilation.7, 9, 10 Loss of S1P1 signaling in ECs destabilizes adherens junctions, reduces endothelial nitric oxide synthase (eNOS) activity, and increases the expression of leukocyte adhesion molecules.10-12 S1P1 thus plays a critical role in sustaining hallmark endothelial functions. S1P is abundant in circulation, where it associates primarily with high density lipoproteins and albumin.7 Erythrocytes and ECs are the main sources of blood and lymph S1P under homeostasis, while platelets store large amounts of S1P that is only released upon activation.7, 9, 13 S1P is exported from ECs by spinster homolog 2 (Spns2) and from platelets and erythrocytes by major facilitator superfamily domain containing 2B (Mfsd2b).7, 14
The multiple sclerosis (MS) drug fingolimod (FTY720) is a functional antagonist of S1P1 that also activates S1P3,4&5 and has shown promise for the treatment of ischemic and hemorrhagic stroke in both experimental models and small-scale clinical trials.15-24 Similar efficacy of S1P1-selective agonists argues that FTY720 protection is mediated by S1P1.6, 19, 21 Loss of efficacy in lymphocyte-deficient mice and correlation between efficacy of lymphocyte depletion and infarct reduction has indicated that S1P1 modulators provide protection by impairing lymphocyte trafficking and, thereby, lymphocyte-driven thromboinflammation.19, 20 Yet the endothelial receptor pool is also engaged, and the risks and benefits of targeting EC S1P1 have not been specifically evaluated.
In this study we have addressed the impact of tissue-specific deficiency of S1P1 and sources of its activating ligand on the development, anatomy, and function of the naïve brain vasculature and on the outcome of transient and permanent MCA occlusion in mice. This revealed an important role for EC S1P1 in cerebrovascular homeostasis and a critical protective role for EC-autonomous engagement of S1P1 signaling during cerebral ischemia. When we addressed the capacity of pharmacological agonists to access S1P1 on the brain endothelium, we found that BBB penetration is required for engagement of EC S1P1 and that this receptor pool is an important target for the protective effects of S1P1 agonism in ischemic stroke.
METHODS
Detailed Methods are available in the Data Supplement.
Data Availability.
The authors declare that all supporting data are available within the article and its online supplementary files.
RESULTS
Endothelial S1P1 plays a critical protective role during cerebral ischemia.
In order to define endogenous roles of EC and leukocyte S1P1 in ischemic stroke, we generated EC S1pr1 knockout (KO) mice with Cdh5-iCreERT2 or Pdgfb-iCreERT2 (S1pr1ECKO) and pan-hematopoietic (HC) S1pr1 KO mice with Mx1-Cre or Vav1-Cre (S1pr1HCKO).9, 11, 25 The Pdgfb-iCreERT2 allele induced near complete recombination of an mTmG reporter in ECs in the cerebral cortex after neonatal tamoxifen administration (Online Fig I A). We have previously reported efficient S1pr1 excision in target tissues with the other Cre drivers.25, 26 In a permanent (p) MCAO model, in which thermocoagulation and subsequent dissection of a section of the MCA distal to the lenticulostriate arteries yields small and cortically confined infarcts but no overt neurological deficits,27, 28 mean 24 hour infarct volumes were on average 71 % larger in neonatally induced S1pr1ECKO males and 65 % larger in S1pr1ECKO females than in littermate controls (Fig 1A). The relative increase was greater three days after pMCAO with 148 % larger infarcts in S1pr1ECKO males (Fig 1B). Although the Pdgfb-iCreERT2 allele used for EC-selective gene deletion has off-target effects in megakaryocytes (MKs),25, 29 the phenotype could be attributed to ECs as it was reproduced in S1pr1ECKO males generated with the EC-selective Cdh5-iCreERT2 (Fig 1C), but not with S1pr1MKKO males generated with the MK-selective Pf4-Cre (Online Fig I B).25 In the same experimental model, hematopoietic S1P1 deficiency induced profound lymphopenia but had no impact on infarct size (Fig 1D, Online Fig I C). Similar results were obtained in a filament-based proximal transient (t) MCAO model, in which infarct volumes measured 24 hours after 60 minutes occlusion were on average more than twice as large and neurological deficits exacerbated in S1pr1ECKO mice (Fig 1E). Induction of gene deletion 10 days prior to surgery in this experiment argued that sensitization to MCAO did not reflect upon developmental consequences of EC S1pr1 deletion. Yet as control infarcts were smaller than typically observed with proximal MCAO – an observation that may be attributed to protective effects of tamoxifen 30, 31 - neonatal deletion was employed in all subsequent experiments. When neonatally-induced S1pr1ECKO males were subjected to a severe tMCAO model in which reperfusion was delayed to 90 minutes, infarcts covered a substantial portion of the MCA territory in controls, and S1pr1ECKO mice exhibited considerable post-reperfusion mortality (68% vs. 32%, respectively, P=0.022; Online Fig I D) that manifested >8 hours after occlusion. Post-mortem analysis did not reveal bleeding, and hemorrhagic transformation was not increased in S1pr1ECKO survivors (minor bleeds were observed in 1/6 S1pr1ECKO vs. 5/14 control infarcts). Loss of the most affected animals may explain a lack of significant difference in infarct volumes observed in survivors (Online Fig I D) and is further addressed below. Unlike the pMCAO model, hematopoietic S1P1 deficiency afforded some neuroprotection 24 hours after 60 minutes tMCAO (Fig 1F; P=0.072). These observations reveal a critical role for endothelial S1P1 in limiting neuronal injury in ischemic stroke irrespective of whether or not the occluded artery is reperfused (Fig 1G). They also support the notion that S1P1 plays a disruptive role in ischemic stroke by supporting lymphocyte egress,20 yet the effect of specifically impairing this pathway was modest and observed only in the context of reperfusion (Fig 1G).
Figure 1: Endothelial S1P1 signaling limits brain injury after permanent and transient MCA occlusion.

A-C. Infarct volumes 24 (A,C) or 72 (B) hours after pMCAO in S1pr1ECKO and littermate males (A, middle panel, B) and females (A, right panel) generated by neonatal S1pr1 deletion with Pdgfb-iCreERT2 (A,B) or Cdh5-iCreERT2 (C). Left panel of A: representative images. D. Basal peripheral blood lymphocyte counts and infarct volumes 24 hours after pMCAO in male mice lacking S1P1 in hematopoietic cells (S1pr1HCKO; Vav1-Cre). E. Infarct volumes and neurological deficits 24 hours after 60 minutes tMCAO in male mice lacking S1P1 in endothelial cells (S1pr1ECKO; Cdh5-iCreERT2; adult deletion). Left panel: representative images. F. Infarct volumes 24 hours after 60 minutes tMCAO in males lacking S1P1 in hematopoietic cells (S1pr1HCKO; Mx1-Cre). Lymphocyte counts pre-MCAO in right panel. G. Schematic representation of the net cell type specific contribution of S1P1 signaling to stroke outcome. Bar graphs show mean ± SEM. Statistical significance assessed by Mann-Whitney test (A, males) or unpaired t-test (all other).
Ischemia mobilizes EC-autonomous S1P provision for S1P1 signaling and stroke protection.
S1P1 drives lymphocyte egress and sustains lung vascular integrity in response to circulating ligand.13, 32 Surprisingly however, postnatal deletion of Sphk1&2 in Mx1-Cre sensitive cells did not significantly impact infarct volumes after permanent or transient MCAO (Fig 2A), even if the same deletion strategy nearly abolishes S1P provision to plasma, resulting in lymphopenia and constitutive vascular leak in the lung.9, 13, 25 This could reflect compound effects of loss of S1P production also in tissue resident cells or of loss of circulating S1P on the activation of S1P1 and other S1PRs, notably S1P2, 33 in several cellular compartments.34 However, lymphocyte S1P1 was dispensable in the pMCAO model (Fig 1D), and deletion of S1pr2 or S1pr3 did not impact outcome in the pMCAO model (Online Fig II A). Selective impairment of S1P release from platelets, which could potentially increase local S1P levels in stroke and change signaling bias,9, 35 did not impact infarct size in the tMCAO model (Fig 2B). Single cell RNA sequencing analysis suggests that brain ECs express the necessary machinery for de novo S1P synthesis and export and may thus constitute a local source of S1P in the neurovascular unit.36 Strikingly, impairment of the production (Sphk1&2ECKO) or the export (Spns2ECKO) of S1P in ECs both exacerbated outcome to a similar degree as EC S1P1 deficiency in the pMCAO and/or the tMCAO model, pointing to a critical role for cell-autonomous S1P provision for EC S1P1 activation (Fig 2C, D and Online Fig II B, C). Excision efficiency was confirmed to be >85% for Sphk1 and Sphk2 in isolated brain endothelial cells from Sphk1&2ECKO (Online Fig II D). In order to address where cell-autonomous S1P1 signaling is engaged after MCAO, we first asked where S1P1 is expressed. Single cell RNA sequencing of brain microvascular fragments show enrichment of S1pr1 transcripts in ECs as well as in astrocytes (Online Fig II E).36 Accordingly, protein expression, which initially appeared widespread and diffuse in the cerebral cortex (Fig 2E), could be resolved by selective deletion of S1pr1 in ECs (S1pr1ECKO) or astrocytes (S1pr1ACKO) (Fig 2E, Online Fig II F, G). The level of EC S1P1 expression is maintained in old mice (Online Fig II G, H).37 We then generated S1P1 GFP signaling (S1P1GS) mice – which leave a nuclear GFP signal after S1P1-β-arrestin coupling 38 - with or without the capacity for EC S1P production (S1pr1Ki/+:H2B-GfpTg/+:Sphk1&2ECWT/KO). Despite widespread receptor expression in astrocytes and ECs throughout the vascular tree, S1P1 signaling was highly restricted to a subset of arteriolar ECs in both young and old mice (Fig 2F, Online Fig II I, J). S1P1-independent H2B-GFP activity was also observed in perivascular cells of arterioles and venules but not in ECs (Online Fig II K). After pMCAO, S1P1 signaling in the infarct region expanded to capillary and venous ECs, but not to astrocytes (Fig 2G, H). While redundant ligand sources9 or ligand-independent activation11 sustained homeostatic signaling in arterioles (Fig 2H), the expansion of S1P1 signaling after MCAO was driven principally by EC-autonomous S1P production (Fig 2H). In order to address the mechanistic basis for this expansion, we evaluated the expression of Sphks, Spns2 and S1P1 in brain microvessels 6 hours after tMCAO. Consistent with single cell RNA sequencing data,36 Sphk2, Spns2, and S1pr1 and to a lesser degree Sphk1 were all expressed in cerebral microvessels of naïve mice. Ischemia triggered robust induction of Sphk1 expression in the ipsilateral hemisphere but had little impact on the expression of the other transcripts (Fig 2I, Online Fig II L). Thus, while circulating S1P sustains lymphocyte trafficking and may contribute to homeostatic signaling in cerebral arterioles, S1P1-dependent neuroprotection in ischemic stroke is driven primarily by engagement of EC-autonomous S1P provision, possibly by transcriptional activation of Sphk1.
Figure 2: Endothelial cell autonomous S1P provision sustains S1P1 activation during cerebral ischemia.

A-D. Infarct volumes 24 hours after pMCAO or 60 minutes tMCAO in male mice lacking S1P production in hematopoietic cells (Sphk1&2HCKO; A), platelets (Sphk1&2MKKO; B), or endothelial cells (Sphk1&2ECKO; C), or deficient in S1P export from blood endothelial cells (Spns2ECKO; D) and respective littermate controls. E. S1P1 expression in the naïve cerebral cortex of wild-type mice (left) and mice lacking S1P1 in astrocytes (S1pr1ACKO, Gfap-Cre, right). Note expression of S1P1 in all vessels. Green, S1P1; white, ECs (CD31); red, vascular smooth muscle cells (ASMA); blue, all cell nuclei (Hoechst). Scale bar: 50 μm. F, G. S1P1 signaling visualized in S1P1 signaling mice (S1P1GS) in a naïve cerebral cortex (F) and 48 hours after pMCAO (G) assessed in the contralateral (left) and ipsilateral (right) cerebral cortex. Note that S1P1 signaling is highly restricted to arteries in the naïve and contralateral cerebral cortex and more widespread but still predominantly endothelial in the ipsilateral cortex. Red, EC nuclei (Erg); green, S1P1 signaling cells (GFP); yellow, S1P1 signaling ECs (GFP/Erg double positive nuclei); white, ECs (CD31); blue, vascular smooth muscle cells (ASMA). Scale bar: 100 μm. H. Quantification of GFP positive arterial and non-arterial ECs in the ipsilateral and contralateral hemisphere of S1P1GS mice with (Sphk1&2ECWT) and without (Sphk1&2ECKO) the capacity for EC S1P production 48 hours after pMCAO. I. Relative expression of S1pr1, Sphk1, Sphk2 and Spns2 in cerebral microvessels isolated from naïve cerebral cortex or 6 hours after tMCAO, normalized to Tjp1 transcript. Bar graphs show mean ± SEM. Statistical significance assessed by ANOVA with Tukey multiple comparisons test (H, I), Mann-Whitney test (C, pMCAo) or unpaired t-test (all other).
Postnatal impairment of EC-autonomous S1P signaling does not impact cerebrovascular anatomy.
Although recombination of loxP-flanked alleles in ECs in neonatal mice in most experiments in this study overcame confounding effects of tamoxifen protection, it also introduced potential confounding effects of deregulated vascular development on stroke outcome. As previously reported for Cdh5-iCreERT2-mediated neonatal deletion of S1pr1,11 Pdgfb-iCreERT2-mediated deletion induced vascular hyper-sprouting and delayed outgrowth of the retinal vasculature (Fig 3A, B). However, this phenotype was not replicated with Pdgfb-iCreERT2-mediated deletion of Sphk1&2 (Fig 3A, B). This suggests that postnatal angiogenesis, like embryonic angiogenesis,9 is sustained by redundant S1P sources and that abnormal vascular patterning is unlikely to explain sensitivity to MCAO in Sphk1&2ECKO mice. Consistent with prenatal development of the cerebral vasculature, we also did not observe significant differences in the number of collateral connections between the MCA and branches of anterior cerebral artery (ACA) extending laterally from the midline between S1pr1ECKO and littermate controls (Fig 3C, D). Vascular density in the cerebral cortex was also unaltered by Pdgfb-iCreERT2-mediated S1pr1 excision, as has previously been reported for Cdh5-iCreERT2-mediated deletion (Fig 3E).26 Thus, sensitivity to MCAO in mice deficient in EC autonomous S1P1 signaling cannot be explained by underlying differences in vascular anatomy.
Figure 3: Loss of EC-autonomous S1P1 signaling does not impact cerebrovascular patterning.

A. Isolectin B4 staining shows vascularization of the mouse retina at postnatal day (P)5 after neonatal Pdgfb-driven S1pr1 or Sphk1&2 deletion. Note delayed expansion of the vascular network (top panel, arrow) and abundant filopodia at the vascular front (bottom panel, arrowheads) of S1pr1ECKO but not Sphk1&2ECKO retinas. Scale bar: 1 mm (top panel), 50 μm (bottom panel) B. Quantification of outgrowth of the retinal vasculature at P5, normalized to weight of pups. C,D. Collateral connections between the MCA and branches of the posterior CA (PCA, white asterisk) and of the anterior CA (ACA, yellow asterisk) extending laterally from the midline between S1pr1ECKO and littermate controls. C, quantification of ACA-MCA connections. D, representative images. E. Vascular density in the cortex of S1pr1ECKO and littermate controls assessed by collagen IV (Coll IV) or Glut1 staining. Representative images of Coll IV staining (left panels) and quantification of both (right panel), scale bar: 100 μm. Bar graphs show mean ± SEM. Statistical significance assessed by one-way ANOVA with Tukey multiple comparisons test (B) and unpaired t-test (all other).
Endothelial S1P1 maintains BBB function.
Both genetic strategies for EC S1pr1 deletion employed in this study result in constitutive vascular leak in the lung that can be replicated by plasma but not by EC S1P deficiency.9, 39 Cdh5-iCreERT2-mediated S1pr1 deletion results in subtler and size-selective permeability of the brain endothelium, 26 which was also observed with Pdgfb-iCreERT2-mediated S1pr1 deletion (Fig 4A). Endotoxin challenge (10 mg/kg, 8 hours) increased 4 kD dextran accumulation to the same degree in S1pr1ECKO and littermate controls (Fig 4B). Thus, S1P1 deficiency does not critically impair the stability of EC junctions at the BBB. Intriguingly, even if S1pr1ECKO mice do not show increased paracellular permeability to dextrans ≥10 kD,26 the cortex of naïve S1pr1ECKO mice did show increased permeability to Evans Blue/albumin, which crosses the BBB primarily by transcellular transport (Fig 4C).40 Neither basal phenotype was replicated in mice lacking EC S1P production, nor was Evans Blue/albumin extravasation affected by lack of plasma S1P (Fig 4A, C), again suggesting source redundancy or ligand-independence of homeostatic EC S1P1 signaling at the BBB. Twenty-four hours after pMCAO, Evans Blue/albumin accumulation in the ipsilateral hemisphere exceeded the relative increase in infarct size in S1pr1ECKO mice with a more diffuse and widespread appearance, and was also significantly higher in the contralateral hemisphere, consistent with results in naïve mice (Fig 4D). In the acute phase after 90 minutes tMCAO – which is associated with high mortality of S1pr1ECKO mice (Online Fig I D) - full T2 weighted magnetic resonance imaging (MRI) revealed severe edema in S1pr1ECKO mice as early as 2.5 hours after reperfusion with a clear shift in the midline 2 hours later (Fig 4E). Image analysis confirmed significantly larger T2 lesions 2.5 hours after reperfusion, demonstrating a clear impact of EC S1P1 deficiency also after 90 minutes of tMCAO despite no significant increase in infarct volumes in the few S1pr1ECKO mice that survived for 24 hours (Online Fig I D). Transtentorial herniation may be followed by cerebellar tonsil herniation and could explain increased mortality in S1pr1ECKO mice in this model (Online Fig I D). Thus, S1P1 preserves BBB integrity in ischemic stroke most likely by restricting vesicular transport, which underlies BBB dysfunction in the acute phase.40
Figure 4. Endothelial S1P1 sustains BBB function.

A. Effect of Pdgfb-iCreERT2-mediated deletion of S1pr1 and Sphk1&2 on the accumulation of 4 kD TRITC-Dextran in the cerebral cortex of naïve mice. B. Effect of Pdgfb-iCreERT2-mediated deletion of S1pr1 on the accumulation of 4 kD TRITC-Dextran in the cerebral cortex 8 hours after challenge with 10 mg/kg LPS i.p. C. Effect of Pdgfb-iCreERT2-mediated deletion (ECKO) of S1pr1 and Sphk1&2 as well as Mx1-Cre-mediated deletion (HCKO) of Sphk1&2 on the accumulation of Evans Blue/albumin in the brain of naïve mice. D. Effect of Pdgfb-iCreERT2-mediated deletion of S1pr1 on Evans Blue/albumin leak 24 hours after pMCAO in ipsilateral and contralateral hemispheres. Left, representative brains. Right, corrected absorbance of full hemisphere extracts. E. Full T2-weighted magnetic resonance imaging (MRI) 4 and 6 hours after 90 min tMCAO in S1pr1ECKO and littermate controls. Left: Representative axial sections from level of the mid-olfactory bulb from the same animal at the two time points. Hatched line indicates midline, asterisk affected MCA territory, and arrow contralateral ventricle. Right: T2 lesion ratios calculated from MRI images based on axial plane images at the mid-olfactory bulb. Bar graphs show mean ± SEM. Statistical significance assessed by Mann-Whitney test (D) and unpaired t-test (all other).
S1P1 supports cerebral vasoreactivity and promotes tissue perfusion after MCA occlusion.
We next addressed if EC S1P signaling regulates vessel diameter and thereby the redistribution of blood to the ischemic penumbra through existing cortical collateral anastomoses. Significantly reduced blood flow responses assessed by Doppler ultrasonography in the somatosensory cortex in response to acetylcholine superfusion (Fig 5A) and in the basilar trunk (BT) in response to CO2 inhalation (Fig 5B) both argued a critical role for EC S1P1 in cerebral blood flow regulation. Blood flow responses in the somatosensory cortex in response to whisker stimulation were nevertheless unaltered, suggesting normal neurovascular coupling, as was mean arterial pressure observed during these recordings (Fig 5A, Online Fig III A). Flow-mediated dilation was also significantly reduced in posterior cerebral artery segments isolated from S1pr1ECKO mice (Fig 5C). A similar phenotype in mesenteric arteries from both S1pr1ECKO and Sphk1&2ECKO mice (Online Fig III B) argued that shear forces can engage EC S1P1 through cell-autonomous S1P release. Impairment of vascular reactivity in S1pr1ECKO mice did not however impact central blood pressure (assessed by telemetry; Fig 5D) or basal brain perfusion (assessed by arterial spin labeling MRI; Fig 5E).41 We next monitored mean blood flow velocities (mBFVs) in the left and right internal carotid arteries (ICA) and in the basilar trunk (BT) before, 50 and 120 minutes after pMCAO in S1pr1ECKO and littermate controls (Fig 5F). No significant genotype-dependent difference was observed in baseline mBFVs (Online Fig III C). Permanent left MCAO decreased mBFVs in the left ICA to 77 % of pre-occlusion values 50 minutes after occlusion in both S1pr1ECKO and control mice (Fig 5F). Consistent with this decrease reflecting upon the reduction in MCA territory downstream of the left ICA, we observed no significant change in mBFVs in the right ICA or the BT (Online Fig III C). A subsequent recovery to near 90 % of pre-occlusion values at 120 minutes after occlusion in controls was interpreted to reflect upon a downstream increase in peri-infarct reflow through branches of the MCA originating upstream from the occlusion and through the distal branches of the ACA and posterior cerebral artery (PCA) (Fig 5F, Online Fig III C). This recovery at 120 minutes after occlusion was absent in S1pr1ECKO mice (Fig 5F, Online Fig III D), and a significant inverse correlation was observed between relative ICA blood flow at 120 minutes and infarct volumes at 24 hours (Fig 5G). To address perfusion directly in the affected cortex after pMCAO, we next visualized red blood cell flux in the area of collateral anastomoses in the leptomeningeal arteries between the MCA and ACA by sidestream dark field imaging (Fig 5H, supplemental videos (SV)1-3). Two hours after pMCAO, unidirectional flow towards the MCA territory was observed in all ACA-MCA collaterals, allowing perfusion of the territory normally supplied by branches of the MCA downstream of, but distal to the occlusion site (SV1&2). Blood moving retrograde switched anterograde up other MCA branches when encountering coagulated blood in the ischemic core (SV3). While the same general pattern was observed in mice of both genotypes, microvascular perfusion in MCA territory proximal to the ACA border was significantly reduced in S1pr1 ECKO mice (Fig 5H, SV2). No significant genotype-dependent difference in ICA blood flow reduction at 50 minutes argued that protective effects of S1P1 signaling take time to establish, possibly because of the need to engage EC-autonomous S1P production through Sphk1 induction (Fig 2I). Accordingly, when occlusion time was reduced to 35 minutes in the tMCAO model, EC S1P1 deficiency no longer influenced infarct size (Fig 5I). These observations are consistent with the notion that EC S1P1 limits the expansion of the necrotic core in the acute phase of stroke by supporting local vasodilation so as to promote retrograde perfusion of affected MCA territories.
Figure 5. Endothelial S1P1 regulates cerebral blood flow and supports tissue perfusion in the acute phase of stroke.

A. Somatosensory cortex blood flow (CBF) assessed by laser Doppler flowmetry in mice equipped with a cranial window in response to whisker stimulation or superfusion of the endothelium-dependent vasodilator acetylcholine (ACh; 10 μmol/L) on exposed neocortex (left). Mean arterial blood pressures monitored in the femoral arteries during the CBF measurements (right). B. Mean blood flow velocities (mBFVs) measured by ultrasound in the basilar trunk of S1pr1ECKO mice and littermate controls before and 2-5 minutes after exposure to a gas mixture of 16% O2, 5% CO2, 79% N2 (normoxia-hypercapnia) and the relative change in velocities presented. Doppler image shows vessels analyzed in B and F. (BT: basilar artery, PCA: posterior cerebral artery, ICA: internal carotid artery, ACA: anterior cerebral artery, AzA: azygos artery). C. Flow-mediated dilation in posterior cerebral artery segments of S1pr1ECKO and littermate control mice assessed by arteriography. D. Blood pressures of non-sedated S1pr1ECKO and littermate control mice recorded for 72 hours by telemetry. Average day and night systolic blood pressure (SBP) is shown; diastolic blood pressure and heart rates also did not differ between the groups. E. Basal perfusion of the cerebral cortex and hippocampus in S1pr1ECKO mice assessed by arterial spin labeling MRI. F. mBFVs measured by ultrasound imaging in left and right intra cranial ICA and BT under 0.5% isoflurane anesthesia before, 50 minutes and 2 hours after electrocoagulation-induced pMCAO. Normalized values in ipsilateral ICA shown, absolute values for all arteries in Online Fig III C. G. mBFVs in the left ICA 2 hours after left MCAO expressed as % of mean pre-occlusion velocities are plotted against infarct volumes in the same mice determined 24 hours after occlusion. H. Blood flow in the leptomeningeal vasculature in the ipsilateral hemisphere was imaged by sidestream dark field imaging through a cranial window 2-2.5 hours after pMCAO. Left panel illustrates approximate positions of regions monitored at the MCA/ACA border. Right panel shows results of automated analysis of microvascular perfusion in the MCA/ACA and MCA/PCA border regions. Representative videos in Data Supplement. I. Infarct volumes (left) 3 days after 35 minutes of tMCAO in S1pr1ECKO males and littermate controls. Graphs show mean ± SEM. Statistical significance was assessed by repeated measures (C, D, F) two-way ANOVA with Bonferroni (C, F) or Sidak’s (D) multiple comparisons test, one-way ANOVA (A CBF, E), linear regression analysis (G), Mann-Whitney test (H) or unpaired t-test (all other).
Endothelial S1P1 signaling maintains microvascular patency in the ischemic penumbra.
In addition to the active redistribution of blood from neighboring vascular territories, perfusion of the ischemic penumbra is highly dependent on microvascular patency within the affected zone. The recruitment of leukocytes to an activated endothelium in the ischemic penumbra can directly impair capillary blood flow and propagate microvascular coagulation and thrombosis.42, 43 S1P1 helps sustain the anti-inflammatory status of the aortic endothelium,12 and we observed a significant increase in ICAM-1 in brain homogenates and in postcapillary venules in the cerebral cortex of naïve S1pr1ECKO mice (Fig 6A, Online Fig IV A). This increase was not observed with selective impairment of S1P production in endothelial or hematopoietic cells (Fig 6A, Online Fig IV A). In the acute phase after pMCAO (2.5 hours), however, an increase in ICAM-1 expression in the ipsilateral hemisphere of control but no S1pr1ECKO mice overcame the genotype-dependent difference (Fig 6B). Myeloperoxidase levels were increased in S1pr1ECKO mice (Fig 6B), albeit not beyond the increase in infarct size (Fig 1A). We next stained for erythrocytes, neutrophils, platelets, and fibrin(ogen) in sections of brains perfused transcardially with heparinized saline three hours after pMCAO. Plasma serotonin was slightly increased at this time independent of genotype, arguing against a significant difference in platelet activation (Online Fig IV B). We nevertheless observed a striking reduction in the penetration of tomato lectin, infused 15 minutes prior to transcardial perfusion, into the MCA territory superior/distal to the core in S1pr1ECKO mice (Fig 6C, Online Fig IV C). This again points to collateral failure. Platelets and fibrin(ogen) were observed within both perfused and non-perfused capillaries only superior to the core and correlated with more intense staining for PECAM1/CD31 (Fig 6C, Online Fig. IV D, E). Fibrin deposition was more widespread and significantly more abundant in S1pr1ECKO mice (Fig 6C, Online Fig IV F). Occasional neutrophils were observed within capillaries on both sides of the infarct core at similar frequency in both genotypes (Fig 6C, Online Fig IV D,F,G). These observations confirm that EC S1P1 signaling promotes the perfusion of the ischemic penumbra and suggest that this involves the maintenance of microvascular patency.
Figure 6: Endothelial S1P1 suppresses tissue perfusion and fibrin formation in MCA territories.

A. ICAM-1 protein in homogenates of cerebral cortex from naïve S1pr1ECKO, Sphk1&2ECKO (Pdgfb-Cre) and Sphk1&2HCKO (Mx1-Cre) mice and littermate controls analyzed by Western Blot and normalized to the vascular marker VE-Cadherin. B. ICAM-1 and myeloperoxidase (MPO) protein in homogenates of contra- and ipsilateral hemispheres 2.5 hours after pMCAO from S1pr1ECKO mice and littermate controls. Top panel, Western Blot. Bottom panel, quantification. C. Female S1pr1ECWT and S1pr1ECKO mice were analyzed three hours after pMCAO. Representative images of brain sections from S1pr1ECWT (left) and S1pr1ECKO (middle & right) mice. Dashed line indicates the perfusion border assessed by tomato lectin infusion 15 minutes prior to sacrifice (top panel). Fibrin(ogen) (red); endothelial cells (CD31, green), platelets (CD41, blue/white). Square indicates area of higher magnification to the right. Note the extended area of poor perfusion superior to the infarct core and fibrin deposition both within and beyond the non-perfused regions. Scale bar: 500 μm for low magnification image and 100 μm for high magnification images. Lectin perfusion was assessed superior and inferior to the infarct core in an area of 600 μm x 1200 μm (“total”), and fibrin(ogen) only superior to the infarct core in an area of 800 μm x 1200 μm (“total”) or only within the distal part in an area of 800 μm x 600 μm (“distal’). Bar graphs show mean ± SEM. Statistical significance assessed by one-way ANOVA with Holm-Sidak multiple comparisons test (B, left), Mann Whitney (B, right) or unpaired t-test (all other).
Receptor polarization restricts S1P1 signaling and ligand access at the blood-neural barrier.
The need for EC autonomous S1P provision to sustain vascular protective S1P1 signaling in the ischemic brain could be explained by depletion of circulating S1P, as observed in myocardial infarction and during systemic inflammation.9, 44 However, plasma and platelet S1P levels were unchanged in the early acute phase of stroke with or without reperfusion (Fig 7A). An alternative explanation could be that receptor polarization restricts access of S1P1 to circulating ligand at the BBB. To address this possibility, we first used confocal microscopy to evaluate the subcellular localization of S1P1. Imaging across one or two nuclei allowed us to distinguish the EC plasma membranes, and S1pr1ACKO mice allowed us to discriminate between the abluminal plasma membrane and astrocyte end-feet in the cerebral cortex. In the developing retina, S1P1 was present primarily on the luminal surface of capillary ECs, where it co-localized with ICAM-2 (Fig 7B, Online Fig V A). By contrast, expression was predominantly abluminal on capillary ECs in the adult retina (Fig 7C, Online Fig V B), although a subset of arteriolar ECs retained luminal expression (Fig 7D, Online Fig V C). S1P1 expression was also predominantly abluminal on capillary ECs in the adult brain (Fig 7E, Online Fig V D). Polarization was retained after tMCAO (Online Fig V E).
Figure 7. Receptor polarization restricts S1P1 signaling and ligand access at the blood-neural barrier.

A. S1P levels in plasma (left) and platelets (right) after permanent and transient filament-induced MCAO relative to sham. B-E. Assessment of S1P1 polarization in capillaries (B, C, E) and artery (D) of the developing retina (B), adult retina (C, D) and adult cerebral cortex (E) of wild-type mice (B, C, D, E upper panel) and mice lacking S1pr1 in astrocytes (S1pr1ACK0; E middle panel) or in ECs (S1pr1ECKO; E lower panel). Note that S1P1 expression (red) colocalizes with the luminal EC marker ICAM-2 (green) in capillaries of the developing retina but not in the mature retina and brain. Luminal expression remains in some arterial ECs in the mature retina. The non-polarized EC marker isolectin B4 (B, C) or GLUT1 (D, E) is shown in white, and the nuclear marker Hoechst in blue. Arrowheads indicate luminal and arrows abluminal side of the endothelium. Scale bars: 2 μm. Plots of fluorescence intensity are provided in Online Fig V. F. S1P1 signaling in the cerebral cortex studied in S1P1 GFP signaling reporter mice (S1P1GS) after systemic (0.6 mg/kg i.v.) or local (0.06 mg/kg intraparenchymal) administration of RP-001 or systemic FTY720 (2x5mg/kg p.o.) or vehicle control by the same route. Note that signaling (GFP positive nuclei, green), usually restricted to ASMA positive arterioles (blue), is widespread in Erg-positive EC (red) after local administration of RP-001. GFP/Erg double positive EC nuclei were counted in arterial (ASMA positive, lower panel) and other ECs (ASMA negative, upper panel) and expressed as a percentage of all ECs in the same category. Upper panel, representative images, lower panel, image-based quantification (only statistical analysis for S1P1GS mice is shown and was done within and across treatment groups, respectively). Scale bar: 100 μm. GFP induction in other tissues in Online Fig VI, high resolution image and quantification of non-EC GFP in Online Fig. VII. G. Evolution of blood flow velocities in the cerebral cortex after injection of RP-001 i.v. (0.6 mg/kg; left panel) or into the cerebrospinal fluid (0.06 mg/kg; right panel) of S1pr1ECKO and littermate control mice. Error bars: SEM. H. S1P1 signaling in the cerebral cortex after systemic administration of CYM-5442 (10 mg/kg) or vehicle control. Left panel: Representative images, scale bar: 100 μm; right panel: Image-based quantification of GFP/Erg double-positive ECs as a fraction of total arterial and non-arterial ECs, respectively. Bar graphs show mean ± SEM. Statistical analyses by Mann Whitney (F, local administration) or by one or two-way ANOVA with Dunnett (A) or Tukey (all other) multiple comparisons test (all other).
To confirm our impression that S1P1 is surface expressed but luminally excluded in the majority of ECs in the cerebral cortex, we again made use of the S1P1 signaling reporter. In naïve S1P1GS mice, hepatocytes show no nuclear GFP although they express S1P1 (Online Fig VI A).38 Systemic administration of the potent S1P1-selective agonist RP-001 (0.6 mg/kg)38, 45 induced robust S1P1 signaling in hepatocytes as well as ECs of skeletal muscle and lung, but not cerebral cortex (Fig 7F, Online Fig VI). In striking contrast, when injected directly into the brain parenchyma, RP-001 (0.06 mg/kg) substantially increased S1P1 signaling in arteries, capillaries and veins of the cerebral cortex (Fig 7F). Astrocytes remained GFP negative despite S1P1 expression (Online Fig VII),36 suggesting together with mostly punctate staining (Fig 7E) that the receptor is not expressed on the astrocyte surface under homeostasis. Further attesting to polarization and a role for S1P1 in blood flow regulation, we observed a significant EC S1P1-dependent increase in cortical blood flow by laser Doppler when RP-001 was administered directly into the cerebrospinal fluid for paravascular access46 but not systemically (Fig 7G). Surprisingly, induction of S1P1 signaling in brain ECs, although detectable, was also minimal after systemic administration of high dose FTY720 (2x5 mg/kg), despite strong activation of the S1P1 reporter in other organs (Fig 7F, Online Fig VI). It should be noted that although FTY720 is known to cross the BBB and desensitize S1P1 on the brain endothelium, this has been demonstrated with high doses over extended time.26, 47 CYM-5442 is an S1P1-selective agonist reported to distribute rapidly and preferentially to the brain after systemic injection.48 Accordingly, at a dose required to induce S1P1 signaling equivalent to RP-001 (0.6 mg/kg) and FTY720 (2x5 mg/kg) in hepatocytes, CYM-5442 (10 mg/kg) induced signaling also in brain ECs after systemic administration (Fig 7H, Online Fig VI A).
Collectively, these observations argue that abluminal polarization shields S1P1 from circulating endogenous and synthetic ligands in capillary, venous, and most arterial ECs once the blood-neural barrier is established, and that BBB penetration is required for agonists to harness this receptor pool. Gradual receptor polarization in all but a small subset of arterial ECs with maturation of the blood-neural barrier provides an explanation for why cell autonomous S1P provision is required for full endothelial S1P1 activation during cerebral ischemia (Fig 2A, C, D, H), although it is dispensable both for S1P1 activation in the developing retina (Fig 3A) and for homeostatic signaling in cerebral arterioles (Fig 2F, H).
A BBB-penetrating S1P1-selective agonist limits cortical infarct expansion after both permanent and transient MCA occlusion.
Our results so far argue that optimal therapeutic S1P1 targeting for stroke would transiently suppress lymphocyte trafficking and activate but not desensitize EC S1P1, and that CYM-5442 may be better suited than RP-001 and FTY720. CYM-5442 distributes preferential to brain and has a relatively short plasma half-life (3 hours).48 Accordingly, CYM-5442 induced lymphopenia 3 hours after administration that was evident at 1 mg/kg and as profound as RP-001 (0.6 mg/kg) and FTY720 (1 mg/kg) at 3 mg/kg (Fig 8A). Twenty-four hours later, lymphocyte counts remained low in mice receiving FTY720, but returned to normal in mice receiving CYM-5442 and RP-001 (Fig 8A). In the pMCAO model, CYM-5442 modestly reduced 24 hour infarct volumes at 1 mg/kg (Online Fig 8A) and provided substantial benefit when administered both immediately and up to 6 hours after occlusion at 3 mg/kg (Fig 8B). In a modified pMCAO model that included permanent ligation of the ipsilateral CCA, infarct volumes were also reduced at 7 days with daily CYM-5442 (3 mg/kg) administration (Online Fig VIII B). Consistent with the engagement of and dependence on EC S1P1, CYM-5442 (3 mg/kg) reversed sensitivity to stroke in Sphk1&2ECKO mice (Fig 8C vs. Online Fig II B), but not in S1pr1ECKO mice (Fig 8D vs. 1A). CYM-5442 (3 mg/kg) also afforded significant protection when administered at reperfusion 60 minutes after MCAO (Fig 8E). Consistent with a mechanism involving collateral blood supply 28, protection in tMCAO was delimited to the cortex, where an infarct reduction of 70% mirrored protection achieved in the cortically restricted pMCAO model (Fig 8E). RP-001, which did not efficiently cross the BBB (Fig 7F), did not provide protection in the pMCAO model despite inducing equivalent lymphopenia and strong S1P1 signaling in other organs (Fig 8A, F, Online Fig VI A). Thus, optimal S1P1 targeting for ischemic stroke requires BBB penetration for engagement of endothelial receptors (Fig 8G), and can provide substantial protection against cortical infarct expansion independent of reperfusion and in a therapeutically relevant time window.
Figure 8. A BBB penetrating S1P1 agonist limits cortical infarct expansion in ischemic stroke.

A. Effects of CYM-5442, RP-001 and FTY720 at indicated concentrations on lymphocyte counts 3 and 24 hours after bolus administration. Values normalized to pre-bleed. Statistical significance assessed in comparison to vehicle control at indicated time points. B. Effect of CYM-5442 (3 mg/kg i.p. 0-6 hours after occlusion) on infarct size 24 hours after pMCAO in wild-type males. C, D. Effect of CYM-5442 (3 mg/kg i.p. immediately after occlusion) on the impact of EC Sphk1&2 (C) and S1P1 (D) deficiency (Pdgfb-iCreERT2) on infarct size 24 hours after pMCAO. E. Effect of CYM-5442 (3 mg/kg i.p. immediately before reperfusion) on infarct size 24 hours after 60 minutes tMCAO (left panel, representative infarcts; right panel, total and regional infarct size). F. Effect of RP-001 (0.6 mg/kg i.p. immediately after occlusion) on infarct size 24 hours after pMCAO. G. Protective actions of CYM-5442 are accounted for mostly by engagement of endothelial S1P1, which promotes vasodilation and BBB integrity and limits fibrin deposition. By inducing transient lymphopenia through modulation of lymphocyte receptors, CYM-5442 may further reduce thromboinflammation, as has been previously described for FTY720. These distinct actions will act in concert to limit inflammation and edema, and promote microvascular patency and perfusion of affected brain regions. PC, pericytes; AC, astrocytes; RBC, red blood cell; PLT, platelet; LY, lymphocyte; Alb, albumin. Bar graphs show mean ± SEM. Statistical significance assessed by repeated measures two-way ANOVA (A), Kruskall-Wallis test with Dunn’s multiple comparisons test (B), unpaired t-test (C, F) or Mann-Whitney test (D, E).
DISCUSSION
S1P1 modulators have shown promise in experimental models and small-scale trials of ischemic and hemorrhagic stroke.6, 22 Yet as their mechanisms of action are not fully elucidated, optimal drug properties and targeting strategies remain to be defined. As protection has been attributed to suppression of lymphocyte-mediated thromboinflammation, most current strategies focus on inhibiting the function of lymphocyte S1P1.6, 19, 20, 24 Employing a variety of genetic and experimental murine models to address endogenous roles of S1P1 signaling in cerebral ischemia, we confirm the potential benefit of targeting lymphocyte receptors but also reveal a critical role for EC S1P1 in cerebrovascular homeostasis and endogenous protection against ischemic brain damage. Compromised BBB function, reduced vasodilatory capacity, and fibrin deposition in the ischemic penumbra provoked collateral failure and rapid expansion of the infarct core in mice with selective deficiency of S1P1 in ECs. This provides genetic evidence to support the critical and multifaceted neuroprotective roles of the endothelium in ischemic stroke and the importance of S1P signaling in coordinating these functions. Addressing mechanisms of S1P1 engagement, we uncover that the receptor is abluminally polarized and insensitive to circulating ligands in most ECs at the blood-neural barrier, imposing a need for cell autonomous ligand provision for stroke protection and BBB penetration for therapeutic receptor engagement. This suggested optimal benefit of joint targeting of lymphocyte and in particular EC receptor pools with BBB penetrating S1P1 agonists, a strategy that we demonstrate to limit cortical infarct spreading in mouse models of MCAO independent of reperfusion.
Our findings argue that endothelial S1P1 is dynamically engaged to limit infarct expansion, as neonatal deletion of S1P1 did not visibly alter vascular anatomy in the adult brain and as exacerbation of infarct size was also observed when S1P1 deficiency was induced in adulthood. Moreover, EC-selective deficiency in sphingosine kinases sensitized to MCAO to a similar degree as S1P1 deficiency even if it did not reproduce defects in retinal angiogenesis or in cerebrovascular homeostasis observed in S1pr1ECKO mice, and sensitization was overcome by compensating for the lack of endogenous ligand with a pharmacological S1P1 agonist.
S1P1 supports hallmark functions of the endothelium in 1) mediating smooth muscle relaxation, 2) maintaining vascular integrity, and 3) suppressing inflammation.7 We provide evidence that S1P1 exerts all these functions in the brain:
1) A role for EC S1P1 in control of cerebral blood flow in the naïve brain was suggested by impaired acetylcholine- and hypercapnia-induced blood flow responses S1pr1ECKO mice, impaired flow-mediated dilation in S1pr1ECKO cerebral arteries ex vivo, and the capacity of an S1P1 agonist to stimulate cerebral blood flow in wild-type but not in S1pr1ECKO mice. Impaired blood flow recovery and microvascular perfusion in S1pr1ECKO mice in the acute phase after MCAO argued that S1P1 also actively supports endothelial function during cerebral ischemia. This was substantiated by the impact of S1P1 deficiency on injury to the cortex, in which MCA branches are well connected to contiguous vascular territories, but not the striatum, in which they are not.28, 49, 50 S1P1 coordinates developmental angiogenesis by promoting perfusion of the nascent vasculature,11 and supports flow-mediated dilation of mesenteric arteries.10 Endothelial NOS, which is important for the vasoactive functions of S1P1,10, 11 also promotes the early establishment of collateral supply, thus counteracting infarct expansion in ischemic stroke.51 It is therefore likely that S1P1 acts at least in part through eNOS. Blunted vasodilation in response to acetylcholine, hypercapnia, and flow, but not whisker stimulation, with EC S1P1 deficiency is intriguing and argues against a role for S1P1 in the mechanisms through which the endothelium contributes to retrograde propagation of vasoactive signals generated by neural activity. The dissociation between endothelium-dependent responses and functional hyperemia has also been observed in mice fed a high salt diet, which display marked suppression of the eNOS-driven response to acetylcholine, but not of functional hyperemia.52, 53 While the role of eNOS and other effectors downstream of S1P1 in this context remains to be specifically addressed, our results suggest that one important mechanism by which EC S1P1 limits infarct expansion is by facilitating retrograde perfusion of the affected MCA territory through collateral anastomoses with contiguous arterial branches.
2) Further attesting to the importance of S1P1 in supporting the functions of the brain endothelium, naïve S1pr1ECKO mice also displayed impaired BBB integrity. Although subtler in the naïve brain than defects in lung vascular integrity observed in the same genetic model,9, 54 dramatic and early onset edema after MCAO nevertheless argues for an important role for S1P1 in supporting BBB function during ischemia. Evans Blue/albumin leak more than doubled in S1pr1ECKO 24 hours after pMCAO, and MRI revealed profound edema within hours after tMCAO. This suggests a possible role for S1P1 in regulating vesicular transport, as edema in ischemic stroke involves transcellular transport in the early phase and tight junction impairment in the late phase.40 Minimal impact of S1P1 deficiency on 4 kDa dextran leak in a model of septic encephalopathy in this study also argued against a critical role for S1P1 in maintaining tight junctions. This is consistent with reports that ApoM-S1P regulates vesicular transport in brain arterioles through S1P1 signaling.55 A second important mechanism by which EC S1P1 limits infarct expansion is therefore through the regulation of BBB integrity, suggesting that benefit afforded by S1P1 agonists on edema formation in ischemic and hemorrhagic stroke may involve direct actions on the BBB.6, 21, 24
3) EC S1P1 deficiency was associated with increased ICAM-1 expression in the naïve brain, suggestive of a role for S1P1 in suppressing endothelial activation and leukocyte adhesion. However, expression was not further increased in the context of ischemia, and we observed no significant difference in the early recruitment of neutrophils to the ischemic penumbra in S1pr1ECKO mice, nor in hemorrhagic transformation, which depends on leukocyte-mediated BBB destruction at later stages. Circulating markers of platelet activation and platelet recruitment to ischemic capillaries were also not substantially affected by EC S1P1 deficiency. On the other hand, the deposition of fibrin, which extended well beyond the boundary of no perfusion 3 hours after pMCAO, reached significantly further into distal MCA territories in S1pr1ECKO mice than in littermate controls. This suggests that microvascular coagulation contributes to rapid deterioration of collateral supply in S1pr1ECKO mice. Whether this reflects the loss of direct actions of S1P1 signaling on the anticoagulant or profibrinolytic status of the endothelium or is a consequence of reduced perfusion remains to be determined. Regardless, it highlights the critical dynamic role of the endothelium in maintaining microvascular patency.
Endothelial S1P1 signaling thus counteracts the expansion of the ischemic core by concerted actions on hallmark endothelial functions in the regulation of smooth muscle tone, vesicular trafficking, and intravascular coagulation.
Highly restricted S1P1 signaling in the naïve cerebral cortex and the need for EC autonomous S1P provision during cerebral ischemia both argue that S1P signaling is tightly controlled at the BBB. Characterization of S1P1 expression and signaling indicates that S1P1 may remain silent in most ECs after maturation of the blood-neural barrier due to S1P1 polarization away from circulating S1P in all but a subset of arteriolar endothelial cells. As homeostatic functions of S1P1 in the naïve brain did not all depend on EC S1P production, this subset of cells may sense circulating or other S1P sources. Although it is possible that the S1P1GS reporter mouse under-represents homeostatic S1P1 signaling, limited GFP accumulation in both ECs and perivascular cells suggests that brain ECs may export S1P only when stressed. In the resting state, most brain ECs express Sphk2, Spns2 and S1pr1, but not Sphk1.36 Induction of Sphk1 transcription as an acute response to ischemia observed in this study could thus represent a trigger for EC S1P1 activation in stroke and explain the limited effect of S1P1 deficiency observed immediately after MCA occlusion. High expression of lipid phosphate phosphatase-3 in pericytes, smooth muscle cells, and astrocytes of the neurovascular unit suggests that local S1P availability is also regulated by dephosphorylation.36, 56 Limiting local S1P availability may serve both to maintain EC S1P1 responsiveness and to prevent S1P-mediated activation of astrocytes.56-58 Restricted S1P1 signaling also in peripheral organs suggests that polarization is not unique to the brain, although it has greater impact on the access of synthetic ligands at the BBB. Thus, homeostatic S1P1 signaling in the cerebral cortex appears to be maintained by a small subset of primarily arteriolar ECs that may have access to circulating ligand, while broader receptor engagement in ischemic stroke depends on the activation of EC-autonomous S1P production, possibly through the expression of Sphk1.
What are the implications of our study for therapeutic targeting of S1P1 in stroke? While modest protection observed in S1pr1HCKO mice in the tMCAO model supports the previously suggested benefit of targeting lymphocyte receptors,20 increased stroke severity and loss of efficacy of S1P1 agonists in S1pr1ECKO mice point to the endothelial receptor pool as their principal therapeutic target. This argues against the use of competitive antagonists or strong functional antagonists, which induce prolonged lymphopenia and may disable EC receptors. Most S1P1 agonists also induce lymphopenia, suggesting that these could provide benefit through both cellular targets. Our results argue that BBB penetration is needed to reach the EC receptor pool. FTY720, the S1P1 modulator employed in most experimental and clinical studies thus far, is nonspecific, did not efficiently penetrate the BBB in this study, desensitizes EC S1P1 at high doses, and induces long lasting lymphopenia.26, 54 While FTY720 is probably mostly activating on EC S1P1 at therapeutic doses, our study argues that a more specific drug could be not only safer but also more efficacious. CYM-5442, an S1P1-selective agonist used in our study, is also desensitizing at high doses but rapidly distributes to brain, triggers only transient immunosuppression, and is mostly washed out from plasma and brain 24 hours after administration.48 The suitability of other second-generation S1PR modulators needs to be defined. It is promising that recently FDA-approved Ozanimod and Siponimod both improve BBB function in models of intracerebral hemorrhage.21, 24 New biased agonists have been developed that are minimally desensitizing on EC receptors and do not induce lymphopenia; 59, 60 protection afforded with ApoM-Fc in a tMCAO model is consistent with our findings and suggest that targeting EC S1P1 alone may provide considerable benefit.59 The S1P1GS mouse used in this study and an analogous firefly split luciferase based reporter provide valuable tools to test BBB penetration of S1P1 agonists that elicit ß-arrestin recruitment.38, 61
In conclusion, this study supports a key role for S1P1 signaling in regulating endothelial function and vascular reactivity in the brain that expands with the engagement of EC-autonomous S1P provision to become a critical determinant of neuronal survival during cerebral ischemia. Although EC S1P1 signaling is partially sustained by cell-autonomous S1P provision, it can be boosted with BBB penetrating pharmacological agonists. Joint targeting of lymphocyte20 and EC S1P1 with BBB penetrating agonists is feasible and provides protection in transient and permanent ischemic stroke models in a therapeutically relevant time frame. Mechanistically, S1P1 targeting may promote regional blood flow, microvascular patency, and BBB integrity, independent of the prolonged immunosuppression induced by some S1P1 modulators. This is consistent with observed efficacy of FTY720 on downstream microvascular perfusion even in patients with failed recanalization to altapase,22 and suggests that S1P1 agonists may preserve microvascular function and the recruitment of the collateral circulation even when recanalization is either not possible or not successful. This therapeutic strategy could therefore be envisioned in patients as soon as stroke is diagnosed, without waiting for the outcome of thrombolysis, which provides measurable benefit in less than 1/3 of patients treated.62 Sustained S1P1 expression during ageing in mice (this study) and humans (Online Fig IX),63 and efficacy of S1P1 agonist on vascular parameters in murine models of diabetes and hypertension suggest that S1P1 remains a viable target in the typical stroke patient.10, 64 This is underscored by efficacy of FTY720 in small scale human trials for ischemic and hemorrhagic stroke, which also do not report an unexpected increase in bleeding, cardiac adverse events, or post-stroke infections.6, 65, 66 Our findings suggest that this strategy may be refined with focus on minimally desensitizing, BBB penetrating S1P1 agonists. The critical protective roles for EC S1P1 and its mechanisms of engagement observed in this study may also be relevant to hemorrhagic stroke, vascular dementia, and ischemic disease of other organs.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
Immune suppression and stimulation of vascular function have been investigated as adjunct therapies to limit the expansion of stroke and reduce hemorrhagic transformation
Sphingosine 1-phosphate (S1P) signaling through S1P receptor-1 (S1P1) plays an important role in immune cell trafficking and vascular homeostasis.
S1P receptor modulators including fingolimod have shown promise for the treatment of ischemic and hemorrhagic stroke in both experimental models and small-scale clinical trials.
Mechanistically, these modulators have been considered to act principally by inhibiting S1P1-mediated sensing of circulating S1P, thus blocking lymphocyte trafficking and immunothrombosis.
What New Information Does This Article Contribute?
Disruption of homeostatic S1P1 signaling in the vascular endothelium, but not lymphocytes, profoundly impacts the outcome in mouse models of ischemic stroke.
Receptor polarization shields endothelial S1P1 from circulating endogenous and synthetic ligands and imposes a need for cell-autonomous S1P provision in the ischemic brain.
A blood-brain penetrating S1P1-selective agonist prevents infarct expansion in permanent and transient models of ischemic stroke during a therapeutically relevant period of time.
Therapies for stroke are currently limited to mechanical and thrombolytic recanalization of the occluded artery. Immune suppression and stimulation of vascular function have both been explored as adjunct therapies to limit the expansion of the infarct core and reduce hemorrhagic transformation. S1P1 has emerged as a potential therapeutic target for both ischemic and hemorrhagic stroke, yet immunosuppression induced by S1P1 modulation could potentially increase the risk of post-stroke infection. By demonstrating that the main protective action of S1P1 modulation is to the endothelial receptor, we demonstrate that it is possible to obtain protection without the prolonged immunosuppression induced by some S1P1 modulators currently being explored for stroke therapy. Coupled with the need for blood brain barrier penetration, this finding has implications as to the choice of S1P1 modulators for stroke therapy. Our study also highlights the potential of targeting endothelial function as a therapeutic approach for the treatment of ischemic stroke.
ACKNOWLEDGEMENTS
We are very grateful for input from all members of the Leducq SphingoNet network and for support from the technical and administrative platforms at the contributing institutions.
SOURCES OF FUNDING
This work was supported by Fondation Leducq (SphingoNet; E.C., T.S., R.L.P, C.I., T.H.), Fondation pour la Recherche Medicale (DCP20171138945-EC; E.C., A.E., B.T.), the French National Research Agency (ANR-19-CE14-0028-01, E.C.), Fondation de France (E.C.), Marie Curie Actions (PRESTIGE-2016-3-0011, A.N.), Lefoulon Delalande (A.C., A.N.), the Intramural Research Program of the National Institutes of Health, the National Institute of Diabetes and Digestive and Kidney Diseases (R.L.P), National Institutes of Health, National Institute of Neurological Disorders and Stroke (NS114561, T.S.) and Fondation Grace de Monaco (P.L.L., C.C.M.).
Nonstandard Abbreviations and Acronyms:
- ACA
anterior cerebral artery
- ACKO
astrocyte selective knockout
- AzA
azygos artery
- BBB
blood-brain barrier
- BT
basilar trunk
- CBF
cerebral blood flow
- EC
endothelial cell
- ECKO
endothelial cell selective knockout
- eNOS
endothelial nitric oxide synthase
- GPCR
G protein-coupled receptor
- HCKO
hematopoietic cell selective knockout
- HDL
high density lipoprotein
- ICA
internal carotid artery
- mBFV
mean blood flow velocity
- MCA
middle cerebral artery
- NO
nitric oxide
- NVU
neurovascular unit
- PBS
phosphate-buffered saline
- PCA
posterior cerebral artery
- PFA
paraformaldehyde
- pMCAO
permanent middle cerebral artery occlusion
- S1P
sphingosine 1-phosphate
- S1P1
sphingosine 1-phosphate receptor-1
- Sphk
sphingosine kinase
- Spns2
spinster homolog 2
- tMCAO
transient middle cerebral artery occlusion
- TTC
triphenyltetrazolium chloride
- WT
wild-type
Footnotes
Publisher's Disclaimer: This article is published in its accepted form. It has not been copyedited and has not appeared in an issue of the journal. Preparation for inclusion in an issue of Circulation Research involves copyediting, typesetting, proofreading, and author review, which may lead to differences between this accepted version of the manuscript and the final, published version.
DISCLOSURES
None.
REFERENCES
- 1.Moskowitz MA, Lo EH, Iadecola C. The science of stroke: Mechanisms in search of treatments. Neuron. 2010;67:181–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Powers WJ, Rabinstein AA, Ackerson T, et al. 2018 guidelines for the early management of patients with acute ischemic stroke: A guideline for healthcare professionals from the american heart association/american stroke association. Stroke. 2018;49:e46–e110 [DOI] [PubMed] [Google Scholar]
- 3.Manning NW, Campbell BC, Oxley TJ, Chapot R. Acute ischemic stroke: Time, penumbra, and reperfusion. Stroke. 2014;45:640–644 [DOI] [PubMed] [Google Scholar]
- 4.Bonnin P, Mazighi M, Charriaut-Marlangue C, Kubis N. Early collateral recruitment after stroke in infants and adults. Stroke. 2019;50:2604–2611 [DOI] [PubMed] [Google Scholar]
- 5.Shuaib A, Butcher K, Mohammad AA, Saqqur M, Liebeskind DS. Collateral blood vessels in acute ischaemic stroke: A potential therapeutic target. Lancet Neurol. 2011;10:909–921 [DOI] [PubMed] [Google Scholar]
- 6.Dreikorn M, Milacic Z, Pavlovic V, Meuth SG, Kleinschnitz C, Kraft P. Immunotherapy of experimental and human stroke with agents approved for multiple sclerosis: A systematic review. Ther Adv Neurol Disord. 2018;11:1756286418770626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Proia RL, Hla T. Emerging biology of sphingosine-1-phosphate: Its role in pathogenesis and therapy. J Clin Invest. 2015;125:1379–1387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cyster JG, Schwab SR. Sphingosine-1-phosphate and lymphocyte egress from lymphoid organs. Annu Rev Immunol. 2011 [DOI] [PubMed] [Google Scholar]
- 9.Gazit SL, Mariko B, Therond P, et al. Platelet and erythrocyte sources of s1p are redundant for vascular development and homeostasis, but both rendered essential after plasma s1p depletion in anaphylactic shock. Circ Res. 2016;119:e110–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cantalupo A, Gargiulo A, Dautaj E, Liu C, Zhang Y, Hla T, Di Lorenzo A. S1pr1 (sphingosine-1-phosphate receptor 1) signaling regulates blood flow and pressure. Hypertension. 2017;70:426–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jung B, Obinata H, Galvani S, Mendelson K, Ding BS, Skoura A, Kinzel B, Brinkmann V, Rafii S, Evans T, Hla T. Flow-regulated endothelial s1p receptor-1 signaling sustains vascular development. Dev Cell. 2012;23:600–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Galvani S, Sanson M, Blaho VA, Swendeman SL, Obinata H, Conger H, Dahlback B, Kono M, Proia RL, Smith JD, Hla T. Hdl-bound sphingosine 1-phosphate acts as a biased agonist for the endothelial cell receptor s1p1 to limit vascular inflammation. Sci Signal. 2015;8:ra79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pappu R, Schwab SR, Cornelissen I, Pereira JP, Regard JB, Xu Y, Camerer E, Zheng YW, Huang Y, Cyster JG, Coughlin SR. Promotion of lymphocyte egress into blood and lymph by distinct sources of sphingosine-1-phosphate. Science. 2007;316:295–298 [DOI] [PubMed] [Google Scholar]
- 14.Vu TM, Ishizu AN, Foo JC, et al. Mfsd2b is essential for the sphingosine-1-phosphate export in erythrocytes and platelets. Nature. 2017;550:524–528 [DOI] [PubMed] [Google Scholar]
- 15.Czech B, Pfeilschifter W, Mazaheri-Omrani N, Strobel MA, Kahles T, Neumann-Haefelin T, Rami A, Huwiler A, Pfeilschifter J. The immunomodulatory sphingosine 1-phosphate analog fty720 reduces lesion size and improves neurological outcome in a mouse model of cerebral ischemia. Biochem Biophys Res Commun. 2009;389:251–256 [DOI] [PubMed] [Google Scholar]
- 16.Wei Y, Yemisci M, Kim HH, Yung LM, Shin HK, Hwang SK, Guo S, Qin T, Alsharif N, Brinkmann V, Liao JK, Lo EH, Waeber C. Fingolimod provides long-term protection in rodent models of cerebral ischemia. Ann Neurol. 2011;69:119–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hasegawa Y, Suzuki H, Sozen T, Rolland W, Zhang JH. Activation of sphingosine 1-phosphate receptor-1 by fty720 is neuroprotective after ischemic stroke in rats. Stroke. 2010;41:368–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ichijo M, Ishibashi S, Li F, Yui D, Miki K, Mizusawa H, Yokota T. Sphingosine-1-phosphate receptor-1 selective agonist enhances collateral growth and protects against subsequent stroke. PLoS One. 2015;10:e0138029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brait VH, Tarrason G, Gavalda A, Godessart N, Planas AM. Selective sphingosine 1-phosphate receptor 1 agonist is protective against ischemia/reperfusion in mice. Stroke. 2016;47:3053–3056 [DOI] [PubMed] [Google Scholar]
- 20.Kraft P, Gob E, Schuhmann MK, Gobel K, Deppermann C, Thielmann I, Herrmann AM, Lorenz K, Brede M, Stoll G, Meuth SG, Nieswandt B, Pfeilschifter W, Kleinschnitz C. Fty720 ameliorates acute ischemic stroke in mice by reducing thrombo-inflammation but not by direct neuroprotection. Stroke. 2013;44:3202–3210 [DOI] [PubMed] [Google Scholar]
- 21.Sun N, Shen Y, Han W, Shi K, Wood K, Fu Y, Hao J, Liu Q, Sheth KN, Huang D, Shi FD. Selective sphingosine-1-phosphate receptor 1 modulation attenuates experimental intracerebral hemorrhage. Stroke. 2016;47:1899–1906 [DOI] [PubMed] [Google Scholar]
- 22.Tian DC, Shi K, Zhu Z, et al. Fingolimod enhances the efficacy of delayed alteplase administration in acute ischemic stroke by promoting anterograde reperfusion and retrograde collateral flow. Ann Neurol. 2018;84:717–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu Z, Fu Y, Tian D, Sun N, Han W, Chang G, Dong Y, Xu X, Liu Q, Huang D, Shi FD. Combination of the immune modulator fingolimod with alteplase in acute ischemic stroke: A pilot trial. Circulation. 2015;132:1104–1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bobinger T, Manaenko A, Burkardt P, et al. Siponimod (baf-312) attenuates perihemorrhagic edema and improves survival in experimental intracerebral hemorrhage. Stroke. 2019:STROKEAHA119027134 [DOI] [PubMed] [Google Scholar]
- 25.Niazi H, Zoghdani N, Couty L, et al. Murine platelet production is suppressed by s1p release in the hematopoietic niche, not facilitated by blood s1p sensing. Blood Adv. 2019;3:1702–1713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yanagida K, Liu CH, Faraco G, Galvani S, Smith HK, Burg N, Anrather J, Sanchez T, Iadecola C, Hla T. Size-selective opening of the blood-brain barrier by targeting endothelial sphingosine 1-phosphate receptor 1. Proc Natl Acad Sci U S A. 2017;114:4531–4536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Poittevin M, Deroide N, Azibani F, Delcayre C, Giannesini C, Levy BI, Pocard M, Kubis N. Glatiramer acetate administration does not reduce damage after cerebral ischemia in mice. J Neuroimmunol. 2013;254:55–62 [DOI] [PubMed] [Google Scholar]
- 28.Prinz V, Endres M. Modeling focal cerebral ischemia in rodents: Introduction and overview In: Dirnagl U, ed. Rodent models of stroke. Humana Press: Springer Protocols; 2016:19–30. [Google Scholar]
- 29.Claxton S, Kostourou V, Jadeja S, Chambon P, Hodivala-Dilke K, Fruttiger M. Efficient, inducible cre-recombinase activation in vascular endothelium. Genesis. 2008;46:74–80 [DOI] [PubMed] [Google Scholar]
- 30.Zhang Y, Jin Y, Behr MJ, Feustel PJ, Morrison JP, Kimelberg HK. Behavioral and histological neuroprotection by tamoxifen after reversible focal cerebral ischemia. Exp Neurol. 2005;196:41–46 [DOI] [PubMed] [Google Scholar]
- 31.Wakade C, Khan MM, De Sevilla LM, Zhang QG, Mahesh VB, Brann DW. Tamoxifen neuroprotection in cerebral ischemia involves attenuation of kinase activation and superoxide production and potentiation of mitochondrial superoxide dismutase. Endocrinology. 2008;149:367–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Camerer E, Regard JB, Cornelissen I, Srinivasan Y, Duong DN, Palmer D, Pham TH, Wong JS, Pappu R, Coughlin SR. Sphingosine-1-phosphate in the plasma compartment regulates basal and inflammation-induced vascular leak in mice. J Clin Invest. 2009;119:1871–1879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim GS, Yang L, Zhang G, Zhao H, Selim M, McCullough LD, Kluk MJ, Sanchez T. Critical role of sphingosine-1-phosphate receptor-2 in the disruption of cerebrovascular integrity in experimental stroke. Nat Commun. 2015;6:7893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kuhn R, Schwenk F, Aguet M, Rajewsky K. Inducible gene targeting in mice. Science. 1995;269:1427–1429 [DOI] [PubMed] [Google Scholar]
- 35.Frej C, Andersson A, Larsson B, Guo LJ, Norstrom E, Happonen KE, Dahlback B. Quantification of sphingosine 1-phosphate by validated lc-ms/ms method revealing strong correlation with apolipoprotein m in plasma but not in serum due to platelet activation during blood coagulation. Anal Bioanal Chem. 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vanlandewijck M, He L, Mae MA, et al. A molecular atlas of cell types and zonation in the brain vasculature. Nature. 2018;554:475–480 [DOI] [PubMed] [Google Scholar]
- 37.Ximerakis M, Lipnick SL, Innes BT, et al. Single-cell transcriptomic profiling of the aging mouse brain. Nat Neurosci. 2019;22:1696–1708 [DOI] [PubMed] [Google Scholar]
- 38.Kono M, Tucker AE, Tran J, Bergner JB, Turner EM, Proia RL. Sphingosine-1-phosphate receptor 1 reporter mice reveal receptor activation sites in vivo. J Clin Invest. 2014;124:2076–2086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Blaho VA, Galvani S, Engelbrecht E, Liu C, Swendeman SL, Kono M, Proia RL, Steinman L, Han MH, Hla T. Hdl-bound sphingosine-1-phosphate restrains lymphopoiesis and neuroinflammation. Nature. 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Knowland D, Arac A, Sekiguchi KJ, Hsu M, Lutz SE, Perrino J, Steinberg GK, Barres BA, Nimmerjahn A, Agalliu D. Stepwise recruitment of transcellular and paracellular pathways underlies blood-brain barrier breakdown in stroke. Neuron. 2014;82:603–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jackman KA, Zhou P, Faraco G, Peixoto PM, Coleman C, Voss HU, Pickel V, Manfredi G, Iadecola C. Dichotomous effects of chronic intermittent hypoxia on focal cerebral ischemic injury. Stroke. 2014;45:1460–1467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Desilles JP, Syvannarath V, Di Meglio L, Ducroux C, Boisseau W, Louedec L, Jandrot-Perrus M, Michel JB, Mazighi M, Ho-Tin-Noe B. Downstream microvascular thrombosis in cortical venules is an early response to proximal cerebral arterial occlusion. J Am Heart Assoc. 2018;7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.von Bruhl ML, Stark K, Steinhart A, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med. 2012;209:819–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Knapp M, Zendzian-Piotrowska M, Blachnio-Zabielska A, Zabielski P, Kurek K, Gorski J. Myocardial infarction differentially alters sphingolipid levels in plasma, erythrocytes and platelets of the rat. Basic Res Cardiol. 2012;107:294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cahalan SM, Gonzalez-Cabrera PJ, Sarkisyan G, Nguyen N, Schaeffer MT, Huang L, Yeager A, Clemons B, Scott F, Rosen H. Actions of a picomolar short-acting s1p(1) agonist in s1p(1)-egfp knock-in mice. Nat Chem Biol. 2011;7:254–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rangroo Thrane V, Thrane AS, Plog BA, Thiyagarajan M, Iliff JJ, Deane R, Nagelhus EA, Nedergaard M. Paravascular microcirculation facilitates rapid lipid transport and astrocyte signaling in the brain. Sci Rep. 2013;3:2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Foster CA, Howard LM, Schweitzer A, Persohn E, Hiestand PC, Balatoni B, Reuschel R, Beerli C, Schwartz M, Billich A. Brain penetration of the oral immunomodulatory drug fty720 and its phosphorylation in the central nervous system during experimental autoimmune encephalomyelitis: Consequences for mode of action in multiple sclerosis. J Pharmacol Exp Ther. 2007;323:469–475 [DOI] [PubMed] [Google Scholar]
- 48.Gonzalez-Cabrera PJ, Cahalan SM, Nguyen N, Sarkisyan G, Leaf NB, Cameron MD, Kago T, Rosen H. S1p(1) receptor modulation with cyclical recovery from lymphopenia ameliorates mouse model of multiple sclerosis. Mol Pharmacol. 2012;81:166–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang H, Prabhakar P, Sealock R, Faber JE. Wide genetic variation in the native pial collateral circulation is a major determinant of variation in severity of stroke. J Cereb Blood Flow Metab. 2010;30:923–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Seyman E, Shaim H, Shenhar-Tsarfaty S, Jonash-Kimchi T, Bornstein NM, Hallevi H. The collateral circulation determines cortical infarct volume in anterior circulation ischemic stroke. BMC Neurol. 2016;16:206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang Z, Huang PL, Ma J, Meng W, Ayata C, Fishman MC, Moskowitz MA. Enlarged infarcts in endothelial nitric oxide synthase knockout mice are attenuated by nitro-l-arginine. J Cereb Blood Flow Metab. 1996;16:981–987 [DOI] [PubMed] [Google Scholar]
- 52.Faraco G, Brea D, Garcia-Bonilla L, et al. Dietary salt promotes neurovascular and cognitive dysfunction through a gut-initiated th17 response. Nat Neurosci. 2018;21:240–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Faraco G, Hochrainer K, Segarra SG, Schaeffer S, Santisteban MM, Menon A, Jiang H, Holtzman DM, Anrather J, Iadecola C. Dietary salt promotes cognitive impairment through tau phosphorylation. Nature. 2019;574:686–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Oo ML, Chang SH, Thangada S, Wu MT, Rezaul K, Blaho V, Hwang SI, Han DK, Hla T. Engagement of s1p(1)-degradative mechanisms leads to vascular leak in mice. J Clin Invest. 2011;121:2290–2300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mathiesen Janiurek M, Soylu-Kucharz R, Christoffersen C, Kucharz K, Lauritzen M. Apolipoprotein m-bound sphingosine-1-phosphate regulates blood-brain barrier paracellular permeability and transcytosis. Elife. 2019;8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gomez-Lopez S, Martinez-Silva AV, Montiel T, Osorio-Gomez D, Bermudez-Rattoni F, Massieu L, Escalante-Alcalde D. Neural ablation of the park10 candidate plpp3 leads to dopaminergic transmission deficits without neurodegeneration. Sci Rep. 2016;6:24028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Choi JW, Gardell SE, Herr DR, Rivera R, Lee CW, Noguchi K, Teo ST, Yung YC, Lu M, Kennedy G, Chun J. Fty720 (fingolimod) efficacy in an animal model of multiple sclerosis requires astrocyte sphingosine 1-phosphate receptor 1 (s1p1) modulation. Proc Natl Acad Sci U S A. 2011;108:751–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gril B, Paranjape AN, Woditschka S, et al. Reactive astrocytic s1p3 signaling modulates the blood-tumor barrier in brain metastases. Nat Commun. 2018;9:2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Swendeman SL, Xiong Y, Cantalupo A, et al. An engineered s1p chaperone attenuates hypertension and ischemic injury. Sci Signal. 2017;10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Poirier B, Briand V, Kadereit D, Schafer M, Wohlfart P, Philippo MC, Caillaud D, Gouraud L, Grailhe P, Bidouard JP, Trellu M, Muslin AJ, Janiak P, Parkar AA. A g protein-biased s1p1 agonist, sar247799, protects endothelial cells without affecting lymphocyte numbers. Sci Signal. 2020;13 [DOI] [PubMed] [Google Scholar]
- 61.Kono M, Conlon EG, Lux SY, Yanagida K, Hla T, Proia RL. Bioluminescence imaging of g protein-coupled receptor activation in living mice. Nat Commun. 2017;8:1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Saver JL, Gornbein J, Grotta J, Liebeskind D, Lutsep H, Schwamm L, Scott P, Starkman S. Number needed to treat to benefit and to harm for intravenous tissue plasminogen activator therapy in the 3- to 4.5-hour window: Joint outcome table analysis of the ecass 3 trial. Stroke. 2009;40:2433–2437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Uhlen M, Fagerberg L, Hallstrom BM, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347:1260419. [DOI] [PubMed] [Google Scholar]
- 64.Whetzel AM, Bolick DT, Srinivasan S, Macdonald TL, Morris MA, Ley K, Hedrick CC. Sphingosine-1 phosphate prevents monocyte/endothelial interactions in type 1 diabetic nod mice through activation of the s1p1 receptor. Circ Res. 2006;99:731–739 [DOI] [PubMed] [Google Scholar]
- 65.Campos F, Qin T, Castillo J, Seo JH, Arai K, Lo EH, Waeber C. Fingolimod reduces hemorrhagic transformation associated with delayed tissue plasminogen activator treatment in a mouse thromboembolic model. Stroke. 2013;44:505–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Salas-Perdomo A, Miro-Mur F, Gallizioli M, Brait VH, Justicia C, Meissner A, Urra X, Chamorro A, Planas AM. Role of the s1p pathway and inhibition by fingolimod in preventing hemorrhagic transformation after stroke. Sci Rep. 2019;9:8309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pitulescu ME, Adams RH. Eph/ephrin molecules--a hub for signaling and endocytosis. Genes Dev. 2010;24:2480–2492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. A global double-fluorescent cre reporter mouse. Genesis. 2007;45:593–605 [DOI] [PubMed] [Google Scholar]
- 69.Bonnin P, Leger PL, Deroide N, Fau S, Baud O, Pocard M, Charriaut-Marlangue C, Renolleau S. Impact of intracranial blood-flow redistribution on stroke size during ischemia-reperfusion in 7-day-old rats. J Neurosci Methods. 2011;198:103–109 [DOI] [PubMed] [Google Scholar]
- 70.Poittevin M, Bonnin P, Pimpie C, Riviere L, Sebrie C, Dohan A, Pocard M, Charriaut-Marlangue C, Kubis N. Diabetic microangiopathy: Impact of impaired cerebral vasoreactivity and delayed angiogenesis after permanent middle cerebral artery occlusion on stroke damage and cerebral repair in mice. Diabetes. 2015;64:999–1010 [DOI] [PubMed] [Google Scholar]
- 71.Faraco G, Moraga A, Moore J, Anrather J, Pickel VM, Iadecola C. Circulating endothelin-1 alters critical mechanisms regulating cerebral microcirculation. Hypertension. 2013;62:759–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Garcia-Bonilla L, Racchumi G, Murphy M, Anrather J, Iadecola C. Endothelial cd36 contributes to postischemic brain injury by promoting neutrophil activation via csf3. J Neurosci. 2015;35:14783–14793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jackman K, Kunz A, Iadecola C. Modeling focal cerebral ischemia in vivo. Methods Mol Biol. 2011;793:195–209 [DOI] [PubMed] [Google Scholar]
- 74.Poittevin M, Bonnin P, Pimpie C, Riviere L, Sebrie C, Dohan A, Pocard M, Charriaut-Marlangue C, Kubis N. Diabetic microangiopathy: Impact of impaired cerebral vasoreactivity and delayed angiogenesis after permanent middle cerebral artery occlusion on stroke damage and cerebral repair in mice. Diabetes. 2014 [DOI] [PubMed] [Google Scholar]
- 75.Leger PL, Bonnin P, Moretti R, Tanaka S, Duranteau J, Renolleau S, Baud O, Charriaut-Marlangue C. Early recruitment of cerebral microcirculation by neuronal nitric oxide synthase inhibition in a juvenile ischemic rat model. Cerebrovasc Dis. 2016;41:40–49 [DOI] [PubMed] [Google Scholar]
- 76.Henrion D, Dechaux E, Dowell FJ, Maclour J, Samuel JL, Levy BI, Michel JB. Alteration of flow-induced dilatation in mesenteric resistance arteries of l-name treated rats and its partial association with induction of cyclo-oxygenase-2. Br J Pharmacol. 1997;121:83–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Franco CA, Jones ML, Bernabeu MO, Geudens I, Mathivet T, Rosa A, Lopes FM, Lima AP, Ragab A, Collins RT, Phng LK, Coveney PV, Gerhardt H. Dynamic endothelial cell rearrangements drive developmental vessel regression. PLoS Biol. 2015;13:e1002125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lamprecht MR, Sabatini DM, Carpenter AE. Cellprofiler: Free, versatile software for automated biological image analysis. Biotechniques. 2007;42:71–75 [DOI] [PubMed] [Google Scholar]
- 79.Lee YK, Uchida H, Smith H, Ito A, Sanchez T. The isolation and molecular characterization of cerebral microvessels. Nat Protoc. 2019;14:3059–3081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ruck T, Bittner S, Epping L, Herrmann AM, Meuth SG. Isolation of primary murine brain microvascular endothelial cells. J Vis Exp. 2014:e52204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Regard JB, Sato IT, Coughlin SR. Anatomical profiling of g protein-coupled receptor expression. Cell. 2008;135:561–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Assmann JC, Muller K, Wenzel J, Walther T, Brands J, Thornton P, Allan SM, Schwaninger M. Isolation and cultivation of primary brain endothelial cells from adult mice. Bio Protoc. 2017;7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.He L, Vanlandewijck M, Mae MA, et al. Single-cell rna sequencing of mouse brain and lung vascular and vessel-associated cell types. Sci Data. 2018;5:180160. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that all supporting data are available within the article and its online supplementary files.