

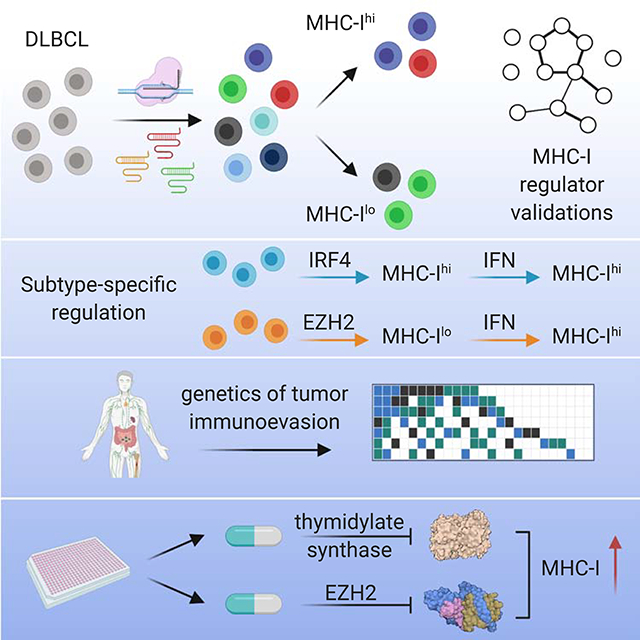
Summary
Tumors frequently subvert MHC class I (MHC-I) peptide presentation to evade CD8+ T cell immunosurveillance, though how this is accomplished is not always well-defined. To identify the global regulatory networks controlling antigen presentation, we employed genome-wide screening in human diffuse large B cell lymphomas (DLBCLs). This approach revealed dozens of genes that positively and negatively modulate MHC-I cell surface expression. Validated genes clustered in multiple pathways including cytokine signaling, mRNA processing, endosomal trafficking, and protein metabolism. Genes can exhibit lymphoma subtype- or tumor-specific MHC-I regulation, and a majority of primary DLBCL tumors displayed genetic alterations in multiple regulators. We established SUGT1 as a major positive regulator of both MHC-I and MHC-II cell surface expression. Further, pharmacological inhibition of two negative regulators of antigen presentation, EZH2 and thymidylate synthase, enhanced DLBCL MHC-I presentation. These and other genes represent potential targets for manipulating MHC-I immunosurveillance in cancers, infectious diseases, and autoimmunity.
Keywords: diffuse large B-cell lymphoma, antigen presentation, MHC class I, HLA class I, MHC class II, immunotherapy, immunoevasion, EZH2, SUGT1, thymidylate synthase
eTOC
MHC class I complexes provide the basis for CD8+ T cell immunosurveillance. Using genome wide screens, Dersh et al. identify novel genes regulating MHC-I surface expression in human B cell lymphomas. This enabled discovery of drugs that enhance tumor antigen presentation to T cells and can potentially improve immunotherapies.
Graphical Abstract
Introduction
CD8+ T cells surveil for malignant or pathogen-infected cells by probing MHC class I (MHC-I), a cell surface heterotrimer constitutively expressed on virtually all nucleated cells in jawed vertebrates. MHC-I complexes are formed in the endoplasmic reticulum (ER) by association of polymorphic heavy chains with β2 microglobulin, subsequently loaded with high affinity peptides transported from the cytoplasm by the peptide transporter TAP. Peptide-loaded MHC-I is trafficked to the plasma membrane where it can be scanned by T cells via their clonally restricted T cell receptors (TCRs), which are selected for their ability to recognize self MHC bound to peptides perceived as non-self.
Debate had raged for decades regarding the ability of T cells to recognize cancer cells as non-self. The success of T cell-based immunotherapy has ended the debate, clearly establishing the relevance of pioneering work defining tumor-specific peptides (Van den Eynde et al., 1995; van der Bruggen et al., 1991). Immunogenic cancer-associated peptides can arise from mutations in tumor cell genes (Yadav et al., 2014), enhanced transcription or translation of non-tolerized reading frames (Laumont et al., 2018), and post-translational modifications of peptides (Liepe et al., 2019).
One of the most compelling findings supporting the clinical potential of immunotherapy has been the relatively facile identification of tumors that downregulate components of the antigen processing and presentation (APP) pathway (Esteban et al., 1990; Hellstrom, 1960; Tran et al., 2016). Since such immunoevasion is one of the principal reasons for immunotherapy failure (Dersh et al., 2020; Sade-Feldman et al., 2017), it is critical to identify gene products that enable or counteract immunoevasion.
To more fully understand APP in cancer, we explored the regulation of MHC complexes in diffuse large B cell lymphoma (DLBCL). DLBCL is the most common adult lymphoid malignancy, the most frequent form of non-Hodgkin lymphoma, and often relapses under standard immunochemotherapy treatments. With frequent genetic alterations, common localization in lymph nodes, and expression of both MHC-I and MHC-II, DLBCLs are likely subjected to intense immunosurveillance. Indeed, immune evasion is common in DLBCL, with an estimated ~40–75% of biopsies showing aberrant MHC-I expression or localization (Challa-Malladi et al., 2011; Ennishi et al., 2019; Nijland et al., 2017).
Rational deployment of immunotherapies will ultimately require deeper understanding of tumor-specific genetic variation, both in antigen presentation (e.g. tumor specific antigens, HLA allotypes) and in oncogenic signaling in general. In DLBCL, for example, genetic analyses combined with survival trends from immunochemotherapies has identified clear genetic subtypes (Alizadeh et al., 2000). The “cell-of-origin” model categorizes tumors based on the differentiation state of their origin B cell. Activated B cell-like (ABC) DLBCL displays gene expression patterns similar to that of activated peripheral B cells and is clinically aggressive. Germinal center B cell-like (GCB) DLBCL likely derives from B cell centrocytes and is more responsive to standard immunochemotherapy (Read et al., 2014; Rosenwald et al., 2002). Even cells of the same classification can be widely heterogenous (Nissen et al., 2019), highlighting a need for deeper personalization in treatment plans, including immunotherapies.
In the present study, we used genome-wide CRISPR screening to investigate the regulation of MHC-I in both ABC and GCB DLBCL patient-derived tumor cells. We identified and validated dozens of genes not previously implicated in APP, demonstrating a complex coordination of cellular pathways. Our findings revealed that control of antigen presentation and immunoevasion varied between tumor lineages and even tumors of the same lineage. We describe examples of cellular complexes that can be targeted with clinically approved small molecules to enhance tumor cell immunogenicity. The work represents a comprehensive global analysis of antigen presentation and points to diverse modes of regulation that may be utilized in human cancers.
Results
Genome-wide CRISPR/Cas9 screens identify regulators of MHC-I in DLBCL
We identified genes that regulate surface MHC-I in DLBCL using four patient-derived lymphoma cell lines representing both GCB- and ABC-classified tumors. Cas9-engineered cells (Phelan et al., 2018) were transduced with the Brunello genome-wide CRISPR library (Doench et al., 2016) (Figure 1A). Through two rounds of flow cytometry-based sorting, we separated populations with low and high MHC-I surface expression using the pan human MHC-I-specific W6/32 antibody. The multi-sort strategy was critical in filtering variation inherent in MHC-I expression in normal distributions of cell populations (Figure 1B).
Figure 1. Genome-wide screens identify regulators of MHC-I in DLBCL.

(A) Overview of the CRISPR/Cas9 screens used to identify regulators of MHC-I in diffuse large B-cell lymphoma tumor lines. (B) Example flow cytometry histograms for sorted MHC-Ilo and MHC-Ihi cells (1°, primary sort; 2°, secondary sort). (C) STARS statistical confidence is plotted against segregation score of each gene in the sorted populations relative to input controls. Negative segregation score: sgRNAs enriched in MHC-Ilo population; positive segregation score: sgRNAs enriched in MHC-Ihi population. (D) (left) Venn diagram of top gene deletions from the MHC-Ilo analyses in four tumor lines. (right) Schematic depicting some genes known in APP and their identifications in the CRISPR screens – coloring refers to Venn diagram colors. (E) Venn diagram of top gene deletions from the MHC-Ihi analyses.
Counts of sgRNA sequences were used to calculate gene-specific segregation scores – Z-score-based measurements of the ratio of sgRNA sequences in MHC-I high vs. low populations. STARS software was also used to score genes via ranking and quantity of enriched sgRNAs in sorted populations relative to input controls (Doench et al., 2016) (Figure 1C; Table S1).
Importantly, nearly all genes previously described as APP regulators were highly ranked, including all components of the peptide loading complex and known MHC-I transcription factors (Figures 1C and 1D). We also identified dozens of putative positive and negative regulators not previously recognized for roles in APP – some tumor-specific and others shared between two or more tumors (Figures 1D and 1E; Table S2).
Validation and pathway analysis of screen hits
To validate positive MHC-I regulators, we cloned sgRNAs for each of 130 potential hits revealed by the genetic screens. After delivery of sgRNAs, cells were selected and mixed with fluorescently distinguishable control cells expressing non-targeting sgRNA for internally controlled flow cytometry analysis of surface MHC-I (Figure 2A). We simultaneously measured expression of CD147, which is not known to participate in APP and therefore controls for non-specific effects on protein trafficking.
Figure 2. Validation of positive regulators of MHC-I.

(A) Example dot plots of how individual genetic ablations were tested for MHC-I effects. Cells were independently infected with lentiviruses encoding targeting or non-targeting sgRNAs in GFP+ or GFP− backbones. Post-selection, cells were mixed and analyzed together by flow cytometry. GFP− and GFP+ populations were used to quantify remaining surface MHC-I. (B) Validations of positive regulators of MHC-I. 60 of the top performing sgRNAs across four parental tumor lines are displayed as a heatmap of surface MHC-I expression relative to non-targeting sgRNAs. Also shown are quantifications of MHC-I per CD147, an unrelated surface protein. Note that not every previously known regulator was reanalyzed (e.g., TAP2, RFXAP). (C) Cumulative loss in MHC-I per CD147 upon the indicated genetic ablation across the four tumor lines. GCBs orange; ABCs blue. (D) STRING analysis of the validated positive regulators of MHC-I in DLBCL. See also Figure S1.
We tested the positive regulator validation panel on each of the four DLBCL cell lines from the CRISPR screens (top 60 validated genes shown in Figure 2B; see also Table S3). Dozens of genetic alterations resulted in consistent loss of surface MHC-I across different lymphoma models (Figure 2C). These genes play roles in MHC-I antigen presentation, ER quality control, interferon signaling, trafficking, and RNA processing and splicing, among others (Figure 2D).
We similarly validated putative APP negative regulators using a panel of sgRNAs based on top screen hits, the majority of which were confirmed (Figure 3A; Table S3). The negative regulators cluster into functional pathways that include mTOR regulation, mRNA capping and translation, the PRC2 complex, the ubiquitin system, as well as numerous endolysosomal trafficking factors likely important for the internalization of MHC-I and its recycling and/or lysosomal degradation (Figure 3C).
Figure 3. Validation of negative regulators of MHC-I.

(A) Validations of negative regulators of MHC-I. 69 sgRNAs across four parental tumor lines are displayed as a heatmap of surface MHC-I expression relative to non-targeting sgRNAs. The B2M sgRNA was included as a control for Cas9 activity and antibody staining. Also shown are quantifications of MHC-I per CD147, an unrelated surface protein. (B) Cumulative gain in MHC-I per CD147 upon the indicated genetic ablation across the four tumor lines. GCBs orange; ABCs blue. (C) STRING analysis of the validated negative regulators of MHC-I in DLBCL. See also Figure S1.
MHC-I polymorphism potentially contributes to variability in the effects of a gene ablation (Table S4); however, we did not observe major differences in the behavior of HLA-A2 in HBL1 cells compared to total MHC-I expression across a variety of cell lines (Figure S1A). Separately, we observed that both segregation score (Figure S1B) and STARS p-values (Figure S1C) from the CRISPR screens correlated with the magnitude of MHC-I alteration in validation experiments, confirming the power of the screens.
While most regulators of APP affected MHC-I surface expression similarly across multiple tumors, examples of lineage or tumor specificity were also clear. For instance, ABC tumors were much more resistant to enhancement of surface MHC-I (see Figure 5). We even identified genes whose deletion exerted opposite effects on MHC-I expression in different tumor lines (Figure 4A).
Figure 4. Specificity of MHC regulation and the role of SUGT1 in antigen presentation.

(A) Opposing effects of genetic deletions on surface MHC-I of different tumor lines measured by flow cytometry. (B) Top performing sgRNAs for the loss of surface HLA-DR, quantified cumulatively across different tumor cells. GCBs orange; ABCs blue. Each gene is also classified with its MHC-I regulator status (bottom boxes). (C) Same as B, but for cumulative gains in HLA-DR. (D) Effects of RFX5 or SUGT1 loss on the surface expression of MHC-I and MHC-II in TMD8. (E) (left) Schematic of T cell co-culture assay. HLA-A2+ HBL1 cells, which natively express the cancer testis antigen NY-ESO-1, were co-cultured with primary human T-cells transduced with a TCR recognizing the NY-ESO-1 peptide 157–165 restricted by HLA-A2. (right) T-cells were monitored for activation by 4–1BB upregulation after 12–14 hours with the indicated HBL1 cells. NT, non-targeting sgRNA. Peptide, exogenously added SLLMWITQV. T cells grown without target cells were manually set to 0% (1.14% average donor 1, 5.03% average donor 2). (F) NLRC5 or SUGT1 were deleted in HBL1 or TMD8 cells, and whole cell lysates were subjected to immunoblot analysis. Arrow, NLRC5. *, undetermined band from anti-NLRC5 antibody. See also Figure S2. (G) qPCR analysis of the indicated transcripts in TMD8 cells modified with NT or SUGT1 sgRNA. (H) Same as G, but with RFX5 deletion. For entire figure, bar graphs represent mean with standard deviations, minimum n = 3.
In conclusion, forward genetic CRISPR screens identified both known and previously undescribed regulators of MHC-I across multiple DLBCLs. Informed by these screens, we individually ablated ~1% of human genes to measure the effects on APP and to validate bona fide regulators. This genome-wide analysis of the MHC-I pathway highlights first, how multiple cellular processes coordinate in immunosurveillance, and second, that individual genes can participate in MHC-I function either globally or at a lineage- or tumor-specific manner.
Multiple genes co-regulate MHC-I and MHC-II
B cells express MHC-II, the target of anti-tumor immunosurveillance mediated by CD4+ T cells, which can exhibit potent anti-tumor activity (Alspach et al., 2019; Sledzinska et al., 2020). On our panel of engineered cell lines, we examined the effect of genetic deletion on MHC-II cell surface expression, using a pan-HLA-DR specific antibody.
Although the validation panel was selected for predicted effects on MHC-I, we identified genes that regulated expression of HLA-DR, including RFX5, which is known to participate in the transcription of both MHC-I and MHC-II (Meissner et al., 2012; Steimle et al., 1995), and CREBBP, which plays a role in maintaining MHC-II expression in lymphomas (Figures 4B and 4C) (Green et al., 2015; Jiang et al., 2017).
Of these genes, we identified SUGT1 as an MHC-I and MHC-II co-regulator that mimics RFX5 function. In TMD8 cells, for example, SUGT1 and RFX sgRNAs had essentially identical effects, reducing surface expression of MHC-I by 50% and MHC-II by 80% (Figure 4D, p < 0.0001). To confirm the importance of SUGT1 silencing on immunosurveillance, we examined HBL1 cells, which endogenously express HLA-A2 and NY-ESO-1, a defined human cancer testis antigen (Robbins et al., 2015). SUGT1-deleted cells impaired activation of primary human T-cells expressing an HLA-A2-restricted TCR specific for the NY-ESO-1157−165 peptide (Figure 4E, p < 0.0001), confirming its requirement for T cell-mediated recognition of tumor cells.
The SUGT1 homolog in plants is an HSP90 co-chaperone required for the biogenesis of NLR gene family members (Mayor et al., 2007; Zhang et al., 2010). Could SUGT1 assist in the folding of NLRC5 and CIITA, both members of the NLR family and master transcription factors, respectively, for MHC-I and MHC-II? Immunoblots show that SUGT1 deletion decreased steady state NLRC5 protein and total MHC-I by ~50% (Figure 4F). Control over NLRC5 was post-transcriptional, as SUGT1 ablation did not affect NLRC5 transcripts (Figure 4G). Loss of SUGT1 did, however, significantly reduce transcription of HLA-A (p = 0.0017) and HLA-B (p = 0.0005), similar to the effect of losing the RFX5 transcription factor (Figures 4G and 4H). SUGT1 deletion also significantly decreased HLA-DRA1 transcription (Figure 4G, p = 0.0002), with a coordinate loss in MHC-II protein expression (Figure S2). We did not observe an alteration in the expression of CIITA protein in SUGT1-deleted cells, though it is possible that CIITA may be misfolded or hypofunctional.
Together, these data demonstrate that previously unidentified gene products such as SUGT1 make significant contributions to MHC presentation, that genes can simultaneously co-regulate MHC-I and MHC-II, and that genetic alterations of these regulators can have major implications for T cell recognition of tumors.
Oncogenic signaling enforces robust antigen presentation in activated B cell-like lymphomas
Our work validating regulators of MHC-I unveiled a clear cell-of-origin correlation: GCBs were much more responsive than ABCs to ablation of negative regulators (Figure 5A, Figure 2C, Figure 3B). We hypothesized that this may be due to ABC cells having higher cell surface expression of MHC-I than GCBs. Indeed, this was observed on a panel of 20 DLBCL tumor lines (Figure 5B, p = 0.0015) and could not be attributed simply to cell surface area (Figure S3A). In these analyses, we only included cells with measurable surface MHC-I and excluded tumor lines with mutations that completely prevented expression (e.g., B2M null).
Figure 5. Activated B cell-like DLBCLs drive MHC-I antigen presentation.

(A) The fold change in surface MHC-I of a gene ablation is plotted as an average between GCBs (SUDHL5, BJAB) and between ABCs (HBL1, TMD8). The diagonal line indicates an equivalent response between the subtypes. (B) A panel of 20 DLBCL cell lines were measured for surface MHC-I by flow cytometry, and MFI were normalized to fluorescent beads. For entire figure: GCBs orange, ABCs blue. (C) mRNA was isolated from the indicated cells and subjected to RNAseq. Each transcript is normalized to the highest FPKM value of the four lines. (D) RNAseq was performed on DLBCL patient tumor biopsies, and NLRC5 gene expression is displayed as log transformed normalized read counts (n = 574 patients). (E) Cells were treated with 0 or 500U/mL IFNγ for 48 hours, at which point RNA was harvested for qPCR of HLA-B transcripts. Bottom boxes represent fold increase. (F) Cells were treated with 0 or 500U/mL of IFNβ or IFNγ and stained for surface MHC-I by flow cytometry after 48 hours. (G) Same as E, but whole cell lysates were extracted and analyzed by immunoblotting. (H) TMD8, HBL1, and SUDHL5 cells were transduced with sgRNA for the indicated genes, and surface MHC-I complexes were measured compared to non-targeting controls while cells retained viability. (I) DLBCL whole cell lysates were blotted with antibodies to the indicated proteins. (J) HLA-B, TLR4, and SUB1 transcriptional start site (tss) occupancy by IRF4 was examined by ChIP. For entire figure, means with standard deviations are plotted; minimum n = 3. See also Figure S3.
Using RNAseq, we found that representative GCB tumor cells had lower expression of transcripts encoding heavy chains, β2m, TAP1, calreticulin, and NLRC5 than ABC tumors, and conversely higher relative amounts of MHC-II-associated mRNAs (Figure 5C). Tumor biopsies from ABC patients also displayed higher mRNA expression of the master transcriptional coordinator NLRC5 than those from GCB patients (Figure 5D, p < 2.22 * 10−16).
In contrast to the interferon (IFN)-mediated induction of MHC-I transcripts (Figure 5E), surface expression (Figure 5F), and protein (Figure 5G) observed in most GCB cells, ABC lines were generally poorly responsive to IFNγ or IFNβ. This was not attributable to a lack of IFNγ receptors or an inability to activate the JAK-STAT pathway (Figures S3B–S3D) and is consistent with the idea that ABC-specific signaling constitutively drives APP gene transcription to near saturation.
We speculated that ABC-specific factors might be relevant to APP pathways. However, sgRNAs targeting essential genes are eliminated from CRISPR screens, masking these relevant regulators from identification. Therefore, to uncover factors in ABC tumors that promote MHC-I gene expression, we deleted a panel of genes known to be important for ABC signaling and survival and measured MHC-I surface expression prior to cell death (Figure 5H; Figure S3E). Silencing of the IRF4-BATF complex substantially reduced surface MHC-I complexes in both HBL1 and TMD8 cells but not in a control GCB tumor line. TNFAIP3 and GRB2 ablation displayed a similar phenotype; a recent study identified TNFAIP3 alterations as significantly more frequent in MHC-Ineg and MHC-IIneg DLBCL tumors compared to tumors that stained positively for the complexes (Ennishi et al., 2019).
IRF4 and BATF are highly expressed in ABCs and required for cell survival (Yang et al., 2012); they are also expressed in a subset of GCB tumors. For example, the GCB WSU-FSCCL expresses IRF4, which correlates with its relatively high expression of MHC-I (Figure 5I). We identified potential binding sites for IRF4 at the promoter of HLA-B using IRF4 ChIP-seq datasets (Figure S3F). Using ChIP, we confirmed that IRF4 associated with the HLA-B transcriptional start site in both an ABC line and an IRF4+ GCB but not in an IRF4− GCB (Figure 5J). DNA enrichment was similar to that of genes known to be regulated by IRF4 (Yang et al., 2012).
Taken together, these findings indicate that oncogenic signaling inherent to ABC tumors can drive high expression of APP genes relative to GCB tumors. Such signaling is at least in part due to the activity of IRF4, which can associate directly with the HLA-B locus, the major source of MHC-I molecules in B cells (Figure S3G). IRF4 may therefore be a pan-DLBCL predictor of immunogenicity when considered in combination with other genetic alterations identified by our screens.
Genetics of antigen presentation in patient tumors
APP genes are commonly altered in cancers, including DLBCL (Chapuy et al., 2018; Schmitz et al., 2018). We therefore examined the alteration status of the validated MHC-I regulators from our genetic screens in a cohort of 574 DLBCL patient biopsy samples.
Exome and RNA sequencing of DLBCL patient tumors confirmed the presence of mutations and copy number alterations in the positive MHC-I regulators (Figure 6A). While mutations occurred most frequently in MHC-I genes themselves (i.e. HLA-A,B,C, and B2M), other now-validated MHC-I regulators also displayed frequent genetic alterations. We also identified genetic mutations and chromosomal alterations in the validated negative regulators in DLBCL patient tumors (Figure 6B). In particular, GCB patients had a high likelihood of EZH2 gain-of-function mutations (Yap et al., 2011). Genetic gains in copy number were also commonly identified.
Figure 6. Genetic analyses of DLBCL cohort and pan-cancer T cell correlations.

(A) DLBCL biopsies were analyzed by whole exome-seq, RNA-seq, and targeted amplicon deep sequencing (Schmitz et al., 2018). Validated MHC-I positive regulators are indicated with their frequency of genetic alterations. (B) Same as A, but with validated negative regulators of MHC-I. (C) Oncoprinter diagram of DLBCL patient biopsy samples, indicating genetic alterations in each patient, their cell-of-origin (COO) subtype, and LymphGen classification (Wright et al., 2020). Right, −log(p-values) indicate likelihood of a non-zero slope from a linear regression of DNA copy number to gene expression (see Figure S4). (D) Frequency of genetic alterations in the indicated genes, separated by tumor COO categorization. Mut, mutation; HD, homozygous deletion; HL, heterozygous loss; gain, single copy gain; amp, multiple copy gain. (E) Co-occurrence and mutual exclusivity of genetic alterations. (F) Correlation of gene expression with CD8+ T cell signatures across TCGA cohorts. Red, positive T cell signature correlation. Blue, negative correlation. (G) Same as F, but for DLBCL negative regulator genes. See also Figure S5.
Keeping our analysis to those genes that showed a genetic alteration consistent with the screen phenotype and with allele frequencies >10%, we identified seven MHC-I regulators that were recurrently altered in DLBCL and not part of the canonical APP pathway (Figure 6C). The identification of these validated genes may help explain previous findings of MHC-Ineg DLBCL tumors that did not display characteristic mutations in classical MHC-I genes (Challa-Malladi et al., 2011; Ennishi et al., 2019). In total, ~81% of DLBCL samples showed at least one form of genetic alteration, though most tumors displayed hits in multiple genes. Importantly, MHC-I regulators were significantly more likely to be specifically targeted, with an average mutation rate of 1.75% versus an average 0.52% for all other genes (p = 2 * 10−18).
Copy number variations generally correlated with gene expression, implying the genetic losses and gains are functionally relevant (Figure S4). Loss of heterozygosity in HLA genes is also a well-characterized immune evasion strategy (McGranahan et al., 2017), and individuals with HLA homozygosity are at enhanced risk for DLBCL (Wang et al., 2018). Additionally, chromosomal losses in tumors were clearly focused around genes relevant to APP and were not solely the byproduct of large deletion events (Figures S5A and S5B).
Mutations in canonical class I pathway genes have been recently characterized to be particularly common in some DLBCL subtypes (Schmitz et al., 2018; Wright et al., 2020). Our lineage analysis revealed that ABC tumors were most likely to have MHC-I pathway-related genetic alterations (Figure 6D). Of ABC tumors, the MCD subtype (MYD88L265P/CD79B mutant) was most enriched in mutations in the canonical APP genes (Figure S5C). This is likely explained by our findings that ABC tumors – and especially MCD cell models – highly express APP machinery and are critically dependent upon IRF4 expression, which is activated by MYD88 and BCR signaling (Figure 5). Thus, oncogenic signaling required in these tumors leads to high immunogenicity, which is bypassed by inactivating mutations in the major components of the class I pathway.
GCB-classified tumors display high frequencies of mutations in B2M and EZH2. Hyperactive EZH2 has been recently correlated with MHCneg tumors (Ennishi et al., 2019) and is consistent with our data finding EZH2 as a negative regulator via unbiased screens. These tumors also show high frequency of IRF8 mutations and B3GNT2 amplifications, mostly stemming from EZB-subtype patients (Figure S5C).
Genetic co-occurrences were also characterized (Figure 6E). In some cases, this reflected gene proximity, such as the common loss of CTDSPL2 and PDIA3 in tumors with B2M deletions, all located on chromosome 15 (Figure S5B). Conversely, some genetic alterations were mutually exclusive – loss of the classical HLA genes were rarely observed with activating EZH2 mutations, highlighting subtype-specific immunoevasion strategies.
We next asked whether the MHC-I regulators discovered in DLBCL might function similarly in other cancers. Using data from The Cancer Genome Atlas (TCGA), we correlated gene expression with tumor CD8+ T cell signatures. Each cancer showed a variable pattern of correlation, but ~30 positive regulators showed consistent significant positive correlations with CD8+ T cell infiltration across multiple malignancies (green highlighted genes, Figure 6F). Similarly, ~10 negative MHC-I regulators displayed consistent negative correlation between expression and T cell signatures (green highlighted genes, Figure 6G). A number of these genes correlated with CD8+ T cell signatures in DLBCL biopsies (Figure S5D). A similar approach with MHC-II regulators across the TCGA showed a weaker overall relationship with CD4+ T cell signatures, but several genes demonstrated significant correlations across two or more cohorts (Figure S5E).
Overall, these data demonstrate the prevalence of genetic alterations in APP machinery in DLBCL and that MHC-I regulators discovered via unbiased screening in one tumor type are favored to play a similar role in other cancers, thus expanding the repertoire of putative evasion factors.
EZH2 and thymidylate synthase are therapeutic targets for DLBCL
Immunotherapy research has largely focused on manipulating T cells, with relatively less attention on promoting tumor cell immunogenicity. To identify small molecules that enhance surface MHC-I, we conducted a targeted small molecule screen. Compounds were selected by cross referencing validated negative regulators identified by the genetic screens with small molecule consensus gene targets; we also selected a number of other known immunity-modulating drugs. 48 compounds were tested on two MHC-Ilo GCB DLBCL cell lines. Flow cytometry-based analysis of MHC-I revealed that several compounds targeting EZH2 or thymidylate synthase (TS/TYMS) enhanced surface expression of MHC-I in a dose-dependent manner (Figure S6; Table S5). These were both validated, top hits in from the genetic screens (Figure 3A).
EZH2 is the catalytic core of the PRC2 complex, a general transcriptional repressor involved in methylating K27 of histone H3; small molecule inhibitors (EZH2i) are already being pursued in the clinic to treat a variety of malignancies (Italiano et al., 2018; Lue and Amengual, 2018). Indeed, inhibiting EZH2 with GSK126 enhanced surface MHC-I in approximately half of 32 DLBCLs tested, whereas MHC-II responses were variable (Figure 7A; Figure S7A). Confirming the functional relevance of enhanced MHC-I expression, we expressed the cancer testis antigen NY-ESO-1 in SUDHL4 cells (Figure 7B; Figure S7B) and found that either ablation of EZH2 (Figure 7C, p < 0.0001) or pretreatment with the EZH2 inhibitor tazemetostat (Figure 7D, p < 0.0001) enhanced primary CD8+ T cell recognition of the target tumor cells. Mechanistically, inhibition of EZH2 led to significant loss of repressive H3K27me3 marks at the promoters of NLRC5 and HLA-B, similar to the loss at the known PRC2-regulated gene MYT1 (Kirmizis et al., 2004) (Figure 7E, p < 0.0001). These results suggest that at least NLRC5 and HLA-B are directly controlled by the PRC2 complex.
Figure 7. Pharmacological targeting of EZH2 and TS enhances tumor antigen presentation.

(A) Tumor lines were treated with the EZH2 inhibitor GSK126 for 7 days, and average fold increases of MHC-I and MHC-II are plotted by heatmap. Cells are categorized by their COO and EZH2 mutational status. (B) Schematic of T cell activation assay. NY-ESO-1 was transduced into the HLA-A2+ SUDHL4 tumor line. Primary human T cells were transduced with a TCR targeting the NY-ESO-1 peptide 157–165 bound to HLA-A2, and activation was measured after co-culture by 4–1BB upregulation. (C) SUDHL4-NY-ESO-1 cells were transduced with sgRNAs targeting AAVS1 (safe harbor, negative control), EZH2, or B2M; 9 days later, they were co-cultured with anti-NY-ESO-1 T cells. T cells grown without target cells were set to 0% (9.28% average donor 1; 2.19% average donor 2). (D) SUDHL4-NY-ESO-1 cells were treated with either DMSO or 5μM tazemetostat for 5 days, followed by washout of drug and subsequent co-culture with anti-NY-ESO-1 T cells. T cells grown without target cells were set to 0% (5.58% average donor 1, 13.27% average donor 2). (E) Chromatin immunoprecipitation (ChIP) for total histone H3, H3K27me3, and H3K4me3 was conducted in SUDHL4 cells +/− GSK126 pretreatment. Fold change is plotted for the indicated promoter amplicons. Heatmap at top right depicts the percent of transcript isolated compared to input. (F) Cells were treated with subtoxic doses of GSK126 for 4 days prior to RNA extraction and RNAseq. Plotted are transcript changes of positive MHC-I regulators. (G) DLBCLs were cultured with the TSi raltitrexed to determine growth inhibition after 48 hours. (H) Cells were treated with raltitrexed (RAL) or DMSO and stained for surface markers after 48 hours (Carnaval, 30nM; DB, 20nM; SUDHL4, 75nM). (I) TYMS expression vs. HLA-A or HLA-B expression in DLBCL patient tumor samples (log-transformed normalized read counts). For entire figure, bar graphs represent mean with standard deviations, minimum n = 3. See also Figure S7.
How do we account for the variability in GCB tumor responsiveness to EZH2i? As EZH2 mutations at Y641 are enzymatically activating (Yap et al., 2011), this might be predictive of drug sensitivity. Indeed, most of the highly responsive lines are heterozygous at this position (Figure 7A; Figures S7C and S7D). EZH2 mutational status is not, however, entirely predictive of MHC-I response, as a number of WT tumors responded well, and two mutant tumors were unresponsive. This mimics results in both preclinical studies (McCabe et al., 2012) and clinical trials (Italiano et al., 2018), where growth inhibition responses to EZH2i have been observed in tumors with both mutant and WT EZH2.
Why would a hyperactive EZH2 mutant tumor fail to respond to EZH2 inhibition, especially if PRC2 control of NLRC5 and HLA-B appears to be conserved (Burr et al., 2019; Zingg et al., 2017)? We treated several cell lines with GSK126 and analyzed transcriptional changes by RNAseq (Figure 7F). Responsive lines exhibited increased HLA-B and NLRC5 transcripts, as predicted. An MHC-I non-responsive EZH2 mutant tumor, WSU-DLCL2, mirrored these changes but also exhibited downregulation of other genes involved in productive APP, including a number of the regulators identified by our CRISPR screens. Thus, the effect of EZH2i on MHC-I expression can be complicated by antagonistic effects on APP accessory genes, and global understanding of regulatory pathways is required to fully appreciate tumor-specific differences.
We also pursued inhibitors of thymidylate synthase. TS contributes to the biosynthesis of thymidine and therefore is critical for DNA replication and repair. Inhibitors of TS (TSi) such as pemetrexed and raltitrexed have been shown as effective chemotherapeutic agents across a variety of malignancies (Rose et al., 2002). Indeed, we observed sensitivity of DLBCLs to TSi, as cell division and viability suffered within 2 days at low nM doses (Figure 7G; Figure S7E). We observed an increase in cell size, consistent with a G1/S block previously described (Berg et al., 2001) (Figure 7H; Figures S7F and S7G).
In a number of tumor lines, treatment with pemetrexed or raltitrexed also significantly boosted surface expression of MHC-I, an increase which could not be explained by surface area increase alone (Figure 7H; Figures S7F and S7G). Importantly, TSi could even stimulate surface MHC-I on an EZH2-mutant tumor line that was resistant to induction by EZH2i (Carnaval), suggesting drug specificity in the ability to manipulate MHC-I. TSi-treated ABC lines did not display the preferential induction of class I (Supplemental Figure 7H).
Further, we observed an inverse correlation between TYMS expression and HLA-A or HLA-B in DLBCL patient tumor samples (Figure 7I) and found that TSi could strongly induce the transcription of HLA-A, HLA-B, and NLRC5 in responsive tumor cells (Supplemental Figure 7I). These results imply a link between thymine metabolism and antigen presentation, which may be exploited by tumors and can be targeted pharmacologically.
In conclusion, our data suggest both EZH2 and TS inhibitors may be useful for enhancing DLBCL immunotherapies and further, that their clinical efficacy may result from a combination of both direct cytotoxicity and enhancement of immunogenicity. Importantly, tumors resistant to one treatment may benefit from other MHC-I-augmenting drugs. More generally, given the heterogeneity of tumors and their pharmacological responses, our work predicts the utility of combination therapy regimens to bypass the diverse immune evasion strategies employed by lymphomas.
Discussion
Cancer immunotherapy exhibits tantalizing but inconsistent clinical responses. Efforts have largely focused on enhancing T cell function, but there are also opportunities to improve targetability by increasing tumor peptide presentation. Additionally, TCR-based therapies are likely to be ineffective at eliminating immunologically invisible tumors. Therefore, a deeper understanding of APP regulation is essential for optimizing immunotherapy interventions.
Here, we employed CRISPR/Cas9 genome-wide screening to create a global map of MHC-I regulators in DLBCL. Dozens of genes participate in MHC-I biogenesis and turnover. These genes function in diverse cellular pathways ranging from epigenetic control to protein trafficking and markedly expand the known APP pathway.
It is important to consider several caveats of the screens. First, CRISPR-mediated ablation of essential genes will eliminate these cells and their corresponding sgRNAs (e.g. loss of proteasome subunits). Second, the phenotype screened – total surface MHC-I – does not report on genes important for the selective presentation of specific peptides. For example, TAPBPR is known to play a role in peptide editing (Hermann et al., 2015), but its loss does not impact overall expression of MHC-I (Boyle et al., 2013). Third, our recent Ribo-Seq analyses of DLBCL reveals translation, typically non-canonical, of many non-annotated genes not included in our library (Cuevas et al., 2020).
Although we individually deleted ~1% of human genes across four tumor lines, our list of validated regulators is certain to be incomplete. We focused mainly on genes that looked to be of importance across multiple tumors, often ignoring tumor-specific hits identified at the screen stage. Additionally, determining the relative “importance” of these genes on tumor APP is complicated by sgRNA frameshift efficiency, mRNA and protein half-lives, and genetic redundancies. Because of the vast number of cell lines created, we could not individually validate all sgRNAs; therefore, there is a strong likelihood that some true regulators were not identified simply by way of inefficient DNA cutting or frameshifting.
As an example of an MHC regulator found in our screens, SUGT1 strongly promoted the expression of both MHC-I and MHC-II genes. For class I, this is at least in part by controlling the steady state protein expression of NLRC5. SUGT1 has been shown to interact with ribosome elongation factor eEF1A1 (Novosylna et al., 2015), which binds defective ribosomal products (Yewdell, 2011) and stimulates their degradation (Gandin et al., 2013; Hotokezaka et al., 2002); therefore, a role outside transcription (e.g. peptide supply) is certainly possible. How other positive regulators influence MHC-I biogenesis is of clear interest and creates opportunities for future research in the field.
Relatively less attention has been given to negative regulation of MHC-I in cancer. As tumor MHC-I expression positively correlate with clinical responses (Harel et al., 2019), increasing MHC-I expression provides an obvious, if largely untapped, potential to enhance immunotherapy. We identified dozens of negative regulators in DLBCL that represent potential therapeutic targets. In particular, the large number of clathrin-mediated endocytosis factors and lysosome-directing trafficking proteins strongly suggest mechanisms by which MHC-I is removed from the cell surface in these tumors and warrant further investigation.
We identified a number of genes that co-regulate MHC-I and MHC-II. CD4+ T cells can participate in tumor clearance by directly lysing tumor cells and by enhancing tumor lysis by other immune cell types (Alspach et al., 2019; Sledzinska et al., 2020). As MHC-II is important for DLBCL patient survival (Ennishi et al., 2019), the dual role of some genes we identified is likely relevant. For example, FBXO11 positively regulated MHC-I but negatively regulated MHC-II. In DLBCL, genetic inactivation of FBXO11 stabilizes the proto-oncogene BCL6 (Duan et al., 2012); it would be appropriate to examine MHC-II expression and CD4+ T cell infiltration in these cases.
ABC-type tumors resisted MHC-I induction compared to GCBs through either genetic manipulation or IFN treatment. Indeed, ABC tumors appear to have constitutively heightened expression of APP machinery components. This is partially explained by the ABC tumor transcription factor repertoire, including IRF4, which we showed associates with HLA-B promoters, and addiction to NF-κB signaling, which is known to stimulate transcription via the κB1/2 motifs of the MHC-I promoters. We hypothesize that the MHC-I transcription factor NLRC5 also plays an important oncogenic role in ABCs (Ludigs et al., 2015). High expression and signaling of NF-κB and STAT3 are common in ABC tumors (Ding et al., 2008; Lam et al., 2008), and both can drive expression of NLRC5 (Cui et al., 2010; Lu et al., 2018), which we observe was highly expressed in ABCs. Since NLRC5 is relatively spared of inactivating mutations in ABC tumors, its reported roles in negatively regulating toxic type I interferon responses may explain its selective retention (Cui et al., 2010; Yang et al., 2012).
Our expanded list of MHC-I regulators provides a potential explanation for clinical samples displaying absent or altered MHC-I expression without accompanying mutations in “canonical” APP genes. Even MHC-I regulators with modest effects may be sequentially selected as alterations combine to gradually increase escape in the face of cytolytic NK pressure. Additionally, most patient samples in our analysis cohort displayed genetic alterations in multiple regulators, which may synergize for immunoevasion. CD8+ T cell infiltration signature does not predict prognosis in DLBCL (Ennishi et al., 2019); we were also unable to detect correlation between survival and most APP regulators’ expression or alterations. These observations can be explained by a model where pervasive aberrations in APP genes and regulators in DLBCL render tumors nearly undetectable to CD8+ T cells.
ABC patient tumors, which drive MHC-I expression, are identified as highly immunoedited. Indeed, many ABC model cell lines were unusable in our analyses, as they lack surface MHC-I to study. Tumors without such genetic evasion strategies are likely immunogenic and must escape from T cell pressure in other ways – their frequent development in extranodal (and immune privileged) sites is well established (Bruno et al., 2014; Kraan et al., 2013; Shi et al., 2019). These findings also help explain the weak clinical outcomes in single-agent checkpoint blockade trials (Ansell et al., 2019; Lesokhin et al., 2016), even though ABC tumors display inflamed phenotypes and can express checkpoint inhibitors (Dufva et al., 2020). Ultimately, NK-mediated or CAR-T therapies may be better suited for ABC DLBCL.
GCB tumors employ different strategies for immunoevasion – many repress MHC-I epigenetically, as mRNA and protein are readily induced by interferons. Transcription is impaired in part by the PRC2 complex, identified recently in a genetic analysis of MHClo DLBCL tumor biopsies (Ennishi et al., 2019) and by correlation analysis of head and neck cancer cohorts (Zhou et al., 2020). EZH2 Y641 mutations, common in GCBs, are considered to be hyperactivating (Yap et al., 2011) and are the target for EZH2 inhibitors aimed at starving tumors of PRC2 activity in the clinic (Kim and Roberts, 2016; Lue and Amengual, 2018).
Our identification of the PRC2 complex as a negative MHC-I regulator via unbiased screening supports the role of this complex in immunoevasion. It has been suggested that the use of EZH2 inhibitors to enhance APP would be limited to tumors with activating mutations (Ennishi et al., 2019). However, given that our screens were conducted in WT EZH2 GCB lines, even unmutated EZH2 can repress MHC-I. Consistent with these findings, many WT EZH2 GCBs and unclassified tumor lines respond to EZH2 inhibitors with increased MHC-I surface expression. Conversely, EZH2i failed to stimulate MHC-I in two tumors with EZH2 activating mutations. These findings indicate that first, basal PRC2 activity in WT EZH2 cells can contribute to repression of MHC class I, and second, mutational status of EZH2 does not necessarily predict pharmacological response. This is in line with a recent report of PRC2 regulation in small cell lung cancer and neural progenitors, which do not contain Y641 mutations (Burr et al., 2019).
Although there is evidence that the PRC2 complex can control the expression of IRF4 in germinal centers (Bujisic et al., 2017; Caganova et al., 2013), we were unable to detect IRF4 or BATF in GCB DLBCLs treated with EZH2i. Therefore, it remains unclear whether IRF4-mediated transcription plays a role during EZH2 inhibition of DLBCL.
Another therapeutic target identified by our genetic and small molecule screens is TS, important to the biosynthesis of thymidine by catalyzing the methylation of dUMP to dTMP (Wahba and Friedkin, 1962). As its enzymatic function supports DNA replication and repair, TS is a longstanding target for anticancer therapies. 5-fluorouracil was introduced in 1957 (Heidelberger et al., 1957), and TS inhibitors are currently approved in treatment of a variety of malignancies.
A recent study has described enhanced immune control of tumors with combination pemetrexed and PD-L1 blockade, with evidence of heightened T cell priming, tumoral leukocytes, and T cell inflamed phenotypes (Schaer et al., 2019). Our results are consistent with their observations of tumor immunogenic cell death; importantly, greater MHC-I on malignant cells may yield more tumor specific antigens and even enhance “cross dressing” of antigen presenting cells (Dolan et al., 2006). Therefore, toxic effects of TS inhibitors, combined with enhanced MHC-I surface expression, could further inflame the tumor environment. This class of small molecules warrants further investigation in animal models of lymphoma.
Concordant with this work, Jongsma et al have also examined the global regulatory network behind MHC-I biogenesis (Jongsma et al). Using an insertional mutagenesis screen in HAP1 cells, they uncovered many of the same regulators as described by our analyses. They identified a glycosphingolipid masking phenomenon, mediated by the SPPL3 and B3GNT5 axis. SPPL3 and a downstream enzyme B3GNT2 were identified in HBL1 cells in our screens, highlighting a putative mechanism of lymphoma evasion relying on the impairment of CD8+ T cell recognition by glycosphingolipids.
Though our work was conducted in B cell lymphomas, no doubt some of the identified MHC-I regulators will be active in other tumor types and healthy tissue. These genes may be potentially targeted by viral immunoevasins or dysregulated in autoimmunity. We detected significant correlations of gene expression with CD8+ T cell signatures across numerous other human cancers using TCGA data, providing clear avenues to pursue. Sequencing data in other public databases might also be productively mined for clues of the participation of these genes in other diseases involving CD8+ T cell immunosurveillance.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Devin Dersh (devin.dersh@nih.gov).
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact without restriction.
Data and Code Availability
Normalized counts of sgRNA library sequencing are available in Table S1. Bulk RNAseq of DLBCL cell lines generated during this study is available at Gene Expression Omnibus (GEO) accession GSE160608. Bulk RNAseq of DLBCLs treated with the EZH2 inhibitor GSK126 generated during this study is available at GEO accession GSE160609. The superseries of data is available at GEO GSE160610. All primary genetic data is available through the NIH dbGAP system under accession numbers phs001444, phs001184, and phs000178 (https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs001444.v1.p1). IRF4 ChIP-seq datasets of lymphoblastoid GM12878 were obtained from GEO accession GSM803390.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
All DLBCL cell lines (see Key Resources Table) were cultured at 37°C in 5% CO2-containing humidified incubators using Advanced RPMI (Gibco) containing 5% fetal bovine serum (Hyclone or Seradigm, heat inactivated and tet-tested), 1% penicillin/streptomycin (Gibco), and Glutamax (Gibco). Lines were routinely tested for mycoplasma using either the Mycoalert Mycoplasma Detection Kit (Lonza) or the Universal Mycoplasma Detection Kit (ATCC). Cell identity was confirmed by either STR testing (ATCC) or PCR-based copy number variant fingerprinting of 16 loci from genomic DNA (Jonathan Keats, personal communication). The generation of doxycycline-inducible Cas9 clones is described in (Phelan et al., 2018). 293FT were used to package all lentiviruses and retroviruses and were maintained at 37°C in 9% CO2-containing humidified incubators using DMEM (Gibco) containing 10% fetal bovine serum (Hyclone, not heat inactivated), Glutamax (Gibco), and non-essential amino acids (Gibco).
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| pan-MHC class I | BioXcell | Clone W6/32, Cat# BE0079 |
| CD147 (BV421) | BD Biosciences | Clone HIM6, Cat# 562583 |
| CD147 (PE) | BD Biosciences | Clone HIM6, Cat# 562552 |
| HLA-DR (PE-Cy7) | Invitrogen | Clone L243, Cat# 25-9952-42 |
| HLA-DR (FITC) | BD Biosciences | Clone L243, Cat# 347363 |
| HLA-DR/DP/DQ (PE-Cy7) | Biolegend | Clone Tu39, Cat# 361708 |
| HLA-DR/DP/DQ (FITC) | BD Pharmingen | Clone Tu39, Cat# 555558 |
| CD86 (BV421) | BD Biosciences | Clone 2331 (FUN-1), Cat# 562432 |
| CD20 (BV605) | BD Biosciences | Clone 2H7, Cat# 563780 |
| CD45 (PE) | Invitrogen | Clone 2D1, Cat# 12-9459-42 |
| HLA-A2 (APC) | BD Biosciences | Clone BB7.2, Cat# 561341 |
| CD19 (PerCP-Cy5.5) | Invitrogen | Clone HIB19, Cat# 45-0199-42 |
| IFNGR (PE) | BD Biosciences | Clone GIR-94, Cat# 558937 |
| IgG2b isotype control (PE) | BD Biosciences | Clone 27-35, Cat# 555058 |
| 4-1BB (APC) | BD Pharmingen | Clone 4B4-1, Cat# 550890 |
| CD3 (BV785) | Biolegend | Clone OKT3, Cat# 317330 |
| mTCRbeta (PE) | Invitrogen | Clone H57-597), Cat# 12-5961-83 |
| SUGT1 (Western blot) | Abcam | rabbit polyclonal, Cat# ab104253 |
| NLRC5 (Western blot) | EMD Millipore | Clone 3H8, Cat# MABF260 |
| HSP90 (Western blot) | Santa Cruz | rabbit polyclonal, Cat# sc-7947 |
| HLA-A/B/C (Western blot) | Abcam | Clone EMR8-5, Cat# ab70328 |
| histone H3 (Western blot) | Cell Signaling | Clone 1B1B2, Cat# 14269S |
| B2M (Western blot) | Dako | rabbit polyclonal, Cat# A0072 |
| TAP1 (Western blot) | EMD Millipore | Clone 148.3, Cat# MABF125 |
| GAPDH (Western blot) | Proteintech | Clone 1E6D9, Cat# 60004-1-Ig |
| IRF4 (Western blot) | Cell Signaling | Clone D9P5H, Cat# 15106S |
| MHC-II (Western blot) | Cell Signaling | Clone LGII-612.14, Cat# 68258S |
| CIITA (Western blot) | Santa Cruz | Clone 7-1H, Cat# sc-13556 |
| CIITA (immunoprecipitation) | Cell Signaling | rabbit polyclonal, Cat# 3793S |
| phospho-JAK2 (Western blot) | Cell Signaling | Clone D4A8, Cat# 8082T |
| 14-3-3 (Western blot) | Santa Cruz | rabbit polyclonal, Cat# sc-1019 |
| IRF4 (ChIP) | Abcam | rabbit polyclonal, Cat# ab101168 |
| IgG control (ChIP) | Sigma | rabbit polyclonal, Cat# 12-370 |
| histone H3 (ChIP) | Abcam | rabbit polyclonal, Cat# ab1791 |
| H3K27me3 (ChIP) | Sigma | rabbit polyclonal, Cat# 07-449 |
| H3K4me3 (ChIP) | Sigma | rabbit polyclonal, Cat# 07-473 |
| Spike-in antibody (ChIP) | Active Motif | Cat# AB_2737370 |
| Bacterial and Virus Strains | ||
| Stbl3 bacteria | Invitrogen | Cat# C739601 |
| Stbl4 bacteria | Invitrogen | Cat# 11635018 |
| Biological Samples | ||
| primary human T cells via PBMCs | Department of Transfusion Medicine, NIH | n/a |
| Chemicals, Peptides, and Recombinant Proteins | ||
| doxycycline hyclate | Sigma | Cat# D9891-56 |
| Lenti-X concentrator | Takara | Cat# 631232 |
| puromycin | Gibco | Cat# A11138-03 |
| NY-ESO-1 peptide, SLLMWITQV | American Peptide | Cat# 311266 |
| CountBright beads | Invitrogen | Cat# C36950 |
| human interferon gamma | Peprotech | Cat# AF-300-02 |
| human interferon beta | Peprotech | Cat# 300-02BC |
| 4X LDS sample buffer | Invitrogen | Cat# NP0007 |
| sodium dodecyl sulfate | Quality Biological | Cat# 351-066-721 |
| EDTA-free Protease Inhibitor Cocktail | Millipore Sigma / Roche | Cat# 4693159001 |
| RNase-free DNase I | New England Biolabs | Cat# M0303L |
| DTT | Sigma | Cat# D5545-25G |
| Advanced RPMI | Gibco | Cat# 12633012 |
| DMEM + Glutamax | Gibco | Cat# 10569044 |
| FBS | Seradigm | Cat# 1500-500 |
| FBS | Hyclone | Cat# SH30071.03 |
| Penicillin-Streptomycin | Gibco | Cat# 15140122 |
| Glutamax | Gibco | Cat# 35050-061 |
| Non-essential amino acids | Gibco | Cat# 11140-050 |
| TransIT-293 | Mirus | Cat# MIR-2700 |
| GSK126 | Cayman Chemical | Cat# 15415 |
| raltitrexed | Fisher, Apexbio | Cat# 50-101-1332 |
| pemetrexed | Abcam | Cat# ab142971 |
| tazemetostat | Cayman Chemical | Cat# 16174 |
| Intercept Blocking Buffer | Licor | Cat# 927-70001 |
| Dynabeads Protein G magnetic | Invitrogen | Cat# 10003D |
| DMSO | ATCC | Cat# 4-X-5 |
| BfuAI | New England Biolabs | Cat# R0701L |
| Antarctic Phosphatase | New England Biolabs | Cat# M0289S |
| T4 PNK | New England Biolabs | Cat# M0201S |
| T4 DNA ligase | New England Biolabs | Cat# M0202M |
| Spike-in Chromatin (ChIP) | Active Motif | Cat# 53083 |
| recombinant IL-2 | NCI Frederick | Cat# BULK Ro 23-6019 |
| ExTaq | Takara | Cat# RR006 |
| SYTOX Blue | Invitrogen | Cat# S34857 |
| SYTOX AAdvanced | Invitrogen | Cat# S10274 |
| brefeldin A | Biolegend | Cat# 420601 |
| Critical Commercial Assays | ||
| RNeasy kit | Qiagen | Cat# 74104 |
| PureYield plasmid miniprep | Promega | Cat# A1223 |
| Plasmid Plus midiprep | Qiagen | Cat# 12945 |
| AccuScript Hi-Fi Reverse Transcriptase | Agilent | Cat# 200820 |
| PowerUp SYBR Green Master Mix | Applied Biosystems | Cat# A25742 |
| Gel extraction kit | Qiagen | Cat# 28706 |
| RNase-free DNase kit | Qiagen | Cat# 79254 |
| DNeasy | Qiagen | Cat# 69504 |
| ChIP DNA Clean & Concentrator Kit | Zymo | Cat# D5205 |
| HiSpeed Plasmid Maxi Kit | Qiagen | Cat# 12662 |
| Qubit™ dsDNA HS Assay Kit | Invitrogen | Cat# Q32851 |
| Bioanalyzer High Sensitivity DNA Reagents | Agilent | Cat# 5067-4626 |
| Mycoalert Mycoplasma Detection Kit | Lonza | Cat# LT07-318 |
| Universal Mycoplasma Detection Kit | ATCC | Cat# 30-1012K |
| DC Protein Assay | Bio-Rad | Cat# 5000112 |
| Illumina TruSeq Stranded Library | Illumina | Cat# 20020594 |
| NEBNext Poly(A) mRNA Magnetic Isolation Module | New England Biolabs | Cat# E7490L |
| NEBNext Ultra II Directional RNA Library Prep Kit | New England Biolabs | Cat# E7760L |
| NEBNext Multiplex Oligos | New England Biolabs | Cat# E7335L |
| DNA Clean & Concentrator | Zymo | Cat# D4033 |
| NuPAGE Bis Tris gels | Invitrogen | Cat# NP0321 |
| iBlot2 NC mini stacks | Invitrogen | Cat# IB23002 |
| 2% E-Gel EX Size Select gels | Invitrogen | Cat# G661012 |
| Deposited Data | ||
| GTEx | Consortium, 2020 | https://gtexportal.org/home/ |
| TCGA data via cBioportal | Hoadley et al., 2018 | cgdsr (CRAN) |
| DLBCL cohort sequencing data | Schmitz et al., 2018; Wright et al., 2020 | NIH dbGAP accession numbers phs001444, phs001184 and phs000178 |
| RNAseq DLBCL cell lines | This paper | GEO GSE160608 |
| RNAseq DLBCL +/− GSK126 | This paper | GEO GSE160609 |
| IRF4 ChIP-seq of GM12878 | n/a | GEO GSM803390 |
| Experimental Models: Cell Lines | ||
| BJAB | Lab of David Baltimore | RRID:CVCL_5711 |
| HBL1 | Lab of Martin Dyer | RRID:CVCL_4213 |
| TMD8 | Lab of Shuji Tohda | RRID:CVCL_A442 |
| RIVA | Lab of Martin Dyer | RRID:CVCL_1885 |
| SUDHL4 | Lab of Mark Raffeld | RRID:CVCL_0539 |
| WSU-DLCL2 | DSMZ | DSMZ Cat# ACC-575, RRID:CVCL_1902 |
| OCILY1 | Lab of OCI/Hans Messner | RRID:CVCL_1879 |
| OCILY3 | Lab of OCI/Hans Messner | RRID:CVCL_8800 |
| HLY1 | Lab of Alison Banham | RRID:CVCL_H207 |
| U2932 | Lab of RM Amini | RRID:CVCL_1896 |
| WSU-FSCCL | DSMZ | DSMZ Cat# ACC-612, RRID:CVCL_1903 |
| MC116 | ATCC | ATCC Cat# CRL-1649, RRID:CVCL_1399 |
| SC1 | DSMZ | DSMZ Cat# ACC-558, RRID:CVCL_1888 |
| OCILY19 | Lab of OCI/Hans Messner | RRID:CVCL_1878 |
| DOHH2 | DSMZ | DSMZ Cat# ACC-47, RRID:CVCL_1179 |
| Karpas422 | Lab of Martin Dyer / DSMZ | DSMZ Cat# ACC-32, RRID:CVCL_1325 |
| DB | ATCC | ATCC Cat# CRL-2289, RRID:CVCL_1168 |
| RL | ATCC | ATCC Cat# CRL-2261, RRID:CVCL_1660 |
| Carnaval | DSMZ | DSMZ Cat# ACC-724, RRID:CVCL_D330 |
| SUDHL6 | Lab of Mark Raffeld | RRID:CVCL_2206 |
| SKI | Lab of Owen O’Connor | RRID:CVCL_E053 |
| SUDHL10 | Lab of Mark Raffeld | RRID:CVCL_1889 |
| SR786 | DSMZ | DSMZ Cat# ACC-369, RRID:CVCL_1711 |
| SUDHL2 | Lab of A Epstein | RRID:CVCL_9550 |
| OCILY18 | Lab of OCI/Hans Messner | RRID:CVCL_1880 |
| OCILY8 | Lab of OCI/Hans Messner/Martin Dyer | RRID:CVCL_8803 |
| HF | ATCC | ATCC Cat# CRL-3383, RRID:CVCL_UI84 |
| OZ | Lab of Martin Dyer | RRID:CVCL_M710 |
| NUDUL1 | Lab of A Epstein | RRID:CVCL_1877 |
| HS602 | ATCC | ATCC Cat# HTB-142, RRID:CVCL_0815 |
| Dogkit | DSMZ | DSMZ Cat# ACC-629, RRID:CVCL_2023 |
| Toledo | Lab of Craig Jordan | RRID:CVCL_3611 |
| SUDHL16 | DSMZ | DSMZ Cat# ACC-577, RRID:CVCL_1890 |
| NUDHL1 | Lab of A Epstein | RRID:CVCL_1876 |
| Pfeiffer | ATCC | ATCC Cat# CRL-2632, RRID:CVCL_3326 |
| SUDHL4 / NY-ESO-1 | This paper | n/a |
| dox-inducible Cas9 engineered DLBCLs | Phelan et al., 2018 | n/a |
| 293FT | Invitrogen | Invitrogen Cat# R70007, RRID:CVCL_6911 |
| SUDHL5 | ATCC | ATCC Cat# CRL-2958, RRID:CVCL_1735 |
| Oligonucleotides | ||
| ChIP/qPCR oligonucleotides | See Table S7 | |
| qPCR oligonucleotides | See Table S7 | |
| Recombinant DNA | ||
| Brunello sgRNA library in lentiGuide-puro | Doench et al., 2016 via Addgene | Addgene #73178 |
| pLKO.1-puro | Scot Wolfe via Addgene | Addgene #52628 |
| pLKO.1-puro/GFP | Phelan et al., 2018 | n/a |
| pMD2.G | Didier Trono via Addgene | Addgene #12259 |
| psPAX2 | Didier Trono via Addgene | Addgene #12260 |
| MSCV-IRES-EGFP | Tannishtha Reya via Addgene | Addgene #20672 |
| NY-ESO-1 MSCV-IRES-EGFP | This paper | n/a |
| pUMVC | Stewart et al., 2003 via Addgene | Addgene #8449 |
| Software and Algorithms | ||
| STRING | Szklarczyk et al., 2019 | https://string-db.org/ |
| R v3.6 | R Core Team | https://www.r-project.org/ |
| gplots v3.0.4 | CRAN/Github | https://github.com/talgalili/gplots |
| Prism v8.2 – 8.4.3 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| Flowjo v10 | BD Biosciences | https://www.flowjo.com/ |
| tidyverse 1.3.0 | Wickham et al., 2019 | https://www.tidyverse.org/ |
| Oncoprinter | cBioportal | https://www.cbioportal.org/oncoprinter |
| Illustrator CC | Adobe | https://www.adobe.com/creativecloud.html |
| IGV v2.8 | Broad Institute | http://software.broadinstitute.org/software/igv/ |
| ImageStudio | Licor | https://www.licor.com/bio/ |
| bcl2fastq v2.17 | Illumina | https://support.illumina.com/sequencing/sequencing_software/bcl2fastq-conversion-software.html |
| Trimmomatic v0.36 | Github | https://github.com/timflutre/trimmomatic |
| STAR v2.6.1c | Github | https://github.com/alexdobin/STAR |
| RSEM v1.3.0 | Github | https://deweylab.github.io/RSEM/ |
| MultiQC v1.7 | Ewels et al., 2016 | http://multiqc.info/ |
| Picard v2.4.1 | Broad Institute, Github | https://github.com/broadinstitute/picard |
| FastQC | Babraham Institute | http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| FastQ Screen | Babraham Institute | https://www.bioinformatics.babraham.ac.uk/projects/fastq_screen/ |
| RSeQC | Github | https://github.com/MonashBioinformaticsPlatform/RSeQC |
| CLC Genomics Workbench v12.0 | Qiagen | https://digitalinsights.qiagen.com/ |
| JMP/Genomics v9.1 | JMP | https://www.jmp.com/en_gb/software/genomics-data-analysis-software.html |
| xCell | Aran et al., 2017 | https://xcell.ucsf.edu/ |
| Geneious Prime | Geneious | https://www.geneious.com/ |
| DLBCL LymphGen | Wright et al., 2020 | https://llmpp.nih.gov/lymphgen/index.php |
| STARS v1.2 | Doench et al., 2016 | https://portals.broadinstitute.org/gpp/public/software/stars |
Primary human T cells
Peripheral blood mononuclear cells were obtained from healthy donors with informed consent; all procedures were approved by the institutional review board of the National Cancer Institute. Primary anti-NY-ESO-1157−165 human T-cells were generated as described (Patel et al., 2017). Briefly, primary lymphocytes were stimulated with IL-2 and anti-CD3 and retrovirally transduced with the anti-NY-ESO-1 TCR from clinical grade retroviral supernatants via RetroNectin (Takara Bio). Cells were expanded by a rapid expansion protocol involving soluble OKT3, IL-2, and irradiated feeder cells. T-cells were maintained at 37°C in 5% CO2-containing humidified incubators in RPMI containing 10% FBS (Hyclone), sodium pyruvate (Sigma), non-essential amino acids (Gibco), Glutamax (Gibco), penicillin/streptomycin (Gibco), and 300 IU/mL IL-2.
METHOD DETAILS
sgRNA cloning
pLKO.1-puro (Addgene #52628, a gift from Scot Wolfe) and the modified pLKO.1-puro/GFP vector system (Phelan et al., 2018) were used to deliver individual sgRNAs to Cas9-expressing cells. Empty vector was prepared by digestion with BfuAI (New England Biolabs) at 50°C for 3 hours and heat inactivation at 65°C for 25 minutes. This was followed by dephosphorylation via Antarctic Phosphatase (New England Biolabs) for 1.5 hours at 37°C and heat inactivation for 4 minutes at 80°C. Digested, dephosphorylated vector was gel purified according to manufacturer’s protocol (Qiagen Gel Extraction kit). Overhang DNA oligos corresponding to sgRNA sequences (ACCG flank on 5’ end, and CAAA flank on the reverse complement 3’ end) were synthesized by Eurofins or IDT and mixed equimolar at a final concentration of 100μM. Oligos were phosphorylated by T4 PNK (New England Biolabs), annealed by temperature drop, diluted, and ligated into pLKO.1 vectors using T4 DNA ligase and manufacturer protocols (New England Biolabs). DNA was transformed into Stbl3 bacteria for plasmid preparation (Thermo Fisher).
sgRNA library
The Brunello CRISPR library targeting the human genome was obtained from Addgene (via John Doench and David Root, Addgene #73178), and 400ng was electroporated into Stbl4 bacteria (Thermo Fisher). Colonies were grown by incubation at 30°C on large bioassay plates; bacteria were harvested by scraping colonies in cold LB medium. DNA was purified using HiSpeed Maxi prep kits (Qiagen).
Lentivirus and retrovirus production
293FT cells were plated one day prior such that transfection was conducted at a confluency of 70–90%. For Brunello library virus preparation, library DNA, pMD2.G (Addgene #12259, gift from Didier Trono), and psPAX2 (Addgene #12260, gift from Didier Trono) were transfected at a 4:3:1 ratio using TransIT293 reagent into T225 flasks as indicated by the manufacturer (Mirus Bio). For individual sgRNA preparations, 60mm plates were transfected with pLKO.1 plasmid, pMD2.G, and psPAX2 at a ratio of 2:1.5:1.06 using TransIT293. Virus-containing supernatant was collected at 24 and 48 hours post-transfection, spun at 500g for 10 minutes, and incubated with Lenti-X concentrator (Takara) for at least 24 hours. Virus was concentrated 32x, resuspended in PBS, aliquoted, and frozen at −80°C. Brunello virus was titrated by puromycin selection compared to uninfected controls; cell viability per volume virus was measured by flow cytometry. Production of the MuLV retroviruses was similar to that of lentiviruses, using MSCV-IRES-GFP (Addgene #20672, gift from Tannishtha Reya) as an expression backbone. MSCV vector containing either nothing (empty vector) or the CTAG1A gene were co-transfected in 293FT with pMD2.G and pUMVC ((Stewart et al., 2003), Addgene #8449, gift from Bob Weinberg) at a ratio of ~1.5:1.2:1.8. Retrovirus was collected, purified, and concentrated identically to lentiviruses.
Genome-wide screening for MHC-I
Two replicates of each doxycycline-inducible Cas9 cell line were independently transduced with Brunello library lentivirus such that transduction efficiency was between 15–25%. sgRNA coverage was maintained throughout the experiments at >500 copies of each sgRNA (~40 million cells for the ~77,000 sgRNAs). Puromycin was added after 3 days of infection, and resistant cells were grown out for 8 days. Pre-doxycycline input samples were harvested, and 200ng/mL doxycycline was added for 8–11 days to induce Cas9 and initiate genetic ablations. Cells were passaged every two days with fresh medium containing doxycycline until the first round of sorting, at which point a post-doxycycline input sample was harvested. 65 million cells were stained with the anti-pan-MHC-I antibody W6/32 (purified antibody from BioXcell, directly conjugated with AlexaFluor 647 via Molecular Probes kit), washed 3x in RPMI- and FBS-containing staining buffer, and sorted for the lowest and highest ~5% populations. Sorted cells were placed back into culture in conditioned medium and expanded continually for 1 week prior to resorting. Resulting cells were stained and sorted for the lowest of the low and highest of the high (~15%) MHC-I expressors, yielding ~2–4 million cells for each final population. gDNA was extracted from all input and sorted samples using either DNeasy kits (Qiagen) or QIAmp DNA Blood maxi kit (Qiagen).
CRISPR library preparation and sequencing
Sequencing libraries were prepared as previously described (Phelan et al., 2018; Webster et al., 2019). Briefly, sgRNA sequences were amplified from 240μg of genomic DNA per sample with primers F-5’AATGGACTATCATATGCTTACCGTAACTTGAAAGTATTTCG and R-5’GTAATTCTTTAGTTTGTATGTCTGTTGCTATTATG and ExTaq (Takara) polymerase. In total, 24 PCR replicates were performed per sample in 100μL reactions with 10μg of genomic DNA per replicate. Sorted populations contained fewer cells and thus all recovered genomic DNA (5–40μg) was amplified in 100μL reactions. Primary PCR reactions were pooled, and sequencing adapters and sample indices were added to 5μL of primary PCR product. Reactions were amplified for 24–27 cycles and then size selected using 2% E-Gel EX Size Select gels (Invitrogen). Libraries were quality checked using Agilent 2100 bioanalyzer (Agilent), Kapa qPCR (Kapa Biosystems), and quantitated by QuBit high sensitivity standards (Thermo Fisher Scientific). Libraries were sequenced on an Illumina NextSeq500 achieving an average of >340X coverage in unsorted samples and >80X coverage in flow-sorted samples. Custom perl scripts were used to extract sgRNA sequences from fastq files and bowtie2 was used to align sgRNA sequences allowing for a 1bp mismatch (Phelan et al., 2018).
STARS analysis of screen
To assemble sgRNAs and rank genes from the screening data, we used STARS_v1.2 (Doench et al., 2016). First, we added 250 pseudo “genes” based on four randomized non-targeting guides present in the Brunello library. Additionally, the most poorly expressed sgRNAs in the input samples were removed from analysis. Z-scores for each sgRNA were calculated based on Log2FoldChange of sorted populations to input controls. Segregation scores were generated by the difference in Z-score of a sgRNA in MHC-I high vs MHC-I low. For robustness, STARS score calculation excluded each gene’s first ranking perturbation, and the perturbation percentage threshold was set to 25%, for an increased number of valid gene guides (up to 4 per gene). Guide ranking was based on their duplicate-average segregation score. STARS analysis was carried out on python 2.7. Note that STAR p-values rounded to 0 were manually set at −log(p-value) of 8 for display. We combined all hits showing a p-value of <0.01 or a segregation score outside the range of −1.5 – 1.5 (Table S2). For all STRING analyses, confidence was set to “medium”, with line thickness indicating relative confidence in the genetic interaction (Szklarczyk et al., 2019).
Validation of MHC-I regulators and flow cytometry
DLBCLs were plated in flat-bottom 96-well plates and infected with saturating amounts of concentrated sgRNA-expressing lentiviruses. Non-targeting sgRNAs were delivered to wells of cells separately from targeting sgRNAs (NT or AAVS1 in pLKO.1-puro if the experimental sgRNAs were in pLKO.1-puro/GFP and vice versa). Two days post-infection, cells were split into medium containing 400ng/mL doxycycline and puromycin (0.5–2 μg/mL depending on the line). Cells were passaged in constant doxycycline and puromycin for ~7–9 days (9–11 days total post-infection) prior to analysis. Cells were harvested and NT sgRNA infected cells were mixed with experimental sgRNA-expressing lines as internal controls delineated by GFP expression. Cells were washed into lymphoma staining buffer (RPMI without phenol red supplemented with Glutamax and 1% FBS) and subsequently stained with antibody solutions at 4°C for 30 minutes with slight shaking. Cells were washed 2–3x prior to analysis on a Celesta, Fortessa X-20, or FACSCalibur (BD Biosciences). FCS files were analyzed by Flowjo version 10 (BD Biosciences). Changes in MHC-I surface expression were calculated as either the changes in raw MFI, or the derived parameter of MHC-I per CD147 per cell. All samples were normalized to NT sgRNA-infected cells. Antibodies used throughout this work include the anti-pan-MHC-I antibody W6/32 (purified antibody from BioXcell, directly conjugated with AlexaFluor 647 via Molecular Probes kit); anti-CD147 (clone HIM6, BV421, BD); anti-HLA-DR (clone L243, PE-Cy7, eBioscience); anti-HLA-DR,DP,DQ (clone Tu39, FITC, BD); anti-HLA-A2 (clone BB7.2, APC, BD); anti-IFNGR (clone GIR-94, BD Pharmingen).
Immunoblotting and immunoprecipitation
When indicated, cells were treated with 500 U/mL human IFNγ and human IFNβ for 2 days prior to isolation (Peprotech). For all lysate blots, cells were harvested and washed in PBS prior to direct lysis with SDS lysis buffer at 95°C for 10–15 minutes − 50mM Tris pH7.4, 150mM NaCl, 1mM EDTA, 2% SDS, protease inhibitor cocktail (Roche), and 15U/mL DNase I (New England Biolabs). Protein concentration of all lysates was determined by DC Protein Assay (Bio-Rad), absorbance measured by a Synergy H1 plate reader (BioTek). For samples subjected to immunoprecipitation, cells were washed in PBS and lysed in RIPA buffer (25mM Tris pH7.4, 150mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, protease inhibitor cocktail) with gentle mixing for 15 minutes at 4°C. Debris was pelleted by centrifugation at 15,000g for 15 minutes. 800–900μg protein was typically used per IP with an anti-CIITA antibody (Cell Signaling). After overnight incubation, magnetic protein G particles (ThermoFisher) were used to isolate captured CIITA. Both lysate and IP samples were prepared in 4X NuPAGE LDS sample buffer (Invitrogen) and run on 4–12% NuPAGE Bis-Tris mini or midi gels. Transfer to nitrocellulose was conducted using iBlot or iBlot 2 (Life Technologies) as described by manufacturer. Blots were blocked using Odyssey Blocking Buffer (OBB, Licor) and probed overnight in primary antibodies in OBB with 0.1% Tween-20. Secondary antibodies were incubated for 1 hour at room temperature in OBB with 0.1% Tween-20. Blots were scanned using the Licor Odyssey CLx (Licor) and analyzed using ImageStudio (Licor). Antibodies used for western blotting include: anti-SUGT1 (Abcam); anti-NLRC5 (clone 3H8, EMD Millipore); anti-HSP90 (Santa Cruz); anti-HLA-ABC (clone EMR8–5, Abcam); anti-histone H3 (Cell Signaling); anti-GAPDH (Proteintech); anti-MHC-II (clone LGII-612.14, Cell Signaling); anti-CIITA (clone 7–1H, Santa Cruz); anti-B2M (Dako); anti-TAP1 (EMD Millipore); anti-IRF4 (Cell Signaling); anti-phospho-JAK2 (Cell Signaling); anti-14–3-3 (Santa Cruz).
HLA typing
HLA typing for class I (HLA-A, HLA-B, HLA-C) and class II (DQA1; DQB1, DRB1 3,4,5; DPB1) was performed by an American Society for Histocompatibility and Immunogenetics (ASHI)-accredited laboratory at The Institute for Immunology and Infectious Diseases at Murdoch University Western Australia using locus-specific PCR amplification of genomic DNA. The assay has been adapted from previously published protocol for Barcoded-PCR method (Erlich et al., 2011) with modifications to the primer sequences. Briefly, 11 PCR amplifications per sample targeting the different HLA loci were set up with primers for a given sample tailed with a specific barcode tag sequence. Amplified products were quantitated, normalized, and pooled by subject up to 48 subjects. The pooled and normalized PCR reactions were purified using 1.8x the PCR reaction volume of AMPure XP beads (Beckman Coulter Inc). Samples were prepared for sequencing on Illumina MiSeq using the manufacturer’s standard library preparation protocol. These libraries were quantified using Kapa universal qPCR library quantification kits (Kapa Biosystems). Sequencing was performed on an Illumina MiSeq using the 2 × 300 paired-end chemistry kit (Illumina). Reads were quality-filtered and passed through a proprietary allele calling algorithm and analysis pipeline using the latest IMGT HLA allele database (Robinson et al., 2015) (http://www.ebi.ac.uk/ipd/imgt/hla/) as a reference. See Supplemental Table S4.
RNAseq
RNA was purified by either TRIzol extraction (Invitrogen) or RNeasy kits (Qiagen) according to manufacturers. All samples had an RNA Integrity (RIN) score of >9 as determined by the Agilent 2100 bioanalyzer or Agilent 2200 Tapestation system (Agilent). For RNAseq of cell lines (Figure 5C), polyA selected mRNA libraries were generated using the Illumina TruSeq Stranded Library protocol as described by the manufacturer. Briefly, 100ng to 1μg of total RNA was used as the input to an mRNA capture with oligo-dT coated magnetic beads. The mRNA was fragmented, and then random-primed cDNA synthesis was performed. The resulting double-strand cDNA was used as the input to a standard Illumina library prep with end-repair, adapter ligation and PCR amplification. mRNA samples were pooled and sequenced as paired-end 76 base pair reads on a Nextseq 500 running RTA 1.18.64 software. Demultiplexing was done using bcl2fastq v2.17. Both reads of each sample were trimmed for contaminating adapters and low-quality bases using Trimmomatic v0.36 and aligned to the Human hg38 reference genome and Gencode v30 annotation using STAR v2.6.1c. RSEM v1.3.0 was used for gene-level expression quantification. Read- and alignment-level quality was assessed using MultiQC v1.7 (Ewels et al., 2016) to aggregate QC metrics from FastQC, FastQ Screen, Picard v2.4.1, RSeQC, and Trimmomatic.
For RNAseq of GSK126 treated cells (Figure 7F), lymphoma lines were treated for 4 days in either DMSO or GSK126 (HBL1: 4 μM; WSU-DLCL2: 4 μM; SUDHL4: 4 μM; SUDHL5: 1.5 μM). Libraries were prepared with New England Biolabs product NEBNext Poly(A) mRNA Magnetic Isolation Module, New England Biolabs NEBNext Ultra II Directional RNA Library Prep Kit for Illumina, and NEBNext Multiplex Oligos for Illumina (Dual Index Primers Set 1) using 25ng total RNA input per sample. Library validation was performed on the Agilent 2200 Tapestation System (Agilent) to verify library size and purity. Library quantification via qPCR was performed on the Applied Biosystems 7900HT Fast Real-Time PCR System (ThermoFischer Scientific) using KAPA Biosystems Complete Library Quantification Kit for Illumina. PhiX was added at 1% to serve as an internal control. The resultant final library pool was 1.8pM final concentration with 1% PhiX spike-in. Paired-end sequencing was completed on an Illumina NextSeq 500 system, running Illumina NextSeq Control Software System Suite version 2.2.0 and RTA version 2.4.11. The final library pool was sequenced via 2 × 76 bp run configuration using the NextSeq 500/550 High Output v2.5 kit, 75. Demultiplexed reads were trimmed, aligned, and mapped to reference genome version hg38 using CLC Genomics Workbench software v12.0. Log2 of total exon counts or TPM were normalized by upper quartile scaling and ANOVA statistics computed using JMP/Genomics version 9.1.
ChIP
IRF4 ChIP:
Lysates of crosslinked cells were sonicated in lysis buffer (20 mM Tris-HCl pH8.1, 10 mM EDTA, 0.2% SDS, complete protease inhibitor cocktail, 1 mM NaF, 0.1 mM Na3VO4, and 10 mM glycerophosphate) using a Covaris Sonicator (8 m, 10% duty cycle, 200 burst per cycle, 75 W peak incident power) to obtain DNA fragment averaging 500–700 bp. Soluble chromatin fraction was diluted 1:2.5 (20 mM Tris-HCl pH8.1, 2 mM EDTA, 250 mM NaCl, 1.6% Triton X-100, complete protease inhibitor cocktail, 1 mM NaF, 0.1 mM Na3VO4, and 10 mM glycerophosphate) and immunoprecipitation was conducted using 5 μg antibody [IRF4 (Abcam ab101168) or control IgG (Sigma-Aldrich 12–370)] and 1–1.4 mg of chromatin at 4°C for 16 h. Immunoprecipitates were incubated with 25 μl of Dynabeads Protein G (Invitrogen) for 2 h, washed 2x with Low-salt wash buffer (20 mM Tris-HCl pH8.1, 2 mM EDTA, 150 mM NaCl, 0.08% SDS, 1% Triton X-100), 1x in High-salt buffer (20 mM Tris-HCl pH8.1, 2 mM EDTA, 500 mM NaCl, 0.08% SDS, 1% Triton X-100), 1x in LiCl buffer (10 mM Tris-HCl pH8.1, 1 mM EDTA, 1% NP40, 250 mM LiCl), and 1x in TE (10 mM Tris-HCl pH8.1, 1.2 mM EDTA). Chromatin was eluted for two cycles of 30 m at 65°C (1% SDS, 0.1 M NaHCO3), NaCl added to 0.25 M, and heated for 12 h at 65°C to reverse crosslinks. Eluants were treated with Proteinase K and RNaseA, and the DNA was purified using ChIP DNA Clean & Concentrator Kit (Zymo Research). Recovered DNA was analyzed in triplicate by qPCR.
H3, H3K27me, and H3K4me3 ChIP:
Lysates of crosslinked cells were sonicated in lysis buffer (10 mM Tris-HCl pH8.1, 150 mM NaCl, 5 mM EDTA, 0.5% Sarkosyl, 0.1% sodium deoxycholate, complete protease inhibitor cocktail, 1 mM NaF, 0.1 mM Na3VO4, and 10 mM glycerophosphate) using a Bioruptor Plus (12 cycles of 30s on/off, high power) to obtain DNA fragment averaging 500–700 bp. Soluble chromatin fraction was diluted 1:5 (10 mM Tris-HCl pH8.1, 5 mM EDTA, 150 mM NaCl, 1% Triton X-100, complete protease inhibitor cocktail, 1 mM NaF, 0.1 mM Na3VO4, and 10 mM glycerophosphate) with the addition of 40 ng Spike-in Chromatin per reaction (Active Motif 53083). Immunoprecipitation was conducted using 4 μg of anti-H3 (Abcam ab1791), 4 μg anti-H3K27me3 (Sigma-Aldrich 07–449), 4 μg anti-H3K4me3 (Sigma-Aldrich 07–473), 4 μg control IgG (Sigma-Aldrich 12–370), 2 μg Spike-in Antibody (AB_2737370) and 900 μg of chromatin at 4°C for 16 h. Immunoprecipitates were incubated with 35 μl of Dynabeads Protein G for 2 h, washed 2x with Low-salt wash buffer (20 mM Tris-HCl pH 8.1, 2 mM EDTA, 200 mM NaCl, 1% Triton X-100), 2x in LiCl buffer (10 mM Tris-HCl pH8.1, 2 mM EDTA, 1% NP40, 250 mM LiCl), and 1x in TE. Chromatin was eluted and purified as in the IRF4 ChIP experiments. Primers were designed according to (Liang et al., 2013) (GAPDH) and (Cao et al., 2014) (MYT1). All primer sequences are indicated in Table S7.
qPCR
RNA was harvested from cells using RNeasy kits (Qiagen), including the optional DNase I treatment step. RNA concentration was quantified, and 100ng was used for cDNA synthesis by AccuScript High Fidelity 1st Strand cDNA synthesis kit (Agilent) using the manufacturer’s protocol. qPCR was conducted in triplicate in a total of 20μL using PowerUp 2x SYBR Green master mix (Applied Biosystems), with a 60°C extension temperature, using the Quant Studio 3 instrument (Applied Biosystems). Data was analyzed by standard ΔΔCt method. Pan HLA qPCR primers were described by (Ramsuran et al., 2015). All primer sequences are indicated in Table S7.
Genetic analysis of DLBCL patient cohort
Mutation calls, DNA copy number analysis and gene expression values were generated as previously described (Schmitz et al., 2018). Somatic mutations were included that displayed a mutant allele frequency greater than 10% and were not found in internal laboratory control DNA or in dbSNP (version 138). LymphGen calls were based on (Wright et al., 2020). Mutations per patient and statistical analyses for mutual exclusivity or co-occurrence were calculated using oncoprinter (https://www.cbioportal.org/oncoprinter) (Cerami et al., 2012; Gao et al., 2013). HLA-A, HLA-B, and HLA-C expression was compared across the DLBCL cohort as log2 normalized read counts, and similar analyses were conducted using GTEx Portal data for healthy donor spleen and whole blood samples (Consortium, 2020). To determine the relative mutation frequency of MHC-I regulators, the top 105 validated hits were analyzed for mutation frequency compared to all other genes in the genome (# mutations) / ((# genes) * (# samples)); p-values were calculated with a Mann-Whitney rank sum test.
T-cell signature analysis in pan-cancer
Raw data for cohorts with >50 patients were downloaded from The Cancer Genome Atlas (Hoadley et al., 2018). CD8+ T-cell enrichment scores for each tumor were identified by xCell (Aran et al., 2017). mRNA expression (RNA Seq V2 RSEM) for each gene were calculated by cBioportal (cgdsr_1.2.10) (Cerami et al., 2012; Gao et al., 2013). For each gene, cohorts were split into tertiles based upon gene expression scores. CD8+ T-cell enrichment score was compared between tumors with the highest expression and those with the lowest expression for direction of change and statistical difference using a two-tailed Wilcoxon Rank Sum Test. P-values were adjusted by Bonferroni correction to correct for multiple analyses within the same cohort.
T-cell co-culture assays
SUDHL4 cells, which do not express NY-ESO-1 endogenously, were transduced with MSCV-IRES-GFP retroviruses containing either NY-ESO-1 or no insert. One week post-infection, stable GFP+ cells were sorted and expanded. Co-culture experiments were conducted in round-bottom 96-well plates with 30,000 – 45,000 anti-NY-ESO-1 TCR-transduced primary human T-cells and variable numbers of target DLBCL cells. Target cells were typically labeled with Cell Trace Violet (Invitrogen) for 15 minutes in DPBS at 37°C and washed prior to co-culture. Cells were incubated overnight for 12–15 hours and spun into antibody solutions in staining buffer (RPMI without phenol red supplemented with Glutamax and 1% FBS) at 4°C for 30 minutes with slight shaking. Cells were washed 2x prior to analysis by a Fortessa X-20 (BD). Dead cells were gated out by inclusion of SYTOX Blue or SYTOX AAdvanced dyes (Invitrogen) during the last wash step. DLBCL target cells were detected by either GFP expression (pLKO.1-puro/GFP transduction), staining with anti-HLA-DR,DP,DQ (clone Tu39, FITC, BD), Cell Trace Violet fluorescence, or a combination thereof. T-cells were identified and analyzed by staining for CD3 (clone OKT3, BV785, BioLegend); mTCRβ to detect the population of transduced cells expressing the chimeric mouse/human anti-NY-ESO-1 TCR (clone H57–597, PE, eBioscience); and 4–1BB (CD137) to monitor T-cell activation (clone 4B4–1, APC, BD).
Targeted small molecule screen
The 48 selected small molecules were dry-spotted into ultra-low attachment 384-well plates (Corning) by acoustic dispensing with an Echo 555 Liquid Handler (Labcyte Inc). Briefly, each compound was dry-spotted in a 7-doses, 1:4 dilution series, corresponding to 5.7nM – 23.5 μM final concentrations post cell addition. Each 384-well plate also included columns of 200nL DMSO controls and two empty columns for manually addition of positive and negative controls. Growth tests were first performed for DB and SUDHL4 cells to determine optimal growth conditions in 384-well plates. Cells were then plated in drug-spotted plates in a total volume of 85μL with 33% conditioned medium. For positive controls, 500 U/mL IFNγ (Peprotech) was added to one column of wells one day prior to collection. 5 μg/mL brefeldin A (Biolegend) was added to one column of wells 18 hours prior to collection to serve as negative controls. After a total of 48 hours of drug exposure, cells were stained with anti-pan-MHC-I antibody W6/32-AlexaFluor647, washed 2x with lymphoma staining buffer, and analyzed by a High Throughput Sampler-equipped Fortessa (BD). To calculate fold changes, MHC-I surface expression was normalized to the average of 16 DMSO control wells.
Targeted sequencing of EZH2
Genomic DNA was purified from each DLBCL cell line using DNeasy Blood & Tissue Kit (Qiagen) following manufacturer instructions. The genomic region containing EZH2 Y641 (chromosome 7 genomic coordinates: 148,811,063–148,812,103) was amplified by PCR with the primer pair: Fwd: 5’-TGGTAAAGCTCTTGTTCTCCC-3’, Rev: 5’-AGAGTGATTTGGTGGTGTCC-3’; amplicon size: 1041 bp. 50–100 ng of gDNA was amplified in 50 μl reaction using Q5® Hot Start High-Fidelity 2X Master Mix (New England BioLabs) with initial denaturation at 98°C for 30 sec followed by 35 cycles of 98˚C 10 sec, 64˚C 30 sec, 72˚C 40 sec and final extension at 72°C for 2 min. PCR products were purified with DNA Clean & Concentrator-25 kit (Zymo Research) and 5 μl of purified products were loaded on 2% E-Gel EX (Invitrogen) to verify the reaction performance and amplicon size. The rest of the purified PCR product was sent for Sanger sequencing (Eurofins Genomics) with sequencing primer: 5’-CAGCTTTCACGTTGACTG-3’. Chromatograms were manually evaluated using Geneious Prime software.
EZH2 and TS inhibition assays
DLBCL cell lines plated in 96-well plates were treated with the indicated concentrations of GSK126 (Cayman Chemical), tazemetostat (EPZ-6438, Cayman Chemical), or DMSO vehicle (ATCC) for a total 7 days, splitting every 2–4 days as required with fresh inhibitors. The indicated concentrations of inhibitors were used as they were generally subtoxic doses for most lines. Cells were stained as described above and analyzed by a Fortessa X-20 or FACSCalibur and FlowJo (BD). The fold change compared to DMSO control is an average of 3–4 biological replicates conducted on different days. For TS inhibition assays, pemetrexed (Abcam) or raltitrexed (ApexBio) were used at indicated concentrations for only 2 days prior to flow cytometry analysis. For cell growth inhibition assays, SYTOX Blue was used to distinguish dead cells and live cell numbers were normalized to CountBright Absolute Counting Beads spiked into the cultures (Invitrogen).
Illustration
Figures were assembled with Adobe Illustrator Creative Cloud; the graphical abstract was prepared with Biorender.
QUANTIFICATION AND STATISTICAL ANALYSIS
One-way ANOVA, unpaired t tests, and Mann Whitney rank tests were performed using GraphPad Prism v8 (GraphPad Software LLC). One-way ANOVA tests were adjusted for multiple comparisons by Dunnett’s test. Kruskal-Wallis tests were performed on R version 3.6, and data were displayed using tidyverse packages (Wickham et al., 2019). Statistical tests and p-values are fully summarized in Table S6.
Supplementary Material
Supplemental Table S1. CRISPR screen normalized counts, segregation scores, and STARS analyses (related to Figure 1).
Supplemental Table S2. Top predicted hits from CRISPR screens (related to Figure 1).
Supplemental Table S3. Validations of ~200 genetic ablations in four DLBCLs (related to Figures 2–3).
Supplemental Table S4. HLA typing of common DLBCL tumor lines (related to STAR Methods).
Supplemental Table S5. Targeted drug screen to identify enhancers of antigen presentation (related to Figure 7).
Supplemental Table S6. Summary of statistical analyses (related to STAR Methods).
Supplemental Table S7. Oligonucleotide sequences (related to STAR Methods).
Highlights.
Genome-wide screens dramatically expand known regulators of the MHC class I pathway
Human B cell lymphomas exhibit tumor- and subtype-specific modes of immunoevasion
Genes such as SUGT1 co-regulate MHC-I and MHC-II expression
Thymidylate synthase and EZH2 inhibitors enhance tumor peptide presentation
Acknowledgements
Special thanks to the NIAID Research Technologies Branch, including Tim Myers, the Genomic Technologies facility, Teresa Hawley, and the Flow Cytometry facility. Thanks to the NCI Center for Cancer Research sequencing facility, as well as support personnel in the laboratories of L.M.S and J.W.Y. We also acknowledge the Centre for Clinical Immunology and Biomedical Studies at the Institute for Immunology & Infectious Diseases for HLA typing. This research was supported by the Intramural Research Program of the NIH – NIAID, NCI, and NCATS – and specifically the NCI FLEX award to J.W.Y and L.M.S.
Footnotes
Declaration of Interests
N.P.R. and R.J.K. are now employees of Lyell Immunopharma and hold equity.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, Boldrick JC, Sabet H, Tran T, Yu X, et al. (2000). Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 403, 503–511. [DOI] [PubMed] [Google Scholar]
- Alspach E, Lussier DM, Miceli AP, Kizhvatov I, DuPage M, Luoma AM, Meng W, Lichti CF, Esaulova E, Vomund AN, et al. (2019). MHC-II neoantigens shape tumour immunity and response to immunotherapy. Nature 574, 696–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansell SM, Minnema MC, Johnson P, Timmerman JM, Armand P, Shipp MA, Rodig SJ, Ligon AH, Roemer MGM, Reddy N, et al. (2019). Nivolumab for Relapsed/Refractory Diffuse Large B-Cell Lymphoma in Patients Ineligible for or Having Failed Autologous Transplantation: A Single-Arm, Phase II Study. J Clin Oncol 37, 481–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aran D, Hu Z, and Butte AJ (2017). xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol 18, 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg RW, Werner M, Ferguson PJ, Postenka C, Vincent M, Koropatnick DJ, and Behrend E (2001). Tumor growth inhibition in vivo and G2/M cell cycle arrest induced by antisense oligodeoxynucleotide targeting thymidylate synthase. J Pharmacol Exp Ther 298, 477–484. [PubMed] [Google Scholar]
- Boyle LH, Hermann C, Boname JM, Porter KM, Patel PA, Burr ML, Duncan LM, Harbour ME, Rhodes DA, Skjodt K, et al. (2013). Tapasin-related protein TAPBPR is an additional component of the MHC class I presentation pathway. Proc Natl Acad Sci U S A 110, 3465–3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno A, Boisselier B, Labreche K, Marie Y, Polivka M, Jouvet A, Adam C, Figarella-Branger D, Miquel C, Eimer S, et al. (2014). Mutational analysis of primary central nervous system lymphoma. Oncotarget 5, 5065–5075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bujisic B, De Gassart A, Tallant R, Demaria O, Zaffalon L, Chelbi S, Gilliet M, Bertoni F, and Martinon F (2017). Impairment of both IRE1 expression and XBP1 activation is a hallmark of GCB DLBCL and contributes to tumor growth. Blood 129, 2420–2428. [DOI] [PubMed] [Google Scholar]
- Burr ML, Sparbier CE, Chan KL, Chan YC, Kersbergen A, Lam EYN, Azidis-Yates E, Vassiliadis D, Bell CC, Gilan O, et al. (2019). An Evolutionarily Conserved Function of Polycomb Silences the MHC Class I Antigen Presentation Pathway and Enables Immune Evasion in Cancer. Cancer Cell 36, 385–401 e388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caganova M, Carrisi C, Varano G, Mainoldi F, Zanardi F, Germain PL, George L, Alberghini F, Ferrarini L, Talukder AK, et al. (2013). Germinal center dysregulation by histone methyltransferase EZH2 promotes lymphomagenesis. J Clin Invest 123, 5009–5022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Q, Wang X, Zhao M, Yang R, Malik R, Qiao Y, Poliakov A, Yocum AK, Li Y, Chen W, et al. (2014). The central role of EED in the orchestration of polycomb group complexes. Nat Commun 5, 3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. (2012). The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2, 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Challa-Malladi M, Lieu YK, Califano O, Holmes AB, Bhagat G, Murty VV, Dominguez-Sola D, Pasqualucci L, and Dalla-Favera R (2011). Combined genetic inactivation of beta2-Microglobulin and CD58 reveals frequent escape from immune recognition in diffuse large B cell lymphoma. Cancer Cell 20, 728–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapuy B, Stewart C, Dunford AJ, Kim J, Kamburov A, Redd RA, Lawrence MS, Roemer MGM, Li AJ, Ziepert M, et al. (2018). Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat Med 24, 679–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium GT (2020). The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuevas MVR, Hardy M-P, Holly J, Bonneil E, Durette C, Courcelles M, Lanoix J, Cote C, Staudt LM, Lemieux S, et al. (2020). Most Non-Canonical Proteins Uniquely Populate the Proteome or Immunopeptidome. Available at SSRN: https://ssrncom/abstract=3671737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J, Zhu L, Xia X, Wang HY, Legras X, Hong J, Ji J, Shen P, Zheng S, Chen ZJ, et al. (2010). NLRC5 negatively regulates the NF-kappaB and type I interferon signaling pathways. Cell 141, 483–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dersh D, Holly J, and Yewdell JW (2020). A few good peptides: MHC class I-based cancer immunosurveillance and immunoevasion. Nat Rev Immunol. [DOI] [PubMed] [Google Scholar]
- Ding BB, Yu JJ, Yu RY, Mendez LM, Shaknovich R, Zhang Y, Cattoretti G, and Ye BH (2008). Constitutively activated STAT3 promotes cell proliferation and survival in the activated B-cell subtype of diffuse large B-cell lymphomas. Blood 111, 1515–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, et al. (2016). Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol 34, 184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolan BP, Gibbs KD Jr., and Ostrand-Rosenberg S (2006). Dendritic cells cross-dressed with peptide MHC class I complexes prime CD8+ T cells. J Immunol 177, 6018–6024. [DOI] [PubMed] [Google Scholar]
- Duan S, Cermak L, Pagan JK, Rossi M, Martinengo C, di Celle PF, Chapuy B, Shipp M, Chiarle R, and Pagano M (2012). FBXO11 targets BCL6 for degradation and is inactivated in diffuse large B-cell lymphomas. Nature 481, 90–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufva O, Polonen P, Bruck O, Keranen MAI, Klievink J, Mehtonen J, Huuhtanen J, Kumar A, Malani D, Siitonen S, et al. (2020). Immunogenomic Landscape of Hematological Malignancies. Cancer Cell 38, 380–399 e313. [DOI] [PubMed] [Google Scholar]
- Ennishi D, Takata K, Beguelin W, Duns G, Mottok A, Farinha P, Bashashati A, Saberi S, Boyle M, Meissner B, et al. (2019). Molecular and Genetic Characterization of MHC Deficiency Identifies EZH2 as Therapeutic Target for Enhancing Immune Recognition. Cancer Discov 9, 546–563. [DOI] [PubMed] [Google Scholar]
- Erlich RL, Jia X, Anderson S, Banks E, Gao X, Carrington M, Gupta N, DePristo MA, Henn MR, Lennon NJ, et al. (2011). Next-generation sequencing for HLA typing of class I loci. BMC Genomics 12, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteban F, Concha A, Delgado M, Perez-Ayala M, Ruiz-Cabello F, and Garrido F (1990). Lack of MHC class I antigens and tumour aggressiveness of the squamous cell carcinoma of the larynx. Br J Cancer 62, 1047–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewels P, Magnusson M, Lundin S, and Kaller M (2016). MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandin V, Gutierrez GJ, Brill LM, Varsano T, Feng Y, Aza-Blanc P, Au Q, McLaughlan S, Ferreira TA, Alain T, et al. (2013). Degradation of newly synthesized polypeptides by ribosome-associated RACK1/c-Jun N-terminal kinase/eukaryotic elongation factor 1A2 complex. Mol Cell Biol 33, 2510–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. (2013). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6, pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green MR, Kihira S, Liu CL, Nair RV, Salari R, Gentles AJ, Irish J, Stehr H, Vicente-Duenas C, Romero-Camarero I, et al. (2015). Mutations in early follicular lymphoma progenitors are associated with suppressed antigen presentation. Proc Natl Acad Sci U S A 112, E1116–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harel M, Ortenberg R, Varanasi SK, Mangalhara KC, Mardamshina M, Markovits E, Baruch EN, Tripple V, Arama-Chayoth M, Greenberg E, et al. (2019). Proteomics of Melanoma Response to Immunotherapy Reveals Mitochondrial Dependence. Cell 179, 236–250 e218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidelberger C, Chaudhuri NK, Danneberg P, Mooren D, Griesbach L, Duschinsky R, Schnitzer RJ, Pleven E, and Scheiner J (1957). Fluorinated pyrimidines, a new class of tumour-inhibitory compounds. Nature 179, 663–666. [DOI] [PubMed] [Google Scholar]
- Hellstrom KE (1960). Studies on isoantigenic variation in mouse lymphomas. J Natl Cancer Inst 25, 237–269. [PubMed] [Google Scholar]
- Hermann C, van Hateren A, Trautwein N, Neerincx A, Duriez PJ, Stevanovic S, Trowsdale J, Deane JE, Elliott T, and Boyle LH (2015). TAPBPR alters MHC class I peptide presentation by functioning as a peptide exchange catalyst. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoadley KA, Yau C, Hinoue T, Wolf DM, Lazar AJ, Drill E, Shen R, Taylor AM, Cherniack AD, Thorsson V, et al. (2018). Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 173, 291–304 e296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotokezaka Y, Tobben U, Hotokezaka H, Van Leyen K, Beatrix B, Smith DH, Nakamura T, and Wiedmann M (2002). Interaction of the eukaryotic elongation factor 1A with newly synthesized polypeptides. J Biol Chem 277, 18545–18551. [DOI] [PubMed] [Google Scholar]
- Italiano A, Soria JC, Toulmonde M, Michot JM, Lucchesi C, Varga A, Coindre JM, Blakemore SJ, Clawson A, Suttle B, et al. (2018). Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: a first-in-human, open-label, phase 1 study. Lancet Oncol 19, 649–659. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Ortega-Molina A, Geng H, Ying HY, Hatzi K, Parsa S, McNally D, Wang L, Doane AS, Agirre X, et al. (2017). CREBBP Inactivation Promotes the Development of HDAC3-Dependent Lymphomas. Cancer Discov 7, 38–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KH, and Roberts CW (2016). Targeting EZH2 in cancer. Nat Med 22, 128–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirmizis A, Bartley SM, Kuzmichev A, Margueron R, Reinberg D, Green R, and Farnham PJ (2004). Silencing of human polycomb target genes is associated with methylation of histone H3 Lys 27. Genes Dev 18, 1592–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraan W, Horlings HM, van Keimpema M, Schilder-Tol EJ, Oud ME, Scheepstra C, Kluin PM, Kersten MJ, Spaargaren M, and Pals ST (2013). High prevalence of oncogenic MYD88 and CD79B mutations in diffuse large B-cell lymphomas presenting at immune-privileged sites. Blood Cancer J 3, e139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam LT, Wright G, Davis RE, Lenz G, Farinha P, Dang L, Chan JW, Rosenwald A, Gascoyne RD, and Staudt LM (2008). Cooperative signaling through the signal transducer and activator of transcription 3 and nuclear factor-{kappa}B pathways in subtypes of diffuse large B-cell lymphoma. Blood 111, 3701–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laumont CM, Vincent K, Hesnard L, Audemard E, Bonneil E, Laverdure JP, Gendron P, Courcelles M, Hardy MP, Cote C, et al. (2018). Noncoding regions are the main source of targetable tumor-specific antigens. Sci Transl Med 10. [DOI] [PubMed] [Google Scholar]
- Lesokhin AM, Ansell SM, Armand P, Scott EC, Halwani A, Gutierrez M, Millenson MM, Cohen AD, Schuster SJ, Lebovic D, et al. (2016). Nivolumab in Patients With Relapsed or Refractory Hematologic Malignancy: Preliminary Results of a Phase Ib Study. J Clin Oncol 34, 2698–2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y, Vogel JL, Arbuckle JH, Rai G, Jadhav A, Simeonov A, Maloney DJ, and Kristie TM (2013). Targeting the JMJD2 histone demethylases to epigenetically control herpesvirus infection and reactivation from latency. Sci Transl Med 5, 167ra165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liepe J, Sidney J, Lorenz FKM, Sette A, and Mishto M (2019). Mapping the MHC Class I-Spliced Immunopeptidome of Cancer Cells. Cancer Immunol Res 7, 62–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L, Zhu F, Zhang M, Li Y, Drennan AC, Kimpara S, Rumball I, Selzer C, Cameron H, Kellicut A, et al. (2018). Gene regulation and suppression of type I interferon signaling by STAT3 in diffuse large B cell lymphoma. Proc Natl Acad Sci U S A 115, E498–E505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludigs K, Seguin-Estevez Q, Lemeille S, Ferrero I, Rota G, Chelbi S, Mattmann C, MacDonald HR, Reith W, and Guarda G (2015). NLRC5 exclusively transactivates MHC class I and related genes through a distinctive SXY module. PLoS Genet 11, e1005088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lue JK, and Amengual JE (2018). Emerging EZH2 Inhibitors and Their Application in Lymphoma. Curr Hematol Malig Rep 13, 369–382. [DOI] [PubMed] [Google Scholar]
- Mayor A, Martinon F, De Smedt T, Petrilli V, and Tschopp J (2007). A crucial function of SGT1 and HSP90 in inflammasome activity links mammalian and plant innate immune responses. Nat Immunol 8, 497–503. [DOI] [PubMed] [Google Scholar]
- McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, Liu Y, Graves AP, Della Pietra A 3rd, Diaz E, et al. (2012). EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 492, 108–112. [DOI] [PubMed] [Google Scholar]
- McGranahan N, Rosenthal R, Hiley CT, Rowan AJ, Watkins TBK, Wilson GA, Birkbak NJ, Veeriah S, Van Loo P, Herrero J, et al. (2017). Allele-Specific HLA Loss and Immune Escape in Lung Cancer Evolution. Cell 171, 1259–1271 e1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner TB, Liu YJ, Lee KH, Li A, Biswas A, van Eggermond MC, van den Elsen PJ, and Kobayashi KS (2012). NLRC5 cooperates with the RFX transcription factor complex to induce MHC class I gene expression. J Immunol 188, 4951–4958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijland M, Veenstra RN, Visser L, Xu C, Kushekhar K, van Imhoff GW, Kluin PM, van den Berg A, and Diepstra A (2017). HLA dependent immune escape mechanisms in B-cell lymphomas: Implications for immune checkpoint inhibitor therapy? Oncoimmunology 6, e1295202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissen MD, Kusakabe M, Wang X, Simkin G, Gracias D, Tyshchenko K, Hill A, Meskas J, Hung S, Chavez EA, et al. (2019). Single Cell Phenotypic Profiling of 27 DLBCL Cases Reveals Marked Intertumoral and Intratumoral Heterogeneity. Cytometry A. [DOI] [PubMed] [Google Scholar]
- Novosylna O, Jurewicz E, Pydiura N, Goral A, Filipek A, Negrutskii B, and El’skaya A (2015). Translation elongation factor eEF1A1 is a novel partner of a multifunctional protein Sgt1. Biochimie 119, 137–145. [DOI] [PubMed] [Google Scholar]
- Patel SJ, Sanjana NE, Kishton RJ, Eidizadeh A, Vodnala SK, Cam M, Gartner JJ, Jia L, Steinberg SM, Yamamoto TN, et al. (2017). Identification of essential genes for cancer immunotherapy. Nature 548, 537–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelan JD, Young RM, Webster DE, Roulland S, Wright GW, Kasbekar M, Shaffer AL 3rd, Ceribelli M, Wang JQ, Schmitz R, et al. (2018). A multiprotein supercomplex controlling oncogenic signalling in lymphoma. Nature 560, 387–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsuran V, Kulkarni S, O’Huigin C, Yuki Y, Augusto DG, Gao X, and Carrington M (2015). Epigenetic regulation of differential HLA-A allelic expression levels. Hum Mol Genet 24, 4268–4275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read JA, Koff JL, Nastoupil LJ, Williams JN, Cohen JB, and Flowers CR (2014). Evaluating cell-of-origin subtype methods for predicting diffuse large B-cell lymphoma survival: a meta-analysis of gene expression profiling and immunohistochemistry algorithms. Clin Lymphoma Myeloma Leuk 14, 460–467 e462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins PF, Kassim SH, Tran TL, Crystal JS, Morgan RA, Feldman SA, Yang JC, Dudley ME, Wunderlich JR, Sherry RM, et al. (2015). A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: long-term follow-up and correlates with response. Clin Cancer Res 21, 1019–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson J, Halliwell JA, Hayhurst JD, Flicek P, Parham P, and Marsh SG (2015). The IPD and IMGT/HLA database: allele variant databases. Nucleic Acids Res 43, D423–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose MG, Farrell MP, and Schmitz JC (2002). Thymidylate synthase: a critical target for cancer chemotherapy. Clin Colorectal Cancer 1, 220–229. [DOI] [PubMed] [Google Scholar]
- Rosenwald A, Wright G, Chan WC, Connors JM, Campo E, Fisher RI, Gascoyne RD, Muller-Hermelink HK, Smeland EB, Giltnane JM, et al. (2002). The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med 346, 1937–1947. [DOI] [PubMed] [Google Scholar]
- Sade-Feldman M, Jiao YJ, Chen JH, Rooney MS, Barzily-Rokni M, Eliane JP, Bjorgaard SL, Hammond MR, Vitzthum H, Blackmon SM, et al. (2017). Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat Commun 8, 1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaer DA, Geeganage S, Amaladas N, Lu ZH, Rasmussen ER, Sonyi A, Chin D, Capen A, Li Y, Meyer CM, et al. (2019). The Folate Pathway Inhibitor Pemetrexed Pleiotropically Enhances Effects of Cancer Immunotherapy. Clin Cancer Res 25, 7175–7188. [DOI] [PubMed] [Google Scholar]
- Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, Roulland S, Kasbekar M, Young RM, Shaffer AL, et al. (2018). Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N Engl J Med 378, 1396–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Han Y, Yang J, Liu P, He X, Zhang C, Zhou S, Zhou L, Qin Y, Song Y, et al. (2019). Clinical features and outcomes of diffuse large B-cell lymphoma based on nodal or extranodal primary sites of origin: Analysis of 1,085 WHO classified cases in a single institution in China. Chin J Cancer Res 31, 152–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sledzinska A, Vila de Mucha M, Bergerhoff K, Hotblack A, Demane DF, Ghorani E, Akarca AU, Marzolini MAV, Solomon I, Vargas FA, et al. (2020). Regulatory T Cells Restrain Interleukin-2- and Blimp-1-Dependent Acquisition of Cytotoxic Function by CD4(+) T Cells. Immunity 52, 151–166 e156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steimle V, Durand B, Barras E, Zufferey M, Hadam MR, Mach B, and Reith W (1995). A novel DNA-binding regulatory factor is mutated in primary MHC class II deficiency (bare lymphocyte syndrome). Genes Dev 9, 1021–1032. [DOI] [PubMed] [Google Scholar]
- Stewart SA, Dykxhoorn DM, Palliser D, Mizuno H, Yu EY, An DS, Sabatini DM, Chen IS, Hahn WC, Sharp PA, et al. (2003). Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 9, 493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, et al. (2019). STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res 47, D607–D613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran E, Robbins PF, Lu YC, Prickett TD, Gartner JJ, Jia L, Pasetto A, Zheng Z, Ray S, Groh EM, et al. (2016). T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N Engl J Med 375, 2255–2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Eynde B, Peeters O, De Backer O, Gaugler B, Lucas S, and Boon T (1995). A new family of genes coding for an antigen recognized by autologous cytolytic T lymphocytes on a human melanoma. J Exp Med 182, 689–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Bruggen P, Traversari C, Chomez P, Lurquin C, De Plaen E, Van den Eynde B, Knuth A, and Boon T (1991). A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science 254, 1643–1647. [DOI] [PubMed] [Google Scholar]
- Wahba AJ, and Friedkin M (1962). The enzymatic synthesis of thymidylate. I. Early steps in the purification of thymidylate synthetase of Escherichia coli. J Biol Chem 237, 3794–3801. [PubMed] [Google Scholar]
- Wang SS, Carrington M, Berndt SI, Slager SL, Bracci PM, Voutsinas J, Cerhan JR, Smedby KE, Hjalgrim H, Vijai J, et al. (2018). HLA Class I and II Diversity Contributes to the Etiologic Heterogeneity of Non-Hodgkin Lymphoma Subtypes. Cancer Res 78, 4086–4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster DE, Roulland S, and Phelan JD (2019). Protocols for CRISPR-Cas9 Screening in Lymphoma Cell Lines. Methods Mol Biol 1956, 337–350. [DOI] [PubMed] [Google Scholar]
- Wickham H, Averick M, Bryan J, Chang W, McGown LDA, Franois R, Grolemund G, Hayes A, Henry L, Hester J, et al. (2019). Welcome to the Tidyverse. The Journal of Open Source Software 4. [Google Scholar]
- Wright GW, Huang DW, Phelan JD, Coulibaly ZA, Roulland S, Young RM, Wang JQ, Schmitz R, Morin RD, Tang J, et al. (2020). A Probabilistic Classification Tool for Genetic Subtypes of Diffuse Large B Cell Lymphoma with Therapeutic Implications. Cancer Cell 37, 551–568 e514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav M, Jhunjhunwala S, Phung QT, Lupardus P, Tanguay J, Bumbaca S, Franci C, Cheung TK, Fritsche J, Weinschenk T, et al. (2014). Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 515, 572–576. [DOI] [PubMed] [Google Scholar]
- Yang Y, Shaffer AL 3rd, Emre NC, Ceribelli M, Zhang M, Wright G, Xiao W, Powell J, Platig J, Kohlhammer H, et al. (2012). Exploiting synthetic lethality for the therapy of ABC diffuse large B cell lymphoma. Cancer Cell 21, 723–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap DB, Chu J, Berg T, Schapira M, Cheng SW, Moradian A, Morin RD, Mungall AJ, Meissner B, Boyle M, et al. (2011). Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood 117, 2451–2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yewdell JW (2011). DRiPs solidify: progress in understanding endogenous MHC class I antigen processing. Trends Immunol 32, 548–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Kadota Y, Prodromou C, Shirasu K, and Pearl LH (2010). Structural basis for assembly of Hsp90-Sgt1-CHORD protein complexes: implications for chaperoning of NLR innate immunity receptors. Mol Cell 39, 269–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Mudianto T, Ma X, Riley R, and Uppaluri R (2020). Targeting EZH2 Enhances Antigen Presentation, Antitumor Immunity, and Circumvents Anti-PD-1 Resistance in Head and Neck Cancer. Clin Cancer Res 26, 290–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zingg D, Arenas-Ramirez N, Sahin D, Rosalia RA, Antunes AT, Haeusel J, Sommer L, and Boyman O (2017). The Histone Methyltransferase Ezh2 Controls Mechanisms of Adaptive Resistance to Tumor Immunotherapy. Cell Rep 20, 854–867. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table S1. CRISPR screen normalized counts, segregation scores, and STARS analyses (related to Figure 1).
Supplemental Table S2. Top predicted hits from CRISPR screens (related to Figure 1).
Supplemental Table S3. Validations of ~200 genetic ablations in four DLBCLs (related to Figures 2–3).
Supplemental Table S4. HLA typing of common DLBCL tumor lines (related to STAR Methods).
Supplemental Table S5. Targeted drug screen to identify enhancers of antigen presentation (related to Figure 7).
Supplemental Table S6. Summary of statistical analyses (related to STAR Methods).
Supplemental Table S7. Oligonucleotide sequences (related to STAR Methods).
Data Availability Statement
Normalized counts of sgRNA library sequencing are available in Table S1. Bulk RNAseq of DLBCL cell lines generated during this study is available at Gene Expression Omnibus (GEO) accession GSE160608. Bulk RNAseq of DLBCLs treated with the EZH2 inhibitor GSK126 generated during this study is available at GEO accession GSE160609. The superseries of data is available at GEO GSE160610. All primary genetic data is available through the NIH dbGAP system under accession numbers phs001444, phs001184, and phs000178 (https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs001444.v1.p1). IRF4 ChIP-seq datasets of lymphoblastoid GM12878 were obtained from GEO accession GSM803390.