

Abstract
KNOTTED-like homeodomain (KNOX) genes are transcriptional regulators that play an important role in morphogenesis. In the present study, a comparative analysis was performed to investigate the molecular evolution of the characteristics of the KNOX gene family in 10 different plant species. We identified 129 KNOX gene family members, which were categorized into two subfamilies based on multiple sequence alignment and phylogenetic tree reconstruction. Several segmental duplication pairs were found, indicating that different species share a common expansion model. Functional divergence analysis identified the 15 and 52 amino acid sites with significant changes in evolutionary rates and amino acid physicochemical properties as functional divergence sites. Additional selection analysis showed that 14 amino acid sites underwent positive selection during evolution, and two groups of co-evolutionary amino acid sites were identified by Coevolution Analysis using Protein Sequences software. These sites could play critical roles in the molecular evolution of the KNOX gene family in these species. In addition, the expression profiles of KNOX duplicated genes demonstrated functional divergence. Taken together, these results provide novel insights into the structural and functional evolution of the KNOX gene family.
Keywords: functional divergence, KNOX, phylogenetic tree, positive selection, segmental duplication
1. Introduction
Homeotic genes are the main genes that regulate the development of organisms. They represent a class of transcription factors (TFs) containing a highly conserved homeobox of 183 bp, which encodes a typical DNA-binding domain of 60 amino acids, also known as a homeodomain (HD). The first cloned homeobox gene was from Drosophila [1]. The highly homologous sequence of Knotted-1 (Kn1) to animal homeoboxes was detected in maize by transposon tagging [2]. Homeobox genes are widely found in eukaryotes [3]. Genes encoding homologous proteins are classified into two classes: three amino acid length extension (TALE) and non-TALE [4]. Four types of TALE genes have been identified in animals: MEIS, IRO (Iroquois), TGIF, and PBC. Furthermore, according to differences in characteristic domains and functions, there are two types in plants: KNOX (KNOTTED-like homeobox) and BELL (BEL-Like) [5].
KNOX proteins can form heterodimers with BELL in the TALE superclass [3]. KNOX includes four domains: a C-terminal homeodomain (HD), KNOX1 and KNOX2 at the conserved N-terminal region, and an ELK domain upstream of the homologous domain. Owing to the similarity between the MEIS and KNOX family structures, the KNOX1 and KNOX2 domains are also known as the MEINOX domain, and there are three additional amino acids (P–Y–P) between the first and second helices in their homeobox [5,6]. In addition, the ELK domain, which can function as a nuclear localization signal (NLS), spans ∼21 amino acids rich in glutamic acid (Glu, E), leucine (Leu, L), and lysine (Lys, K) [7]. Between the ELK and KNOX2 domains is the GSE domain, which is rich in proline (Pro, P), glutamic acid (Glu, E), serine (Ser, S), and threonine (Thr, T). The residue sequence (PEST sequence) regulates protein stability and degrades its encoded protein through the ubiquitin degradation pathway [8]. Furthermore, Kerstetter et al. [9] classified the KNOX gene family into class I and class II KNOX subfamilies based on structural features, phylogenetic relationships, and expression patterns.
KNOX genes have been isolated from many plants, such as Nicotiana tabacum [10], Arabidopsis thaliana [11,12], Solanum lycopersicum [13], Medicago truncatula [14], and Physcomitrella patens [15]. In most monocots, the KNOX1 gene is expressed only in shoot apical meristems (SAM) and not in the primordium. In compound-leaf species, KNOX1 are expressed in both SAM and the leaf primordium [16], showing that they may play a significant role in maintaining diversity in leaf morphology [3]. The KNOX2 gene regulates the morphological transformation of haploid to diploid cells in terrestrial plants [17].
In A. thaliana, the class I subfamily includes STM (SHOOT MERISTEMLESS), KNAT1, KNAT2, and KNAT6 [18], and the class II subfamily includes KNAT3, KNAT4, KNAT5, and KNAT7 [3,19], which are widely distributed. STM and KNAT1 are used to establish and maintain SAM. Similarly, KNAT6 has previously been shown to function in the maintenance of borders during SAM and embryogenesis [20]. Furthermore, KNAT1 promotes inflorescence development, while KNAT2 regulates flower type [8,18,21]. The class I gene STM regulates the development of the plant meristem in Arabidopsis [8], and regulation of gene expression leads to the petal spurs rapidly evolving in Antirrhinum [22]. In summary, the class I KNOX gene is involved in the morphogenesis of lateral organs and maintains the function of SAM and the diversity of leaf morphology [3,22].
Meanwhile, the KNOX class homeobox genes Oskn2 and Oskn3 in rice are both expressed in the tissues of the SAM and participate in the regulation of SAM formation. For instance, class II KNOX genes, such as KNAT3, KNAT4, and KNAT5, contribute to the differentiation of tissues in organs in Arabidopsis [9,23,24]. The regulatory network within which KNAT7 functions contributes to the negative regulation of Arabidopsis and Populus secondary cell wall biosynthesis [24,25]. The class II subfamily lacks phenotypic due to mutations; however, there have been relatively few previous studies. In brief, KNOX genes are involved in the growth and development of different tissues and organs in different species [26,27,28]. Plants must constantly adjust their physiological processes to adapt to changes in the external environment [29]. TFs are considered to be key targets for studying the molecular mechanisms of abiotic stress response because they, either alone or collectively, regulate the expression of many downstream target genes [30].
In the present study, we identified KNOX genes in different species and classified them by reconstructing phylogenetic trees. Then, we identified the critical amino acid sites responsible for functional divergence, positive selection, and co-evolution. Together with expression profiles, we present some insights into the molecular evolution of the KNOX gene family, which can be useful for future research on the functions of these genes.
2. Materials and methods
2.1. Identification of plant KNOX gene family
Genes from the plant KNOX gene family were identified from 10 species that represented monocotyledonous, dicotyledonous, and bryophyte plants. The KNOX gene family members from the Arabidopsis genome were obtained from the TAIR database (http://www.arabidopsis.org/) and then BLAST searched as seed sequences in the Phytozome database (http://www.phytozome.org) to obtain homologous sequences from nine other species (Glycine max, Populus trichocarpa, Gossypium raimondii, Solanum lycopersicum, Oryza sativa, Brachypodium distachyon, Sorghum bicolor, Zea mays, and Physcomitrella patens). If the E value of the sequence was ≤1 × 10−5, then it was listed as a candidate sequence. The Pfam (http://pfam.xfam.org) and SMART (http://smart.embl-heidelberg.de/) online tools were used to determine whether the candidate sequence contained the KNOX1, KNOX2, ELK, and HD to ensure that the sequence domain was intact and used for the next analysis. In addition, coding sequences, protein sequences, and genomic sequences of KNOX family members were downloaded from the Phytozome database. The physicochemical properties of the KNOX gene family were obtained from the ExPASy database (https://www.expasy.org/), including the amino acid number, isoelectric point (PI), and molecular weight (MW) of the protein [31].
2.2. Phylogenetic tree construction
Multiple sequence alignment of the sequences from KNOX family members was performed with the MUSCLE program [32,33]. Three methods were used to construct the phylogenetic tree: Bayesian phylogenetic trees in MrBayes 3.2.5 [34] and neighbor-joining (NJ) and maximum-likelihood (ML) trees in MEGA 7.0 [35]. The reliability of interior branches was assessed with 1,000 bootstrap samples [35].
2.3. Exon–intron structure and motif analysis
The exon–intron structure was analyzed using the online tool GSDS (http://gsds.cbi.pku.edu.cn/) with the coding sequences (CDS) and genomic sequences of KNOX family members [36]. Conserved motifs of KNOX family members were identified using the online tool MEME (http://meme-suite.org/tools/meme) [37]. The maximum number of motifs = 10, and the remaining parameters were set to the default settings.
2.4. Duplication event analysis
Tandem duplication and segmental duplication were used to determine the main amplification methods of the KNOX gene family. The synonymous substitution rates (K s) of gene pairs produced by segmental repeat events were identified using the Plant Genome Duplication Database (http://chibba.agtec.uga.edu/duplication) [38]. To avoid the risk of saturation and improve the accuracy of the results, the K s value greater than 1 and anchors less than 3, the approximate age of the segmental duplication event was estimated by the following formula: T = K s/2λ [39]. The synonymous substitutions per year (λ) were 1.5 × 10−8 for Arabidopsis [40], 6.1 × 10−9 for Glycine max, 6.5 × 10−9 for Brachypodium distachyon [41], 9.1 × 10−7 for Populus trichocarpa [42], 1.5 × 10−8 for Gossypium raimondii [43], 6.5 × 10−9 for Oryza sativa, 6.1 × 10−9–6.5 × 10−9 for Sorghum bicolor [44], and 6.5 × 10−9 for Zea mays [45].
2.5. Functional divergence analysis
DIVERGE 3.0 was used to detect the functional divergence between clusters of the KNOX gene family [46]. The extent of divergence can be measured using the type I (site-specific altered selective constraints) and type II (radical shift in amino acid physiochemical properties) functional divergence coefficients (θI and θII) between subfamilies [47,48,49]. Moreover, Bayesian posterior probability (Q k) can detect specific amino acid sites where functional divergence has occurred. In our study, the threshold of Q k was set to 0.9.
2.6. Positive selection analysis
Positive selection was investigated using the maximum likelihood approach in the CODEML procedure in PAML [50,51]. Site models, including null models (M0 and M3) and alternative hypothesis models (M7 and M8), were implemented in this program. A detailed description of the positive selection site test method can be found in Wang et al. [52].
2.7. Coevolution analysis
Coevolution analysis using protein sequences (CAPS) was performed with PERL-based software [53]. A detailed description of the coevolution sites test method can be found in Song et al. [54].
2.8. Protein structure prediction
The 3D structure of the KNOX protein was predicted using online software PHYRE2 (http://www.sbg.bio.ic.ac.uk/phyre2/html/page.cgi?id=index) [55]. We used protein sequences to construct the 3D structure of KNOX family member AT4G08150 and then to screen important amino acids sites that were labeled on the 3D structure.
2.9. Expression analysis of KNOX genes
RNA-Seq data were introduced to further analyze the expression of plant KNOX genes. The Arabidopsis eFP Browser (http://bar.utoronto.ca/efp/cgi-bin/efpWeb.cgi) tool and the rice eFP Browser (http://www.bar.utoronto.ca/efprice/cgi-bin/efpWeb.cgi) tool were used to search data from Arabidopsis and rice, respectively. A heat map was generated using the TBtools program [56].
3. Results
3.1. Identification of KNOX gene family
Nine KNOX genes of Arabidopsis were obtained from the TAIR database. The gene AT1G14760 was not analyzed because it contains only two KNOX domains, which belong to the KNATM class, that were not included in this analysis. Furthermore, 121 candidate KNOX gene sequences from nine other species were obtained through BLAST searches in the Phytozome database (Table A1). The Pfam [57] and SMART [58] online tools were used to ensure the wholeness of the domains (KNOX1, KNOX2, ELK, and HD). The results showed that the domains were complete, and a total of 129 typical KNOX family members were identified. The relevant information groups, gene IDs, protein lengths, isoelectric point of the deduced polypeptides, and molecular weight of the KNOX genes are listed in Table A2. The average number of amino acid residues ranged from 215 to 636 (average 345), and the isoelectric point of the KNOX gene family ranged from 5.12 to 9.53 (average 6.62). Except for GRMZM2G433591, all other members were weakly acidic. The average molecular mass of the KNOX family ranged from 28,353.8 to 72,405.5 Da (average 38,715.4 Da).
3.2. Phylogenetic analysis of the KNOX gene family
To investigate the phylogenetic relationships of KNOX genes in 10 plant species (Arabidopsis thaliana, Glycine max, Populus trichocarpa, Gossypium raimondii, Solanum lycopersicum, Oryza sativa, Brachypodium distachyon, Sorghum bicolor, Zea mays and Physcomitrella patens), we used the MUSCLE software [32,33] to perform multiple sequence alignments of 129 protein sequences and then used three methods to construct phylogenetic trees: neighbor-joining (Figure A1), maximum-likelihood (data not displayed), and Bayesian inference [34,35] (Figure 1a). According to the results, the topology of the three methods was consistent; the subsequent study uses the Bayesian phylogenetic tree. The 129 homeobox genes from 10 species, including monocots and dicots and P. patens, were divided into two subfamilies: class I and class II [59]. The sequence analysis and expression pattern analysis supported this result, which was also consistent with other previous research [9]. The class I subfamily includes 80 members, while the class II subfamily includes 49 members. This difference may be due to the method of gene amplification, which leads to the difference in the number of subfamily members.
Figure 1.

Phylogenetic relationship, exon–intron structure, and motif structure of plant KNOX gene family members. (a) The rooted Bayesian phylogenetic tree. The branches of the two different colors represent different subfamilies: blue represents the class I KNOX gene and red represents the class II KNOX gene. (b) Exon and intron structure of KNOX genes. Yellow boxes, exons; lines, introns. The lengths of boxes and lines are scaled according to gene length. (c) MEME motif structures. Numbers and different colors were used to represent conservative motifs.
Figure 1b shows the exon–intron structure of KNOX gene family members analyzed by the GSDS online system [60]. Most members of the subfamily contained five exons. The number of exons was conservative within the subfamily, and there was no significant difference in the number of exons in the same subfamily. Of the class I members, 78.75% contained five exons, whereas 13 members contained four exons (Figure 1b), and 91.84% of the members of the class II subfamily contained five exons. The number of exons in P. patens was significantly different. For example, Ppls154_83V6 contained 10 exons, indicating that exons may have been lost during evolution to adapt to the environment.
The conserved domains of the KNOX gene family were analyzed using the MEME online tool [37], and 10 conserved motifs were obtained that were named motif1–motif10. The structure and order of these motifs in the two subfamilies are shown in Figure 1c. Motifs 8, 4, 3, 2, and 1 were widely distributed among species in that order, except for P. patens, which did not contain motif 8. However, there were also slight differences between different members of the same plant. Most members of the class I subfamily contained motifs 8, 4, 3, 6, 2, and 1 arranged in that order, and most members of the class II subfamily contained motifs 8, 4, 10, 3, 5, 9, 7, 2, and 1 arranged in that order. Differences in motifs may be an important cause of functional divergence in the two subfamilies.
3.3. Expansion analysis of plant KNOX gene family
Gene duplication is a major driving force of adaptive evolution in species [61]. In this study, we investigated the gene duplication mode of the KNOX gene family and mainly studied tandem duplication and segmental duplication. Tandem duplication gene pairs were detected in only three species; all of which were members of the monocotyledonous of the class I subfamily. Furthermore, segmental duplication genes were clearly detected in eight species (Table A3). Of the segmental duplication gene pairs, 93% were detected in dicotyledons, whereas only four KNOX segmental duplication gene pairs were detected in monocotyledons. We found that the class I subfamily contained both segmental and tandem duplication genes, which may explain the higher number of genes in class I than class II. In dicotyledonous plants, genes are mainly amplified through segmental duplication; in monocotyledonous plants, tandem duplication and segmental duplication coexist. To estimate the approximate time of segmental duplication events, the base synonymous mutation rate (K s-values) was used [38] (Table A3). The results showed that the segmental duplication events of most species were consistent with the large-scale duplication events, and segmental duplications were preserved after genome duplication.
3.4. Functional divergence analysis of KNOX gene family
To determine the difference in the evolutionary rates and physicochemical properties of amino acid sites, the type I and II functional divergence of the two subfamilies was estimated using DIVERGE [48,62]. Key amino acid sites for functional divergence were determined based on posterior probability (Q k). The results in Table 1 show that the divergence coefficients of type I of the two subfamilies were significant (θI = 0.442 ± 0.052; LRT = 71.696; P < 0.01), indicating that the amino acid sites between the two subfamilies have different evolutionary rates. Meanwhile, the type II coefficients of the two subfamilies were also significant (θII = 0.106 ± 0.186; P < 0.01), indicating the possible presence of type II divergence sites during evolution between the two subfamilies. Furthermore, the amino acid sites were analyzed between groups under stringent conditions (Q k > 0.9) to confirm the amino acid sites where functional divergence had occurred [48].
Table 1.
Functional divergence between subfamilies of the plant KNOX gene family
| Group1 | Group2 | Type I | Type II | |||
|---|---|---|---|---|---|---|
| θI ± s.e. | LRT | Q k > 0.9 | θII ± s.e. | Q k > 0.9 | ||
| Class I | Class II | 0.442 ± 0.052 | 71.696** | 15 | 0.106 ± 0.186 | 52 |
Note: θI and θII, the coefficients of type-I and type-II functional divergence between class I and class II. LRT: likelihood ratio test. **P < 0.01, highly significant. Q k: posterior probability.
The results identified 15 sites with a high probability of being associated with type I functional divergence. There were 52 type II functional divergence sites (Table 2), more than twice the sites identified for type I, of which eight points (140A, 155Q, 170A, 223I, 224R, 283H, 286K, and 345Q) occurred in both type I and type II functional divergence, indicating that they underwent changes in evolutionary rates and physicochemical properties simultaneously. Therefore, these sites are expected to play an important role in functional differences during evolution. Apart from this, the number of type I and type II functional divergence sites was different, and more critical amino acid sites were identified as type II functional divergence within each subfamily. Hence, the functional divergence between genes of the two subfamilies was attributed primarily to rapid changes in amino acid physiochemical properties, followed by a shift in evolutionary rates.
Table 2.
Functional divergence sites between subfamilies of the plant KNOX gene family
| Amino acid sites | |
|---|---|
| Type I | 138I, 140A, 155Q, 164U, 170A, 198M, 213T, 222F, 223I, 224R, 229Q, 283H, 286K, 345Q, 362P |
| Type II | 133A, 137K, 140A, 145S, 146T, 151Y, 153D, 155Q, 158G, 159A, 161P, 163V, 166R, 169A, 170A, 171R, 175E, 196Q, 211E, 214R, 215P, 217Q, 220M, 221E, 223I, 224R, 225R, 253S, 256E, 257E, 278R, 282N, 283H, 285L, 286K, 287K, 300S, 305K, 310K, 312A, 313R, 317L, 318T, 322L, 324Y, 332S, 336A, 340S, 344D, 345Q, 359H, 372D |
Note: amino acid sites in bold font indicate that they were responsible for both type I and type II functional divergence.
3.5. Positive selection and co-evolution in KNOX gene family
The site model was selected to determine the selection pressure on different amino acid codon sites [51]. The results are shown in Table 3. The selection pressure was significantly different between M0 (one-ratio) and M3 (discrete; P < 0.01). The M3 model was better than the M0 model, indicating that different sites experience different selection pressures. The 2Δ ln L of M7 (beta) vs. M8 (beta & ω > 1) was 5795.66, the likelihood ratio test result was extremely significant (df = 2, P < 0.01), and the M8 model had an ω value of 2.63459, much >1, indicating that 14 amino acid positions were strongly affected by positive selection. Table 3 shows the positive selection sites with a posterior probability >95%. Among them, 143H, 171R, and 228S were significant positive selection sites, and 130D, 133A, 134M, 140A, 149Q, 165D, 172Q, 232M, 315K, 318T, and 322L were extremely significant positive selection sites.
Table 3.
Positive selection analysis among KNOX genes using site-specific models
| Model | InLa | 2Δl | Estimate of parameters | Positively selected sitesb |
|---|---|---|---|---|
| M0 | −25111.04 | 632.77**(M0 vs. M3) | ω = 0.07564 | Not allowed |
| M3 | −24478.27 | p 0 = 041377, ω 0 = 0.00271, p 1 = o.37838, ω 1 = 0.05236, p 2 = 0.20785, ω 2 = 0.20938 | None | |
| M7 | −24434.60 | 5795.66**(M7 vs. M8) | p = 0.63964, q = 6.96441 | Not allowed |
| M8 | −30230.26 | p 0 = 0.99999, p = 0.98088, q = 1.39062, p 1 = 0.00001, ω = 2.63459 | 130D**, 133A**, 134M**, 140A**, 143H*, 149Q**, 165D**, 171R*, 172Q**, 228S*, 232M**, 315K**, 318T**, 322L** |
Note: a log likelihood. b positive selection sites are inferred at posterior probabilities >95%. *P < 0.05; **P < 0.01. Amino acid sites in bold font also found to be involved in the functional divergence.
We used CAPS, which is significantly more sensitive than other methods, to analyze coevolved amino acid residues in the KNOX gene family [53]. We found two groups of coevolved sites: 248S and 249D, and 382L and 383Y. All sites were labeled according to their 3D structure to further investigate their interdependence (Figure 3).
Figure 3.
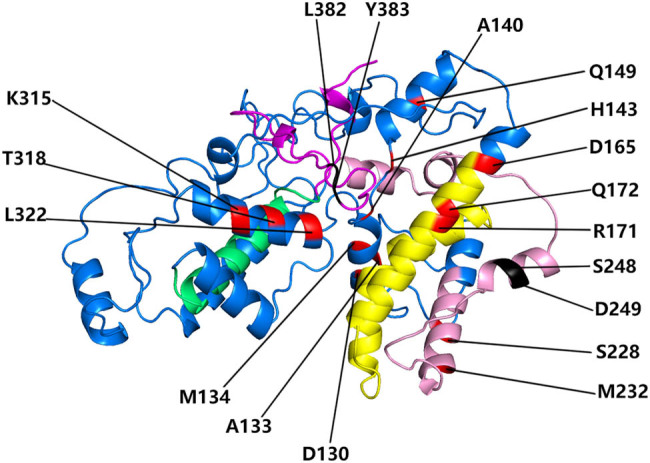
Model building of KNOX protein 3D structure. This figure was produced using Chimera software, and amino acids refer to the AT4G08150 sequence. The HD, ELK, KNOX2, and KNOX1 domains are in yellow, pink, lime green, and magenta, respectively. The red indicates those that had undergone positive selection, and black indicates amino acid sites identified by the co-evolution analysis.
3.6. Three-dimensional structure prediction and critical amino acid site identification of plant KNOX proteins
We used PHYRE2 to predict the 3D structure of the KNOX family member AT4G08150 [55,63]. The critical amino acid sites were displayed by the multiple sequence alignment and 3D structure (Figures 2 and 3). These 14 sites were mainly dispersed on the KNOX1 domain, two positive selection sites were distributed on the KNOX2 domain, and three positive selection sites were distributed on the HD first alpha helix. The results indicated that the KNOX1 domain was more susceptible to positive selection pressure during the evolution of the KNOX gene family. Amino acid position 140A has undergone both functional divergence and positive selection and was located in the KNOX1 domain and at the C-terminus of motif 8 (Figure 2). The two pairs of co-evolutionary sites we detected were marked on the 3D structure (Figure 3). We found that two sets of positive selection sites were located on the C-terminal non-functional domain and were close to each other, showing that they may play a certain role in maintaining the spatial structural stability of KNOX proteins.
Figure 2.

Multiple sequence alignment of Arabidopsis KNOX sequences. Typical domains KNOX1, KNOX2, ELK, and HD of KNOX protein are marked by yellow, purple, blue, and grey shadows, respectively. Motifs 1–4 and motif 8 are indicated with brown arrows above sequences. The amino acid sites of type I and II functional divergences, positive selection, and co-evolution are labeled, respectively, with blue circles, red circles, green triangles, and black boxes. Blue and green frames indicate the first α-helix and PYP loop, respectively.
3.7. Expression analysis of KNOX gene family
To investigate the expression patterns of homologous KNOX genes in subgroups involved in plant growth and development, a heat map was constructed using TBtools (Figures 4 and 5). Members of the same subfamily exhibited similar transcription abundance profiles; however, there were also members that had similar expression profiles but unique phylogenies, such as AT1G62990. It should be noted that AT4G08150 and AT1G70510 belong to the same subfamily (class I). They are expressed at high levels in the pedicels, hypocotyls, and stem but at lower levels in cotyledons and leaves. From the overall expression level, the higher expression levels of AT1G23380 and AT1G62360 in shoots may be related to their indispensability for the formation and maintenance of SAM [29]. These results suggested that members in the same subfamily may play similar roles in the same organization. AT5G11060, AT5G25220, and AT4G32040 are from the class II subfamily. Their overall transcription was richer than that of the class I subfamily. The expression level was higher in senescing leaves. AT4G32040 was highly expressed in dry seeds. AT5G11060 was more highly expressed in leaves and different stages of flowers (Figure 4). LOC_Os02g08544 and LOC_Os06g43860 are also from the class II subfamily, and both of them were highly expressed in all tissues and organs (Figure 5). Three members, LOC_Os02g08544, LOC_Os06g43860, and LOC_Os08g19650, were highly expressed in mature and young leaves, whereas the other members exhibited relatively low expression levels. In addition, the expression of LOC_Os03g51690, LOC_Os07g03770, and LOC_Os05g03884 was higher in the seeds but lower in the leaves, which indicates that sub-functionalization had occurred.
Figure 4.

Expression profiles of Arabidopsis thaliana KNOX genes. The expression level is represented by a color: dark red indicates the highest expression level and dark blue indicates the lowest expression level. Other colors indicate medium levels of expression.
Figure 5.
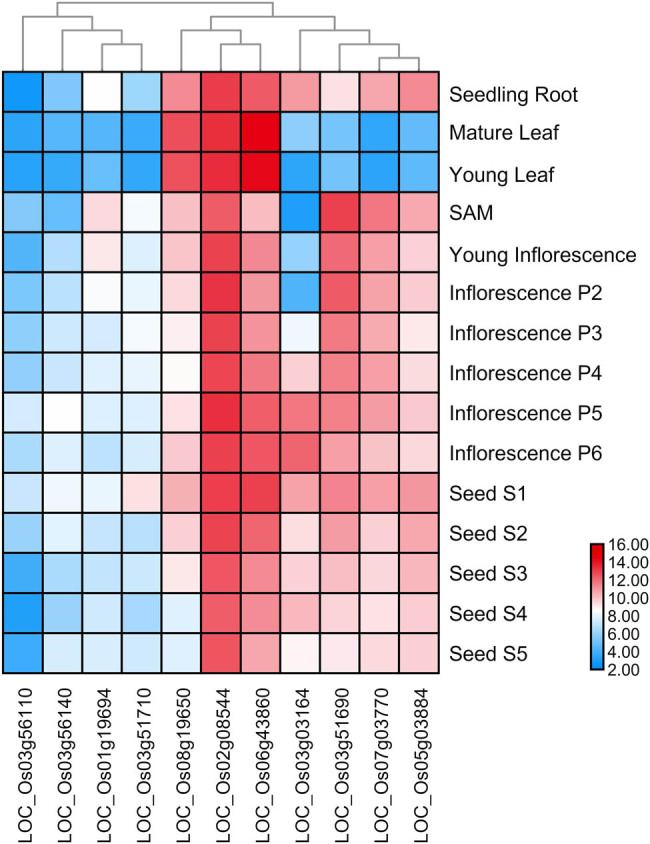
Expression profiles of rice KNOX genes. The expression level is represented by a color: dark red indicates the highest expression level, and dark blue indicates the lowest expression level. Other colors indicate medium levels of expression.
4. Discussion
4.1. Genomic analysis of the KNOX gene family
In the present study, we isolated 129 candidate KNOX gene sequences after removing incomplete and redundant sequences from 10 different species. There were nine from Arabidopsis [64] and eight from Solanum lycopersicum [65], which are consistent with the results of previous studies. Genome-wide analysis showed that the KNOX gene family was divided into two subfamilies: class I and class II (Figure 1). Both subfamilies contain monocots, dicots, and P. patens. Class I subfamily KNOX genes are similar to zmkn1 and are mainly expressed on the SAM of monocots and dicots [3,66]. According to previous studies, only one KNOX gene had evolved before the emergence of terrestrial plants, indicating that KNOX genes originated during the divergence of the last common ancestor of moss and vascular plants. KNOX genes are divided into four domains: KNOX1, KNOX2, ELK, and HD [64,67]. The KNOX1 domain has negative regulatory effects on the transcription of target genes. The KNOX2 domain mediates the interaction between KNOX and members of the BELL gene family. The HD consists of three helices and is conserved in eukaryotes and is involved in DNA binding [9,68]. Members in the same subfamily contain similar numbers of exons and introns, except for the number of exons in P. patens, which may also be due to the absence of exons for functional adaptation during evolution. Intriguingly, most members of the KNOX gene family contained five identical conserved motifs, except that P. patens does not contain motif 8. A high degree of sequence identity and similar exon–intron structures of KNOX genes across families suggests that the KNOX family has undergone gene duplication events throughout evolution.
Although repeated genes may have evolved few novel functions, they play an important role in the origin of species and the evolution of biological functions [42,69]. Gene duplication plays a significant role not only in the process of genome rearrangement and expansion but also in the diversification of gene functions and the large number of gene families [70]. Segmental duplication, tandem duplication, and transposition events, such as retro and replicative transposition, are the three main forces that drive the expansion of gene families [61,70]. Transposition events are difficult to identify based on sequence analysis alone; therefore, we focused on segmental and tandem duplication events. The results of the present study indicated that tandem duplication was detected in three species from the class I subfamily, indicating that the genes produced by tandem duplication did not undergo functional divergence (Table A3). Segmental duplication was detected in eight species from both subfamilies. Monocotyledonous plants had both tandem and segmental duplication, and dicotyledonous plants had only segmental duplication, especially, most of the KNOX genes in soybean and cotton. Therefore, the main amplification method of soybean and cotton is segmental duplication. Segmental duplication is likely to have played a pivotal role in KNOX gene expansion in dicots. In addition, the number of segmental duplication events in the two subfamilies was similar, and only segmental duplication occurred in the class II subfamily, which may be the reason for the larger number of class I subfamily members. Class I subfamily members are isolated from different angiosperms. They are expressed in meristems and not in differentiated tissues or organs that are related to maintaining the properties of meristems [9]. Large-scale duplication may have also been involved in the expansion of the KNOX gene family.
4.2. Functional divergence, positive selection, and co-evolution analysis
We selected the 3D structure of the Arabidopsis KNOX protein AT4G08150 for observation and marked the detected sites on the predicted 3D structure. The functional diversity between different subfamilies was mainly determined by specific amino acids in the subfamily, and the major reason for the functional divergence in repeated genes was possibly owing to the accumulation of amino acid mutation sites [42,71,72]. Type I and type II functional divergences between gene clusters of KNOX subfamilies were estimated by posterior probability analysis. By analyzing the functional divergence of the KNOX gene family, we identified a total of 15 type I functional divergence sites and 52 type II functional divergence sites from two subfamilies (Table 2). This result indicates diversification in the evolutionary rates of specific amino acid sites or significant changes in the physicochemical properties of amino acids. There are far more type II than type I functional divergence sites, which indicated that type II functional divergence is dominant. Besides, eight amino acid sites were identified as both type I and type II functional divergence sites, indicating that they had undergone simultaneous shifts in evolutionary rates and physicochemical properties. However, the lack of significant differences in the degree of functional divergence between most subfamily pairs suggests that genes belonging to these subfamilies might perform similar functions.
Positive selection has been associated with gene duplication and functional divergence. Here, we used computer simulations to evaluate the performance of Bayesian predictions for amino acids under positive selection [50]. However, the functions of most amino acid residues were conserved, and only a few amino acid sites can function in molecular adaptation [73]. In the present study, we detected 14 positive selection sites, three significant positive selection sites, and 11 extremely significant selection sites (Table 3). The positive selection sites were mainly distributed in the KNOX1 domain, indicating that the KNOX gene family had been subjected to different selection pressures during evolution. Interestingly, we found that the site 140A experienced both type I and type II functional divergences and positive selection and may have an important role.
The complexity of protein evolution is directly proportional to the potential function and structure of interactions between co-evolving sites within the molecule, and co-evolving amino acid sites interact between complex functional domains of a protein [74]. Detection of co-evolving sites will provide important evidence for the study of the mechanisms underlying molecular evolution. A co-evolution analysis of the KNOX gene family detected two sets of adjacent co-evolving sites: 248S and 249D, and 382L and 383Y, both of which were in the c-terminal domain. Based on the analysis of the 3D structure (Figure 3), the co-evolving sites are closer in the 3D structure. Thus, the interaction of these sites may have a stabilizing effect on the spatial structure of KNOX proteins.
4.3. Expression analysis of KNOX gene family
Most KNOX gene families exhibited variable expression levels in different tissues and organs (Figures 4 and 5). Class I KNOX genes are mainly expressed in the SAM and the class II genes are more widely expressed [9], as can be seen in the heatmap, which shows members of the class II subfamily expressed in most tissues and organs. The expression of KNOX genes mainly in the shoot may be related to important role within the SAM [29]. For example, the expression of KNOX genes in the shoot was upregulated in Arabidopsis. In addition, during the evolution of angiosperms, the KNOX1 gene was involved in the control of leaf shape. The expression pattern of KNOX1 in the primordium of a leaf is highly related to the shape of the leaf. We suspect that the key amino acid sites in the KNOX1 domain may be related to the expression of KNOX1 [16]. The expression of genes in different tissues reflects the diversity of functions. In summary, expression profiles of KNOX family members are largely organ specific, indicating that KNOX genes are differentially expressed in different groups and that regulatory regions of KNOX genes may have diverged. Importantly, the results also demonstrate divergence in the expression of KNOX duplicated genes during evolution.
5. Conclusions
In the present study, a total of 129 KNOX family members were identified from 10 species through extensive analysis of gene families, which were divided into two subfamilies by phylogenetic analysis. Monocots and dicots were amplified differently. Both tandem and segmental duplication are found in monocotyledonous plants, whereas dicotyledonous plants only have segmental duplication. Gene replication provides the main driving force for adaptive evolution of species. The large proportion of type II functional divergence that occurred indicated that the mode of functional divergence for KNOX proteins mainly relates to changes in the physicochemical properties of amino acids. The site-specific model analysis revealed that the KNOX gene family contains 14 positive selection sites, mainly located in the KNOX1 domain, which suffers from strong positive selection pressure. Two pairs of amino acid sites close to each other in 3D structure were identified by co-evolutionary analysis, indicating that they may play a key role in the stability of KNOX protein structure and function. Furthermore, KNOX genes exhibited different expression profiles in different organs as well as different functions. Our study provides a deeper understanding of the structural and functional evolution of the KNOX gene family and provides a basis for further research on KNOX proteins.
Acknowledgments
This work was supported by the Natural Science Foundation of Beijing, China (6192002) and the Science and Technology Development Project of the Beijing Education Commission (KM201710028010).
Appendix
Figure A1.
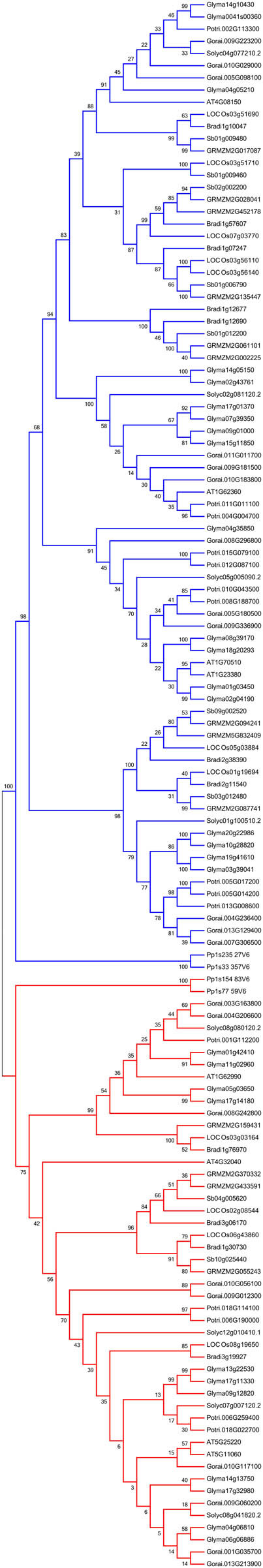
Neighbor-joining phylogenetic tree of the KNOX gene family. Two branches of different colors represent different subfamilies: blue represents class I KNOX genes, and red represents class II KNOX genes.
Table A1.
Number of the KNOX genes in ten plant species
| Plant taxa | Species | Number of KNOX genes |
|---|---|---|
| Dicotyledonous angiosperms | Arabidopsis thaliana | 8 |
| Glycine max | 29 | |
| Populus trichocarpa | 15 | |
| Gossypium raimondii | 21 | |
| Solanum lycopersicum | 8 | |
| Monocotyledonous angiosperms | Oryza sativa | 11 |
| Brachypodium distachyon | 11 | |
| Sorghum bicolor | 9 | |
| Zea mays | 13 | |
| Bryophyte | Physcomitrella patens | 4 |
Table A2.
Identified KNOX genes and their related information
| Group | Gene ID | ORF (aa) | pIa | Mw (Da) |
|---|---|---|---|---|
| I | AT4G08150 | 398 | 6.02 | 45,835.5 |
| I | AT1G70510 | 310 | 4.9 | 35,638 |
| II | AT5G25220 | 431 | 5.86 | 47,599.8 |
| II | AT5G11060 | 393 | 5.87 | 44,385 |
| II | AT4G32040 | 383 | 6.03 | 43,283.6 |
| I | AT1G23380 | 329 | 4.92 | 37,189.9 |
| I | AT1G62360 | 382 | 6.19 | 42,753.1 |
| II | AT1G62990 | 291 | 6.1 | 32,908.1 |
| I | Glyma14g10430 | 385 | 6.39 | 44,100 |
| I | Glyma0041s00360 | 387 | 6.43 | 44,458.4 |
| I | Glyma04g05210 | 361 | 6.55 | 42,059.2 |
| I | Glyma17g01370 | 269 | 6.46 | 30,750.9 |
| I | Glyma09g01000 | 379 | 6.07 | 42,374.1 |
| I | Glyma15g11850 | 375 | 5.83 | 41,992.8 |
| I | Glyma07g39350 | 362 | 6.18 | 40,816.2 |
| I | Glyma01g03450 | 309 | 5.22 | 35,125.5 |
| I | Glyma02g04190 | 308 | 5.37 | 34,903.3 |
| I | Glyma08g39170 | 321 | 4.99 | 36,284.8 |
| I | Glyma18g20293 | 324 | 5.25 | 36,864.4 |
| I | Glyma14g05150 | 311 | 6.72 | 35,759.5 |
| I | Glyma20g22986 | 307 | 4.97 | 34,913.5 |
| I | Glyma10g28820 | 296 | 5.06 | 33,654 |
| I | Glyma19g41610 | 311 | 5.62 | 35,503.1 |
| I | Glyma03g39041 | 307 | 5.6 | 35,332 |
| I | Glyma04g35850 | 278 | 5.96 | 31,949.2 |
| II | Glyma13g22530 | 346 | 5.59 | 39,797.2 |
| II | Glyma17g11330 | 345 | 5.59 | 39,683.1 |
| I | Glyma02g43761 | 295 | 5.7 | 33,383.3 |
| II | Glyma04g06810 | 411 | 5.73 | 45,982 |
| II | Glyma09g12820 | 369 | 5.51 | 41,611.2 |
| II | Glyma06g06886 | 398 | 5.9 | 44,607.6 |
| II | Glyma01g42410 | 281 | 6.43 | 32,145.4 |
| II | Glyma11g02960 | 279 | 6.36 | 31,898.1 |
| II | Glyma14g13750 | 412 | 5.67 | 45,926.9 |
| II | Glyma05g03650 | 293 | 6.34 | 33,241.4 |
| II | Glyma17g14180 | 292 | 6.24 | 33,080.3 |
| II | Glyma17g32980 | 411 | 5.94 | 46,083.5 |
| I | Potri.002G113300 | 368 | 5.98 | 42,459.4 |
| I | Potri.011G011100 | 373 | 6.13 | 41,549.5 |
| I | Potri.004G004700 | 369 | 6.14 | 41,189.2 |
| I | Potri.015G079100 | 347 | 5.03 | 38,787 |
| I | Potri.010G043500 | 309 | 4.88 | 35,200.6 |
| I | Potri.012G087100 | 340 | 4.95 | 37,951.3 |
| I | Potri.008G188700 | 341 | 5.51 | 38,685.4 |
| I | Potri.005G017200 | 316 | 5.31 | 36,277.5 |
| I | Potri.005G014200 | 317 | 5.31 | 36,324.5 |
| I | Potri.013G008600 | 320 | 5.15 | 36,167.5 |
| II | Potri.006G259400 | 432 | 5.91 | 48,185.5 |
| II | Potri.018G114100 | 334 | 5.74 | 38,045.4 |
| II | Potri.018G022700 | 426 | 5.88 | 47,762.8 |
| II | Potri.001G112200 | 301 | 6.23 | 33,920.2 |
| II | Potri.006G190000 | 338 | 5.81 | 38,563.1 |
| I | Gorai.009G223200 | 369 | 5.8 | 42,516.5 |
| I | Gorai.010G029000 | 364 | 6.14 | 41,729.5 |
| I | Gorai.005G098100 | 310 | 7.1 | 35,620.9 |
| I | Gorai.011G011700 | 351 | 6.01 | 39,746.5 |
| I | Gorai.010G183800 | 355 | 6.21 | 39,951.8 |
| I | Gorai.005G180500 | 314 | 5.01 | 35,440.9 |
| I | Gorai.009G181500 | 353 | 6 | 39,924.9 |
| I | Gorai.009G336900 | 313 | 4.73 | 35,622.3 |
| I | Gorai.013G129400 | 312 | 5.17 | 35,341.7 |
| I | Gorai.008G296800 | 303 | 5.47 | 34,206.5 |
| I | Gorai.007G306500 | 320 | 4.73 | 36,277.4 |
| I | Gorai.004G236400 | 289 | 5.66 | 33,046 |
| II | Gorai.009G060200 | 425 | 5.79 | 46,817.2 |
| II | Gorai.010G117100 | 434 | 5.89 | 48,085.5 |
| II | Gorai.008G242800 | 303 | 6.5 | 34,634.2 |
| II | Gorai.001G035700 | 443 | 5.94 | 48,698 |
| II | Gorai.003G163800 | 299 | 5.9 | 33,596.7 |
| II | Gorai.010G056100 | 290 | 6.2 | 32,792.9 |
| II | Gorai.004G206600 | 300 | 6.23 | 33,765 |
| II | Gorai.013G213900 | 448 | 6.01 | 49,635.2 |
| II | Gorai.009G012300 | 295 | 5.35 | 33,294.4 |
| I | Solyc04g077210.2 | 355 | 5.86 | 40,093.7 |
| I | Solyc02g081120.2 | 354 | 5.72 | 39,654.6 |
| I | Solyc05g005090.2 | 320 | 4.79 | 36,695.8 |
| I | Solyc01g100510.2 | 335 | 5.3 | 37,912.2 |
| II | Solyc08g041820.2 | 349 | 5.37 | 39,649.4 |
| II | Solyc08g080120.2 | 310 | 6.06 | 35,307.6 |
| II | Solyc07g007120.2 | 431 | 5.67 | 48,015.7 |
| II | Solyc12g010410.1 | 329 | 5.58 | 38,098.1 |
| I | LOC_Os03g51690 | 361 | 6.37 | 39,898 |
| I | LOC_Os07g03770 | 355 | 6.5 | 38,590.4 |
| I | LOC_Os03g56110 | 341 | 6.31 | 37,235.7 |
| I | LOC_Os01g19694 | 301 | 6.13 | 32,735.8 |
| I | LOC_Os05g03884 | 311 | 5.21 | 33,348.2 |
| I | LOC_Os03g51710 | 377 | 5.67 | 41,382.8 |
| I | LOC_Os03g56140 | 385 | 6.41 | 41,456.7 |
| II | LOC_Os08g19650 | 278 | 5.8 | 30,891.6 |
| II | LOC_Os02g08544 | 313 | 5.72 | 33,875.9 |
| II | LOC_Os06g43860 | 323 | 6.02 | 35,106.2 |
| II | LOC_Os03g03164 | 314 | 5.73 | 34,607 |
| I | Bradi1g10047 | 372 | 6.38 | 41,486.1 |
| I | Bradi1g57607 | 321 | 6.2 | 35,556 |
| I | Bradi1g07247 | 345 | 6.13 | 38,136.9 |
| I | Bradi2g11540 | 290 | 5.6 | 32,008 |
| I | Bradi2g38390 | 300 | 5.61 | 32,991.1 |
| I | Bradi1g12690 | 313 | 5.49 | 34,969.6 |
| I | Bradi1g12677 | 251 | 6.61 | 28,353.8 |
| II | Bradi3g19927 | 368 | 5.49 | 39,693.3 |
| II | Bradi1g30730 | 317 | 5.69 | 34,713.7 |
| II | Bradi3g06170 | 316 | 5.72 | 34,253.3 |
| II | Bradi1g76970 | 314 | 5.82 | 34,834.4 |
| I | Sb02g002200 | 356 | 6.23 | 38,884.6 |
| I | Sb01g009480 | 360 | 6.56 | 39,863.8 |
| I | Sb01g006790 | 349 | 5.96 | 38,713.5 |
| I | Sb01g009460 | 372 | 6.29 | 40,540.6 |
| I | Sb03g012480 | 294 | 5.6 | 32,458.4 |
| I | Sb01g012200 | 334 | 5.53 | 36,797.8 |
| I | Sb09g002520 | 303 | 5.34 | 32,961.1 |
| II | Sb10g025440 | 319 | 5.79 | 34,605.6 |
| II | Sb04g005620 | 444 | 6.34 | 48,081.1 |
| I | GRMZM2G028041 | 351 | 6.4 | 38,800.6 |
| I | GRMZM2G452178 | 360 | 6.23 | 39,215 |
| I | GRMZM2G017087 | 359 | 6.41 | 39,826.6 |
| I | GRMZM2G135447 | 364 | 5.91 | 40,276.2 |
| I | GRMZM2G061101 | 352 | 6.53 | 38,968.6 |
| I | GRMZM2G002225 | 328 | 5.25 | 36,438.7 |
| I | GRMZM2G094241 | 307 | 5.27 | 33,326.4 |
| I | GRMZM2G087741 | 295 | 5.51 | 32,493.4 |
| I | GRMZM5G832409 | 298 | 5.32 | 32,662.6 |
| II | GRMZM2G055243 | 316 | 5.69 | 34,522.6 |
| II | GRMZM2G370332 | 310 | 5.84 | 33,791.9 |
| II | GRMZM2G433591 | 363 | 9.78 | 39,958.3 |
| II | GRMZM2G159431 | 310 | 5.73 | 34,336.7 |
| I | Pp1s235_27V6 | 508 | 5.55 | 57,539.1 |
| I | Pp1s33_357V6 | 445 | 5.44 | 50,715.3 |
| II | Pp1s154_83V6 | 636 | 5.59 | 72,405.5 |
| II | Pp1s77_59V6 | 518 | 5.24 | 58,858.8 |
ORF = open reading frame; aa = amino acids; Mw = molecular weight.
Isoelectric point of the deduced polypeptide.
Table A3.
Estimates of the dates for the segmental duplication events of KNOX gene family in eight plants
| Segmental pairs | K s (mean ± SD) | Estimated time (MYA) | WGD (MYA) | |
|---|---|---|---|---|
| AT5G11060 | AT5G25220 | 0.765 ± 0.129 | 25.5 | 28–48 |
| Glyma04g05210 | Glyma0041s00360 | 0.664 ± 0.171 | 54.4 | 13, 59 |
| Glyma14g10430 | Glyma0041s00360 | 0.125 ± 0.039 | 10.2 | |
| Glyma01g03450 | Glyma02g04190 | 0.181 ± 0.165 | 14.8 | |
| Glyma01g03450 | Glyma08g39170 | 0.696 ± 0.169 | 57 | |
| Glyma01g42410 | Glyma05g03650 | 0.709 ± 0.111 | 58.1 | |
| Glyma01g42410 | Glyma11g02960 | 0.147 ± 0.142 | 12 | |
| Glyma01g42410 | Glyma17g14180 | 0.711 ± 0.127 | 58.3 | |
| Glyma02g04190 | Glyma08g39170 | 0.664 ± 0.146 | 54.4 | |
| Glyma02g04190 | Glyma18g20293 | 0.605 ± 0.035 | 49.6 | |
| Glyma02g43761 | Glyma14g05150 | 0.237 ± 0.220 | 19.4 | |
| Glyma03g39041 | Glyma10g28820 | 0.606 ± 0.136 | 49.7 | |
| Glyma03g39041 | Glyma19g41610 | 0.144 ± 0.071 | 11.8 | |
| Glyma03g39041 | Glyma20g22986 | 0.610 ± 0.154 | 50 | |
| Glyma04g05210 | Glyma14g10430 | 0.613 ± 0.170 | 50 | |
| Glyma04g06810 | Glyma06g06886 | 0.159 ± 0.119 | 13 | |
| Glyma04g06810 | Glyma17g32980 | 0.633 ± 0.045 | 51.9 | |
| Glyma05g03650 | Glyma11g02960 | 0.766 ± 0.103 | 62.8 | |
| Glyma05g03650 | Glyma17g14180 | 0.163 ± 0.059 | 13.4 | |
| Glyma06g06886 | Glyma17g32980 | 0.620 ± 0.029 | 50.8 | |
| Glyma07g39350 | Glyma09g01000 | 0.646 ± 0.142 | 53 | |
| Glyma07g39350 | Glyma15g11850 | 0.705 ± 0.164 | 57.8 | |
| Glyma07g39350 | Glyma17g01370 | 0.145 ± 0.120 | 11.9 | |
| Glyma09g01000 | Glyma15g11850 | 0.183 ± 0.157 | 15 | |
| Glyma09g01000 | Glyma17g01370 | 0.659 ± 0.168 | 54 | |
| Glyma10g28820 | Glyma19g41610 | 0.572 ± 0.082 | 46.9 | |
| Glyma10g28820 | Glyma20g22986 | 0.145 ± 0.059 | 11.9 | |
| Glyma11g02960 | Glyma17g14180 | 0.741 ± 0.113 | 60.7 | |
| Glyma13g22530 | Glyma17g11330 | 0.153 ± 0.106 | 12.5 | |
| Glyma14g13750 | Glyma17g32980 | 0.158 ± 0.045 | 13 | |
| Glyma15g11850 | Glyma17g01370 | 0.660 ± 0.178 | 54.1 | |
| Glyma19g41610 | Glyma20g22986 | 0.573 ± 0.101 | 47 | |
| Potri.005G014200 | Potri.013G008600 | 0.313 ± 0.113 | 17.2 | 8–13 |
| Potri.005G017200 | Potri.013G008600 | 0.332 ± 0.111 | 18.2 | |
| Potri.006G190000 | Potri.018G114100 | 0.241 ± 0.065 | 13.2 | |
| Potri.006G259400 | Potri.018G022700 | 0.294 ± 0.115 | 16.2 | |
| Potri.008G188700 | Potri.010G043500 | 0.268 ± 0.094 | 14.7 | |
| Gorai.001G035700 | Gorai.010G117100 | 0.515 ± 0.130 | 17.2 | 13–20 |
| Gorai.001G035700 | Gorai.013G213900 | 0.530 ± 0.100 | 17.7 | |
| Gorai.001G035700 | Gorai.009G060200 | 0.495 ± 0.105 | 16.5 | |
| Gorai.003G163800 | Gorai.004G206600 | 0.535 ± 0.122 | 17.8 | |
| Gorai.003G163800 | Gorai.008G242800 | 0.626 ± 0.153 | 20.9 | |
| Gorai.004G206600 | Gorai.008G242800 | 0.534 ± 0.109 | 17.8 | |
| Gorai.004G236400 | Gorai.013G129400 | 0.420 ± 0.078 | 14 | |
| Gorai.004G236400 | Gorai.007G306500 | 0.519 ± 0.135 | 17.3 | |
| Gorai.005G098100 | Gorai.010G029000 | 0.710 ± 0.127 | 23.7 | |
| Gorai.005G098100 | Gorai.009G223200 | 0.528 ± 0.078 | 17.6 | |
| Gorai.007G306500 | Gorai.013G129400 | 0.425 ± 0.045 | 14.2 | |
| Gorai.009G012300 | Gorai.010G056100 | 0.484 ± 0.124 | 16.1 | |
| Gorai.009G060200 | Gorai.010G117100 | 0.530 ± 0.088 | 17.7 | |
| Gorai.009G060200 | Gorai.013G213900 | 0.526 ± 0.112 | 17.5 | |
| Gorai.009G181500 | Gorai.010G183800 | 0.482 ± 0.132 | 16.1 | |
| Gorai.009G181500 | Gorai.011G011700 | 0.617 ± 0.171 | 20.6 | |
| Gorai.009G223200 | Gorai.010G029000 | 0.637 ± 0.152 | 21.2 | |
| Gorai.010G183800 | Gorai.011G011700 | 0.683 ± 0.233 | 22.8 | |
| LOC_Os02g08544 | LOC_Os06g43860 | 0.660 ± 0.128 | 50.8 | 30–40,66–70 |
| Bradi1g30730 | Bradi3g06170 | 0.863 ± 0.017 | 66.4 | 56–73 |
| Sb04g005620 | Sb10g025440 | 0.685 ± 0.140 | 52.7–56.1 | 70 |
| GRMZM2G135447 | GRMZM2G452178 | 0.597 ± 0.024 | 45.9 | 12, 70 |
MYA = million years ago.
Footnotes
Conflict of interest: The authors state no conflict of interest.
References
- [1].Gehring WJ, Affolter M, Bürglin T. Homeodomain proteins. Annu Rev Biochem. 1994;63:487–526. [DOI] [PubMed]; Gehring WJ, Affolter M, Bürglin T. Homeodomain proteins. Annu Rev Biochem. 1994;63:487–526. doi: 10.1146/annurev.bi.63.070194.002415. [DOI] [PubMed] [Google Scholar]
- [2].Vollbrecht E, Veit B, Sinha N, Hake S. The developmental gene Knotted-1 is a member of a maize homeobox gene family. Nature. 1991;350(6315):241–3. [DOI] [PubMed]; Vollbrecht E, Veit B, Sinha N, Hake S. The developmental gene Knotted-1 is a member of a maize homeobox gene family. Nature. 1991;350(6315):241–3. doi: 10.1038/350241a0. [DOI] [PubMed] [Google Scholar]
- [3].Hake S, Smith HM, Holtan H, Magnani E, Mele G, Ramirez J. The role of KNOX genes in plant development. Annu Rev Cell Dev Biol. 2004;20:125–51. [DOI] [PubMed]; Hake S, Smith HM, Holtan H, Magnani E, Mele G, Ramirez J. The role of KNOX genes in plant development. Annu Rev Cell Dev Biol. 2004;20:125–51. doi: 10.1146/annurev.cellbio.20.031803.093824. [DOI] [PubMed] [Google Scholar]
- [4].Lee JH, Lin H, Joo S, Goodenough U. Early sexual origins of homeoprotein heterodimerization and evolution of the plant KNOX/BELL family. Cell. 2008;133(5):829–40. [DOI] [PubMed]; Lee JH, Lin H, Joo S, Goodenough U. Early sexual origins of homeoprotein heterodimerization and evolution of the plant KNOX/BELL family. Cell. 2008;133(5):829–40. doi: 10.1016/j.cell.2008.04.028. [DOI] [PubMed] [Google Scholar]
- [5].Bürglin TR. Analysis of TALE superclass homeobox genes (MEIS, PBC, KNOX, Iroquois, TGIF) reveals a novel domain conserved between plants and animals. Nucleic Acids Res. 1997;25(21):4173–80. [DOI] [PMC free article] [PubMed]; Bürglin TR. Analysis of TALE superclass homeobox genes (MEIS, PBC, KNOX, Iroquois, TGIF) reveals a novel domain conserved between plants and animals. Nucleic Acids Res. 1997;25(21):4173–80. doi: 10.1093/nar/25.21.4173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Chen H, Rosin FM, Prat S, Hannapel DJ. Interacting transcription factors from the three-amino acid loop extension superclass regulate tuber formation. Plant Physiol. 2003;132(3):1391–404. [DOI] [PMC free article] [PubMed]; Chen H, Rosin FM, Prat S, Hannapel DJ. Interacting transcription factors from the three-amino acid loop extension superclass regulate tuber formation. Plant Physiol. 2003;132(3):1391–404. doi: 10.1104/pp.103.022434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hofer J, Gourlay C, Michael A, Ellis TH. Expression of a class 1 knotted1-like homeobox gene is down-regulated in pea compound leaf primordia. Plant Mol Biol. 2001;45(4):387–98. [DOI] [PubMed]; Hofer J, Gourlay C, Michael A, Ellis TH. Expression of a class 1 knotted1-like homeobox gene is down-regulated in pea compound leaf primordia. Plant Mol Biol. 2001;45(4):387–98. doi: 10.1023/a:1010739812836. [DOI] [PubMed] [Google Scholar]
- [8].Scofield S, Dewitte W, Murray JA. The KNOX gene SHOOT MERISTEMLESS is required for the development of reproductive meristematic tissues in Arabidopsis. Plant J. 2007;50(5):767–81. [DOI] [PubMed]; Scofield S, Dewitte W, Murray JA. The KNOX gene SHOOT MERISTEMLESS is required for the development of reproductive meristematic tissues in Arabidopsis. Plant J. 2007;50(5):767–81. doi: 10.1111/j.1365-313X.2007.03095.x. [DOI] [PubMed] [Google Scholar]
- [9].Kerstetter R, Vollbrecht E, Lowe B, Velt B, Yamaguchi J, Hake S. Sequence analysis and expression patterns divide the maize Knotted1-like homeobox gene into two classes. Plnat Cell. 1994;6(12):1877–87. [DOI] [PMC free article] [PubMed]; Kerstetter R, Vollbrecht E, Lowe B, Velt B, Yamaguchi J, Hake S. Sequence analysis and expression patterns divide the maize Knotted1-like homeobox gene into two classes. Plnat Cell. 1994;6(12):1877–87. doi: 10.1105/tpc.6.12.1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Sakamoto T, Nishimura A, Tamaoki M, Kuba M, Tanaka H, Iwahori S, et al. The conserved KNOX domain mediates specificity of tobacco KNOTTED1-type homeodomain proteins. Plant Cell. 1999;11(8):1419–32. [DOI] [PMC free article] [PubMed]; Sakamoto T, Nishimura A, Tamaoki M, Kuba M, Tanaka H, Iwahori S. et al. The conserved KNOX domain mediates specificity of tobacco KNOTTED1-type homeodomain proteins. Plant Cell. 1999;11(8):1419–32. doi: 10.1105/tpc.11.8.1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ori N, Eshed Y, Chuck G, Bowman JL, Hake S. Mechanisms that control KNOX gene expression in the Arabidopsis shoot. Development. 2000;127(24):5523–32. [DOI] [PubMed]; Ori N, Eshed Y, Chuck G, Bowman JL, Hake S. Mechanisms that control KNOX gene expression in the Arabidopsis shoot. Development. 2000;127(24):5523–32. doi: 10.1242/dev.127.24.5523. [DOI] [PubMed] [Google Scholar]
- [12].Li E, Bhargava A, Qiang W, Friedmann MC, Forneris N, Savidge RA, et al. The Class II KNOX gene KNAT7 negatively regulates secondary wall formation in Arabidopsis and is functionally conserved in Populus. New Phytol. 2012;194(1):102–15. [DOI] [PubMed]; Li E, Bhargava A, Qiang W, Friedmann MC, Forneris N, Savidge RA. et al. The Class II KNOX gene KNAT7 negatively regulates secondary wall formation in Arabidopsis and is functionally conserved in Populus. New Phytol. 2012;194(1):102–15. doi: 10.1111/j.1469-8137.2011.04016.x. [DOI] [PubMed] [Google Scholar]
- [13].Jasinski S, Kaur H, Tattersall A, Tsiantis M. Negative regulation of KNOX expression in tomato leaves. Planta. 2007;226(5):1255–63. [DOI] [PubMed]; Jasinski S, Kaur H, Tattersall A, Tsiantis M. Negative regulation of KNOX expression in tomato leaves. Planta. 2007;226(5):1255–63. doi: 10.1007/s00425-007-0572-5. [DOI] [PubMed] [Google Scholar]
- [14].Peng J, Yu J, Wang H, Guo Y, Li G, Bai G, et al. Regulation of compound leaf development in Medicago truncatula by fused compound leaf1, a class M KNOX gene. Plant Cell. 2011;23(11):3929–43. [DOI] [PMC free article] [PubMed]; Peng J, Yu J, Wang H, Guo Y, Li G, Bai G. et al. Regulation of compound leaf development in Medicago truncatula by fused compound leaf1, a class M KNOX gene. Plant Cell. 2011;23(11):3929–43. doi: 10.1105/tpc.111.089128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Frangedakis E, Saint-Marcoux D, Moody LA, Rabbinowitsch E, Langdale JA. Nonreciprocal complementation of KNOX gene function in land plants. New Phytol. 2017;216(2):591–604. [DOI] [PMC free article] [PubMed]; Frangedakis E, Saint-Marcoux D, Moody LA, Rabbinowitsch E, Langdale JA. Nonreciprocal complementation of KNOX gene function in land plants. New Phytol. 2017;216(2):591–604. doi: 10.1111/nph.14318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Tsuda K, Hake S. Diverse functions of KNOX transcription factors in the diploid body plan of plants. Curr Opin Plant Biol. 2015;27:91–6. [DOI] [PubMed]; Tsuda K, Hake S. Diverse functions of KNOX transcription factors in the diploid body plan of plants. Curr Opin Plant Biol. 2015;27:91–6. doi: 10.1016/j.pbi.2015.06.015. [DOI] [PubMed] [Google Scholar]
- [17].Sakakibara K, Ando S, Yip HK, Tamada Y, Hiwatashi Y, Murata T, et al. KNOX2 genes regulate the haploid-to-diploid morphological transition in land plants. Science. 2013;339(6123):1067–70. [DOI] [PubMed]; Sakakibara K, Ando S, Yip HK, Tamada Y, Hiwatashi Y, Murata T. et al. KNOX2 genes regulate the haploid-to-diploid morphological transition in land plants. Science. 2013;339(6123):1067–70. doi: 10.1126/science.1230082. [DOI] [PubMed] [Google Scholar]
- [18].Byrne ME, Simorowski J, Martienssen RA. ASYMMETRIC LEAVES1 reveals KNOX gene redundancy in Arabidopsis. Development. 2002;129(8):1957–65. [DOI] [PubMed]; Byrne ME, Simorowski J, Martienssen RA. ASYMMETRIC LEAVES1 reveals KNOX gene redundancy in Arabidopsis. Development. 2002;129(8):1957–65. doi: 10.1242/dev.129.8.1957. [DOI] [PubMed] [Google Scholar]
- [19].Reyes-Rivera J, Rodríguez-Alonso G, Petrone E, Vasco A, Vergara-Silva F, Shishkova S, et al. Expression of the knotted homeobox genes in the cactaceae cambial zone suggests their involvement in wood development. Front Plant Sci. 2017;8:218. [DOI] [PMC free article] [PubMed]; Reyes-Rivera J, Rodríguez-Alonso G, Petrone E, Vasco A, Vergara-Silva F, Shishkova S. et al. Expression of the knotted homeobox genes in the cactaceae cambial zone suggests their involvement in wood development. Front Plant Sci. 2017;8:218. doi: 10.3389/fpls.2017.00218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Belles-Boix E, Hamant O, Witiak SM, Morin H, Traas J, Pautor V. KNAT6: an Arabidopsis homeobox gene involved in meristem activity and organ separation. Plant Cell. 2006;18(8):1900–7. [DOI] [PMC free article] [PubMed]; Belles-Boix E, Hamant O, Witiak SM, Morin H, Traas J, Pautor V. KNAT6: an Arabidopsis homeobox gene involved in meristem activity and organ separation. Plant Cell. 2006;18(8):1900–7. doi: 10.1105/tpc.106.041988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ragn L, Belles-Boix E, Gün M, Pautot V. Interaction of KNAT6 and KNAT2 with BREVIPEDICELLUS and PENNYWISE in Arabidopsis inflorescences. Plant Cell. 2008;20(4):888–900. [DOI] [PMC free article] [PubMed]; Ragn L, Belles-Boix E, Gün M, Pautot V. Interaction of KNAT6 and KNAT2 with BREVIPEDICELLUS and PENNYWISE in Arabidopsis inflorescences. Plant Cell. 2008;20(4):888–900. doi: 10.1105/tpc.108.058230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Golz JF, Keck EJ, Hudson A. Spontaneous mutations in KNOX genes give rise to a novel floral structure in Antirrhinum. Curr Biol. 2002;12(7):515–22. [DOI] [PubMed]; Golz JF, Keck EJ, Hudson A. Spontaneous mutations in KNOX genes give rise to a novel floral structure in Antirrhinum. Curr Biol. 2002;12(7):515–22. doi: 10.1016/s0960-9822(02)00721-2. [DOI] [PubMed] [Google Scholar]
- [23].Furumizu C, Alvarez JP, Sakakibara K, Bowman JL. Antagonistic roles for KNOX1 and KNOX2 genes in patterning the land plant body plan following an ancient gene duplication. PLoS Genet. 2015;11(2):e1004980. [DOI] [PMC free article] [PubMed]; Furumizu C, Alvarez JP, Sakakibara K, Bowman JL. Antagonistic roles for KNOX1 and KNOX2 genes in patterning the land plant body plan following an ancient gene duplication. PLoS Genet. 2015;11(2):e1004980. doi: 10.1371/journal.pgen.1004980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wang S, Yamaguchi M, Grienenberger E, Martone PT, Samuels AL, Mansfield SD. The Class II KNOX genes KNAT3 and KNAT7 work cooperatively to influence deposition of secondary cell walls that provide mechanical support to Arabidopsis stems. Plant J. 2020;101(2):293–309. [DOI] [PubMed]; Wang S, Yamaguchi M, Grienenberger E, Martone PT, Samuels AL, Mansfield SD. The Class II KNOX genes KNAT3 and KNAT7 work cooperatively to influence deposition of secondary cell walls that provide mechanical support to Arabidopsis stems. Plant J. 2020;101(2):293–309. doi: 10.1111/tpj.14541. [DOI] [PubMed] [Google Scholar]
- [25].Liu Y, You S, Taylor-Teeples M, Li WL, Schuetz M, Brady SM, et al. BEL1-LIKE HOMEODOMAIN6 and knotted Arabidopsis THALIANA7 interact and regulate secondary cell wall formation via repression of revoluta. Plant Cell. 2014;26(12):4843–61. [DOI] [PMC free article] [PubMed]; Liu Y, You S, Taylor-Teeples M, Li WL, Schuetz M, Brady SM. et al. BEL1-LIKE HOMEODOMAIN6 and knotted Arabidopsis THALIANA7 interact and regulate secondary cell wall formation via repression of revoluta. Plant Cell. 2014;26(12):4843–61. doi: 10.1105/tpc.114.128322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Cheng X, Li M, Abdullah M, Li G, Zhang JY, Manzoor MA, et al. In Silico genome-wide analysis of the pear (Pyrus bretschneideri) KNOX family and the functional characterization of PbKNOX1, an Arabidopsis BREVIPEDICELLUS orthologue gene, involved in cell wall and lignin biosynthesis. Front Genet. 2019;10:623. [DOI] [PMC free article] [PubMed]; Cheng X, Li M, Abdullah M, Li G, Zhang JY, Manzoor MA. et al. In Silico genome-wide analysis of the pear (Pyrus bretschneideri) KNOX family and the functional characterization of PbKNOX1, an Arabidopsis BREVIPEDICELLUS orthologue gene, involved in cell wall and lignin biosynthesis. Front Genet. 2019;10:623. doi: 10.3389/fgene.2019.00632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Yoon J, Cho LH, Antt HW, Koh HJ, An G. KNOX Protein OSH15 induces grain shattering by repressing lignin biosynthesis genes. Plant Physiol. 2017;174(1):312–25. [DOI] [PMC free article] [PubMed]; Yoon J, Cho LH, Antt HW, Koh HJ, An G. KNOX Protein OSH15 induces grain shattering by repressing lignin biosynthesis genes. Plant Physiol. 2017;174(1):312–25. doi: 10.1104/pp.17.00298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Jia P, Zhang CG, Xing LB, Li YM, Shah K, Zuo XY, et al. Genome-wide identification of the MdKNOX gene family and characterization of its transcriptional regulation in malus domestica. Front Plant Sci. 2020;11:128. [DOI] [PMC free article] [PubMed]; Jia P, Zhang CG, Xing LB, Li YM, Shah K, Zuo XY. et al. Genome-wide identification of the MdKNOX gene family and characterization of its transcriptional regulation in malus domestica. Front Plant Sci. 2020;11:128. doi: 10.3389/fpls.2020.00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Denison FC, Paul AL, Zupanska AK, Ferl RJ. 14-33 proteins in plant physiology. Semin Cell Dev Biol. 2011;22(7):720–7. [DOI] [PubMed]; Denison FC, Paul AL, Zupanska AK, Ferl RJ. 14-33 proteins in plant physiology. Semin Cell Dev Biol. 2011;22(7):720–7. doi: 10.1016/j.semcdb.2011.08.006. [DOI] [PubMed] [Google Scholar]
- [30].Bhattacharjee A, Jain M. Homeobox genes as potential candidates for crop improvement under abiotic stress. plant acclimation to environmental. Stress. 2012;7:163–76.; Bhattacharjee A, Jain M. Homeobox genes as potential candidates for crop improvement under abiotic stress. plant acclimation to environmental. Stress. 2012;7:163–76. [Google Scholar]
- [31].Bjellqvist B, Basse B, Olsen E, Celis JE. Reference points for comparisons of two-dimensional maps of proteins from different human cell types defined in a pH scale where isoelectric points correlate with polypeptide compositions. Electrophoresis. 1994;15(3-4):529–39. [DOI] [PubMed]; Bjellqvist B, Basse B, Olsen E, Celis JE. Reference points for comparisons of two-dimensional maps of proteins from different human cell types defined in a pH scale where isoelectric points correlate with polypeptide compositions. Electrophoresis. 1994;15(3-4):529–39. doi: 10.1002/elps.1150150171. [DOI] [PubMed] [Google Scholar]
- [32].Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113. [DOI] [PMC free article] [PubMed]; Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–7. [DOI] [PMC free article] [PubMed]; Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–7. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ronquist F, Teslenko M, Pvander M, Ayres DL, Darling A, Höhna S, et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61(3):539–42. [DOI] [PMC free article] [PubMed]; Ronquist F, Teslenko M, Pvander M, Ayres DL, Darling A, Höhna S. et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61(3):539–42. doi: 10.1093/sysbio/sys029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33(7):1870–4. [DOI] [PMC free article] [PubMed]; Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33(7):1870–4. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Hu B, Jin J, Guo AY, Zhang H, Luo J, Gao G. GSDS 2.0: an upgraded gene feature visualization server. Bioinformatics. 2015;31(8):1296–7. [DOI] [PMC free article] [PubMed]; Hu B, Jin J, Guo AY, Zhang H, Luo J, Gao G. GSDS 2.0: an upgraded gene feature visualization server. Bioinformatics. 2015;31(8):1296–7. doi: 10.1093/bioinformatics/btu817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, et al. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37(Web Server issue):W202–8. [DOI] [PMC free article] [PubMed]; Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L. et al. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37(Web Server issue):W202–8. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Tang H, Wang X, Bowers JE, Ming R, Alam M, Paterson AH. Unraveling ancient hexaploidy through multiply-aligned angiosperm gene maps. Genome Res. 2008;18(12):1944–54. [DOI] [PMC free article] [PubMed]; Tang H, Wang X, Bowers JE, Ming R, Alam M, Paterson AH. Unraveling ancient hexaploidy through multiply-aligned angiosperm gene maps. Genome Res. 2008;18(12):1944–54. doi: 10.1101/gr.080978.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Yin G, Xu H, Xiao S, Qin Y, Li Y, Yan Y, et al. The large soybean (Glycine max) WRKY TF family expanded by segmental duplication events and subsequent divergent selection among subgroups. BMC Plant Biol. 2013;13:148. [DOI] [PMC free article] [PubMed]; Yin G, Xu H, Xiao S, Qin Y, Li Y, Yan Y. et al. The large soybean (Glycine max) WRKY TF family expanded by segmental duplication events and subsequent divergent selection among subgroups. BMC Plant Biol. 2013;13:148. doi: 10.1186/1471-2229-13-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Bowers JE, Chapman BA, Rong J, Paterson AH. Unravelling angiosperm genome evolution by phylogenetic analysis of chromosomal duplication events. Nature. 2003;422(6930):433–8. [DOI] [PubMed]; Bowers JE, Chapman BA, Rong J, Paterson AH. Unravelling angiosperm genome evolution by phylogenetic analysis of chromosomal duplication events. Nature. 2003;422(6930):433–8. doi: 10.1038/nature01521. [DOI] [PubMed] [Google Scholar]
- [41].Blanc G, Wolfe KH. Widespread paleopolyploidy in model plant species inferred from age distributions of duplicate genes. Plant Cell. 2004;16(7):1667–78. [DOI] [PMC free article] [PubMed]; Blanc G, Wolfe KH. Widespread paleopolyploidy in model plant species inferred from age distributions of duplicate genes. Plant Cell. 2004;16(7):1667–78. doi: 10.1105/tpc.021345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Lynch M, Conery JS. The evolutionary fate and consequences of duplicate genes. Science. 2000;290(5494):1151–5. [DOI] [PubMed]; Lynch M, Conery JS. The evolutionary fate and consequences of duplicate genes. Science. 2000;290(5494):1151–5. doi: 10.1126/science.290.5494.1151. [DOI] [PubMed] [Google Scholar]
- [43].Wang K, Wang Z, Li F, Ye W, Wang J, Song G, et al. The draft genome of a diploid cotton Gossypium raimondii. Nat Genet. 2012;44(10):1098–103. [DOI] [PubMed]; Wang K, Wang Z, Li F, Ye W, Wang J, Song G. et al. The draft genome of a diploid cotton Gossypium raimondii. Nat Genet. 2012;44(10):1098–103. doi: 10.1038/ng.2371. [DOI] [PubMed] [Google Scholar]
- [44].Vandepoele K, Simillion C, Van de Peer Y. Evidence that rice and other cereals are ancient aneuploids. Plant Cell. 2003;15(9):2192–202. [DOI] [PMC free article] [PubMed]; Vandepoele K, Simillion C, Van de Peer Y. Evidence that rice and other cereals are ancient aneuploids. Plant Cell. 2003;15(9):2192–202. doi: 10.1105/tpc.014019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Fan K, Wang M, Miao Y, Ni M, Bibi N, Yuan S, et al. Molecular evolution and expansion analysis of the NAC transcription factor in Zea mays. PLoS One. 2014;9(11):e111837. [DOI] [PMC free article] [PubMed]; Fan K, Wang M, Miao Y, Ni M, Bibi N, Yuan S. et al. Molecular evolution and expansion analysis of the NAC transcription factor in Zea mays. PLoS One. 2014;9(11):e111837. doi: 10.1371/journal.pone.0111837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Gu X, Zou Y, Su Z, Huang W, Zhou Z, Arendsee Z, et al. An update of DIVERGE software for functional divergence analysis of protein family. Mol Biol Evol. 2013;30(7):1713–9. [DOI] [PubMed]; Gu X, Zou Y, Su Z, Huang W, Zhou Z, Arendsee Z. et al. An update of DIVERGE software for functional divergence analysis of protein family. Mol Biol Evol. 2013;30(7):1713–9. doi: 10.1093/molbev/mst069. [DOI] [PubMed] [Google Scholar]
- [47].Lichtarge O, Bourne HR, Cohen FE. An evolutionary trace method defines binding surfaces common to protein families. J Mol Biol. 1996;257(2):342–58. [DOI] [PubMed]; Lichtarge O, Bourne HR, Cohen FE. An evolutionary trace method defines binding surfaces common to protein families. J Mol Biol. 1996;257(2):342–58. doi: 10.1006/jmbi.1996.0167. [DOI] [PubMed] [Google Scholar]
- [48].Gaucher EA, Gu X, Miyamoto MM, Benner SA. Predicting functional divergence in protein evolution by site-specific rate shifts. Trends Biochem Sci. 2002;27(6):315–21. [DOI] [PubMed]; Gaucher EA, Gu X, Miyamoto MM, Benner SA. Predicting functional divergence in protein evolution by site-specific rate shifts. Trends Biochem Sci. 2002;27(6):315–21. doi: 10.1016/s0968-0004(02)02094-7. [DOI] [PubMed] [Google Scholar]
- [49].Gu X. A simple statistical method for estimating type-II (cluster-specific) functional divergence of protein sequences. Mol Biol Evol. 2006;23(10):1937–45. [DOI] [PubMed]; Gu X. A simple statistical method for estimating type-II (cluster-specific) functional divergence of protein sequences. Mol Biol Evol. 2006;23(10):1937–45. doi: 10.1093/molbev/msl056. [DOI] [PubMed] [Google Scholar]
- [50].Anisimova M, Bielawski JP, Yang Z. Accuracy and power of Bayes prediction of amino acid sites under positive selection. Mol Biol Evol. 2002;19(6):950–8. [DOI] [PubMed]; Anisimova M, Bielawski JP, Yang Z. Accuracy and power of Bayes prediction of amino acid sites under positive selection. Mol Biol Evol. 2002;19(6):950–8. doi: 10.1093/oxfordjournals.molbev.a004152. [DOI] [PubMed] [Google Scholar]
- [51].Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24(8):1586–91. [DOI] [PubMed]; Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24(8):1586–91. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- [52].Wang L, Wu N, Zhu Y, Song W, Zhao X, Li Y, et al. The divergence and positive selection of the plant-specific BURP-containing protein family. Ecol Evol. 2015;5(22):5394–5412. [DOI] [PMC free article] [PubMed]; Wang L, Wu N, Zhu Y, Song W, Zhao X, Li Y. et al. The divergence and positive selection of the plant-specific BURP-containing protein family. Ecol Evol. 2015;5(22):5394–5412. doi: 10.1002/ece3.1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Fares MA, McNally D. CAPS: coevolution analysis using protein sequences. Bioinformatics. 2006;22(22):2821–2. [DOI] [PubMed]; Fares MA, McNally D. CAPS: coevolution analysis using protein sequences. Bioinformatics. 2006;22(22):2821–2. doi: 10.1093/bioinformatics/btl493. [DOI] [PubMed] [Google Scholar]
- [54].Song W, Qin Y, Zhu Y, Yin G, Wu N, Li Y, et al. Delineation of plant caleosin residues critical for functional divergence, positive selection and coevolution. BMC Evol Biol. 2014;14:124. [DOI] [PMC free article] [PubMed]; Song W, Qin Y, Zhu Y, Yin G, Wu N, Li Y. et al. Delineation of plant caleosin residues critical for functional divergence, positive selection and coevolution. BMC Evol Biol. 2014;14:124. doi: 10.1186/1471-2148-14-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Kelley LA, Sternberg MJ. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc. 2009;4(3):363–471. [DOI] [PubMed]; Kelley LA, Sternberg MJ. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc. 2009;4(3):363–471. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- [56].Cao Y, Meng D, Chen Y, Abdullah M, Jin Q, Lin Y, et al. Comparative and expression analysis of ubiquitin conjugating domain-containing genes in two pyrus species. Cells. 2018;7:77. [DOI] [PMC free article] [PubMed]; Cao Y, Meng D, Chen Y, Abdullah M, Jin Q, Lin Y. et al. Comparative and expression analysis of ubiquitin conjugating domain-containing genes in two pyrus species. Cells. 2018;7:77. doi: 10.3390/cells7070077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL, et al. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 2016;44(D1):D279–85. [DOI] [PMC free article] [PubMed]; Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL. et al. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 2016;44(D1):D279–85. doi: 10.1093/nar/gkv1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Letunic I, Doerks T, Bork P. SMART: recent updates, new developments and status in 2015. Nucleic Acids Res. 2015;43(Database issue):D257–60. [DOI] [PMC free article] [PubMed]; Letunic I, Doerks T, Bork P. SMART: recent updates, new developments and status in 2015. Nucleic Acids Res. 2015;43(Database issue):D257–60. doi: 10.1093/nar/gku949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Hamant O, Pautot V. Plant development: a TALE story. C R Biol. 2010;333(4):371–81. [DOI] [PubMed]; Hamant O, Pautot V. Plant development: a TALE story. C R Biol. 2010;333(4):371–81. doi: 10.1016/j.crvi.2010.01.015. [DOI] [PubMed] [Google Scholar]
- [60].Guo AY, Zhu QH, Chen X, Luo JC. GSDS: a gene structure display server. Hereditas. 2007;29(8):1023–6. [PubMed]; Guo AY, Zhu QH, Chen X, Luo JC. GSDS: a gene structure display server. Hereditas. 2007;29(8):1023–6. [PubMed] [Google Scholar]
- [61].Kong H, Landherr LL, Frohlich MW, Leebens-Mack J, Ma H, dePamphilis CW. Patterns of gene duplication in the plant SKP1 gene family in angiosperms: evidence for multiple mechanisms of rapid gene birth. Plant J. 2007;50(5):873–85. [DOI] [PubMed]; Kong H, Landherr LL, Frohlich MW, Leebens-Mack J, Ma H, dePamphilis CW. Patterns of gene duplication in the plant SKP1 gene family in angiosperms: evidence for multiple mechanisms of rapid gene birth. Plant J. 2007;50(5):873–85. doi: 10.1111/j.1365-313X.2007.03097.x. [DOI] [PubMed] [Google Scholar]
- [62].Gu X. Statistical methods for testing functional divergence after gene duplication. Mol Biol Evol. 1999;16(12):1664–74. [DOI] [PubMed]; Gu X. Statistical methods for testing functional divergence after gene duplication. Mol Biol Evol. 1999;16(12):1664–74. doi: 10.1093/oxfordjournals.molbev.a026080. [DOI] [PubMed] [Google Scholar]
- [63].Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 2015;10(6):845–58. [DOI] [PMC free article] [PubMed]; Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 2015;10(6):845–58. doi: 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Mukherjee K, Brocchieri L, Bürglin TR. A comprehensive classification and evolutionary analysis of plant homeobox genes. Mol Biol Evol. 2009;26(12):2775–94. [DOI] [PMC free article] [PubMed]; Mukherjee K, Brocchieri L, Bürglin TR. A comprehensive classification and evolutionary analysis of plant homeobox genes. Mol Biol Evol. 2009;26(12):2775–94. doi: 10.1093/molbev/msp201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Ye SG, Zai WS, Xiong ZL, Zhang HL, Ma YR. Genome-wide identification of KNOX gene family in tomato and their evolutionary relationship in Solanaceae. J Nucl Agric Sci. 2017;31(7):1263–71.; Ye SG, Zai WS, Xiong ZL, Zhang HL, Ma YR. Genome-wide identification of KNOX gene family in tomato and their evolutionary relationship in Solanaceae. J Nucl Agric Sci. 2017;31(7):1263–71. [Google Scholar]
- [66].Jackson D, Veit B, Hake S. Expression of maize KNOTTED1 related homeobox genes in the shoot apical meristem predicts patterns of morphogenesis in the vegetative shoot. Development. 1994;120:405–13.; Jackson D, Veit B, Hake S. Expression of maize KNOTTED1 related homeobox genes in the shoot apical meristem predicts patterns of morphogenesis in the vegetative shoot. Development. 1994;120:405–13. [Google Scholar]
- [67].Gao J, Yang X, Zhao W, Lang T, Samuelsson T. Evolution, diversification, and expression of KNOX proteins in plants. Front Plant Sci. 2015;6:882. [DOI] [PMC free article] [PubMed]; Gao J, Yang X, Zhao W, Lang T, Samuelsson T. Evolution, diversification, and expression of KNOX proteins in plants. Front Plant Sci. 2015;6:882. doi: 10.3389/fpls.2015.00882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Byrne ME, Groover AT, Fontana JR, Martienssen RA. Phyllotactic pattern and stem cell fate are determined by the Arabidopsis homeobox gene BELLRINGER. Development. 2003;130(17):3941–50. [DOI] [PubMed]; Byrne ME, Groover AT, Fontana JR, Martienssen RA. Phyllotactic pattern and stem cell fate are determined by the Arabidopsis homeobox gene BELLRINGER. Development. 2003;130(17):3941–50. doi: 10.1242/dev.00620. [DOI] [PubMed] [Google Scholar]
- [69].Kondrashov FA, Rogozin LB, Wolf YI, Koonin EV. Selection in the evolution of gene duplications. Genome Biol. 2002;3(2):research0008. [DOI] [PMC free article] [PubMed]; Kondrashov FA, Rogozin LB, Wolf YI, Koonin EV. Selection in the evolution of gene duplications. Genome Biol. 2002;3(2):research0008. doi: 10.1186/gb-2002-3-2-research0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Cannon SB, Mitra A, Baumgarten A, Young ND, May G. The roles of segmental and tandem gene duplication in the evolution of large gene families in Arabidopsis thaliana. BMC Plant Biol. 2004;4:10. [DOI] [PMC free article] [PubMed]; Cannon SB, Mitra A, Baumgarten A, Young ND, May G. The roles of segmental and tandem gene duplication in the evolution of large gene families in Arabidopsis thaliana. BMC Plant Biol. 2004;4:10. doi: 10.1186/1471-2229-4-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Moore RC, Purugganan MD. The early stages of duplicate gene evolution. Proc Natl Acad Sci U S A. 2003;100(26):15682–7. [DOI] [PMC free article] [PubMed]; Moore RC, Purugganan MD. The early stages of duplicate gene evolution. Proc Natl Acad Sci U S A. 2003;100(26):15682–7. doi: 10.1073/pnas.2535513100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Ha M, Kim ED, Chen ZJ. Duplicate genes increase expression diversity in closely related species and allopolyploids. Proc Natl Acad Sci U S A. 2009;106(7):2295–300. [DOI] [PMC free article] [PubMed]; Ha M, Kim ED, Chen ZJ. Duplicate genes increase expression diversity in closely related species and allopolyploids. Proc Natl Acad Sci U S A. 2009;106(7):2295–300. doi: 10.1073/pnas.0807350106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Golding GB, Dean AM. The structural basis of molecular adaptation. Mol Biol Evol. 1998;15(4):355–69. [DOI] [PubMed]; Golding GB, Dean AM. The structural basis of molecular adaptation. Mol Biol Evol. 1998;15(4):355–69. doi: 10.1093/oxfordjournals.molbev.a025932. [DOI] [PubMed] [Google Scholar]
- [74].Fares MA, Travers SA. A novel method for detecting intramolecular coevolution: adding a further dimension to selective constraints analyses. Genetics. 2006;173(1):9–23. [DOI] [PMC free article] [PubMed]; Fares MA, Travers SA. A novel method for detecting intramolecular coevolution: adding a further dimension to selective constraints analyses. Genetics. 2006;173(1):9–23. doi: 10.1534/genetics.105.053249. [DOI] [PMC free article] [PubMed] [Google Scholar]