

Abstract
In rapid proliferating cancer cells, there is a need for fast ATP and lactate production, therefore cancer cells turn off oxidative phosphorylation and turn on the so called "Warburg effect". This regulating the expression of genes involved in glycolysis. According to many studies, glucose transporter 1, which supplies glucose to the cell, is the most abundantly expressed transporter in cancer cells. Hexokinase 2, is one of four hexokinase isoenzymes, is also another highly expressed enzyme in cancer cells and it functions to enhance the glycolytic rate. The up-regulation of these two proteins has been established as an important factor in promoting development and metastasis in many types of cancer. Furthermore, other enzymes involved in glycolysis pathway such as phosphoglucose isomerase and glyceraldehyde 3-phosphate dehydrogenase, exhibit additional functions in promoting tumor growth in a non-glycolytic way. This review demonstrates the pivotal role of GLUT1, HK2, PGI and GAPDH in cancer development. In particular, we look at how the multifunctional proteins, PGI and GAPDH, affect cancer cell survival. We also present various clinical cancer cases in terms of the overexpression of selected proteins, which may be considered as a therapeutic target.
Keywords: Glucose transporter 1, hexokinase 2, phosphoglucose isomerase, glyceraldehyde 3-phosphate dehydrogenase, cancer development
Introduction
The Warburg effect is associated with various types of cancer and causes an uptake of glucose into the cell. Thus, anaerobic glycolysis is characteristic cancer metabolism [1, 2]. Studies have shown that alterations in the metabolism of cancer cells plays a critical role in the cells survival and proliferation. With the growth of the neoplasm, blood flow to cells located inside the tumor decreases, which leads to hypoxia, and a decrease of ubiquitin-dependent degradation of the HIF-1 α transcription factor. This results in transcription which adapts cancer cells to hypoxic stress. Due to the decrease of aerobic respiration, the expression of glycolysis pathway enzymes and glucose transporters increases in response to HIF-1 (hypoxia inducible factor 1) [3, 4].
The increased expression of glucose transporter 1 (GLUT1) occurs as a result of hypoxic conditions. Furthermore, transport of vesicles with the GLUT1 transporter and its incorporation into the cell membrane increases under hypoxia [5]. The overexpression of the GLUT1 transporter contributes to the increase of glucose up-take, lactate production, and cancer development. This demonstrates that GLUT1 plays a critical role in carcinogenesis [6].
Hexokinase 2 (HK2) overexpression is another factor which contributes to the enhanced glycolytic rate and tumor progression. In tumors, when activated by HIF-1, HK2 interacts with the voltage dependent anion channel (VDAC) [7, 8]. This inhibits proapoptotic proteins and the initiation of apoptosis. The initial overexpression of HK2 under hypoxic conditions may confer a resistance to apoptosis to cancer cells [9].
PGI maintains equilibrium in glucose related processes such as glycolysis, gluconeogenesis, pentose phosphate pathway, and non-glycolytic processes. Its overexpression, which occurs in cancer, affects the glycolysis rate and also processes independent with glucose metabolism, with both functions involved in promoting cancer development [10].
GAPDH is regulated by many different factors, as well as by the enzyme itself and it can control various cellular processes. In the nucleus, the enzyme binds to nucleic acids, facilitating RNA export to the cytoplasm, and protects mRNA from degradation. Furthermore, GAPDH stabilizes telomere structure, increases genome integrity and is involved in cancer cell proliferation [11, 12]. The, multifunctional GAPDH contributes to cancer development in a more complex way compared to the previous proteins.
The purpose of this review is to describe the expression and function of the selected proteins within the glycolysis pathway and how they contribute to carcinogenesis. We show that GLUT1 transporter, hexokinase 2, phosphoglucose isomerase and glyceraldehyde 3-phosphate dehydrogenase is differentially regulated in cancers. We also list how these proteins mediate cancer development in both a glycolytic and non-glycolytic way within clinical and in vitro cases.
Functional analysis of GLUT1, HK2, PGI and GAPDH
GLUT1 interacts with glucose by transporting sugar through the membrane to the cell, while hexokinase 2 transforms glucose into glucose 6-phosphate. As the initiators of glycolysis, these two may be considered as the most important proteins within the pathway. However, the role of phosphoglucose isomerase (PGI) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) are also essential for maintaining an equilibrium in glucose catabolism. Many studies demonstrate that PGI and GAPDH are multifunctional enzymes and are involved in non-glycolytic processes to maintain cell homeostasis.
GLUT1
Glucose transporter 1 belongs to the GLUT transporter family which is in charge of facilitating the influx of hexose sugars to the cell. The class I transporters includes four glucose transporters designated 1 to 4, which have been extensively analyzed with regards to their structure, properties and tissue localization [5, 13]. Glucose transporter 1 was the first facilitative sugar transporter to be identified in the HepG2 cell line, but the real research began when GLUT1 was purified from the human erythrocyte membrane. GLUT1 is prominently expressed in the brain and erythrocytes, but a smaller amount is also present in muscle, liver and adipocytes [13, 14]. Additionally, apart from glucose, GLUT1 is also involved in the transport of galactose and ascorbic acid [15].
Hexokinase
Hexokinases (HKs) (EC 2.7.1.1) catalyze the first and most important step in the process of glucose catabolism. In the cell, glucose is phosphorylated by HKs, with concomitant dephosphorylation of ATP, to produce glucose-6-phosphate (G-6-P). G-6-P itself acts as a forerunner for glycolysis (ATP), but also for glycogenesis, pentose phosphate pathway, and biosynthesis of hexosamine. Thus, HK plays an essential role in the synthesis and break-down of metabolic processes [16]. Four isoforms of hexokinase have been characterized in humans: HK 1, 2, 3 and 4. While they possess similarities in their structure, all isoforms vary from each other significantly through expression pattern, regulatory properties, and cellular localisation. Hexokinase 2 (HK2), is abundantly expressed during embryo development, but has also been found to be elevated primarily in tissues such as adipose tissue, skeletal muscles, cardiac muscles, and in lungs [17, 18]. It has been shown that hexokinase 1 and 2 are able to bind to mitochondria via VDAC, the outer mitochondrial membrane protein, which is essential for mitochondria homeostasis. This binding to the VDAC occurs through the mitochondrial binding motif at the N-terminus. This induces a release of cytochrome c from the mitochondrial intermembrane space, leading to apoptosis [19].
Phosphoglucose isomerase
Phosphoglucose isomerase (EC 5.3.1.9) (PGI) is a ubiquitous, cytosolic enzyme, which catalyzes the second step in glycolysis, the reversible conversion of glucose-6-phosphate (G6P) to fructose-6-phosphate (F6P). Apart from glycolysis, PGI is also engaged in the pentose phosphate pathway and glucose synthesis [20]. It plays a role in glucose metabolism, and has demonstrated activities similar to a cytokine. Molecular cloning and sequencing have characterized PGI as sperm antigen-36, myofibril-bound serine proteinase inhibitor (MBSPI), neuroleukin (NLK), maturation factor (MF), and autocrine motility factor (AMF) [21]. NLK is known as a support of spinal and sensory neuron survival, as it is able to bind to a surface component of the sensory neuron [22, 23]. The cDNA sequence has shown that the neuroleukin gene is highly homologous to human PGI [24]. Maturation factor is a 54.3-kD peptide which functions to induce differentiation of myeloid leukemia HL-60 cells to terminal monocytic cells [25]. The amino acid sequence showed a 100% homology to neuroleukin and phosphoglucose isomerase and further data show that the 54.3-kD maturation factor has PGI enzymatic activity as well [26]. Extracellularly, PGI behaves like a potent cytokine and is known as an autocrine motility factor. It functions to induce the mitogenic, motogenic pathway, cell differentiation, tumor progression and the stimulation of metastasis [27]. Amino acid sequencing of purified mouse AMF showed PGI, NLK and AMF sequence compatibility [23]. Interestingly, murine AMF possesses the enzymatic features of PGI and was able to induce motility of mouse PGI-stimulated fibrosarcoma cells, which was further confirmed by using specific PGI inhibitors [28]. AMF/PGI exhibits its multifunctional growth factor-like activity via a unique cognate 78 kDa (gp78) seven-transmembrane glycoprotein receptor (autocrine motility factor receptor, AMFR) [20]. Moreover, some studies have shown that the human PGI/AMF is a phosphoprotein, which is phosphorylated at Ser185 by casein kinase 2 (CK2) and that phosphorylation inhibits its enzymatic activity [21]. It was also confirmed, that in normal and cancer cells, PGI, AMF, NLK, MF exhibit the identical cDNA sequences. This is surprising as enzyme secretion is completely unrelated to alternative splicing or maturation process in cancer cells [23].
Fig. 1.
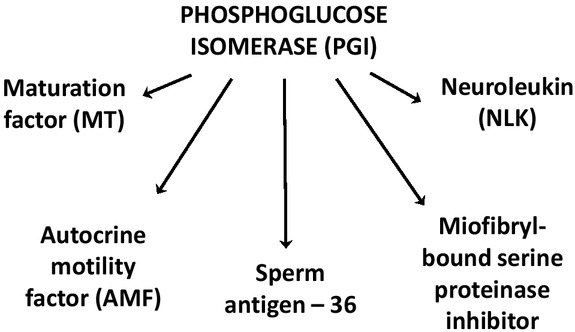
Diverse roles of phosphoglucose isomerase (PGI).
GAPDH
Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) is an enzyme within the glycolytic pathway and is responsible for the oxidation of 3-phosphoglycerate aldehyde (GAP) to 1,3-bisphosphoglycerate (1.3BPG), this results in the simultaneous production of NADH [11, 29]. GAPDH as a protein may undergo post-translational modification like S-nitrosylation, acetylation, phosphorylation, as well as O-linked N-acetyl glucosamine (O-GlcNAc) and oxidative modifications. As a result of these modifications, the functioning of the protein changes. Interestingly, as a gene encoding a single 38-kDa protein without alternate splicing, GAPDH displays various functions independent of its role in energy generation [30, 31]. It is involved in vesicular tubular complex (VTC) formation and membrane-trafficking from the endoplasmic reticulum (ER) to the Golgi compartment. Another function is maintaining the integrity of DNA through transcriptional regulator Apurinic/Apyrimidinic Endonuclease (APE-1) and is necessary for maintaining the stability of colony-stimulating factor (CSF-1) mRNA. It has been shown that elevated mRNA and protein levels of CSF-1, occurs within ovarian cancer cells, and this promotes its proliferation and invasive differentiation [32]. Another regulatory function of GAPDH is controlling the activity of angiotensin II, an essential cardiovascular protein. The enzyme can also bind to phosphatidylserine (PS) and through this, inhibits the formation of the nuclear membrane [31, 32]. Another interaction of GAPDH is the binding to telomerase RNA component (TERC), which is able to control genome and chromosome integrity as well as maintain telomere homeostasis [12, 30]. Beside these varied processes, GAPDH is involved in nuclear tRNA transport, export of RNA from the nucleus, protecting mRNA from degradation [11], apoptosis, DNA repair, DNA replication in the nucleus, vesicular transport, membrane fusion, microtubule bundling, cell invasion, carcinogenesis and iron metabolism [33, 34]. Not surprisingly, many studies present GAPDH as a contributor in the development of various human pathologies. It has been confirmed, that reciprocal action between GAPDH and proteins like LAPP (L-amyloid precursor protein), huntingtin, atrophin, ataxin and AR (androgen receptor) occurs; where each protein is involved in varying degrees in the development or progression of these disorders [35].
Changes in gene expression
Carcinogenesis is a process whereby normal cells transform into cancerous cells. The genesis of this transformation lies in the mutation of DNA in the cell. These mutations lead to changes in gene expression and can disrupt the balance between cell proliferation and apoptosis pathways. Proliferating cancer cells require an increased amount of energy. This can be obtained by increasing glycolysis, and as a result, leads to the upregulation of glycolytic proteins, such as GLUT1, HK2, PGI and GAPDH. Numerous studies have confirmed that changes in specific gene expression profiles, promote cancer cell development, tumor growth and metastasis. Proteins within the glycolysis pathway are involved in a number of functions, GLUT1 is involved in increasing up-take of glucose by cancer cells. On the other hand, beyond converting glucose to G-6-P, Hexokinase 2, is engaged in inhibition of apoptosis. While both PGI and GAPDH possess functions which contribute to cancer cell survival.
Fig. 2.
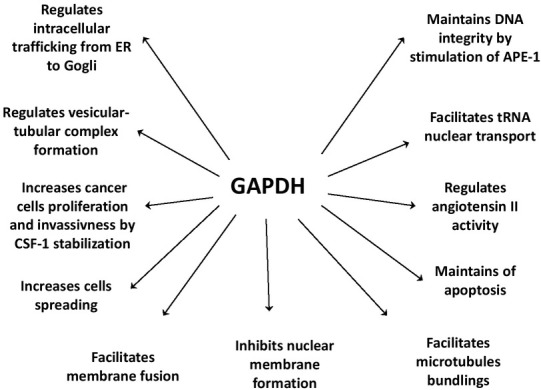
Diverse functions of glyceraldehyde 3-phosphate dehydrogenase (GAPDH).
GLUT1 causes an up-regulation in glucose up-take
It has been demonstrated that expression of glucose transporter 1 is notably enhanced at both the transcriptional (mRNA) and posttranscriptional (protein) level in response to hypoxia [5]. The control at the transcriptional level engages transactivation via Sp1 and suppression through Sp3. The ratio of these two transcriptional factors establishes the final levels of GLUT1 mRNA and protein [15]. Moreover, glucose transporter 1 has been shown to be regulated not only by HIF-1, but also through c-Myc and protein kinase B (PKB) [36]. Tumor cells enhance glucose uptake via induction of GLUT1 and sodium/ glucose cotransporter 1 (SGLT1) and coordinate the increased entry of glucose with increased glycolysis [37]. The elevated levels of GLUT1, which occurs primarily in hypoxic tumors, results in an enhanced influx of glucose to the cell. As a result this leads to an increase of the glycolysis rate. This aids in tumor progression and a subsequent increased risk of metastasis [6, 38]. The isoform 1 of glucose transporter has been shown to be overexpressed in various types of human cancers and this is usually associated with poor patient outcomes. In contrast, the insulin-sensitive GLUT4 expression is significantly decreased in cancer cells. The disruption in balance between glucose transporter 1 and 4 expression may lead to insulin independent sugar uptake in cancer cells [36, 38]. There are also studies that have reported associations between GLUT expression and proliferative indices, whilst others suggest that GLUT expression may be of prognostic significance [39].
Hexokinase 2 targets apoptosis
Hexokinase 2 isoform is a unique glycolytic enzyme in tumor metabolism. Under conditions when there is limited access to oxygen in cancer cells or in solid tumors, when there is a complete lack of oxygen, hexokinase 2 mRNA and protein level is significantly increased. In some cases, HK2 expression is increased by as much as 10-times. This then causes an increase of the glycolytic pathway and, subsequently, enhanced glucose uptake by GLUT1 [40, 41]. It has been demonstrated that oxygen deprivation results in the activation of HIF-1, which binds to c-Myc to transactivate HK2. Then, the phosphorylated hexokinase 2 binds to VDAC1 (voltage-dependent anion channel 1) at the outer mitochondrial membrane. The interaction between HK2 and VDAC1, inhibits the binding of the proapoptotic protein, Bax to VDAC1. This prevents the formation of the channel through which cytochrome c (cyt c) can escape from the mitochondria to trigger apoptosis. The increased expression of HK2 in cancer cells induces a specific shift from HK1 to HK2, which brings a huge metabolic advantage by granting protection from apoptosis [36]. Through mitochondrial binding, HK2 is constantly active and directly stimulated by ATP from the oxidative phosphorylation (OXPHOS). This allows the enzyme to conduct glucose phosphorylation more efficiently, independent of ATP supply from glycolysis. Apart from regulation of glucose flux to the cell, the mitochondrial location of hexokinase 2 plays a major role in regulating life and death of cancer cells [42]. In cancer cells the amount of HK2 binding to the outer mitochondrial membrane is significantly elevated. Some studies have established that in liver tumor cells, hexokinase 2 protein level was elevated more that 500-times compared to normal cells [17]. Furthermore, another study confirmed that in tumor cells, hexokinase 2 is associated with the mitochondrial surface even in 80% of its total amount [43]. In cancerous cells where there is a significant increase in HK2 expression, chemical agents that down-regulate HK2 activity and prevent its binding to the voltage dependent anion channel 1 are able to kill off cancer cells [42].
Fig. 3.
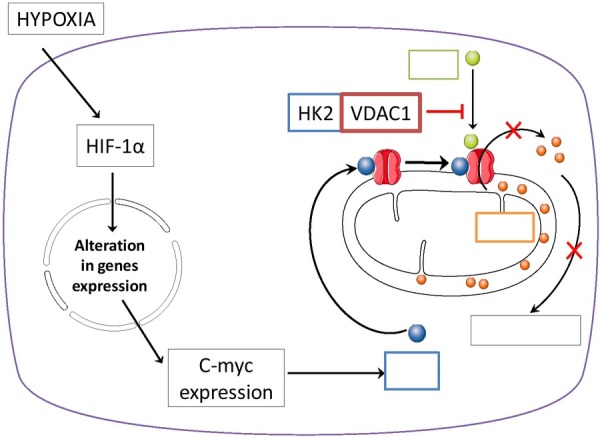
Hexokinase 2 (HK2) transactivation by c-myc (via HIF-1) leads to HK2 binding to VADC1, and Bax protein binactivation arrests the release of cytochrome c (cyt c) to inhibit apoptosis.
Factors affecting PGI gene up-regulation
The enzyme, PGI/AMF, is involved in stimulating cell motility and the growth of cancer cells [44]. This stimulation is achieved through PGI/AMF attachment to the cell surface receptor, gp78. The binding by PGI/AMF activates the protein kinase C (PKC) pathway which promotes cell locomotion by enhancement arachidonic acid synthesis. Numerous studies confirm, that elevated expression of AMF and gp78 is associated with tumor malignancy [28, 45]. The upregulation of PGI/AMF is known to activate the ERK signaling pathway and this leads to an elevated expression of Cyclin D1. This results in increased metastasis and a decrease in patient survival [46, 47, 48]. The up-regulation of PGI/AMF induces activation of the SNAIL promoter and an increased expression of the transcription factor SNAIL. This causes a decrease in E-cadherin expression and an increase in cell motility [20, 27, 49]. Moreover, regulating SNAIL expression contributes to EMT (ephitelial to mesenchymal transition), MET (mesenchymal to ephitelial transition) and cancer invasion [50]. HER2 receptor overexpression is known to enhance AMF secretion in breast cancer, which increases tumor aggressiveness and drug resistance [51]. The upregulation of PGI/AMF is also associated with an increase in DNA-binding activity of NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells), which increases ZEB1 and ZEB2 expression, resulting in the induction of EMT [52].
Under hypoxic conditions, the mRNA level of PGI/AMF expression increases in different human cancer cell lines and appears to be controlled via the vascular endothelial growth factor (VEGF), which is mediated by HIF-1α [10, 53, 54]. Some studies indicate that HIF-1α induces PGI/AMF expression during oxygen deprivation, in a PI3K-dependent way. Under hypoxic conditions a dramatic increase of PGI/AMF levels are observed in the nucleus [10]. The secretion may be caused by PGI/AMF phosphorylation at serine 185 by CK2, which is likely due to conformational changes [22].
Other studies suggest that AMF stimulation activates matrix metalloproteinase-3 (MMP3) expression through a MAPK signaling pathway and this results in increased cell motility [55, 56]. The PGI/AMF overexpression contributes to new micro vessel formation and cells treated with AMF express gp78, which is associated with motility [45]. It has been shown that gp78 expression was significantly increased in neoplastic cells, which may lead to increased efflux [23]. Moreover, the inactivation of the gp78 receptor, diminishes the activation of the transduction pathways that are associated with the onset of EMT/migration/ invasion [57].
Fig. 4.
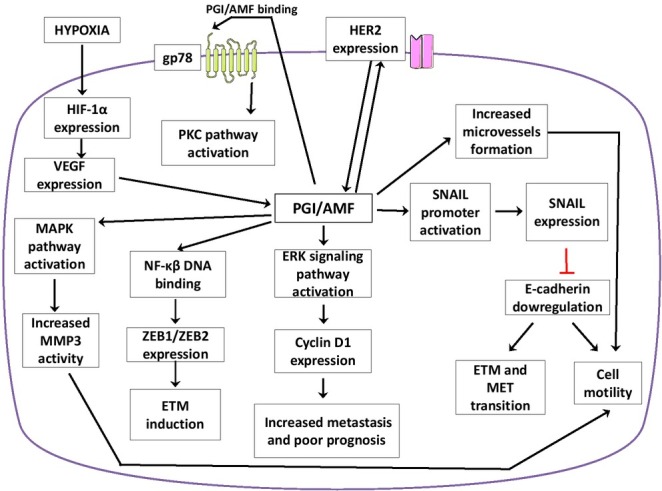
PGI/AMF/NLK/MF implications in cancer cell.
GAPDH role in cancer development
It has been established that in rapidly dividing neoplastic cells, expression of GAPDH is strongly dependent on the proliferative phase of the cell [36]. During oxygen deprivation, mRNA levels of GAPDH are significantly elevated. This up-regulation is associated with enhanced HIF-1α activity, which has been observed in vascular endothelial cells (EC) and in alveolar epithelial cells [29, 58]. GAPDH up-regulation in EC appears in the nucleus, particulate, and cytosolic fractions [59]. Hypoxia activates the PI3K/AKT pathway, phosphorylating and activating AKT (p-AKT). This leads to an increase in HIF-1α expression causing an upregulation of glycolytic enzymes. It was also shown that insulin mediated AKT activation, leading to GAPDH phosphorylation and the promotion of glycolysis. Interestingly, in neoplastic cells, GAPDH acts together with activated PKB and inhibits its dephosphorylation, which leads to Bcl-xl overexpression and as a result, protects cancer cells from CICD (caspase-independent cell death) [34].
It is known that GAPDH undergoes regulation via endogenous NO and p53 transcription factor. NO and p53 contribute to GAPDH overexpression, translocation and enhances its binding to SIAH1, which leads to the induction of apoptosis [42]. It was recently demonstrated that GAPDH increases its activity in response to ethanol stimulation. It translocates to the nucleus and interacts with TIEG2, which leads to TIEG2- mediated gene transcription for MAO B and the neuron apoptosis [60]. This demonstrates the important role that GAPDH plays in apoptosis. Another apoptotic pathway involves TNF-related apoptosis-inducing ligand (TRAIL). TRAIL is cytotoxic to most thyroid cancer cells and induces GAPDH NO-mediated S-nitrosylation. This leads to GAPDH nuclear translocation and initiation of apoptosis [61]. The Sp1 transcription factor is known to bind to the GC-box in breast cancer cells in the presence and lack of oxygen. The knockdown of Sp1 decreases hypoxia-increased promoter activity and GAPDH expression in MCF-7 cells [62]. It was also shown that in MCF7 human breast cancer cells, estradiol induced a statistical increase in GAPDH expression [63]. GAPDH is also engaged in the up-regulation of Sp-1 mediated SNAIL expression, leading to epithelial to mesenchymal transition (ETM) [64].
The suppression of c-Jun/AP-1 or Ca2+/calmodulin-dependent protein kinases activity causes a downregulation of GAPDH mRNA levels. The increased concentration of intracellular Ca2+ elevates expression of the GAPDH gene via the AP-1-dependent pathway [59]. The enzyme is also known to promote mRNA stability of colony-stimulating factor-1 (CSF-1) by binding to its AU-rich terminal 144nt region. This cytokine is associated with carcinogenesis and leads to a worse prognosis among patients with ovarian carcinoma [65]. It also has been proven, that GAPDH elicits aging in cancer cells, causing a down-regulation of telomerase activity and repression of the telomere length shortening process. Interaction between enzymes is down-regulated via NAD+ and G3P, best known as GAPDH substrates. This demonstrates that its substrates act as a significant check to stop GAPDH shifting from glucose breakdown process to telomere signaling [30]. When GAPDH binds to telomeres, it regulates telomerase activity. This results in suppression of the telomere length shortening process. GAPDH also functions to stabilize the structure of chromosomes and the integrity of the genome, and thus maintains equilibrium of cancer cells proliferation [12]. The overexpression of GAPDH interacts with the polyQ tract of the androgen receptor (AR) which mediates prostate cancer development and growth. GAPDH elevates AR transactivation, by initiating endogenous PSA (prostate-specific antigen) expression. For this reason, enhanced GAPDH expression in prostate cancer can be one of the many reasons why anti-androgens are unsuccessful in tumor growth suppression and instead has the opposite effect [33].
Appearance in cancer and clinical cases
Many studies indicate that GLUT1 and HK2 are considered to be the most important proteins in the Embden-Meyerhof-Parnas pathway. This is because many clinical cancer cases have established the presence and overexpression in various cancer types. In most cases, GLUT1 and hexokinase 2 up-regulation is caused by tumor hypoxia and HIF-1α overactivity. Notwithstanding, phosphoglucose isomerase and GAPDH seem to be involved in many clinical cases as well, but their appearance is not always dependent on hypoxia.
GLUT1 expression increases via HIF-1α
Various studies have demonstrated that GLUT1 expression is regulated in many types of cancers throughout the body [66, 67]. Glucose transporter 1 was found to be involved in enhancing the malignant potential and invasiveness, in colorectal, gastric, lung, ovarian and urinary bladder cancers [66, 68]. An altered level of GLUT1 has also been observed in breast cancer [67]. Interestingly, both endometrial and breast poorly differentiated tumors had significantly increased levels of GLUT1 and GLUT3 expression when compared to well-differentiated tumors. GLUT1 and GLUT3 seem to be important markers in endometrial and breast tumor differentiation [68].
Additionally, enhanced expression of GLUT1 is related with negative estrogen receptor (ER), progesterone receptor (PR), and higher nuclear grade. Moreover, breast cancers with positive GLUT1 expression were found to act more aggressively and have increased malignant features [66]. Thus, glucose transporter 1 may be taken into consideration as a predictor of poorer prognosis among breast cancer patients [66]. Glucose transporter 1 expression in squamous cell carcinomas differs from adenocarcinomas, both in localization and cellular occurrence.
It has been estimated that significant cytoplasmic expression was found in gastric, breast, prostate, thyroid papillary and follicular adenocarcinomas. The positive GLUT1 cytoplasmic pattern has been shown in gastric adenocarcinomas, including diffuse and intestinal tumors. Membranous expression was found in cervix uterine squamous cell carcinomas as well as in neck and head carcinoma [15]. Nonetheless, sarcomas, lymphomas, hepatoblastomas, and melanomas have shown no observable GLUT1 expression, suggesting, that in these particular types of cancers, a different GLUT family member controls sugar transportation, or that GLUT1 expression appears at a specific phase of cancer development [15].
Hexokinase 2 in clinical reports
Hexokinase 2 was found to be up-regulated in many types of cancer, resulting in an increased glycolytic rate, which is a phenotype for cancer cells [69]. HK2 overexpression is seen in many tumors, for example, breast and cervical cancer, as well as pancreatic. Furthermore, there is potential for it to be involved in the metastatic cascade. Other studies have shown that increased expression of HK2 is involved in the progression of CCA (cholangiocellular carcinoma) and HCC (hepatocellular carcinoma) [70, 71]. The HK2-immunohistochemistry of CCA patients demonstrated that the HK2 gene was expressed at low levels in normal stromal tissue. In contrast, the expression is significantly elevated in hyperplasia, dysplasia and CCA. In this particular study, HK2 was observed in the cytoplasm however the enzyme’s irregular expression was perceived in precancerous and CCA samples, suggesting HK2 plays a critical role in development of this specific cancer [72]. Hexokinase 2 expression is also up-regulated in lung carcinomas and is required by proliferating cancer cells caused by the impairment of tumor progression following its downregulation [73]. Furthermore, it has been found to be highly expressed in laryngeal carcinoma and oral squamous cell carcinoma. Moreover, the expression of HIF-1α was observed to influence hexokinase gene expression in gastric adenocarcinoma [34]. The increased expression of HK2 occurs in ovarian, liver, and colon carcinoma as well as in non-Opisthorchis viverrini (Ov) associated CCA [70-72]. Elevated levels of HK2 are correlated with poor outcomes, with respect to tumor size and increased metastatic potential [72]. Hexokinase 2 expression is increased in human glioblastoma multiforme (GBM), the most common malignant brain tumor [19]. It also has been established that elevated expression of hexokinase 2 occurs in metastatic neuroblastoma forms and in samples from stage 4 neuroblastoma patients when compared to local variants and samples from stage 1 patients. This increased HK2 expression is clearly associated with tumor malignancy and gives neuroblastoma, metastatic features [74]. The disruption of HK2 activity in GBM cells restores oxidative glucose metabolism and increases apoptosis [19].
PGI/AMF/NLK/MF in clinical cancer cases
PGI/AMF is secreted by tumors and has been shown to be involved in tumor progression [75]. Initially, PGI was characterized as an autocrine motility factor (AMF) from the conditioned medium of human melanoma. It is involved in proliferation, invasion, migration, tumor survival, and neovascularization [76]. The levels of PGI/AMF and its cell surface receptor gp78/AMFR expression are associated with the pathologic stage, grade, and degree of tumor penetration to surrounding tissues. It has been shown to be overexpressed along with PGI/AMF in various metastatic tumors [77]. The presence of both receptor and enzyme in the serum acts as a biomarker as it is associated with cancer progression [21].
However, AMF has not yet been observed as a secretory protein in normal cells. Many clinical reports indicate that the enzymatic activity of AMF was present in the serum and urine of patients with malignant tumors like breast, lung, colorectal, kidney and gastrointestinal carcinomas [77]. Moreover, abnormally elevated activity of PGI/AMF was observed in the plasma of patients with acute myelogenous leukemia [26]. In metastatic cancer biopsies it was found that there were elevated levels of PGI/AMF. [57]. Another study has shown that the PGI/AMF concentration in urine is increased in patients with transitional cell bladder carcinoma [78]. Autocrine motility factor expression has also been detected in tissue specimens from 119 patients with pulmonary adenocarcinoma. AMF expression was correlated with lymph node metastasis. It was found that the prognosis of AMF-positive patients was poorer than that of AMF-negative patients. Within pulmonary adenocarcinomas, the expression of AMF is associated with tumor aggressiveness [79]. Overexpression of PGI was also found in paraffin sections from 10 colon cancer patients by immunohistochemistry. Strong cytoplasmic staining of the PGI protein was detected in cancer specimens, but weakly expressed in normal colonic epithelium. Strongly increased PGI expression was observed in the cytoplasm of cancer cells but not in that of normal colonic cells [50]. The stage IV malignant breast cancer biopsies also exhibited an elevated amounts of PGI/AMF, at 60-times normal [57]. It was established that oxygen deprivation increases the mRNA expression of the autocrine motility factor in numerous cancer cells and also enhanced the motility of pancreatic cancer cells [20, 53].
The expression of AMF/PGI/NLK mRNA in the pancreatic, colon, lung, hepatoma and ovarian cancer cell lines was elevated more during oxygen deprivation, suggesting that the enhanced AMF/PHI/NLK expression in hypoxia was common in various cancer cells [53]. Moreover, in hepatoma cell lines treated with exogenous PGI/AMF, there was increased migration of Huh7 and HepG2 cells with MMP-3 as a mediatory factor of this process. However PGI/AMF does not affect Hep3B cells [55]. Another report revealed that in the human colon, bladder, fibrosarcoma, prostate and breast cancer cell line, PGI/AMF mRNA presented an increased concentration in hypoxia (10-fold) than in normoxia, which was similar to the increased level of HIF-1α in response to oxygen deprivation [10]. Increased expression of PGI also correlated with the enhancement of tumor formation and metastasis of human colon cancer cells [50]. In breast carcinoma, hypoxia increased expression of the HIF-1α, which led to PGI/AMF overexpression and was down-regulated via phosphatidylinositol 3-kinase signaling pathway inhibitors [10]. This study showed that the cell motility factor PGI/AMF is a hypoxia-inducible gene in various cancer cell lines, resulting in its elevated secretion following induction of cell motility.
GAPDH clinical and in vitro cancer reports
To maintain a fast rate of proliferation, escape from the immune system, and avoidance of apoptosis, cancer cells are required to switch to a high glycolytic rate. As a result, there is an increased expression of proteins involved in glycolysis. With the increase in energy, there is an increase in tumor size, and this leads to hypoxia. It is well documented and confirmed by many studies in cancer cells, that hypoxic conditions contribute to an increase of HIF-1α expression and causing changes of glyceraldehyde 3-phosphate dehydrogenase expression. Thus, GAPDH is overexpressed during a lack of oxygen in many cancer cell lines like AR-positive prostate adenocarcinoma cells [33], alveolar epithelial cells and spontaneous cervical cancer cells and brain capillary endothelial cells [80], prostate [81], and lung cancer cell line [82].
However, it has been observed that GAPDH is not always overexpressed under conditions where there is a lack of oxygen. In many neoplastic cell lines, the enzyme expression seems to be independent from HIF-1α activity. It was present in malignant glioma cells, hepatocellular carcinoma, hepatoma, lung adenocarcinoma epithelial cell lines, and colon cancer cell lines [80]. Independently from HIF-1α expression, GAPDH and Na+/K+-ATPase were observed on the cell surface of breast primary cancer cells and another two breast cancer cell lines [83]. Elevated levels of GAPDH has also been demonstrated in epithelial ovarian carcinoma cells [65]. Additionally, it was shown that S-nuclear transportation of GAPDH and accumulation of protein carbonyls (a marker of oxidative damage) in TRAIL-sensitive thyroid cancer cells [61]. appropriate The regulation of GAPDH under hypoxic condition, suggest that enzyme cannot be consider as an therapeutic. On the other hand, there is still plenty of evidence that confirms GAPDH overexpression in clinical cases.
GAPDH overexpression has been demonstrated in almost 50% of 256 ovarian and fallopian tube cancer samples. Co-expression with CSF-1 has also been found in ovarian carcinoma [65]. Additionally, serum CSF-1 levels were found to be valid tumor marker, with enhanced the likelihood of foreshadowing relapse of disease and its progression. Increased concentration of both serum and ascitic CSF-1 levels are associated with a worse prognosis. GAPDH’s role in prognosis was examined in breast cancer, where the GAPDH mRNA expression in samples correlated with a reduction of patient survival [65]. In human prostate cancer, GAPDH overexpression was observed, in an increased amount in the nuclei of high-grade prostate tumors, when compared to normal prostate tissue. Moreover, bone metastases from prostate cancer exhibited significantly augmented cytoplasmic enzyme expression [81]. According to the prostate cancer study, in 13 prostate cancer samples, diverse nuclear and cytoplasmic GAPDH staining was observed, but it was not noticeable in the normal tissues [84]. Another study demonstrated that, in prostate cancer the expression of GAPDH was dependent on the stage of the disease, and it was particularly increased in advanced disease phases [85]. The same study provided evidence that GAPDH is overexpressed in cervix and breast cancer. In these cervix, breast, and prostate cancer cases, overexpression of GAPDH has been found in membrane, cytoplasmic and particularly in the nuclear compartment [85]. The GAPDH gene was also observed in a huge amount of unselected primary breast cancers, and in MCF7 cells treated with estradiol. In the samples of primary breast cancers, it was found that expression of the enzyme’s gene was statistically significantly related with estradiol and progesterone receptor concentrations. Elevated GAPDH expression was also correlated with decreased general survival and relapse-free survival in patients with primary breast cancer. Nonetheless, statistical estimations revealed that GAPDH cannot be taken into consideration as a useful prognostic factor in breast cancer [63]. It has been confirmed that GAPDH also undergoes up-regulation during melanoma progression [86]. Enzyme’s mRNA expression is notably elevated in thick melanomas when compared to primary thin melanomas. This also occurs in metastatic melanomas when compared to lymph-node metastatic melanomas. However, no specific point-mutations in GAPDH-specific exons were found in any patient [86]. In human colorectal carcinoma tissue GAPDH expression is significantly up-regulated compared to the adjacent normal tissues, and also increased in colon cancer cell lines when compared to the non-tumor colon mucosa cells in culture. The expression of GAPDH was further elevated in the liver metastatic tissues compared to the original colon cancer tissue of the same patients, suggesting that high expression of GAPDH might play an important role in colon cancer development and metastasis [87]. Another study confirms that in colorectal cancer patients there is an increased GAPDH expression. Interestingly, its expression in stage I tumors was more highly elevated than in any other stage of progression. In addition, patients with a more advanced disease stage presented similar GAPDH expression levels, with no meaningful differences between them [88]. However, there was a notable decrease in GAPDH expression when progression of invasion into the intestine wall and the extension to neighboring structures occurred [88]. Nevertheless, GAPDH expression is elevated in colon cancer and significantly enhanced in liver metastasis, which leads to the conclusion that the enzyme may contribute to the induction of colon cancer metastasis [64]. The study included 82 consecutive patients, stage I-III NSCLC provides evidence, that GAPDH gene overexpression in resected tumor samples is an adverse prognostic factor in NSCLC [89].
Conclusion
We presented an important role of GLUT1, HK2, PGI and GAPDH in cancer progression. While, in cancer, GLUT1 enhances glucose up-take and glycolytic rate, HK2 seems to be major regulator of apoptosis. It has been proved that PGI participates in cancer metastasis and invasion and GAPDH affects cancer cell survival. Nevertheless, not all of them are considered to be good prognostic factors in cancer development. According to clinical reports, GLUT1 and hexokinase 2 appeared in most clinical cases and should be taken into consideration in the context of a good target for cancer therapy, but further investigation is necessary.
Table 1.
Alterations in selected glycolytic gene expression.
| Protein | Type | Localization | Expression alterations | Type of cancer | References |
|---|---|---|---|---|---|
| Glucose transporter 1 (GLUT1) | Transporter | Membrane | Overexpressed | Brain, head and neck, thyroid, lung, breast, stomach, colorectal, renal, kidney, urinary bladder, ovarian, endometrial, prostate. | 15, 66, 67, 68 |
| Hexokinase 2 (HK2) | Enzyme | Cytoplasm | Overexpressed | Glioblastoma, neuroblastoma, lung, breast, liver, hepatocellular, cholangiocarcinoma, pancreatic, colon, ovarian. | 19, 70, 71, 72, 73, 74 |
| Phosphoglucoso isomerase (PGI) | Enzyme | Cytoplasm | Overexpressed | Breast, lung, pulmonary adenocarcinoma, gastrointestinal, kidney, colorectal, leukemia. | 26, 50, 57, 77, 78, 79 |
| Glyceraldehyde 3-phosphate | Enzyme | Cytoplasm | Overexpressed | Lung (NSCLC), breast, colorectal, ovarian, fallopian, prostate, melanoma. | 63, 64, 65, 81, 84-89 |
| dehydrogenase (GAPDH) |
Footnotes
Conflict of interest: Authors state no conflict of interest
References
- [1].Alfarouk KO, Muddathir AK, Shayoub MEA.. Tumor Acidity as Evolutionary Spite. Cancers. 2011;3(1):408. doi: 10.3390/cancers3010408. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Hsu PP, Sabatini DM. Cancer Cell Metabolism: Warburg and Beyond. Cell. 2008;134:703. doi: 10.1016/j.cell.2008.08.021. –. [DOI] [PubMed] [Google Scholar]
- [3].Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012;40:109. doi: 10.1093/nar/gkr988. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kim JW, Dang CV. Cancer’s molecular sweet tooth and the Warburg effect. Cancer Res. 2006;66(18):8927. doi: 10.1158/0008-5472.CAN-06-1501. –. [DOI] [PubMed] [Google Scholar]
- [5].Zhang JZ, Alireza B, Faramarz IB. Regulation of glucose transport by hypoxia. Am J Kidney Dis. 1999;34(1):189. doi: 10.1016/s0272-6386(99)70131-9. –. [DOI] [PubMed] [Google Scholar]
- [6].Airley R, Loncaster J, Davidson S, Bromley M, Roberts S, Patterson A. Glucose transporter glut-1 expression correlates with tumor hypoxia and predicts metastasis-free survival in advanced carcinoma of the cervix. Clin Cancer Research. 2001;7(4):928. et al. –. [PubMed] [Google Scholar]
- [7].Rempel A, Mathupala SP, Griffin CA, Hawkins AL, Pedersen PL. Glucose catabolism in cancer cells: amplification of the gene encoding type II hexokinase. Cancer Res. 1996;56:2468. –. [PubMed] [Google Scholar]
- [8].Mathupala SP, Ko YH, Pedersen PL. Hexokinase II: cancer’s double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene. 2006;25:4777. doi: 10.1038/sj.onc.1209603. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Shimizu S, Matsuoka Y, Shinohara Y. Essential role of voltage-dependent anion channel in various forms of apoptosis in mammalian cells. J Cell Biol. 2001;152:237. doi: 10.1083/jcb.152.2.237. et al. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Funasaka T, Yanagawa T, Hogan V, Raz A. Regulation of phosphoglucose isomerase/autocrine motility factor expression by hypoxia. FASEB J. 2005;19(11):1422. doi: 10.1096/fj.05-3699com. –. [DOI] [PubMed] [Google Scholar]
- [11].Bonafé N, Gilmore-Hebert M, Folk NL, Azodi M, Zhou Y, Chambers SK. Glyceraldehyde-3-phosphate dehydrogenase binds to the AURich 3′ untranslated region of colony-stimulating factor-1 (CSF-1) messenger RNA in human ovarian cancer cells: Possible role in CSF-1 posttranscriptional regulation and tumor phenotype. Cancer Res. 2005;65:3762. doi: 10.1158/0008-5472.CAN-04-3954. –. [DOI] [PubMed] [Google Scholar]
- [12].Pariona-Llanos R, Pavani RS, Reis M, Noël V, Silber AM, Armelin HA, Elias MC. Glyceraldehyde 3-Phosphate Dehydrogenase-Telomere Association Correlates with Redox Status in Trypanosoma cruzi. PLoS ONE. 2015;10(3) doi: 10.1371/journal.pone.0120896. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wood IS, Trayhurn P. Glucose transporters (GLUT and SGLT): expanded families of sugar transport proteins. Br J Nutr. 2003;89(1):3. doi: 10.1079/BJN2002763. –. [DOI] [PubMed] [Google Scholar]
- [14].Vrhovac I, Breljak D, Sabolic I. Glucose transporters in the mammalian blood cells. Period Biol. 2014;116:61. –. [Google Scholar]
- [15].Carruthers A, DeZutter J, Ganguly A, Devaskar SU. Will the original glucose transporter isoform please stand up! Am J Physiol Endocrinol Metab. 2009;297:836. doi: 10.1152/ajpendo.00496.2009. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Roberts DJ, Miyamoto S. Hexokinase II integrates energy metabolism and cellular protection: Akting on mitochondria and TORCing to autophagy. Cell Death Differ. 2015;22:248. doi: 10.1038/cdd.2014.173. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Chen Z, Zhang H, Lu W, Huang P. Role of mitochondria-associated hexokinase II in cancer cell death induced by 3-bromopyruvate. Biochim Biophys Acta. 2009;1787(5):553. doi: 10.1016/j.bbabio.2009.03.003. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Wilson JE. Isozymes of mammalian hexokinase: structure, subcellular localization and metabolic function. J Exp Biol. 2003;206:2049. doi: 10.1242/jeb.00241. –. [DOI] [PubMed] [Google Scholar]
- [19].Wolf A, Agnihotri S, Micallef J, Mukherjee J, Sabha N, Cairns R. Hexokinase 2 is a key mediator of aerobic glycolysis and promotes tumor growth in human glioblastoma multiforme. J. Exp. Med. 2011;208(2):313. doi: 10.1084/jem.20101470. et al. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Tsutsumi S, Yanagawa T, Shimura T, Kuwano H, Raz A. Autocrine Motility Factor Signaling Enhances Pancreatic Cancer Metastasis. Clin Cancer Res. 2004;10:7775. doi: 10.1158/1078-0432.CCR-04-1015. –. [DOI] [PubMed] [Google Scholar]
- [21].Yanagawa T, Funasaka T, Tsutsumi S, Raz T, Tanaka N, Raz A. Differential regulation of phosphoglucose isomerase/autocrine motility factor activities by protein kinase CK2 phosphorylation. J Biol Chem. 2005;280(11):10419. doi: 10.1074/jbc.M409457200. –. [DOI] [PubMed] [Google Scholar]
- [22].Haga A, Niinaka Y, Raz A. Phosphohexose isomerase/autocrine motility factor/neuroleukin/maturation factor is a multifunctional phosphoprotein. Biochim et Biophys Acta. 2000;1480:235. doi: 10.1016/s0167-4838(00)00075-3. –. [DOI] [PubMed] [Google Scholar]
- [23].Niinaka Y, Paku S, Haga A, Watanabe H, Raz A. Expression and Secretion of Neuroleukin/Phosphohexose Isomerase/Maturation Factor as Autocrine Motility Factor by Tumor Cells. Cancer Research. 1998;58:2667. –. [PubMed] [Google Scholar]
- [24].Chaput M, Claes V, Portetelle D, Cludts I, Cravador A, Burny A. The neurotrophic factor neuroleukin is 90% homologous with phosphohexose isomerase. Nature. 1988;332(6163):454. doi: 10.1038/332454a0. et al. –. [DOI] [PubMed] [Google Scholar]
- [25].Cordeiro AT, Godoi PHC, Silva CHTP, Garratt RC, Oliva G, Thiemann OH. Crystal structure of human phosphoglucose isomerase and analysis of the initial catalytic steps. Biochimica et Biophysica Acta. 2003;1645:117. doi: 10.1016/s1570-9639(02)00464-8. –. [DOI] [PubMed] [Google Scholar]
- [26].Xu W, Seiter K, Feldman E, Ahmed T, Chiao JW. The differentiation and maturation mediator for human myeloid leukemia cells shares homology with neuroleukin or phosphoglucose isomerase. Blood. 1996;87(11):4502. –. [PubMed] [Google Scholar]
- [27].Funasaka T, Hogan V, Raz A. Phosphoglucose isomerase/autocrine motility factor mediates epithelial and mesenchymal phenotype conversions in breast cancer. Cancer Res. 2009;69:5349. doi: 10.1158/0008-5472.CAN-09-0488. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Watanabe H, Takehana K, Date M, Shinozaki T, Raz A. Tumor Cell Autocrine Motility Factor Is the Neuroleukin/Phosphohexose Isomerase Polypeptide. Cancer Res. 1996;56:2960. –. [PubMed] [Google Scholar]
- [29].Graven KK, McDonald RJ, Farber HW. Hypoxic regulation of endothelial glyceraldehyde-3-phosphate dehydrogenase. Am J Physiol Cell Physiol. 1998;274(2):347. doi: 10.1152/ajpcell.1998.274.2.C347. –. [DOI] [PubMed] [Google Scholar]
- [30].Nicholls C, Pinto AR, Lia H, Li L, Wang L, Simpson R. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) induces cancer cell senescence by interacting with telomerase RNA component. Proc Natl Acad Sci USA. 2012;109(33):13308. doi: 10.1073/pnas.1206672109. et al. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Sirover MA. Structural analysis of glyceraldehyde-3-phosphate dehydrogenase functional diversity. Int J Bichem Cell Biol. 2014;57:20. doi: 10.1016/j.biocel.2014.09.026. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Sirover MA. On the functional diversity of glyceraldehyde-3-phosphate dehydrogenase: Biochemical mechanisms and regulatory control. Biochim et Biophys Acta. 2011;1810:741. doi: 10.1016/j.bbagen.2011.05.010. –. [DOI] [PubMed] [Google Scholar]
- [33].Harada N, Yasunaga R, Higashimura Y, Yamaji R, Fujimoto K, Moss J. Glyceraldehyde-3-phosphate Dehydrogenase Enhances Transcriptional Activity of Androgen Receptor in Prostate Cancer Cells. JBiol Chem. 2007;282(31):22651. doi: 10.1074/jbc.M610724200. et al. –. [DOI] [PubMed] [Google Scholar]
- [34].Zhang JY, Zhang Hong CQ, Giuliano AE, Cui XJ, Zhou GJ. Critical protein GAPDH and its regulatory mechanisms in cancer cells. Cancer Biol Med. 2003;12:10. doi: 10.7497/j.issn.2095-3941.2014.0019. et al. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Sirover MA. New insights into an old protein: the functional diversity of mammalian glyceraldehyde-3-phosphate dehydrogenase. Biochim et Biophys Acta. 1999;1432:159. doi: 10.1016/s0167-4838(99)00119-3. –. [DOI] [PubMed] [Google Scholar]
- [36].Zheng L, Li J, Luo Y. Deniz Ekinci. Glucose Metabolism and Cancer. Biochemistry, Prof. In Tech; 2012. [Google Scholar]
- [37].Ganapathy V, Thangaraju M, Prasad PD. Nutrient transporters in cancer: relevance to Warburg hypothesis and beyond. Pharmacol Ther. 2009;121(1):29. doi: 10.1016/j.pharmthera.2008.09.005. –. [DOI] [PubMed] [Google Scholar]
- [38].Szablewski L. Expression of glucose transporters in cancers. Biochim Biophys Acta. 2013;1835(2):164. doi: 10.1016/j.bbcan.2012.12.004. –. [DOI] [PubMed] [Google Scholar]
- [39].Airley RE, Mobasheri A. Hypoxic regulation of glucose transport, anaerobic metabolism and angiogenesis in cancer: novel pathways and targets for anticancer therapeutics. Chemotherapy. 2007;53(4):233. doi: 10.1159/000104457. –. [DOI] [PubMed] [Google Scholar]
- [40].Yasuda S, Arii S, Mori A, Isobe N, Yang W, Oe H, Fujimoto A, Yonenaga Y, Sakashita H, Imamura M. Hexokinase II and VEGF expression in liver tumors: correlation with hypoxia-inducible factor-1α and its significance. J Hepatology. 2004;40(1):117. doi: 10.1016/s0168-8278(03)00503-8. –. [DOI] [PubMed] [Google Scholar]
- [41].Natsuizaka M, Ozasa M, Darmanin S, Miyamoto M, Kondo S, Kamada S. Synergistic up-regulation of Hexokinase-2, glucose transporters and angiogenic factors in pancreatic cancer cells by glucose deprivation and hypoxia. ExpCell Res. 2007;313(15):3337. doi: 10.1016/j.yexcr.2007.06.013. et al. –. [DOI] [PubMed] [Google Scholar]
- [42].Cardaci S, Desideri M, Ciriolo MR. Targeting aerobic glycolysis: 3-bromopyruvate as a promising anticancer drug. J. Bioenerg. Biomembr. 2012;44:17. doi: 10.1007/s10863-012-9422-7. –. [DOI] [PubMed] [Google Scholar]
- [43].Woldetsadik AD, Vogel MC, Rabeh WM, Magzoub M. Hexokinase II–derived cell penetrating peptide targets mitochondria and triggers apoptosis in cancer cells. FASEB J. 2017;31(5):2168. doi: 10.1096/fj.201601173R. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Pouysségur J, Franchi A, Salomon JC, Silvestre P. Isolation of a Chinese hamster fibroblast mutant defective in hexose transport and aerobic glycolysis: its use to dissect the malignant phenotype. Proc Nat Acad Sci USA. 1980;77(5):2698. doi: 10.1073/pnas.77.5.2698. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Funasaka T, Haga A, Raz A, Nagase H. Tumor Autocrine Motility Factor Is an Angiogenic Factor That Stimulates Endothelial Cell Motility. Biochem Biophys Res Commun. 2001;284:1116. doi: 10.1006/bbrc.2001.4912. –. [DOI] [PubMed] [Google Scholar]
- [46].Thomas GR, Nadiminti H, Regalado J. Molecular predictors of clinical outcome in patients with head and neck squamous cell carcinoma. Int J Exp Pathol. 2005;86(6):347. doi: 10.1111/j.0959-9673.2005.00447.x. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Jares P, Colomer D, Campo E. Genetic and molecular pathogenesis of mantle cell lymphoma: perspectives for new targeted therapeutics. Nat Rev Cancer. 2007;7(10):750. doi: 10.1038/nrc2230. –. [DOI] [PubMed] [Google Scholar]
- [48].Zong M, Lu T, Fan S, Zhang H, Gong R, Sun L. Glucose-6-phosphate isomerase promotes the proliferation and inhibits the apoptosis in fibroblast-like synoviocytes in rheumatoid arthritis. Arthritis Res Ther. 2015;17(1):100. doi: 10.1186/s13075-015-0619-0. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Funasaka T, Raz A. The role of autocrine motility factor in tumor and tumor microenvironment. Cancer Metastasis Rev. 2007;26:725. doi: 10.1007/s10555-007-9086-7. –. [DOI] [PubMed] [Google Scholar]
- [50].Tsutsumi S, Fukasawa T, Yamauchi H, Kato T, Kigure W, Morita H. Phosphoglucose isomerase enhances colorectal cancer metastasis. Int J Oncol. 2009;35:1117. doi: 10.3892/ijo_00000427. et al. –. [DOI] [PubMed] [Google Scholar]
- [51].Kho DH, Makker NP, Balan V, Hogan V, Tait L, Wang Y. Autocrine Motility Factor Promotes HER2 Cleavage and Signaling in Breast Cancer Cells. American Association for Cancer Res. 2013;73:1411. doi: 10.1158/0008-5472.CAN-12-2149. et al. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Ahmad A, Aboukameel A, Kong D, Wang Z, Sethi S, Chen W. Phosphoglucose Isomerase/Autocrine Motility Factor mediates epithelial-mesenchymal transition regulated by miR-200 in breast cancer cells. Cancer Res. 2011;71(9):3400. doi: 10.1158/0008-5472.CAN-10-0965. et al. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Niizeki H, Kobayashi M, Horiuchi I, Akakura N, Chen J, Wang J. Hypoxia enhances the expression of autocrine motility factor and the motility of human pancreatic cancer cells. Br J Cancer. 2002;86(12):1914. doi: 10.1038/sj.bjc.6600331. et al. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Brahimi-Horn CM, Pouyssegur J. Harnessing the hypoxia-inducible factor in cancer and ischemic disease. Biochem Pharmacol. 2006;73:450. doi: 10.1016/j.bcp.2006.10.013. –. [DOI] [PubMed] [Google Scholar]
- [55].Yu FL, Liao MH, Lee JW, Shihb WL. Induction of hepatoma cells migration by phosphoglucose isomerase/autocrine motility factor through the upregulation of matrix metalloproteinase-3. Biochem Biophys Res Commun. 2004;314:76. doi: 10.1016/j.bbrc.2003.12.056. –. [DOI] [PubMed] [Google Scholar]
- [56].Haga A, Funasaka T, Deyashiki Y, Raz A. Autocrine motility factor stimulates the invasiveness of malignant cells as well as up-regulation of matrix metalloproteinase-3 expression via a MAPK pathway. FEBS Lett. 2008;582:1877. doi: 10.1016/j.febslet.2008.05.005. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Gallardo-Pérez JC, Rivero-Segura NA, Marín-Hernández A, Moreno-Sánchez R, Rodríguez-Enríquez S. GPI/AMF inhibition blocks the development of the metastatic phenotype of mature multi-cellular tumor spheroids. BiochimBiophys Acta. 2014;1843:1043. doi: 10.1016/j.bbamcr.2014.01.013. –. [DOI] [PubMed] [Google Scholar]
- [58].Escoubet B, Plane`s C, Clerici C. Hypoxia increases glyceraldehyde-3-phosphate dehydrogenase transcription in rat alveolar epithelial cells. Biochem Biophys. Res. Commun. 1999;266(1):156. doi: 10.1006/bbrc.1999.1798. –. [DOI] [PubMed] [Google Scholar]
- [59].Yamaji R, Fujita K, Takahashi S, Yoneda H, Nagao K, Masuda W. Hypoxia up-regulates glyceraldehyde-3-phosphate dehydrogenase in mouse brain capillary endothelial cells: involvement of Na+/Ca2+ exchanger. Biochim Biophys Acta Mol Cell Res. 2003;1593(2–3):269. doi: 10.1016/s0167-4889(02)00397-x. et al. –. [DOI] [PubMed] [Google Scholar]
- [60].Ou XM, Stockmeier CA, Meltzer HY, Overholser JC, Jurjus GJ, Dieter L. A Novel Role for Glyceraldehyde-3-Phosphate Dehydrogenase and Monoamine Oxidase B Cascade in Ethanol-Induced Cellular Damage. Biol. Psychiatry. 2010;67:855. doi: 10.1016/j.biopsych.2009.10.032. et al. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Du ZX, Wang HQ, Zhang HY, Gao DX. Involvement of Glyceraldehyde-3-Phosphate Dehydrogenase in Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand-Mediated Death of Thyroid Cancer Cells. Endocrinology. 2007;148(9):4352. doi: 10.1210/en.2006-1511. –. [DOI] [PubMed] [Google Scholar]
- [62].Higashimura Y, Nakajima Y, Yamaji R, Harada N, Shibasaki F, Nakano Y. Up-regulation of glyceraldehyde-3-phosphate dehydrogenase gene expression by HIF-1 activity depending on Sp1 in hypoxic breast cancer cells. Arch Biochem Biophys. 2011;509:1. doi: 10.1016/j.abb.2011.02.011. et al. –. [DOI] [PubMed] [Google Scholar]
- [63].Révillion F, Pawlowski V. Hornez L, Peyrat JP. Glyceraldehyde-3-phosphate dehydrogenase gene expression in human breast cancer. Eur JCancer. 2000;36:1038. doi: 10.1016/s0959-8049(00)00051-4. –. [DOI] [PubMed] [Google Scholar]
- [64].Liu K, Tang Z, Huang A, Chen P, Liu P, Yang J. Glyceraldehyde-3-phosphate dehydrogenase promotes cancer growth and metastasis through upregulation of SNAIL expression. Int J Oncol. 2017;50:252. doi: 10.3892/ijo.2016.3774. et al. –. [DOI] [PubMed] [Google Scholar]
- [65].Zhou Y, Yi X, Stoffer JB, Bonafe N, Gilmore-Hebert M, McAlpine J. The multifunctional protein glyceraldehyde-3-phosphate dehydrogenase is both regulated and controls CSF-1 mRNA stability in ovarian cancer. Mol. Cancer Res. 2008;6(8):1375. doi: 10.1158/1541-7786.MCR-07-2170. et al. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Kang SS, Chun YK, Hur MH, Lee HK, Kim YJ, Hong SR. Clinical Significance of Glucose Transporter 1 (GLUT1) Expression in Human Breast Carcinoma. Jpn. J. Cancer Res. 2002;93:1123. doi: 10.1111/j.1349-7006.2002.tb01214.x. et al. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Macheda ML, Rogers S, Best JD. Molecular and cellular regulation of glucose transporter (GLUT) proteins in cancer. J Cell Physiol. 2005;202:654. doi: 10.1002/jcp.20166. –. [DOI] [PubMed] [Google Scholar]
- [68].Krześlak A, Wojcik-Krowiranda K, Forma E, Jozwiak P, Romanowicz H, Bienkiewicz A. Expression of GLUT1 and GLUT3 Glucose Transporters in Endometrial and Breast Cancers. Pathol Oncol Res. 2012;18(3):721. doi: 10.1007/s12253-012-9500-5. et al. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Gandham SM, Talekar M, Singh A, Amiji MM. Inhibition of hexokinase-2 with targeted liposomal 3-bromopyruvate in an ovarian tumor spheroid model of aerobic glycolysis. Int J Nanomedicine. 2015;10:4405. doi: 10.2147/IJN.S82818. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Botzer LE, Maman S, Sagi-Assif O, Meshel T, Nevo I, Yron I. Hexokinase 2 is a determinant of neuroblastoma me tastasis. Br J Cancer. 2016;114:759. doi: 10.1038/bjc.2016.26. et al. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Zhong JT, Zhou SH. Warburg effect, hexokinase-II, and radioresistance of laryngeal carcinoma. Oncotarget. 2017;8(8):14133. doi: 10.18632/oncotarget.13044. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Thamrongwaranggoon U, Seubwai W, Phoomak Ch, Sangkhamanon S, Cha’on U, Boonmars T. Targeting hexokinase II as a possible therapy for cholangiocarcinoma. Biochem Biophys Res Commun. 2017;484:409. doi: 10.1016/j.bbrc.2017.01.139. et al. –. [DOI] [PubMed] [Google Scholar]
- [73].Salani B, Del Rio A, Marini C, Sambuceti G, Cordera R, Maggi D. Metformin, cancer and glucose metabolism. Endocr Relat Cancer. 2014;21:461. doi: 10.1530/ERC-14-0284. –. [DOI] [PubMed] [Google Scholar]
- [74].Nevo I, Oberthuer A, Botzer E, Sagi-Assif O, Maman S, Pasmanik-Chor M. Gene-expression-based analysis of local and metastatic neuroblastoma variants reveals a set of genes associated with tumor progression in neuroblastoma patients. Int J Cancer. 2010;126(7):1570. doi: 10.1002/ijc.24889. et al. –. [DOI] [PubMed] [Google Scholar]
- [75].de Padua MC, Delodi G, Vučetić M, Durivault J, Vial V, Bayer P. Disrupting glucose-6-phosphate isomerase fully suppresses the “Warburg effect” and activates OXPHOS with minimal impact on tumor growth except in hypoxia. Oncotarget. 2017;8(50):87623. doi: 10.18632/oncotarget.21007. et al. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Liotta LA, Mandler R, Murano G, Katz DA, Gordon RK, Chiang PK. Tumor cell autocrine motility factor. Proc Natl Acad Sci USA. 1986;83:3302. doi: 10.1073/pnas.83.10.3302. et al. – 3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Funasaka T, Hu H, Yanagawa T, Hogan V, Raz A. Down-Regulation of Phosphoglucose Isomerase/Autocrine Motility Factor Results in Mesenchymal-to-Epithelial Transition of Human Lung Fibrosarcoma Cells. Cancer Res. 2007;67:4236. doi: 10.1158/0008-5472.CAN-06-3935. –. [DOI] [PubMed] [Google Scholar]
- [78].Furuta E, Okuda H, Kobayashi A, Watabe K. Metabolic genes in cancer: Their roles in tumor progression and clinical implications. Biochim Biophys Acta. 2010;1805:141. doi: 10.1016/j.bbcan.2010.01.005. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Takanami I, Takeuchi K, Naruke M, Kodaira S, Tanaka F, Watanabe H. Autocrine motility factor in pulmonary adenocarcinomas: results of an immunohistochemical study. Tumour Biol. 1998;19(5):384. doi: 10.1159/000030031. et al. –. [DOI] [PubMed] [Google Scholar]
- [80].Said HM, Polat B, Hagemann C, Anacker J, Flentje M, Vordermark D. Absence of GAPDH regulation in tumor-cells of different origin under hypoxic conditions in–vitro. BMC Res Notes. 2008;2:8. doi: 10.1186/1756-0500-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Epner DE, Coffey DS. Glyceraldehyde-3-Phosphate Dehydrogenase in Prostate Cancer Cells and Normal Prostate Tissue. Prostate. 1996;28:372. doi: 10.1002/(SICI)1097-0045(199606)28:6<372::AID-PROS6>3.0.CO;2-C. –. [DOI] [PubMed] [Google Scholar]
- [82].Tokunaga K, Nakamura Y, Sakata K, Fujimori K, Ohkubo M, Sawada K. Enhanced Expression of a Glyceraldehyde-3-phosphate Dehydrogenase Gene in Human Lung Cancers. Cancer Res. 1987;47:5616. et al. –. [PubMed] [Google Scholar]
- [83].Correa CR, Bertollo CM, Zouain CS, Goes AM. Glyceraldehyde-3-phosphate dehydrogenase as a surface associated antigen on human breast cancer cell lines MACL-1 and MGSO-3. OncolRep. 2010;24:677. doi: 10.3892/or_00000907. –. [DOI] [PubMed] [Google Scholar]
- [84].Epner DE, Coffey DS. There are multiple forms of glyceraldehyde-3-dehydrogenase in prostate cancer cells and normal prostate tissue. Prostate. 1999;28:372. doi: 10.1002/(SICI)1097-0045(199606)28:6<372::AID-PROS6>3.0.CO;2-C. –. [DOI] [PubMed] [Google Scholar]
- [85].Elkhalfi B, Senhaji N, Benomar H. Soukri A Study of glyceraldehyde-3-phosphate dehydrogenase expression in the tumor process of: Breast, cervix and prostate cancers. AdvBiol Chem. 2012;2:335. –. [Google Scholar]
- [86].Ramos D, Pellin-Carcelen A, Agusti J, Murgui A, Jorda E, Pellin A. Deregulation of Glyceraldehyde-3-Phosphate Dehydrogenase Expression During Tumor Progression of Human Cutaneous Melanoma. Anticancer Res. 2015;35:439. et al. –. [PubMed] [Google Scholar]
- [87].Tang Z, Yuan S, Hu Y, Zhang H, Wu W, Zeng Z. Over-expression of GAPDH in human colorectal carcinoma as a preferred target of 3-Bromopyruvate Propyl Ester. J Bioenerg Biomembr. 2012;44:117. doi: 10.1007/s10863-012-9420-9. et al. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Tarrado-Castellarnau M, Diaz-Moralli S, Polat IH, Sanz-Pamplon R, Alenda C, Moreno V. Glyceraldehyde-3-phosphate dehydrogenase is overexpressed in colorectal cancer onset. Transl Med Commun. 2017;2:6. et al. [Google Scholar]
- [89].Puzone R, Savarino G, Salvi S, Dal Bello MG, Barletta G, Genova C. Glyceraldehyde-3-phosphate dehydrogenase gene overexpression correlates with poor prognosis in non-small cell lung cancer patients. Mol Cancer. 2013;12(1):97. doi: 10.1186/1476-4598-12-97. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]