

Abstract
The main purpose of our study is to optimize the formulation for Ginkgolide B (GB) solid dispersion in order to improve its dissolution in water. For the preparation of GB solid dispersion, we use a solvent method. The optimized formulation consists of GB : PVPK30 = 1:10, PVPK30 as the carrier, and ethanol : dichloromethane = 1:1 as the solvent. They were treated ultrasonically for 10 min at 60°C. The results from scanning electron microscopy (SEM), differential scanning calorimetry (DSC), X-ray diffraction (XRD), and particle size analysis show that the morphological appearance of GB changed significantly in solid dispersion. The original crystal form of GB no longer existed, but it was uniformly dispersed within PVPK30 in a non-crystalline form. It is probably because the -C=O in GB forms hydrogen bonds with the -OH of PVPK30 or urea; this produces a nice solid dispersion and significantly improves the dissolution of GB in water. When GB is in a solid dispersion system, GB’s dissolution in water could be enhanced from 30% to 80%. Furthermore, it may even be produced as a solid agent.
Keywords: Ginkgolide B, Solid dispersion, Rate of dissolution, Crystal
1. Introduction
Ginkgolide B (GB) is a Ginkgo biloba extract in the terpenoid class. GB is a very fat-soluble and physiologically active chemical in Ginkgo leaves. It is also famous for its platelet activation, and has important implications in the treatment of cardiovascular diseases and phlegm-stasis during ischemic strokes [1, 2, 3, 4]. In addition, it can be applied in the clinical treatment of acute pancreatitis, thrombosis, and metastatic cancer, demonstrates anti-oxidation and anti-aging effects, and has a protective effect on neuronal injuries. However, due to its diterpenoid ingredient with a unique 20-carbon “cage” structure, it has a low solubility in water, which leads to a slow dissolution speed for oral administration, and low bioavailability, which affects the efficacy of its oral formulations.
Currently, there are sustained-release tablets, soft capsules, dripping pills, emulsions, and freeze-dried powders for the injection of GB preparation, and each formulation has a different evaluation. Among these applications, sustained-release tablets have accurate dose, but they also have the disadvantages of low bioavailability and being hard to swallow for some patients. Soft capsules can disguise the unpleasant taste of GB and provide good stability, but if the temperature is high, GB will decompose easily. The dripping pills have a stable quality and high bioavailability, but the drug loading is small. Injectable GB has a single component, significant efficacy with high safety, and good market acceptance; however, its clinical efficacy is limited due to its poor solubility in water [5, 6]. In addition to the abovementioned formulations, there are many new formulations yet to be developed. It is possible to further improve efficacy and drug safety, simplify quality control, and provide broad prospects for the clinical application of the drug.
In order to improve the solubility, dissolution rate, and bioavailability of GB in water, this study employed solid dispersion technology, which is seldom applied to GB [7, 8, 9]. Solid dispersion (SD) is a method for dispersing a drug in solid excipient to form a dispersal system in a solid state [10, 11]. The drug particle size in the excipient is between 0.001 to 0.1 mm, in order to increase the dissolution rate of a poorly soluble drug in water and further improve its utilization in vivo. First, a drug and a hydrophilic excipient are dissolved in a co-solvent or melted by heating, and then the solvent is cooled or evaporated; after that, the solid dispersion of the drug and its excipient are obtained. There are many methods to obtain solid dispersions. These dispersions can be divided into the following standards: (1) By dissolution behaviors, including immediate-release, sustained release, or enteric solid dispersions. (2) By preparation methods, such as eutectic solution, solid solution, co-precipitate, or glass solution. In solid dispersions, the dispersed forms of drugs include microcrystalline, colloidal, molecular, and amorphous states, which can exist alone or together. Generally, a drug existing as a microcrystalline, molecular state, or a eutectic mixture in a solid dispersion system, has good dispersion [12]. After a drug contacts with gastrointestinal fluid, the dissolution speed increases, which improves the effects of the absorbed drug and reduces adverse drug reactions, making it adapt better to the patient’s compliance and dosage level [13, 14]. There are also many preparation methods for SD such as melting, solvent, solvent-melting, grinding, solvent spray-drying, and lyophilisation. SD have high dispersion and low stability levels [15, 16]. By forming an amorphous or partially amorphous state, or a supersaturated solid solution of a drug, a solid dispersion is thus solid dispersed. During the preparation process, whichever method changes the physical structure of the drug itself changes its current state. Thermodynamically, these solid dispersed states are unstable and tend to transform into a stable crystallization state. Improper ratios of a drug and its excipient, improper excipient materials during the preparation process, too-high storage temperatures and humidity, and extended storage times can all affect the stability of a solid dispersion, potentially causing drug aging such as higher hardness, crystallization, roughening, and decreased drug dissolution [17]. The most common problem for solid dispersions is aging. There have been many studies conducted in recent years with the goal of prolonging the shelf life of drugs to maintain relative stability by means of improving preparation technology, finding appropriate additives, and choosing a suitable or polyol excipient.
Using GB as the original drug and PVPK30, urea, and HPMC as the excipients, this study prepared GB solid dispersion drugs using the solvent method [18, 19, 20]. The formulation design consisted of orthogonal and single factor experiments. Next, the solid dispersion was analyzed, determined, and observed with SEM, DSC, XRD, and particle size analysis. Finally, the dissolution of GB in water was investigated.
2. Materials and methods
2.1. Drugs and equipment
The following items were employed in this study: GB with content ≥98%, purchased from Shanghai Jinsui Biotechnology Co., Ltd.; urea purchased from Beijing Xingye Chengxin Chemical Industrial Co., Ltd.; polyvinylpyrroliolone purchased from Beijing Xinya Huayang Technology Development Co., Ltd.; hydroxypropyl methyl cellulose purchased from Beijing Gaohua WeiyeFood Additives Co., Ltd.; ethanol (HPLC grade) purchased from Honeywell Trading (Shanghai) Co., Ltd.; dichloromethane (HPLC grade) purchased from Beijing Huibao Lianhe Chemical Technology Co., Ltd.; methanol (HPLC grade) purchased from Honeywell Trading (Shanghai) Co., Ltd.; methanol (AR) purchased from Beijing Orly Chemical Reagent Company; isopropanol purchased from Beijing Chemical Works; distilled water; and deionized water.
Equipment: Lark LA series electronic balance from Changshu Lark Balance Co., Ltd.; LK-Q1036 ultrasonic cleaner from Zhuhai Lingke Automation Technology Co., Ltd.; magnetic stirrer SH-2 from Qun’an Laboratory Instrument Co., Ltd.; multi-head magnetic stirrer from Shanghai Qige Industrial Co., Ltd.; oven from Daxiang Electronic Machinery Co., Ltd.; TU-1901 Ultraviolet visible spectrophotometer from Beijing Persee General instrument Co., Ltd.; ZRS-8G intelligent dissolution tester from Tianjin University Radio Factory; and TD6M desk centrifuge from Gaoke Instrument factory.
2.2. GB analytical method
Ultraviolet visible spectrophotometry was used to determine the concentration of GB in solid dispersion. A sample of 10.00 mg GB reference was transferred into a 10 ml volumetric flask with an appropriate amount of water added and heated to promote dissolution. Then, it was cooled down to room temperature to make a 1 mg/ml reference solution. Further gradient solutions of 0.2, 0.4, 0.6, and 0.8 mg/ml were prepared. The maximum absorption wavelength was 216 nm. The regression equation was Y = 0.3145x + 0.1011, with R = 0.99994, and a linear range of 0.2-1mg/ml.
2.3. Preparation of GB solid dispersions
Appropriate amounts of GB, PVPK30, urea, and HPMC were weighed precisely. The mixed solvents of dichloromethane and ethanol were prepared according to Table 1 for the orthogonal experiment. After the GB was mixed with each excipient, the corresponding amount of mixed solvent was added for dissolution, sonicated, and then stirred at 60°C until completely mixed. After heating to evaporate the solvent, the resulting product was placed in a vacuum oven at 60°C to remove residual solvents. The obtained white block of product was ground in a mortar, filtered with an 80 mesh sieve, and finally, the white GB solid dispersion was obtained.
Table 1.
Orthogonal experimental design for GB solid dispersions
| Number | Excipient | Ratio of GB to excipient | Ratio of solvent mixture |
|---|---|---|---|
| P1 | PVPK30 | 1:2 | 0:1 |
| P2 | PVPK30 | 1:6 | 1:1 |
| P3 | PVPK30 | 1:10 | 1:2 |
| P4 | Urea | 1:2 | 1:1 |
| P5 | Urea | 1:6 | 1:2 |
| P6 | Urea | 1:10 | 0:1 |
| P7 | HPMC | 1:2 | 1:2 |
| P8 | HPMC | 1:6 | 0:1 |
| P9 | HPMC | 1:10 | 1:1 |
2.4. Orthogonal experimental design for GB solid dispersions
An orthogonal experiment was performed to optimize the formulation for the GB solid dispersion. Three factors were chosen: excipient, the ratio of GB to excipient, and the ratio of solvent mixture. For each factor, three levels were tested. Using the dissolution rate in vitro as the dependent variable, a L9 (34) orthogonal table was used to study the factors affecting the solid dispersion formulation.
2.5. Phase identification of GB solid dispersions
In this experiment, we used XRD and DSC to identify the original GB drug, excipients, processing mixtures, and solid dispersions.
The DSC used an empty aluminum crucible as the reference; a 5mg sample was kept in another aluminum crucible, with a scanning speed of 10K·min-1 (K is the symbol used for the Kelvin unit of temperature), and a scanning range of 50-260°C. The melting point of GB is 280°C, so the scanning range needed to avoid GB decomposing.
XRD conditions were as follows: Cu Kα target, voltage 40kV, current 60mA, scanning speed 4°-min-1, and scanning range 3-50°C.
2.6. Dissolution determination of GB solid dispersions
After the original drug and solid dispersions were separately loaded into the capsules, the dissolution rates of GB in vitro were determined according to the 3rd method (small cup method) in Appendix XC of “Chinese Pharmacopoeia” 2015 edition. The dissolution medium was distilled water, speed was set to 100 r·min-4, and temperature was 37 ± 1°C. At 10, 15, 20, 30, 45 and 60 min, a 2 ml sample was taken. After samples were taken, the same volume of distilled water at the same temperature was immediately added back to the solution. The samples were then centrifuged at 5,000 r·min-1 for 15 min, and its supernatant was used to determine the cumulative percentage of drug dissolution (Q%).
An is the dissolution amount measured at each time point.
V1 is a fixed sampling volume at each point in time.
V2 is the volume of dissolution medium.
2.7. Qualitative study of dissolution
Excessive amounts of drug and solid dispersions were added to an appropriate amount of distilled water and stirred at room temperature with a magnetic stirrer. The dissolution phenomena were observed at 10, 15 and 30 min.
3. Results
3.1. Formulations of GB solid dispersions and optimization of the preparation process
3.1.1. Screening of excipients
PVPK30, HPMC, and urea were the excipients for GB solid dispersions and their effects on drug dissolution were investigated. Results are shown in Figure 1.
Fig. 1.

Effects of different excipients on drug dissolution. Each value represents the mean ± S.D. (n = 3).
From an economic point of view, the excipients we selected are cheap and easy to get. As for the performance of excipients, not only does PVPK30 have excellent physiological inertia (not participate in human metabolism) but also has good biocompatibility of skin, mucous membranes, and eyes. It is one of the latest medicinal materials that are internationally advocated. Urea has the ability to increase the solubility of organic matter, and it can also improve the stability of the material. HPMC has good physiological inertia too: no oral absorption, safe and nontoxic, dimensional stability, excellent film-forming properties, and a wide range of resistance to enzyme, dispersion and adhesion characteristics.
As shown in the results, after the preparation of the solid dispersions with either urea or PVPK30, the cumulative dissolution rate of the drug significantly increased compared with the original drug, with at least a 2.4-fold increase. In one hour, the accumulative dissolution rate of the GB solid dispersion using PVPK30 as the excipient was greater than 65%, which was better than using urea as the excipient. The experimental results showed that, among the three excipients, only PVPK30 and urea improved the drug dissolution. Because the preparation of solid dispersion with HPMC was not successful, the dissolution effect could not be determined. Furthermore, the enhancement of the dissolution with PVPK30 was better than with urea.
3.1.2. Screening of an optimal formulation for GB solid dispersion
The result for the screening of an optimal formulation according to the orthogonal experimental table by dissolution experiments is shown in Table 2.
Table 2.
L9(34) orthogonal experiment results
| Number | Excipient | Ratio of GB to excipient | Ratio of solvent mixture (dichloromethane: ethanol) | Dissolution Rate (mg/mL) |
|---|---|---|---|---|
| P1 | PVPK30 | 1:2 | 0:1 | 0.5265 |
| P2 | PVPK30 | 1:6 | 1:1 | 0.6536 |
| P3 | PVPK30 | 1:10 | 1:2 | 0.8330 |
| P4 | Urea | 1:2 | 1:1 | 0.4228 |
| P5 | Urea | 1:6 | 1:2 | 0.6622 |
| P6 | Urea | 1:10 | 0:1 | 0.8220 |
| P7 | HPMC | 1:2 | 1:2 | 0 |
| P8 | HPMC | 1:6 | 0:1 | 0 |
| P9 | HPMC | 1:10 | 1:1 | 0 |
| K1 | 0.6710 | 0.4360 | 0.4495 | |
| K2 | 0.6357 | 0.4386 | 0.3588 | |
| K3 | 0 | 0.5517 | 0.4984 | |
| R | 0.6710 | 0.1157 | 0.1396 | |
| Optimal Formulation | PVPK30 | 1:10 | 1:2 |
From Table 2, it can be seen that the order of the effects of each factor on the dissolution rate are as follows: excipient type > ratio of solvent mixture > ratio of GB to excipient. The optimal combination is PVPK30 as the excipient, with a ratio of solvent mixture of 1:2 and a ratio of GB to excipient of 1:10.
3.1.3. Relationship between drug and excipients
Solid dispersions with the ratios of GB to PVPK30 of 1:4, 1:8, and 1:12 were prepared. The in vitro dissolution of the solid dispersions was observed, and the results are shown in Figure 2.
Fig. 2.

Effects of different ratios of GB to PVPK30 on drug dissolution. Each value represents the mean ± S.D. (n = 3).
As the excipient proportion gradually increased, the dissolution rate increased, reaching a maximum at three times higher than the original drug. This indicated that the excipient had a significant solubilizing effect on the original drug in a highly dispersed system.
3.1.4. Relationship between the drug and the ratio of solvent mixture
During preparation of solid dispersions with the solvent method, the solvent types may affect drug dissolution as well. According to the solubility properties of the excipient and the drug, this experiment prepared solid dispersions with four types of solvents (mixtures) including dichloromethane/ethanol of 1:1, 1:2, and 1:3, and ethanol only. The dissolutions are shown in Figure 3.
Fig. 3.

Effects of different ratios of dichloromethane to ethanol on drug dissolution. Each value represents the mean ± S.D. (n = 3).
As the proportion of dichloromethane increased, the dissolution rate of GB increased. However, the drug and the excipient were hardly soluble in dichloromethane, thus 1:1 was chosen for the dichloromethane : ethanol ratio.
3.1.5. Verification of optimized formulation
According to the results of formulation optimization, the final formulation was as follows: PVPK30 as the excipient, ethanol : dichloromethane = 1:1 as the solvent, the ratio of drug to excipient of 1:10, ultrasonic time of 10 min, and bath temperature of 60°C . The dissolution experiment was repeated three times using the optimized formulation, and the results are shown in Figure 4.
Fig. 4.

Verification of optimized formulation
The dissolution profiles were highly consistent for the three repeats, exhibiting no significant differences, which indicated good reproducibility of this formulation. The cumulative dissolution rates were more than 85% for all three repeats, which was a significant improvement of the dissolution rate.
3.2. Qualitative study of dissolution
Compared with the original drug, the GB solid dispersions dissolved almost completely, whereas most GB is not dissolved in water (Table 3). With respect to the dissolution among the P1, P2, and P3 formulations, the GB solid dispersions in P1 and P2 were completely dissolved with a clear solution. However, there were some residual substances remaining in P3, possibly due to the improper ratio of solvent mixture.
Table 3.
Qualitative table of dissolution phenomena
| Formulation | Dissolution |
|---|---|
| P1 (excipient: PVPK30; ratio of GB to excipient: 1:2; ratio of solvent mixture: 0:1) | Completely dissolved, clear solution. |
| P2 (excipient: PVPK30; ratio of GB to excipient: 1:6; ratio of solvent mixture: 1:1) | Completely dissolved, clear solution. |
| P3 (excipient: PVPK30; ratio of GB to excipient: 1:10; ratio of solvent mixture: 1:2) | Largely dissolved. |
| GB original drug | Rarely dissolved, cloudy solution. |
3.3. DSC identification
DSC could identify the original drug GB, excipient (PVPK30), physical mixture, and solid dispersion of GB-excipient (PVPK30), shown in Figure 5.
Fig. 5.

DSC identification profiles: 1) GB original drug; 2) Physical mixture of GB-excipient (PVPK30/urea); 3) GB solid dispersion; 4) Excipient PVPK30 (urea)
The GB original drug presented an endothermic peak at 134°C and 232°C; whereas the excipient had no endothermic peak. The physical mixture had an endothermic peak at 113°C; the solid dispersion had an endothermic peak at 131°C, which had a shape consistent with the corresponding excipient. Both the physical mixture and the solid dispersion had endothermic peaks at 232°C, which indicated the formation of non-stationary dispersion, as shown in Figure 6.
Fig. 6.

GB morphology (magnification ×2000)
3.4. SEM observation
SEM was used to observe the changes in the GB morphology before and after the preparation of solid dispersion. Experimental conditions were as follows: FESEM-SIRINO-100 scanning electron microscope, 25 KV accelerating voltage, and magnifications of 100, 200, 400, 500, 2,000 and 5,000. The results are shown in Figures 6-11.
Fig. 11.

Optimized GB solid dispersion (magnification ×5,000)
Fig. 7.

Optimized GB solid dispersion (magnification ×200)
Fig. 8.

Optimized GB solid dispersion (magnification ×400)
Fig. 9.

Optimized GB solid dispersion (magnification ×500)
Fig. 10.

Optimized GB solid dispersion (magnification ×2,000)
Fig. 12.
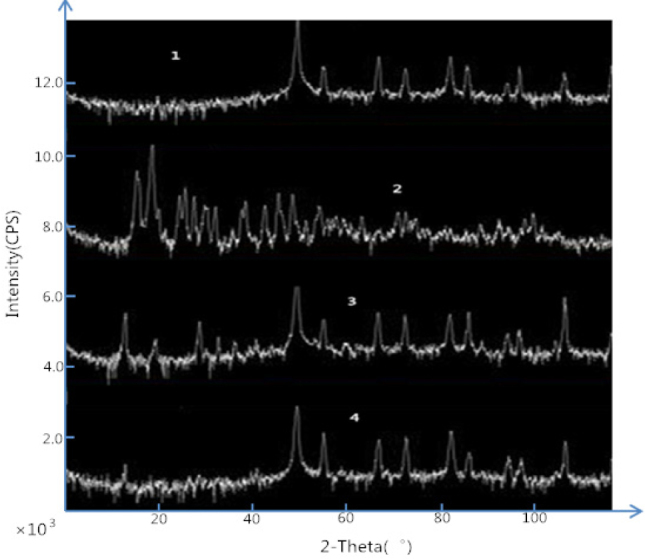
XRD graphs: 1) Excipient (PVPK30); 2) GB original drug; 3) GB solid dispersion; 4) Physical mixture of GB-excipient (PVPK30)
Fig. 13.

Particle size distribution of optimal GB solid dispersion
From Figures 6-11, the morphology of the GB changed significantly among the solid dispersions. The original GB drug primarily demonstrated crystal sheets, whereas the solid dispersions showed no crystals, which indicated the GB was completely dispersed in the excipient.
3.5. XRD analysis
GB crystals have obvious characteristic diffraction peaks and the maximum absorbance is about 22°. The carrier is a nonionic, amorphous polymer powder. It has a diffraction peak at 50°. The diffraction peak of GB crystals is very small in GB solid dispersion and the diffraction peaks of the excipients are also greatly reduced. This shows that the GB and most of the excipients in the SD form amorphous co-precipitating crystals. GB dispersed uniformly in solution in amorphous state.
3.6. Particle size analysis
The particle size of the solid dispersion ranges from 8.13-97.29 μm; the average diameter is 49.32 μm, and peak diameter is 54.96 μm. Most of the particles were distributed in the range of 39.23-88.58 μm, and only a peak appeared at about 50 μm, which indicated that GB had stable distribution in the solid dispersion system. It also showed that PVPK30 as an excipient has better protection and dispersion for GB.
4. Discussion
SD is a method for dispersing drugs in solid excipient and forming dispersal systems in solid state. The drug particle size in the excipient is between 0.001 and 0.1 mm, designed to increase the dissolution rate of a poorly soluble drug in water and to further improve its utilization in vivo [21, 22]. Using GB as the original drug and PVPK30, urea, and HPMC as the excipients, this study prepared a GB solid dispersion drug using the solvent method. The solid dispersion was observed, determined, and analyzed by SEM, DSC, XRD, and particle size analysis.
We find that the GB dissolution rate increases as the proportion of excipient increases. As a nonionic polymer, PVPK30 has good solubility in water and demonstrates dispersion and protection for GB. The unsuccessful preparation of solid dispersions with HPMC might be due to the methoxyl group in HPMC’s structure [23]. The bond between methoxyl groups and HPMC is easily broken at high temperatures. When HPMC loses the methoxyl groups, it will become a gel in the solution, which results in the decreasing water solubility and lazy surface activity. The dissolution rate of GB in solid dispersions also increases as the proportion of dichloromethane goes up. Ultrasonic treatment can make GB structure relatively loose, promote the combining of GB and excipient. Combination of GB and excipient can reduce the surface tension and increase the steric resistance of membranous layers, so the crystal of GB solid dispersion dispersed uniformly in the solution [22, 24]. Identified by SEM, DSC, XRD, and particle size analysis, GB in the solid dispersion exists in a state completely different from the crystalline state of the physical mixture, and is possibly uniformly dispersed in the excipient by hydrogen bonds or polymer states which have no specific crystal shape [25]. As can be seen from Fig. 6-11, the surface structure of GB solid dispersion prepared by the solvent method is dense. It forms a three-dimensional network structure of the polymer, so its stability and solubility could be improved; the original drug shows mainly lamellar crystals while the solid dispersion exhibits a columnar crystal structure. GB in the solid dispersion is completely dispersed in the solid dispersion system by the action of excipient PVPK30, GB no longer had the original crystal characteristics after combined with excipients, which covers the crystal morphological characteristics of GB. It has been proven that the obtained solid dispersion could improve the solubility and dissolution rate of GB in water. The dissolution experiments show that the in vitro dissolution is significantly enhanced. Although the study of solid dispersions is well-developed, we still need further systematic discussion on how to improve the dissolution of drug solid dispersions in vitro, how to study pharmacokinetic behavior in vivo, the mechanism of solid dispersion to improve drug dissolution, and the influence of thermodynamic and kinetic factors on physical stability. Our future research topic is going to be the large scale production of solid dispersions and its commercial application.
Acknowledgement
This work was financially supported by National Natural Science Foundation of China (Grant No. 11475020 and No. 81511140298), project of Chaoyang district collaborative innovation (No. XC1608) and Beijing Union University Graduate Program.
Abbreviations
- GB
Ginkgolide B
- SD
Solid dispersion
- SEM
Scanning electron microscopy
- DSC
Differential scanning calorimetry
- XRD
X-ray diffraction
- PVPK30
polyvinylpyrrolidone K30
- HPMC
hydroxypropyl methyl cellulose
Footnotes
Conflict of interest: Authors state no conflict of interest.
References
- [1].Shi XL, Zhu Y, Yu JN, Xu XM.. Development in New Dosage Forms of Active Components from Ginkgo biloba Extracts. J. China. Pharmacist. 2014;17:1943–1945. [Google Scholar]
- [2].Liu FP.. Prevention and treatment of cardiovascular diseases in middle and old aged. J. Contemporary Medicine. 2011;17:47–48. [Google Scholar]
- [3].Chen WJ, Xie BJ, H WW.. Research Progress on chemical structure and pharmacological action of Ginkgo biloba. J. Chin. Int J.Pharm. 1998;33:516–519. [Google Scholar]
- [4].Yu HZ, Li GZ, Cao Y.. Determination of flavonoids in Ginkgo biloba. J. Forest By-Product and Speciality in China. 2005;4:22–22. [Google Scholar]
- [5].Hiromasa U, Yuichi T, Masaaki I.. Improvement of dissolution and absorption properties of poorly water soluble drug by preparing spray-dried powders with a-glucosyl hesperidin. J. Pharm. 2010;392:101–103. doi: 10.1016/j.ijpharm.2010.03.037. [DOI] [PubMed] [Google Scholar]
- [6].Furqan AM, Sonali JD, Vaishali TT. Improvement of dissolution rate of aceclofenac by solid dispersion technique. J. Powder. Technol. 2011;207:47–48. [Google Scholar]
- [7].Malin Y, Han LM, Zhang ZR, Wang JX.. Preparation and in vitro characterization of solid dispersions of Ginkgo biloba. China journal of Chinese .Materia .Medica. 2009;34:1368–1372. [PubMed] [Google Scholar]
- [8].Wang SH., Wang ., Sun XD, Li ZP.. Application progress of solid dispersion and its technology in pharmaceutical formulations. J. Guiding Journal of Traditional. Chinese Medicine and Pharmacy. 2014;20:54–56. [Google Scholar]
- [9].Wen J. D. College of pharmacy. Vol. 2013. Shan dong University; Study on the preparation of glimepiride solid dispersion and dispersion tablet; pp. 5–12. [Google Scholar]
- [10].Houde L, Grant David JW, Zhang GG.. A calorimetric investigation of thermodynamic and molecular mobility contributions to the physical stability of two pharmaceutical glasses. J. Pharm Sci. 2007;96:71–75. doi: 10.1002/jps.20633. [DOI] [PubMed] [Google Scholar]
- [11].Konno H, Taylor LS.. Influence of different polymers on the crystallization tendency of molecularly dispersed amorphous felodipine. J. Pharm Sci. 2006;95:26–35. doi: 10.1002/jps.20697. [DOI] [PubMed] [Google Scholar]
- [12].Zhang BS, Zhang YQ, Yang LS, Chen J.. Study on Microcrystalline Structure and Properties of Corn, Cassava and Potato Starch Granules. J. Food. Science. 2001;22:11–13. [Google Scholar]
- [13].Abdul-Fattan AM, Bhargava HN.. Preparation and in vitro evaluation of solid dispersion s of halofantrine. J. Int J.Pharm. 2001;235:17–19. doi: 10.1016/s0378-5173(01)00941-3. [DOI] [PubMed] [Google Scholar]
- [14].Damian F, Blaton N, Kinget R.. Physical stability of solid dispersions of the antiviral agent UC-781 with PEG 6000, Geluci-re 44/14 and PVPK30. Int. J. Pharm. 2002;224:87–89. doi: 10.1016/s0378-5173(02)00316-2. [DOI] [PubMed] [Google Scholar]
- [15].Liu D, Zhang ZH, Chen XY, Jia XB.. Study on preparation of ginkgolides component solid dispersions micro pill drug release unit. J. China journal of Chinese. Materia. Medica. 2014;39:1002–1006. [PubMed] [Google Scholar]
- [16].Liu YS, Gao S, Ke X, Sun HZ, Wang YB.. Recent Research Advances in Solid Dispersions of Poorly Soluble Drugs. J. Pharmaceutical. Development. 2013;37:167–172. [Google Scholar]
- [17].Ma Y, Lv ZY, Di LQ, Li JS, An K.. The Choice of Preferred Materials of Ginkgolide Solid Dispersions. J. Nanjing Univ. Tradit Chin. Med. 2015;31:174–178. [Google Scholar]
- [18].Peng X.. Study on the solid dispersion of Simvastatin. D. Southwestern University. 2013:4–20. [Google Scholar]
- [19].Vasconcelos T, Sarmento B, Paulo C.. Solid dispersions as strategy to improve oral bioavailability of poor water soluble drugs. J . Drug. Discov. Today. 2007;12:1068–1071. doi: 10.1016/j.drudis.2007.09.005. [DOI] [PubMed] [Google Scholar]
- [20].Yoshihashi Y.. Estimation of physical stability of amorphous solid dispersion using differential scanning calorimetry. J. Therm. Anal. Calorim. 2006;85:689–693. [Google Scholar]
- [21].Lamas M. C., Leonardi D., Lambri O. A., Bassani G., Barrera M. G., Bolmaro R. E., Salomon C. J.. Preparation, characterization and dissolution studies of fast release diclofenac sodium tablets from PVP solid dispersions. Pharm. Dev. Technol. 2010;15:162–168. doi: 10.1080/10837450903085400. [DOI] [PubMed] [Google Scholar]
- [22].Bley H., Fussnegger B., Bodmeier R.. Characterization and stability of solid dispersions based on PEG/polymer blends. Int. J. Pharm. 2010;390:165–173. doi: 10.1016/j.ijpharm.2010.01.039. [DOI] [PubMed] [Google Scholar]
- [23].Mesnukul A., Yodkhum K., Phaechamud T.. Solid Dispersion Matrix Tablet Comprising Indomethacin-PEG-HPMC Fabricated with Fusion and Mold Technique. Indian J. Pharm. Sci. 2009;71:413–420. doi: 10.4103/0250-474X.57290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Heo M. Y., Piao Z. Z., Kim T. W., Cao Q. R., Kim A., Lee B. J.. Effect of solubilizing and microemulsifying excipients in polyethylene glycol 6000 solid dispersion on enhanced dissolution and bioavailability of ketoconazole. Arch. Pharm. Res. 2005;28:604–611. doi: 10.1007/BF02977766. [DOI] [PubMed] [Google Scholar]
- [25].Raja H. K. P., Suresh B., Raju J., Prabhakar R. V.. Solubility Enhancement and Physicochemical Characterization of Carvedilol Solid Dispersion with Gelucire 50/13. Arch Pharm Res. 2011;34:51–57. doi: 10.1007/s12272-011-0106-3. [DOI] [PubMed] [Google Scholar]