

We developed an approach for the expansion of Mauritian cynomolgus macaque polyspecific regulatory T cells (Tregs) through the combination of MHC‐mismatched CD40L‐engineered B cells. Expanded Tregs expressed high levels of FoxP3 and Helios, a high percentage of TSDR demethylation, and strong suppression of naïve T cell responses in vitro. This approach has the potential to be translated to clinical studies for deceased‐donor transplants.
![]()
Keywords: CD40L‐stimulated B cells, non‐human primates, regulatory T cells, tolerance
Summary
The therapeutic applications of regulatory T cells (Tregs) include treating autoimmune diseases, graft‐versus‐host disease and induction of transplantation tolerance. For ex‐vivo expanded Tregs to be used in deceased donor transplantation, they must be able to suppress T cell responses to a broad range of human leukocyte antigen (HLA). Here, we present a novel approach for the expansion of polyspecific Tregs in cynomolgus macaques that was adapted from a good manufacturing practice‐compliant protocol. Tregs were isolated by fluorescence‐activated cell sorting (FACS) and expanded in the presence of a panel of CD40L‐stimulated B cells (CD40L‐sBc). Prior to Treg culture, CD40L‐sBc were expanded in vitro from multiple major histocompatibility complex (MHC)‐disparate macaques. Expanded Tregs expressed high levels of forkhead box protein 3 (FoxP3) and Helios, a high percentage of Treg‐specific demethylated region (TSDR) demethylation and strong suppression of naïve T cell responses in vitro. In addition, these Tregs produced low levels of inflammatory cytokines and were able to expand post‐cryopreservation. Specificity assays confirmed that these Tregs were suppressive upon activation by any antigen‐presenting cells (APCs) whose MHC was shared by CD40L‐sBc used during expansion, proving that they are polyspecific. We developed an approach for the expansion of highly suppressive cynomolgus macaque polyspecific Tregs through the use of a combination of CD40L‐engineered B cells with the potential to be translated to clinical studies. To our knowledge, this is the first report that uses a pool of MHC‐mismatched CD40L‐sBc to create polyspecific Tregs suitable for use in deceased‐donor transplants.
Introduction
Since the introduction of calcineurin inhibitors, considerable improvement has been made in patient survival after organ transplantation [1]. Currently, the major reasons for graft loss are sequelae of immunosuppression, such as infection and cancer [2, 3]. The induction of donor‐specific tolerance would eliminate the need for immunosuppression and be expected to improve quality of life and patient/graft survival. However, given the good outcomes of transplants performed with the current standard of care, it is critical that tolerance‐inducing strategies are shown to be safe and effective in non‐human primate (NHP) models prior to clinical trials, as they represent the closest approximation to human immunology. Specifically, the results in the Mauritian origin cynomolgus macaque (MCM) model quite accurately reflect the rate of tolerance induction in subsequent human studies [4].
Regulatory T cells (Tregs) represent a lymphocyte subpopulation with immunomodulatory properties characterized by cell surface expression of CD4 and CD25 and expression of the transcription factor forkhead box protein 3 (FoxP3). Tregs have shown promising results for the treatment of autoimmune diseases [5, 6, 7, 8, 9], graft‐versus‐host disease (GVHD) prophylaxis [10, 11, 12, 13, 14, 15, 16, 17] and tolerance induction in transplantation [18, 19]. Several methods for expanding Tregs in vitro have been reported [4, 20]. Donor‐specific Tregs have been described to be more suppressive than polyclonal Tregs [20]. However, in the transplantation setting, they cannot be produced in time for early infusion into deceased‐donor transplant recipients, leaving ‘off‐the‐shelf’ polyclonal Tregs as the only alternative for peritransplant infusion. Therefore, protocols for the production of highly suppressive polyclonal Tregs that can be easily translated into the clinic must be optimized.
CD40L‐stimulated B cells (CD40L‐sBc) have been used as immunostimulatory antigen‐presenting cells (APCs) for in‐vitro expansion of donor‐specific Tregs in patients with autoimmune diseases [8]. As this protocol meets good manufacturing practice (GMP) standards, we sought to translate it to the MCM model to create polyspecific Tregs (psTregs) and study their in‐vitro potency prior to testing their efficacy in vivo. We adapted this approach [20] and combined CD40L‐sBc isolated and expanded from donors with different MHC to produce Tregs with a broad reactivity that could be used in transplantation to prevent graft rejection or as GVHD prophylaxis. In addition, we tested the potency of these cells after cryopreservation as an ‘off‐the‐shelf’ product and after re‐expansion to demonstrate that they are high suppressors post‐thaw and can become more potent suppressors after re‐expansion.
Here, we present three approaches for the culture of MCM CD40L‐sBc in addition to a novel approach for the expansion of MCM psTregs stimulated with a pool of MHC‐mismatched CD40L‐sBc.
Materials and methods
Sample collection and peripheral blood mononuclear cell (PBMC) isolation
Heparinized blood was drawn from MCM (Charles River Primates, Wilmington, MA, USA; Sanofi‐Synthelabo, Bridgewater, NJ, USA; Bioculture Group, Glenmoore, PA, USA). All animals were negative for B virus, simian T lymphotropic virus, simian retrovirus, simian immunodeficiency virus, simian varicella virus and malaria. All macaques were housed at the Institute of Comparative Medicine (Columbia University Medical Center, New York, NY, USA). This facility holds a current United States Department of Agriculture (USDA) registration, public health service (PHS) assurance and is American Association for Accreditation of Laboratory Animal Care (AAALAC)‐accredited. All experimental procedures were approved by the Columbia University Institutional Animal Care and Use Committee.
Diluted (1 : 2) blood with phosphate‐buffered saline (PBS) (Corning, Corning, NY, USA) was overlaid on 60% Percoll (Sigma‐Aldrich, St Louis, MO, USA) and centrifuged. The buffy coat was collected and contaminating red blood cells were lysed [4].
B cell culture
Irradiated (46 Gy) human erythroleukemia cells (K562) expressing CD40L, human leukocyte antigen‐D related (HLA)‐DR and CD64 were used to stimulate B cells [20]. CD40L‐expressing K562 cell growth medium consisted of 89% Iscove’s modified Dulbecco’s medium (IMDM) (Life Technologies, Grand Island, NY, USA), 10% fetal bovine serum (FBS) (Gemini Bio‐Products, Sacramento, CA, USA) and 1% penicillin/streptomycin (Life Technologies).
B cell growth medium consisted of 90% X‐Vivo 15 (Lonza, Basel, Switzerland) or AIM‐V medium (Life Technologies), 10% heat‐inactivated human AB serum (Gemini Bio‐Products) and 100 U/ml of insulin (Eli Lilly, Indianapolis, IN, USA). Cryopreserved or freshly isolated MCM PBMCs or splenocytes were plated with CD40L‐expressing K562 cells at a ratio of 2 : 1. B cell donors were selected in order to obtain a pool of disparate MHC to cover a broad range of alloantigens. Three approaches were investigated for B cell expansion that ranged from 10 to 14 days of culture (Table 1). Interleukin (IL)‐4 (R&D Systems, Minneapolis, MN, USA), ganciclovir (GCV) (Genentech, South San Francisco, CA, USA) and cyclosporin A (CyA) (Sigma‐Aldrich) were added to all cultures. Cells were phenotypically assessed for expression of CD3 (to assess T cell contamination), CD40, CD20, CD80 and MHC class II prior to cryopreservation.
Table 1.
MCM B cell expansion and activation protocols
| Protocol 1 | Protocol 2 | Protocol 3 | |
|---|---|---|---|
| Culture duration | 10 days | 14 days | 14 days |
| Restimulation with CD40L‐expressing K562 cells | Day 7 | Day 10 | Day 7 |
| Medium feeding | Day 2, 4 | Day 2, 4, 7 | Days 2, 4, 10 |
| CyA dose | 2 µg/ml (days 0), 1 µg/ml (days 2, 4) | 2 µg/ml (days 0, 2, 4), 1 µg/ml (days 7, 10) | 2 µg/ml (days 0, 2, 4), 1 µg/ml (days 7, 10) |
| IL‐4 dose | 0.8 ng/ml (days 0), 0.4 ng/ml (days 2, 4, 7) | 0.8 ng/ml (days 0), 0.4 ng/ml (days 2, 4, 7, 10) | 0.8 ng/ml (day 0), 0.4 ng/ml (days 2, 4, 7, 10) |
| GCV dose | 10 µg/ml (days 0), 5 µg/ml (days 2, 4, 7) | 10 µg/ml (days 0), 5 µg/ml (days 2, 4, 7, 10) | 10 µg/ml (day 0), 5 µg/ml (days 2, 4, 7, 10) |
MCM = Mauritian cynomolgus macaque; IL = interleukin; GCV = ganciclovir; CyA = cyclosporin A.
Treg culture
Tregs were stained with CD4 (L200, BD Biosciences, San Jose, CA, USA), CD25 (BC96; BioLegend, San Diego, CA, USA), CD127 (HIL‐7R‐M21; BD Pharmingen, San Diego, CA, USA) and CD8 (RPA‐T8, BD or BW135/80; Miltenyi Biotec, Auburn, CA, USA) monoclonal antibodies. The top 1% of CD25+ cells within the CD4+CD8−CD127− gate were sorted. Fluorescence activated cell sorting (FACS) was performed on the Influx Cell Sorter (BD Biosciences).
Tregs were expanded for a minimum of 14 days for up to 28 days, depending on the rate of cell growth. Treg‐growth medium consisted of 90% X‐Vivo 15 (Lonza) or AIM‐V medium (Life Technologies) with 10% heat‐inactivated human AB serum (Gemini Bio‐Products). Irradiated (30 Gy) CD40L‐sBc from three to four animals representative of diverse MHCs were pooled and plated at a 1 : 4 ratio of Treg : CD40L‐sBc; 300 IU/ml of IL‐2 (PeproTech, Rocky Hill, NJ, USA) [20] were added to the culture on day 0 and at time of restimulation (Table 2). Cells were split when overgrowth was observed, with the addition of Treg medium and 300 IU/ml of IL‐2. Phenotype and suppressive capacity were assessed prior to cryopreservation.
Table 2.
CD40L‐sBc regulatory T cell (Treg) expansion protocol
| Day 0 | Day 7 | Day 14 | Day 21 | Day 28 | |
|---|---|---|---|---|---|
| CD40L‐sBc | ✔ | ✔ | ✔ | ✔ | Harvest |
| Interleukin (IL)‐2 | ✔ | ✔ | ✔ | ✔ |
Suppression assays
PBMC responders were plated in triplicate. Tregs were plated with PBMCs starting at a 1:1 Treg : PBMC ratio and serially diluted up to 1 : 32 Tregs : PBMCs. PBMCs were stimulated with anti‐CD2 CD3 CD28 beads (Miltenyi Biotec); 1 μCi of thymidine (Perkin Elmer, Waltham, MA, USA) was added per well 4 days after plating. Cells were harvested (Tomtec, Chicago, IL, USA) 24 h thereafter. A 1450 MicroBeta plate reader (Perkin Elmer) was used to measure the thymidine uptake [4]. Percentage suppression was calculated with the formula: {[(1 – counts per min (cpm) of Treg‐treated T cells)/cpm of control T cells] × 100}.
Cryopreservation
Cells were cryopreserved in 95% FBS (Gemini) and 5% dimethyl sulfoxide (DMSO) (Sigma‐Aldrich) at 10 million cells/ml in a step‐down cell freezer (CryoMed Freezer; Thermo Fisher Scientific, Fremont, CA, USA) at 1°C/min and stored in a liquid‐nitrogen freezer.
Re‐expansion post‐cryopreservation
Cells were thawed in a 37°C water bath. Group 1 was stimulated with anti‐CD2CD3CD28 beads (Miltenyi Biotec); group 2 with irradiated artificial APCs (aAPCs) [21] consisting of mouse fibroblast cells (L929) transduced with human CD32, CD58 and CD80 [22, 23] preloaded with 100 ng/ml of anti‐CD3; and group 3 with a pool of MHC‐disparate CD40L‐sBc at a 1 : 4 Treg : CD40L‐sBc ratio. The three conditions received Treg medium and 300 IU/ml of IL‐2 and were expanded for 9 days. Treg lines were expanded for 14–21 days before cryopreservation. Absolute numbers, phenotype and suppressive capacity were assessed throughout the culture.
Flow cytometric analysis
The cell surface antibodies used for flow cytometric (FCM) analysis included anti‐CD20 (Miltenyi Biotec), CD40 (BioLegend), CD80 (BD Biosciences), HLA‐DR (BD Biosciences), CD3 (SP34‐2, BD Biosciences), CD4 (L200, BD Biosciences), CD8 (BW135/80, Miltenyi Biotec), CD25 (BC96, BioLegend), CD45RA (T6D11, Miltenyi Biotec) and CD127 (HIL‐7R‐M21, BD Pharmingen). FoxP3 (236A/E7 and PCH101, eBioscience), Helios (BioLegend) and cytotoxic T‐lymphocyte antigen 4 (CTLA‐4) (BD Biosciences) antibodies were used intracellularly for Treg assessment. CTLA‐4 was assessed before and after stimulation with phorbol 12‐myristate 13‐acetate (PMA) and ionomycin for 4 h. Gates were drawn based on isotype controls (IS11‐3B2.2.3; Miltenyi Biotec). Cells were permeabilized with the BioLegend FoxP3 fixation/permeabilization buffer set. Data were collected on LSRFortessa (BD Biosciences) and analyzed using FCS Express (De Novo Software, Glendale, CA, USA).
Specificity assay
Responders (T cells) and stimulators (PBMCs) were plated at a 1 : 2 T cell : PBMC ratio. Tregs were plated at a 1 : 1 Treg : T cell ratio and then serially diluted to 1 : 128. Each condition was plated in triplicate. T cells were isolated through magnetic‐activated cell sorting (MACS) (Miltenyi Biotec) with CD3‐positive selection; 1 μCi/well of thymidine (Perkin Elmer) was added 4 days after plating. Cells were harvested (Tomtec) 24 h after adding thymidine. A 1450 MicroBeta plate reader (Perkin Elmer) was used to measure thymidine uptake.
Treg‐specific demethylated region (TSDR) analysis of MCM Tregs
Quantitative polymerase chain reaction (qPCR) for the FoxP3 region was performed by Epiontis (Berlin, Germany). Using a standard dilution curve, the number of demethylated and methylated genomic regions was determined, referred as TpG and CpG plasmid units, respectively. The Treg content was calculated by dividing the number of TpG plasmid units by the sum of all FoxP3 regions in the sample: TpG/(TpG+CpG) [4].
Assessment of intracellular cytokine production
Tregs and PBMCs (positive control) were stimulated for 4 h with PMA/ionomycin in the presence of brefeldin A (leukocyte activation cocktail; BD Biosciences). Prior to fixation and permeabilization, cells were stained with anti‐CD3 (BD Biosciences), CD4 (BD Horizon), and CD8 (Miltenyi Biotec). Cells were stained intracellularly with antibodies against interferon (IFN)‐γ (BioLegend), IL‐17A) (Miltenyi Biotec) and IL‐13 (BD Bioscience). Cells were read using the Aurora spectral analyzer (Cytek Biosciences, Fremont, CA, USA).
Statistics
Data were analyzed using unpaired Student’s t‐test with GraphPad Prism version 7.05. P‐values of ≤ 0·05 were considered statistically significant (*P < 0·05, **P < 0·01, ***P < 0·001, ****P < 0·0001).
Results
Expansion and activation of MCM B cells
We studied three approaches for the expansion of MCM B cells from PBMCs or splenocytes (Table 1). An average of 6·2 × 106 ± 3·5 × 106 B cells (based on the percentage of CD20+ cells in PBMCs/splenocytes) was plated with CD40L‐expressing K562 cells for up to 14 days (Fig. 1a) with IL‐4 for B cell stimulation, CyA to avoid T cell growth and GCV for the control of Epstein–Barr virus (Table 1). Protocol 1 was based on previous human studies [20]. Cells were cultured for 10 days. Slower cell growth was observed with this approach compared to protocols 2 and 3 (Fig. 1b). At the end of the culture, an average of 40·88 × 106 ± 9·1 × 106 cells was obtained (Fig. 1c). In order to improve the cell yield, culture time was increased to 14 days for protocols 2 and 3, achieving 115·4 × 106 ± 16·34 × 106 and 136·6 × 106 ± 16·78 × 106 cells, respectively, with significant increase in B cell numbers compared to protocol 1 [Fig. 1c; P = 0·0017 (protocol 1 versus 2), P = 0·0004 (protocol 1 versus 3)]. We calculated the fold expansion in order to normalize the cell growth to the number of B cells plated on day 0. The highest average fold expansion was achieved with protocol 3 (22·3‐fold), compared to protocol 2 (16·1‐fold) and protocol 1 (9·5‐fold) (Fig. 1d), with a significant difference between protocols 1 and 3 (P = 0·02).
Fig. 1.
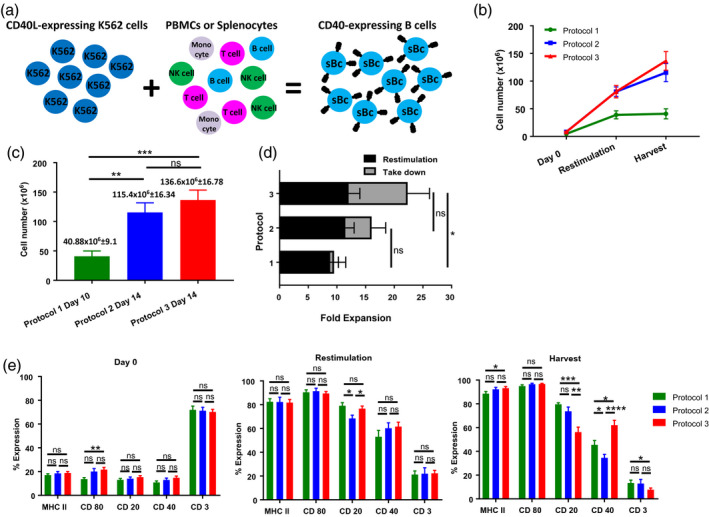
Expansion and activation of cynomolgus macaque (MCM) B cells for regulatory T cell (Treg) expansion. B cell expansion from fresh or cryopreserved peripheral blood mononuclear cells (PBMCs) or splenocytes. Protocol 1 includes 10 lines, protocol 2 includes 14 lines and protocol 3 includes 18 lines from nine animals. (a) Schema for the expansion and activation of MCM B cells. (b) B cell growth (mean and standard error of the mean (s.e.m.) from day 0 until the harvest for each of the B cell expansion protocols. (c) Average cell number at the end of the B cell culture for each protocol. Average and s.e.m. are detailed at the top of each bar. (d) Fold expansion for each B cell protocol. (e) Expression of B cell characteristic markers [major histocompatibility complex (MHC) II, CD80, CD20, CD40] and assessment of T cell contamination for each B cell expansion protocol at different time‐points (day 0, restimulation and harvest).
We analyzed the number of B cells and concurrent T cell contamination in culture over time (Fig. 1e). As expected on day 0, few cells expressed B cell markers and there was a high percentage of T cells (Fig. 1e). Restimulation with fresh CD40L‐K562 cells was performed on day 10 for protocols 1 and 2 and on day 7 for protocol 3. Over time, the expression of B cell markers increased and the T cell counts decreased with all protocols (Fig. 1e). The concentration of CyA was increased in protocols 2 and 3 in an attempt to decrease the number of contaminating T cells. The three approaches produced cells with comparable MHC II and CD80 levels. Cells from protocols 1 and 2 had higher percentages of CD20+ cells (P = 0·0003, protocol 1 versus 3; P = 0·0043, protocol 2 versus 3), but protocol 3 produced cells with the highest CD40 expression (P = 0·0118, protocol 1 versus 3; P < 0·0001, protocol 2 versus 3), and with the lowest T cell contamination [significant difference between protocols 1 and 3 (P = 0·0348)] (Fig. 1e).
Due to the higher yield and the lower T cell contamination observed in CD40L‐sBc cultured under protocol 3, we utilized this approach in the experiments to follow.
Expansion of CD40L‐B cell‐stimulated MCM Tregs (sBc‐Tregs)
Tregs were cultured with MHC‐disparate CD40L‐sBc from multiple donors at a ratio of 1 : 4 Treg : CD40L‐sBc. Tregs were FACS sorted based on the CD4+CD8−CD25highCD127− phenotype [4]. An average of 62 609 (± 34 668) Tregs were plated on day 0 and cultured for a minimum of 14 days, but up to 28 days depending on cell growth (Fig. 2a), reaching an average of 60·6 × 106 ± 9·2 × 106 Tregs at the end of the culture. The largest fold expansion was observed between days 7 and 14, at which time the majority of the Treg lines were cryopreserved (Fig. 2b). On average, greater than 80% FoxP3 expression was detected, with no significant difference between the three harvest time‐points. Therefore, the duration of the cell culture did not affect FoxP3 expression (Fig. 2c). Helios expression on Tregs has been associated with a thymic origin, in addition to a more activated phenotype and a higher suppressive capacity in vitro [24]. We observed that Tregs cultured with CD40L‐sBc expressed high levels of Helios, suggestive of a stable Treg phenotype (Fig. 2d, representative figure). CTLA‐4 is used by Tregs to elicit suppression and it has been reported as an indicator of homeostasis in stable Tregs [25]. We found that CD40L‐sBc Tregs homogeneously expressed CTLA‐4, which was further increased upon additional stimulation (Fig. 2e, representative figure). Treg lines exhibited potent suppressive capacity as, on average, they mediated 50% suppression compared to the positive control (bead‐stimulated PBMCs) at a 1 : 16 ratio of Treg : PBMCs (Fig. 2f). Human CD45RA+ Tregs have been reported to be more homogeneous compared to CD45RA− memory Tregs [26]. We have previously reported no correlation between the level of FoxP3 and CD45RA in MCM Tregs expanded with aAPCs and donor PBMCs [4]. Using CD40L‐sBc as stimulators during Treg expansion also resulted in no correlation between the expression of these two markers (Fig. 2g). To assess their stability, we determined the percentage of demethylation in the TSDR. Tregs expressing high levels of FoxP3 that were potent suppressors showed high levels of Treg‐specific demethylated region (TSDR) demethylation (Fig. 2h,i). In addition, we found no correlation between the level of TSDR demethylation and the duration of the cell culture, with similar levels in Treg lines cultured for 14, 21 or 28 days (Fig. 2j).
Fig. 2.
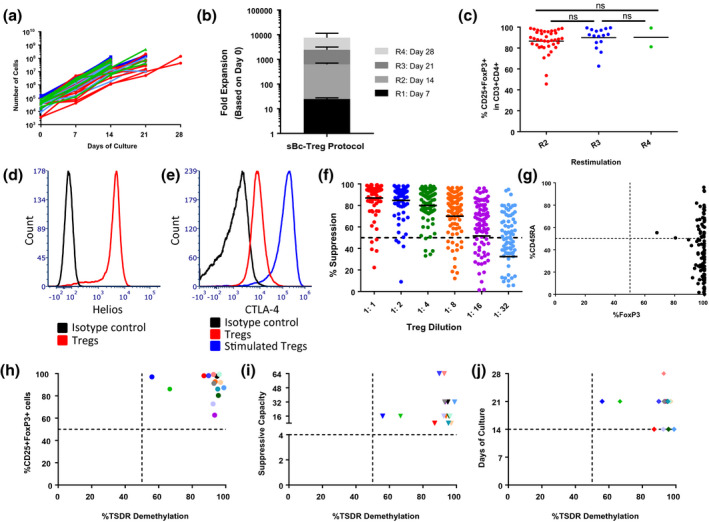
Expansion of cynomolgus macaque (MCM) regulatory T cells (Tregs) with CD40L‐sBc. Expansion of MCM Tregs with CD40L‐sBc stimulation. (a) Treg numbers at each restimulation and take‐down (n = 57). (b) Mean and standard error of the mean (s.e.m.) of the fold expansion of the Treg lines at each restimulation until the take‐down. ‘R’ represents restimulation number. (c) Percentage of CD25+FoxP3+ cells within CD3+CD4+ cells at the take‐down. (d) Representative Helios expression on cultured Tregs (red) (n = 5). (e) Representative cytotoxic T lymphocyte antigen‐4 (CTLA‐4) expression on cultured Tregs (red) and cultured Tregs activated with phorbol myristate acetate (PMA)/ionomycin for 4 h (blue) (n = 5). (f) Percentage of suppressive capacity of the Treg lines (n = 57). The dotted line represents 50% suppression as reference. Mean of the suppressive capacity is represented with a black line for each ratio of Treg : peripheral blood mononuclear cells (PBMCs). (g) Correlation between CD45RA and forkhead box protein 3 (FoxP3) expression. Correlation between the percentage of Treg‐specific demethylated region (TSDR) demethylation and the (h) expression of FoxP3, (i) suppressive capacity (the Treg : responder ratio that achieves 50% suppression of the positive control) and (j) days of culture. Each color represents a different Treg line that is the same throughout Fig. 2h–j (n = 15).
CD4+CD8+ T cells in Treg cultures show Treg‐like characteristics
In our previous studies, Tregs cultured with aAPCs and/or donor PBMCs as stimulators variably showed outgrowth of CD8+ single‐positive (SP) T cells [4]. In this protocol, we observed overgrowth of CD4+CD8+ double‐positive (DP) T cells and very few CD8+ SP T cells (Fig. 3a, P < 0·0001). FoxP3 and Helios expression was similar in CD4+ SP, CD4+CD8+ DP and total CD3+ T cell populations after culture (Fig. 3b,c). Using FACS sorting, we assessed the suppressive capacity of the CD4+CD8+ DP cell population and noted comparable suppressive potency as the purified Tregs from the same population (Fig. 3d). In addition, we did not observe an inverse correlation between the level of CD3+CD4+CD8+ T cells and the level of TSDR demethylation (Fig. 3e). The only Treg line that had higher CD4+CD8+ T cell counts (green triangle) with a lower TSDR demethylation also had a higher percentage of CD8+ T cells (green square), which have previously been shown to cause higher TSDR methylation when contaminating Treg populations [4]. Therefore, CD4+CD8+ DP T cells presented Treg‐like characteristics based on their phenotypical and functional data, and may not be detrimental if administered in vivo.
Fig. 3.
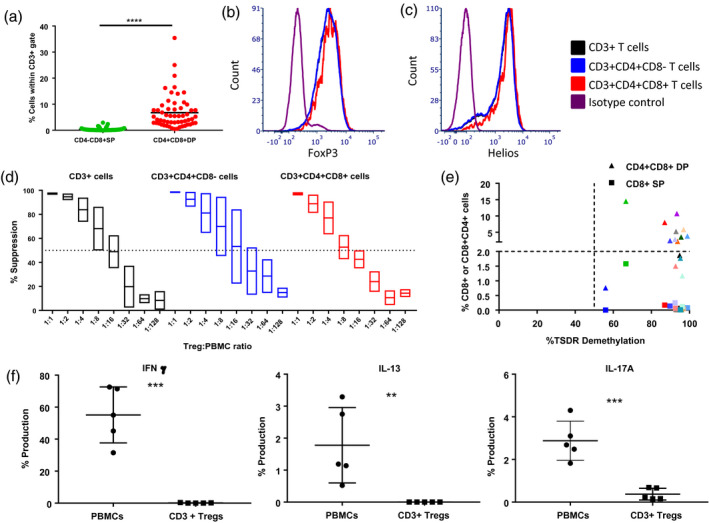
Forkhead box protein 3 (FoxP3) expression and suppressive capacity of CD4+CD8+ DP T cells. (a) Comparison between the percentage of CD3+CD4‐CD8+ SP and CD3+CD4+CD8+ double‐positive (DP) T cells present in the regulatory T cell (Treg) cultures at the harvest time‐point (days 14, 21 or 28) (n = 57). Study of the (b) FoxP3, (c) Helios expression and (d) suppressive capacity [mean and standard error of the mean (s.e.m.)] of total CD3+ T cells (black), CD3+CD4+CD8− SP T cells (blue) and CD3+CD4+CD8+ DP T cells (red) (n = 2). (e) Correlation between the percentage of Treg‐specific demethylated region (TSDR) demethylation in total CD3+ cells and the percentage of CD8+ single‐positive (SP) and CD4+CD8+ (DP) T cells present in culture. Each color represents a different Treg line (n = 15). (f) Assessment of the secretion of interferon (IFN)‐γ, interleukin (IL)‐13 and IL‐17A in peripheral blood mononuclear cells (PBMCs) and cultured sBc‐Tregs (n = 5).
CD40L‐sBc Tregs lack production of inflammatory cytokines upon in‐vitro stimulation
To understand the function and stability of CD40L‐sBc Tregs, we assessed their ability to secrete inflammatory cytokines upon activation. We compared the production of IFN‐γ, IL‐13 and IL‐17A between PBMCs (positive control) and CD40L‐sBc Tregs at different time‐points (days 7, 14 and 21). While PBMCs produced these three cytokines, as expected, there was negligible production from CD40L‐sBc Tregs (Fig. 3f).
These data support the safety of CD4+CD8+ DP T cells, as Treg lines with a high percentage of CD4+CD8+ DP T cells (14%) also lacked production of IFN‐γ, IL‐13 and IL‐17A (Fig. 3f).
Study of the specificity of MCM sBc‐Tregs
We used a combination of CD40L‐sBc from multiple MCM donors with diverse MHC as stimulators to expose the Tregs to a variety of alloantigens (Fig. 4a). To study the polyspecificity of the Tregs, we cultured them with different stimulators and responders and quantified the suppressive effect of the Tregs. We defined cells as MHC‐matched or mismatched to CD40L‐sBc if the MHC was similar or different, respectively, from the CD40L‐sBc used for Treg expansion.
Fig. 4.
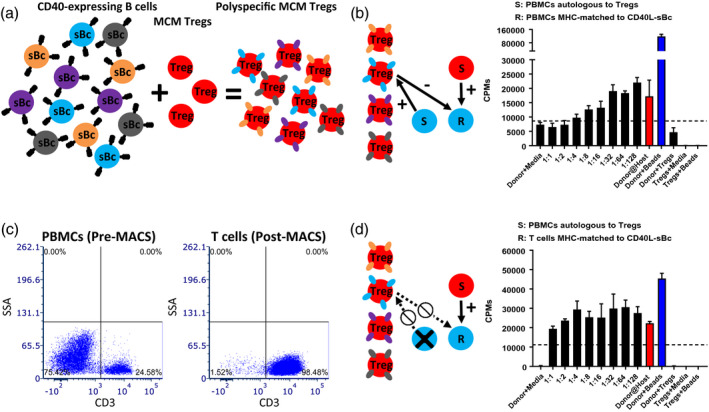
Difference in suppression based on the responder‐type cells. (a) Schema for the expansion protocol of cynomolgus macaque (MCM) regulatory T cells (Tregs) with CD40L‐sBc. (b) Representative figure of a specificity assay with peripheral blood mononuclear cells (PBMCs) as responders. (c) Representative figure of before (left panel) and after (right panel) magnetic‐activated cell sorted (MACS)‐isolated T cells. (d) Representative figure of a specificity assay with MACS‐isolated T cells as responders.
We first assessed if Tregs would suppress responses of PBMCs that were MHC‐matched to CD40L‐sBc when stimulated by PBMCs that were autologous to the Tregs. In this condition, Tregs could be stimulated by APCs from the ‘responder’ PBMCs or the stimulators and efficient suppression was observed (Fig. 4b). Then, to eliminate the potential for Treg activation by ‘responder’ APCs leading to bystander suppression, responder T cells were MACS‐enriched, which allowed for the isolation of highly purified T cells (98·48%) (Fig. 4c). The Treg suppression was eliminated when purified T cells were used as responders (Fig. 4d), suggesting that Tregs had been activated by the APCs from the ‘responder’ PBMCs from the same donors as those used for Treg expansion (Fig. 4b). Therefore, we performed the future specificity studies with MACS‐isolated T cells as responders (Fig. 5).
Fig. 5.
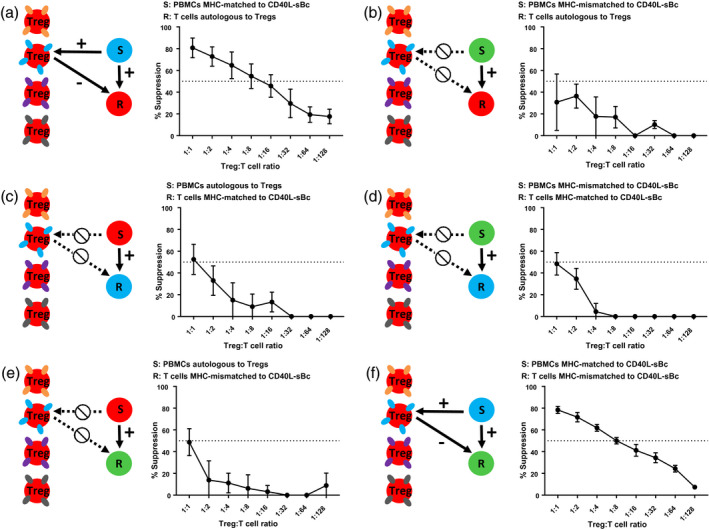
Specificity data for cynomolgus macaque (MCM) CD40L‐sBc‐stimulated regulatory T cells (Tregs). Mean and standard error of the mean (SEM) of the suppressive capacity of CD40L‐sBc Tregs to responders that were (a,b) autologous to Tregs (n = 5, n = 3), (c,d) major histocompatibility complex (MHC)‐matched (n = 4, n = 2) or (e,f) MHC‐mismatched to CD40L‐sBc used for Treg expansion (n = 3, n = 4) with a variety of stimulators. Responders were MACS‐isolated T cells from peripheral blood mononuclear cells (PBMCs) or splenocytes and stimulators were PBMCs.
We then examined the Tregs’ ability to suppress autologous T cell responses stimulated by APCs that were either MHC‐matched or mismatched to the CD40L‐sBc used during Treg expansion (Fig. 5a,b). We found increased suppression by the Tregs when stimulated by APCs that were MHC‐matched to CD40L‐sBc than when stimulated by APCs expressing MHC not present in the Treg expansion phase (Fig. 5a,b). If the stimulator did not express any MHC to which the Tregs were exposed to during expansion, suppression was poor and typically did not exceed 50% suppression at a Treg : responder ratio of 1 : 1 (Fig. 5b). When responders that were MHC‐matched to CD40L‐sBc were stimulated by either APCs autologous to the Tregs or MHC‐mismatched to CD40L‐sBc, Tregs exhibited poor suppressive capacity (Fig. 5c,d). To confirm these findings, we stimulated T cells that were MHC‐mismatched to CD40L‐sBc with PBMCs autologous to the Tregs (Fig. 5e) or MHC‐matched to CD40L‐sBc (Fig. 5f). We again found that the Tregs exhibited good suppression (> 50% at 1 : 8 Treg : responder) only when the stimulator’s MHC was present during Treg expansion (Fig. 5f). Collectively, our data demonstrate that multiple CD40L‐sBc drive the expansion of Tregs specific for alloantigens presented by the CD40L‐sBc irrespective of the responder MHC (Fig. 5a,f). After activation, Tregs could suppress any naive T cell responder.
Expansion after cryopreservation of CD40L‐sBc Tregs
To ensure that CD40L‐sBc Tregs could be banked prior to in‐vivo infusion, we studied the effect of cryopreservation on viability, phenotype and function with and without post‐thaw restimulation. Three approaches were studied: stimulation with anti‐CD2 CD3 CD28 beads, anti‐CD3 preloaded aAPCs [21] or CD40L‐sBc from four donors (with MHC matched to that of the CD40L‐sBc used for prior Treg expansion), all in the presence of IL‐2. There was no Treg expansion with anti‐CD2 CD3 CD28 bead stimulation (Fig. 6a). Restimulation with aAPCs or CD40L‐sBc led to a 10‐fold expansion in Treg numbers after 7 days (Fig. 6b). FoxP3 expression was comparable in Tregs regardless of stimulation approach (Fig. 6c). We determined the percentage of CD4+CD8+ DP T cell contamination to determine if the method of restimulation affected this population. The protocol using aAPCs promoted the CD4+CD8+ DP cell growth more than the other two approaches, but the difference did not reach statistical significance (Fig. 6d). We also studied the functionality of the Tregs after re‐expansion (Fig. 6e). The suppressive capacity of Tregs restimulated with aAPCs was comparable to that of sBc‐restimulated Tregs. Optimal suppression was noted on day 3, followed by day 4 of restimulation. Given these results, we propose to restimulate psT Tregs for 4 days with aAPCs, anti‐CD3 and IL‐2, as this method provides optimal suppressive capacity and a degree of expansion of psTregs, while remaining relatively easy to perform.
Fig. 6.
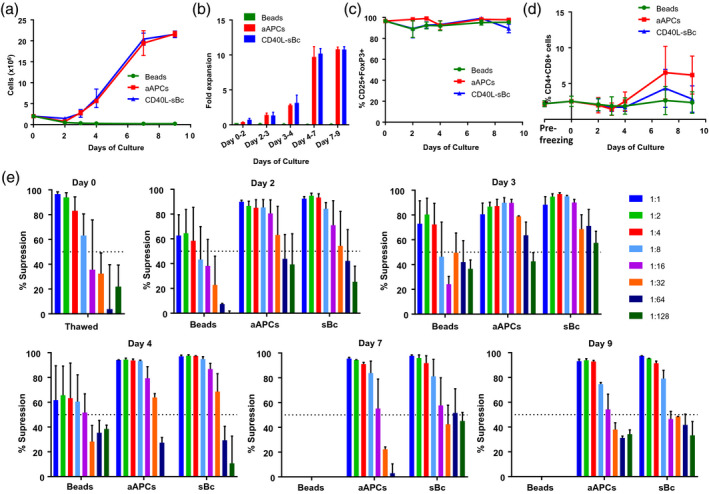
Restimulation of sBc‐regulatory T cells (Tregs) after cryopreservation. Study of cryopreserved and re‐expanded CD40L‐sBc Tregs. Representation of two Treg lines from two different animals recultured under three different conditions. (a) Treg growth after thawing. (b) Fold expansion after 9 days of culture. (c) CD25+forkhead box protein 3 (FoxP3)+ expression in CD3+CD4+ cells. (d) Percentage of CD4+CD8+ T cells. (e) Suppressive capacity of the Treg lines cultured under each condition at the thawing day and days 2, 3, 4, 7 and 9 after reculture.
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Discussion
Treg infusion has been reported safe in patients enrolled in clinical trials for the treatment of autoimmune diseases [27], graft‐versus‐host disease [14] and induction of transplantation tolerance for kidney [28] or liver allografts [19] [Regulatory T Cell Therapy in Liver Transplant Patients (ThRIL) study]. Treg expansion protocols are therefore necessary to obtain sufficient numbers so that an in‐vivo effect may be observed. We have previously reported four approaches for the expansion of polyclonal MCM Tregs for preclinical studies through the stimulation provided by aAPCs and/or donor PBMCs [4]. Here, we report a novel approach using CD40L‐sBc to obtain MCM psTregs. Adapted from clinical studies where CD40L‐sBc were used to grow donor‐specific Tregs [20], our protocol combines CD40L‐sBc from multiple donors with diverse MHC to produce Tregs that react to a broad range of alloantigens. Using this approach, we aimed to obtain Tregs that could suppress HVG as well as GVH responses, and therefore may be useful in combining solid organ and bone marrow transplantation for tolerance induction.
The expansion of NHP B cells has been reported for different purposes, such as the study of B cells, diagnostic tools [i.e. enzyme‐linked immunospot (ELISPOT)], therapeutic cell expansion (i.e. Tregs) or cell therapy (i.e. Bregs) [29]. We report for the first time, to our knowledge, the use of a combination of MHC‐disparate CD40L‐sBc for the expansion of MCM psTregs. We first studied three approaches to engineer B cells through co‐culture with CD40L‐expressing K562 cells. Protocol 3, which had an extended culture time and higher CyA doses compared to the original clinical protocol [20], produced the largest number of CD40L‐sBcs with the lowest T cell contamination. Therefore, this was the optimal protocol for B cell expansion.
We developed a novel approach to obtain psTregs by stimulation with a pool of MHC‐mismatched CD40L‐sBc. Using this approach, we obtained > 600‐fold expansion of Tregs in 14 days. We have previously reported higher Treg yields through the use of aAPCs [4], but the culture time was 26 days instead of 14–21 days for our current approach. If higher yields are desired with the CD40L‐sBc approach, Tregs can also be expanded for longer while maintaining the FoxP3 expression and suppression, although higher CD40L‐sBc numbers would be also required for expansion to maintain the ratio of stimulators with Tregs at 1 : 4 Tregs : CD40L‐sB.
FoxP3 and Helios levels remained high for up to 28 days with minimal secretion of inflammatory cytokines. CTLA‐4 expression was homogeneously expressed in the Treg populations, which demonstrated increased levels upon stimulation. Tregs showed potent suppressive capacity and had high TSDR demethylation levels. These data suggest that Tregs expanded under this approach are a potentially good product for in‐vivo use.
We have previously reported the outgrowth of CD8+ SP T cells with other approaches for MCM Treg expansion [4], raising the concern that we were generating donor‐specific effectors along with Tregs. Based on our previous experience with this CD8+ SP population, we did not find that these CD8+ SP T cells share Treg characteristics [4]. With the CD40L‐sBc Treg expansion protocol, we report minimal CD8+ SP T cell growth that could be easily removed by sorting, if desired. In contrast, we observed the development of CD4+CD8+ DP T cells. FACS‐sorted CD4+CD8+ T cells were able to suppress responders at the same Treg : PBMC ratios as traditional CD4+CD8− Tregs. FoxP3 and Helios expression was comparable to that in CD4+CD25+ cells and minimal secretion of inflammatory cytokines was observed. In humans, CD4+CD8+ DP T cells are a small subpopulation of lymphocytes that have been reported to be activated T cells with memory phenotype, while in animal models they have been reported to have regulatory properties [30]. The results in our model suggest that, in MCM, the CD4+CD8+ DP T cells might be a subpopulation of Tregs that up‐regulate CD8 in addition to CD4. Another explanation could be that a Treg phenotype has been induced in CD4+CD8+ cells isolated from circulation in this model. Regardless, these findings would need further assessment in human Tregs expanded with this approach.
In order to translate this psTreg‐expansion approach to deceased donation protocols, Tregs need to suppress immune responses to a broad array of HLA with high prevalence in the organ donor pool. We therefore assessed the specificity capabilities of Tregs expanded under this approach. We showed that for Tregs to be potent suppressors of the recipient’s response to any given donor, they must be exposed to donor‐HLA‐matched CD40L‐sBc during the cell expansion. This Treg‐expansion strategy would, therefore, be a promising option in deceased donor transplantation where the donor HLA is known only immediately before the transplant.
Cryopreservation is a necessary step in generating Tregs suitable for use with deceased‐donor organs as they would be readily accessible when an organ is accepted. For Tregs to become an ‘off‐the‐shelf’ product, they need to maintain their suppressive properties through the cryopreservation process. While thawed Tregs were able to preserve 50% suppressive capacity at an average of a 1 : 8 Treg : PBMC ratio, after reculturing with aAPCs or CD40L‐sBc, Tregs become more activated and showed higher suppressive potency. We therefore opted to restimulate the Tregs for 4 days with aAPCs for optimal combination of Treg function and expansion for ongoing in‐vivo studies given the ease of using aAPCs, specifically when high Treg numbers are desired.
Other approaches that include the use of aAPCs have been reported for the expansion of NHP and human Tregs that led to polyclonal Tregs [4, 31]. In addition, as in human studies, anti‐CD3CD28 beads have been reported for the expansion of Tregs in NHP models [32]. Although Tregs cultured under these approaches have potential to be used as ‘off‐the‐shelf’ products, antigen‐primed Tregs have been reported to be more suppressive than polyclonal Tregs. Therefore, higher yields would be needed to achieve similar in‐vivo effects. Another approach that has been reported is the use of donor PBMCs, alone or in combination with aAPCs [4]. These Tregs had increased suppression towards the donor, and an overall lower suppression to other allogeneic not‐previously encountered stimulators. Approaches for the expansion of donor‐specific Tregs have included the use of CD40L‐stimulated B cells isolated from the donor that demonstrated superior potency than those stimulated partially with beads [20]. In our protocol, we modified this last approach using a combination of different donors to obtain B cells with disparate MHC in order to create polyspecific Tregs. Although we obtained lower yields than previously reported in this MCM model [4], cells were overall more suppressive, and therefore the total number of Tregs needed for in‐vivo infusion may actually be less. In addition, these cells can be further expanded after thawing since their potency was further improved in this process.
Several clinical trials have reported the infusion of Tregs for the induction of kidney or liver transplantation tolerance [18, 19], with the majority of these studies involving living‐donor transplants and infusion of donor‐specific Tregs. Even though donor‐specific Tregs have been reported to be more potent than polyclonal Tregs [20], they cannot be produced in a timely manner for early infusion into deceased‐donor transplant recipients, leaving polyclonal Tregs as the only alternative for peritransplant infusion. Therefore, protocols for the production of highly suppressive polyclonal Tregs that can be easily translated into the clinic must be optimized. Previous studies performed by our group in a MCM model reported the use of polyclonal Tregs to promote renal allograft tolerance through mixed hematopoietic chimerism across MHC barriers, but success was achieved in only a few recipients [33]. The Tregs generated with the current protocol suppress naive T cell responses at a lower Treg : responder ratio than those in our previous report. Additionally, this protocol does not generate substantial numbers of CD3+CD4‐CD8+ T cells in the culture, decreasing the likelihood of expanding alloreactive effector T cells. Finally, this protocol has the advantage of being adapted from a clinical protocol that meets GMP standards, which could facilitate translation of studies from primates to the clinic.
Conclusions
We have developed an approach for the expansion of MCM psTregs through the use of CD40L‐engineered B cells that may be translated to clinical studies. Significant numbers of highly suppressive Tregs with stable levels of FoxP3, Helios and demethylated TSDR were obtained, which could be further expanded post‐cryopreservation. This is the first report, our knowledge, that uses a pool of MHC‐mismatched CD40L‐sBc to create psTregs suitable for use in deceased‐donor transplantation.
Disclosures
Q. T. is a co‐inventor on two patents on regulatory T cell therapy and a cofounder of Sonoma Biotherapeutics. The rest of the authors declare that they have no competing interests.
Author contributions
P. A. G., M. S. and A. G. performed research, analyzed data and wrote the manuscript. N. L., E. L., S. K., S. H. H., J. S., G. P. and K. B. performed research, collected samples and analyzed data. Q. T. provided the CD40L‐K562 cell line.
Acknowledgements
Funding for this study was provided by National Institutes of Health (NIH) grant R01OD017949 and U19AI131474 as part of the NIH NHP Transplantation Tolerance Cooperative Study Group sponsored by the National Institute of Allergy and Infectious Diseases. Research reported in this publication was performed in the CCTI Flow Cytometry Core, supported in part by the Office of the Director, NIH under the award S10OD020056. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Azzi JR, Sayegh MH, Mallat SG. Calcineurin inhibitors: 40 years later, can't live without. J Immunol 2013; 191:5785–91. [DOI] [PubMed] [Google Scholar]
- 2. Dantal J, Soulillou JP. Immunosuppressive drugs and the risk of cancer after organ transplantation. N Engl J Med 2005; 352:1371–3. [DOI] [PubMed] [Google Scholar]
- 3. Schulz TF. Cancer and viral infections in immunocompromised individuals. Int J Cancer 2009; 125:1755–63. [DOI] [PubMed] [Google Scholar]
- 4. Alonso‐Guallart P, Zitsman JS, Stern J et al Characterization, biology, and expansion of regulatory T cells in the cynomolgus macaque for pre‐clinical studies. Am J Transplant 2019; 19:2186–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Marek‐Trzonkowska N, Mysliwiec M, Dobyszuk A et al Administration of CD4(+)CD25(high)CD127(‐) regulatory T cells preserves beta‐cell function in type 1 diabetes in children. Diabetes Care 2012; 35:1817–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Marek‐Trzonkowska N, Mysliwec M, Siebert J, Trzonkowski P. Clinical application of regulatory T cells in type 1 diabetes. Pediatr Diabetes 2013; 14:322–32. [DOI] [PubMed] [Google Scholar]
- 7. Marek‐Trzonkowska N, Mysliwiec M, Dobyszuk A et al Therapy of type 1 diabetes with CD4(+)CD25(high) CD127‐regulatory T cells prolongs survival of pancreatic islets – results of one year follow‐up. Clin Immunol 2014; 153:23–30. [DOI] [PubMed] [Google Scholar]
- 8. Bluestone JA, Buckner JH, Fitch M et al Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med 2015; 7:315ra189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Desreumaux P, Foussat A, Allez M et al Safety and efficacy of antigen‐specific regulatory T‐cell therapy for patients with refractory Crohn’s disease. Gastroenterology 2012; 143:1207–17 e2. [DOI] [PubMed] [Google Scholar]
- 10. Brunstein CG, Blazar BR, Miller JS et al Adoptive transfer of umbilical cord blood‐derived regulatory T cells and early viral reactivation. Biol Blood Marrow Transplant 2013; 19:1271–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brunstein CG, Miller JS, McKenna DH et al Umbilical cord blood‐derived T regulatory cells to prevent GVHD: kinetics, toxicity profile, and clinical effect. Blood 2016; 127:1044–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Edinger M, Hoffmann P. Regulatory T cells in stem cell transplantation: strategies and first clinical experiences. Curr Opin Immunol 2011; 23:679–84. [DOI] [PubMed] [Google Scholar]
- 13. Theil A, Tuve S, Oelschlagel U et al Adoptive transfer of allogeneic regulatory T cells into patients with chronic graft‐versus‐host disease. Cytotherapy 2015; 17:473–86. [DOI] [PubMed] [Google Scholar]
- 14. Trzonkowski P, Bieniaszewska M, Juscinska J et al First‐in‐man clinical results of the treatment of patients with graft versus host disease with human ex‐vivo expanded CD4+CD25+CD127‐T regulatory cells. Clin Immunol 2009; 133:22–6. [DOI] [PubMed] [Google Scholar]
- 15. Brunstein CG, Miller JS, Cao Q et al Infusion of ex‐vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood 2011; 117:1061–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Di Ianni M, Falzetti F, Carotti A et al Tregs prevent GVHD and promote immune reconstitution in HLA‐haploidentical transplantation. Blood 2011; 117:3921–8. [DOI] [PubMed] [Google Scholar]
- 17. Martelli MF, Di Ianni M, Ruggeri L et al HLA–haploidentical transplantation with regulatory and conventional T‐cell adoptive immunotherapy prevents acute leukemia relapse. Blood 2014; 124:638–44. [DOI] [PubMed] [Google Scholar]
- 18. Geissler EK. The ONE Study compares cell therapy products in organ transplantation: introduction to a review series on suppressive monocyte‐derived cells. Transplant Res 2012; 1:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Safinia N, Vaikunthanathan T, Fraser H et al Successful expansion of functional and stable regulatory T cells for immunotherapy in liver transplantation. Oncotarget 2016; 7:7563–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Putnam AL, Safinia N, Medvec A et al Clinical grade manufacturing of human alloantigen‐reactive regulatory T cells for use in transplantation. Am J Transplant 2013; 13:3010–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Levings MK, Sangregorio R, Roncarolo MG. Human CD25(+)CD4(+) T regulatory cells suppress naive and memory T cell proliferation and can be expanded in vitro without loss of function. J Exp Med 2001; 193:1295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Malefyt RD, Verma S, Bejarano MT, Ranesgoldberg M, Hill M, Spits H. Cd2/Lfa‐3 or Lfa‐1/Icam‐1 but not Cd28/B7 interactions can augment cytotoxicity by virus‐specific CD8+ cytotoxic lymphocytes‐T. Eur J Immunol 1993; 23:418–24. [DOI] [PubMed] [Google Scholar]
- 23. Levings MK, Bacchetta R, Schulz U, Roncarolo MG. The role of IL‐10 and TGF‐beta in the differentiation and effector function of T regulatory cells. Int Arch Allergy Immunol 2002; 129:263–76. [DOI] [PubMed] [Google Scholar]
- 24. Thornton AM, Korty PE, Kim YC, Martens C, Shevach EM. Helios expression defines a phenotypically distinct population of Treg cells. J Immunol 2018; 200:116. [Google Scholar]
- 25. Tang AL, Teijaro JR, Njau MN et al CTLA4 expression is an indicator and regulator of steady‐state CD4(+)FoxP3(+) T cell homeostasis. J Immunol 2008; 181:1806–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hoffmann P, Eder R, Boeld TJ et al Only the CD45RA+ subpopulation of CD4+CD25high T cells gives rise to homogeneous regulatory T‐cell lines upon in vitro expansion. Blood 2006; 108:4260–7. [DOI] [PubMed] [Google Scholar]
- 27. Arellano B, Graber DJ, Sentman CL. Regulatory T cell‐based therapies for autoimmunity. Discov Med 2016; 22:73–80. [PMC free article] [PubMed] [Google Scholar]
- 28. Mathew JM, H.‐Voss J, LeFever A et al A Phase I clinical trial with ex vivo expanded recipient regulatory T cells in living donor kidney transplants. Sci Rep 2018; 8:7428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kim JS, Byun N, Chung H et al Cell enrichment‐free massive ex‐vivo expansion of peripheral CD20(+) B cells via CD40‐CD40L signals in non‐human primates. Biochem Biophys Res Commun 2016; 473:92–8. [DOI] [PubMed] [Google Scholar]
- 30. Clenet ML, Gagnon F, Moratalla AC, Viel EC, Arbour N. Peripheral human CD4(+)CD8(+) T lymphocytes exhibit a memory phenotype and enhanced responses to IL‐2, IL‐7 and IL‐15. Sci Rep 2017; 7:11612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Guo H, Zhang H, Lu L, Ezzelarab MB, Thomson AW. Generation, cryopreservation, function and in vivo persistence of ex vivo expanded cynomolgus monkey regulatory T cells. Cell Immunol 2015; 295:19–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Singh K, Kozyr N, Stempora L et al Regulatory T cells exhibit decreased proliferation but enhanced suppression after pulsing with sirolimus. Am J Transplant 2012; 12:1441–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Duran‐Struuck R, Sondermeijer HP, Buhler L et al Effect of ex vivo‐expanded recipient regulatory T cells on hematopoietic chimerism and kidney allograft tolerance across MHC barriers in cynomolgus macaques. Transplantation 2017; 101:274–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.